


Abstract
The vast majority of the genome is transcribed by RNA polymerases. G+C-rich regions of the chromosomes and negative superhelicity can promote the invasion of the DNA by RNA to form R-loops, which have been shown to block DNA replication and promote genome instability. However, it is unclear whether the R-loops themselves are sufficient to cause this instability or if additional factors are required. We have investigated replisome collisions with transcription complexes and R-loops using a reconstituted bacterial DNA replication system. RNA polymerase transcription complexes co-directionally oriented with the replication fork were transient blockages, whereas those oriented head-on were severe, stable blockages. On the other hand, replisomes easily bypassed R-loops on either template strand. Replication encounters with R-loops on the leading-strand template (co-directional) resulted in gaps in the nascent leading strand, whereas lagging-strand template R-loops (head-on) had little impact on replication fork progression. We conclude that whereas R-loops alone can act as transient replication blocks, most genome-destabilizing replication fork stalling likely occurs because of proteins bound to the R-loops.
INTRODUCTION
Transcription of genes from the leading-strand DNA replication template results in co-directional (CO) replication-transcription collisions. Head-on (HO) encounters, which are generally thought to be more detrimental to genome stability (1–5), result from replisome collisions with RNA polymerases [RNAP(s)] transcribing genes from the lagging-strand template. This is manifested in many organisms where transcription, especially of highly-transcribed genes such as the Escherichia coli rrn operons, is biased to result in CO replication-transcription collisions (6–8). Similarly, polar replication fork barriers exist in eukaryotes to prevent HO replication encounters with highly transcribed rDNA genes (9). Despite these strategies, replication forks regularly collapse, even after CO collisions in vivo (10).
CO replication-transcription collisions with a single RNAP complex were resolved easily in vitro (11–13). However, highly transcribed genes are often bound by many RNAPs at the same time (14). Blockage of a single RNAP can thus generate a more potent replication obstacle if trailing RNAPs collide with the stalled RNAP (15). Sources of RNAP stalling vary, ranging from template damage (16–18) to natural pausing at different sites during transcription (19,20). Extended pausing can lead to RNAP backtracking, with the 3′-OH end of the transcript no longer present in the active site (21). Backtracked RNAPs are very stable, generating increasing problems for replication bypass (22). Displacement or processing of the RNAP complexes is required, as a failure to overcome replication-transcription collisions has detrimental effects on genome stability (2,4,10,22). Thus, multiple mechanisms have evolved to ameliorate the stresses of replication-transcription collisions ranging from removal of RNAPs (4,23–29), to the modulation of transcription elongation and complex stability (22,30,31), and the promotion of transcription termination (32).
Forward translocation of replisomes and RNA polymerases generates positively supercoiled DNA ahead and negatively supercoiled DNA behind the complexes (33). Formation of negatively supercoiled DNA favors hybridization of RNA to DNA, forming R-loops (34). Mapping experiments estimate that 5%-10% of genomes are occupied by R-loops even in wild-type cells (35–38). R-loop formation is essential for some cellular processes, such as the replication of mitochondrial DNA and some plasmids, antisense transcription, telomere maintenance, class switch recombination, and the suppression of transcriptional silencing (reviewed in Garcia-Muse and Aguilera (39) and Crossley et al. (40)). Conversely, R-loops have also been shown to block DNA replication and promote genome instability (38,41–44). Instability because of increased levels of R-loops is largely dependent on active DNA replication (42,45–47), implicating collisions of the replisome with R-loops as the sources of the genomic instability. Levels of R-loops are therefore managed via unwinding of the RNA–DNA hybrid by helicases (25,37,48), suppression by topoisomerases (49–51), and degradation by RNases (35,49). Elevated R-loop formation and genome instability has been observed in several RNA processing mutants (35,52,53), suggesting that cells tightly regulate the ability of transcripts to form R-loops. R-loops have been found at sites of DNA breaks, where a strand nick can nucleate their formation (54–56), and it was shown that persistent R-loops inhibit the repair of the broken DNA (44,57). However, the exact interplay between R-loop formation, DNA nicking or double-stranded break formation, and replication collisions remains elusive. The presence of single-stranded (ss) DNA in the displaced strand of an R-loop provides cleavage sites for ssDNA-specific nucleases (58). Replication of gapped DNA can lead to the formation of a double-stranded DNA break (22), a possible source of genome instability. Alternatively, replication-R-loop collisions may induce fork remodeling, which can be recombinogenic if the R-loop cannot be resolved (59,60).
R-loops are usually probed either using the RNA-DNA specific S9.6 antibody or by overexpressing an inactive (GFP-tagged) RNase H mutant (58,61,62). Neither of these techniques reveals what fraction of R-loops is associated with RNAP complexes. Overexpression of active RNase H is a common tool to reverse R-loop-induced genome instability in vivo (46,63,64). However, it is not clear if such treatment simply prevents the accumulation of R-loops alone. It was shown that R-loop levels are increased in a yeast mutant that has a reduced capacity of reactivating backtracked RNAPs (65). This complex interplay between RNAPs and R-loops in vivo makes it difficult to ascertain whether R-loops by themselves or R-loops associated with either RNAPs or other proteins are the source of genome instability.
Although replication collisions with a single RNAP complex have been investigated previously in vitro (11–13,66,67), it is unknown how a replisome deals with more complex RNAP arrays. It is also unclear how replication-R-loop collisions cause genome instability in vivo (46,47). These studies could not distinguish if the instability resulted from replication fork encounters with R-loops alone, or if R-loops stabilize RNAPs as replication blocks. We have investigated collisions of replisomes with RNAPs and R-loops in either orientation. We show that the potential for replication fork pausing or stalling increases with the number of RNAPs or R-loops and the length of the transcripts. Replication collisions with RNAP arrays were more detrimental than single RNAP collisions. However, a single RNAP encountered HO was a more potent replication obstacle than a CO RNAP array. We show directly that R-loops without an associated RNAP are a transient obstacle to replication fork progression when encountered on the leading-strand template, whereas in the absence of RNAP, R-loops on the lagging-strand template did not cause any significant replication fork blockage.
MATERIALS AND METHODS
Generation of E. coli strains
JGB412
To improve the yield of His10-tagged RNAP core during purification from a greAB mutant background, the rpoC-PPX-His10 allele from pEcrpoABC(-XH)Z was integrated into BL21(DE3). First, the 1.7 kb SmaI and HindIII rpoC-PPX-His10 fragment from pEcrpoABC(-XH)Z was blunt-ended with Klenow and cloned upstream of the <cat> cassette into NdeI-digested and Klenow-treated pKD3. The 1.1 kbp rpoC-PPX-His10 <cat> fragment was amplified using oligonucleotides oFW_rpoC-4177 (Supplementary Table S1) and oRV_<cat>+rpoC5'UTR, introducing 50 nt of homology of the PCR product on both sides of the chromosomal rpoC stop codon. The fragment was inserted into BL21(DE3) using λRed recombinase expressed from pKD46 (68), selecting for chloramphenicol resistance. The insert was confirmed by DNA sequencing. The rpoC-PPX-His10 <cat> allele was re-introduced into a fresh BL21(DE3) isolate by P1 transductions to remove any off-target modifications that resulted from the initial recombineering steps. This strain (JGB406) was then P1 transduced with a JW3148–1 lysate and selected on kanamycin for the introduction of a greA deletion (JGB407). Both antibiotic resistance cassettes were removed with pCP20-expressed FLP recombinase (68), giving strain JGB410. In a final step, greB was deleted by P1 transduction of JGB410 with a lysate from strain JW3369-1 and colonies were selected on kanamycin at 30°C. The rpoC-PPX-His10<> modification and greA and greB deletions were confirmed by PCR and the strain was saved as JGB412 (Supplementary Table S2).
JGB413
Strain JGB413 was generated via CRISPR-Cas9 recombineering, as described in Jiang et al. (69). The guide RNA was expressed from a modified version of pTargetF-cadA where the cadA-N20 containing EcoRI-SpeI fragment was replaced with the EcoRI-SpeI fragment of gBlock gsgRNA (rpoB′3716) that contains an rpoB specific N20 (5′-CGTGCATCTTGTCGTCGACC-3′) directing Cas9-cleavage of wild-type rpoB at position 3716. The rpoB*35 repair template was created by cloning an XbaI- and HindIII-digested ∼2 kbp wild type rpoBC fragment (amplified with oligonucleotides oFW_rpoB-2898 and oRV_rpoC-879 from genomic DNA) into pUC19 digested with the same enzymes. The pUC-2kb_rpoBC plasmid was then digested with BstEII and PstI and the 191 base pair fragment was replaced with the BstEII and PstI fragment of gBlock grpoB*35+PAM- to form pUC-2kb_rpoB*35-C. This gBlock fragment introduced a silent C3712T mutation at the PAM, which conserved rpoB His1237, and a C3732A missense mutation to introduce the H1244Q change of the rpoB*35 allele.
First, the kanamycin resistance gene from XL1-Blue Δtus::<kan> was removed using pCP20 (68), giving JGB399. The rpoB*35 mutation was then introduced into JGB399 using plasmids pCas and pTargetF-rpoB′3716 (for rpoB and suicide sgRNA expression). The repair template was a 1.8 kbp fragment amplified from pUC-2kb_rpoB*35-C using oligonucleotides oFW_rpoB-3261 and oRV_rpoC-756. Isolates were selected on LB medium containing kanamycin and spectinomycin at 30°C. Single colonies were then grown in LB medium at 37°C to lose pCas9. Loss of both plasmids was confirmed by sensitivity to kanamycin and spectinomycin. The presence of the rpoB*35 mutation was confirmed by DNA sequencing and the isolate was saved as JGB413.
Construction of replication templates
Initially, gBlock gT7A1-AGU19-pheA was digested with XbaI and inserted into the XbaI site of M13-JY13 (70), resulting in template CO19(AGU) (Supplementary Table S3). No isolates were found where replication and transcription would occur head-on. We suspect that such an orientation of the promoter insert is not possible, as transcription terminator read-through interferes with the M13 origin that is adjacent to the XbaI site. In order to create a template for head-on replication transcription collisions, the terB2-oriC fragment from CO19(AGU) was amplified with primers oFW_oriC-terB2 and oRV_oriC-terB2, digested with EcoRI and PvuI, and inserted into similarly-digested CO19(AGU) giving plasmid HO19(AGU).
To generate a template that allows RNAP stalling after 19 nt by the addition of only ApC, ATP and GTP, oligonucleotides oT7A1-AG19-top and oT7A1-AG19-bottom containing BstEII and PspOMI overhangs were phosphorylated, annealed and inserted into BstEII- and PspOMI-digested CO19(AGU) to replace the bacteriophage T7 A1 promoter and initial mRNA transcript, resulting in template CO19. HO19 was generated by cloning the T7 A1 promoter-containing 1.2 kb KpnI–SphI fragment from CO19 into the inverted oriC-terB-containing 9.4 kb KpnI–SphI fragment from HO19(AGU).
Templates allowing RNAP stalling at the T7 A1 transcript after 19 nt (ApC, ATP, GTP) or 100 nt (ApC, ATP, GTP, UTP) were generated by inserting the XbaI-digested ds gBlock gT7A1-AG19/AGU100-pheA into XbaI-digested CO19 and HO19(AGU). The resulting plasmids, CO100RNAI+ and HO100RNAI+ carry the RNA I promoter of the pBR322 origin, which can generate a 14 nt transcript/R-loop when ApC, ATP, GTP and UTP are added to transcription reactions. Therefore, CO100RNAI+ and HO100RNAI+ were digested with ApaLI and AcuI, blunt-ended with Klenow, and re-ligated to remove this 228 bp region and form CO100 and HO100, respectively (Supplementary Table S3).
Purification of replication templates
Replication templates were purified as M13 RF DNA from cell pellets of 5–8 l of M13-infected JGB413 cells grown in 2× YT broth supplemented with 5 mM MgCl2 and 12.5 μg/ml tetracycline. Cells were lysed by alkaline lysis, protein was removed by KOAc-SDS precipitation, RNA was digested with RNase A, and the DNA recovered by ethanol precipitation after two extractions with phenol-chloroform-isoamylalcohol (25:24:1). The pellet was resuspended in 10 mM Tris–HCl (pH 7.5), 1 mM EDTA (TE) and supercoiled RF DNA isolated by CsCl (1 g/ml) density gradient centrifugation in the presence of 200 μg/ml ethidium bromide in a Sorvall T-865 rotor at 40 000 rpm for 30–36 h at 15°C. DNA was removed from the gradients using a syringe, ethidium bromide was removed by extraction with CsCl-saturated isopropanol, and the DNA was dialyzed against TE and ethanol precipitated. The DNA was further purified by velocity sedimentation through 15–35% sucrose gradients (20 mM Tris–HCl (pH 7.5), 1 M NaCl, 1 mM EDTA) in a Sorvall Surespin 630 rotor at 27 000 rpm for 18–20 h at 4°C. Gradients were fractionated from the bottom of the tube, supercoiled RF DNA-containing fractions were pooled, dialyzed against TE, and the DNA ethanol precipitated.
Templates containing a single CPD lesion were generated as described previously (70,71).
Replication and transcription proteins
Purified as described: RNA polymerase core (72); σ70 (73); DnaA and HU (74); DnaB, DnaC, and DnaG (75); DnaN (β clamp) (76); Pol III* (77); SSB (78); Tus (79); DNA Gyrase (80); UvrD, gift of T. Lohman.
To remove RNase contamination in SSB and Tus preparations, the proteins were further purified by chromatography on hydroxylapatite-CF11 cellulose (60:17) columns equilibrated in 50 mM Tris–HCl (pH 8), 1 mM EDTA, 1 mM DTT, 5% glycerol (buffer A) and 0.1 M NaCl and eluted with a linear gradient of 0–0.6 M (NH4)2SO4 in buffer A. SSB and Tus eluted at 250 mM and 100 mM, respectively. Proteins were dialyzed against 50 mM Tris–HCl (pH 8), 1 mM EDTA, 1 mM DTT, 35% glycerol and 0.1 M NaCl, and stored in aliquots at −80°C.
An SDS-polyacrylamide gel of all proteins used is shown in Supplementary Figure S1A.
Transcription reactions
Transcription reaction mixtures (60 μl) contained 8 nM template, 50 mM HEPES–KOH (pH 8), 75 mM potassium glutamate, 10 mM DTT, 10 mM Mg(OAc)2, 0.1 mg / ml BSA, 15 μg/ml (3.15 U/ml) heparin, and 250 μM ApC. When required, [α-32P]GTP was added to 0.05 μM. RNAP holoenzyme was formed on ice by mixing RNAP core enzyme with a 5-fold excess of σ70. Reactions were incubated at 37°C for 1 min prior to transcription initiation by the addition RNAP holoenzyme (200 nM RNAP core enzyme and 1 μM σ70).
Transcription reactions to form 19mer R-loops and RNAP complexes (on templates CO19 and HO19) contained 0.5 μM ATP and GTP. For R-loop templates, 3′-dCTP was also added to 250 μM 1 min after the addition of RNAP holoenzyme and incubation was continued for 14 min until further processing (see below).
To form a single 100mer RNAP complex or R-loop on templates CO100 and HO100, transcription reaction mixtures were spin-dialyzed after 8 min of incubation with 0.5 μM ATP and GTP. Spin columns were prepared as described by Kreuzer and Alberts (81). Sepharose 4B was washed three times with an equal volume of TE and formed into a column (500 μl resin) in a 1.5 ml Eppendorf tube that had three holes poked in the bottom with an 18 g needle and contained 50 μl siliconized glass beads at the bottom to support the resin. Spin columns were equilibrated by two washes with 500 μl GF buffer (60 mM HEPES-KOH (pH 8), 90 mM potassium glutamate, 12 mM DTT, 12 mM Mg(OAc)2, 0.5 μM ATP, 0.5 μM GTP and 0.015% IGEPAL CA-630). GF buffer was removed by centrifugation at 735 × g for 2 min before the addition of transcription reaction mixtures. DNA-RNAP complexes, but not unbound RNAP, were eluted by centrifugation at 735 × g for 2 min. ATP, GTP, and UTP (final concentration of 10 μM) were added to the eluent and incubated for 5 min at 37°C to extend the 19mer transcript into a 100mer. R-loop templates were additionally chain terminated with the addition of 3′-dCTP (final concentration of 250 μM). When required, [α-32P]GTP was added back as well.
RNAP or R-loop arrays (on templates CO100 and HO100) were formed by incubation with 10 μM ATP, GTP, and UTP at 37°C. To form R-loop templates, 3′-dCTP was additionally added to 250 μM 1 min after the addition of RNAP holoenzyme and 250 μM 3′-dNTPs were added to the transcription reactions after 10 min of incubation to also incorporate chain terminators into the transcripts of the trailing RNAPs during an additional incubation of 5 min.
Templates on which no transcription complexes had formed were inactivated for subsequent replication initiation by digestion with PspOMI (0.5 U/μl) at 37°C for 10 min. However, with RNAP- or R-loop-free control reactions, where the PspOMI site is not obstructed by an RNAP complex at this stage, this step was omitted. For R-loop templates, after PspOMI digestion, SDS and proteinase K were added to final concentrations of 0.3% and 0.1 mg/ml, respectively, and the incubation continued at 37°C for 10 min. R-loop templates were then separated from free proteins by gel filtration through a 3 mm × 195 mm Sepharose 4B column (71) equilibrated and developed in GF buffer. RNAP-templates were gel filtered without prior proteinase K digestion. Fractions containing DNA were pooled (about 110 μl) and typically contained 1–2 nM RNAP- or R-loop-DNA complexes. RNAP complexes remained bound to template during gel filtration, as estimated by comparing levels of supercoiled DNA without and with PspOMI digestion (101 ± 3% of the supercoiled templates retained the RNAP, averaged for all different RNAP species and templates, n = 24). R-loop occupancy was estimated in the same manned to be 82 ± 7% of the supercoiled templates (averaged for all different R-loop species and templates, n = 18).
Replication reactions
Replication reaction mixtures (25–120 μl) containing gel filtration pools (20–100 μl) of either DNA-R-loop or DNA-RNAP complexes, 50 mM HEPES–KOH (pH 8), 75 mM potassium glutamate, 10 mM DTT, 10 mM Mg(OAc)2, 0.1 mg/ml BSA, 40 μM dNTPs, 0.033 μM [α-32P]dATP, 1 mM ATP, 0.2 mM GTP, 1 μM SSB, 50 nM Tus, and 40 nM DNA gyrase were assembled on ice. Reaction mixtures containing R-loop templates also contained 0.2 mM CTP and UTP. [α-32P]dATP was omitted when R-loops were to be visualized (Figures 4 and 5Aii and Aiii). Reaction mixtures were then shifted to 37°C and replication proteins were added to final concentrations of 140 nM DnaA, 200 nM DnaB, 200 nM DnaC, 320 nM DnaG, 20 nM Pol III*, 30 nM DnaN2 and 25 nM HU. Aliquots (8 μl) were withdrawn at the indicated times and the replication reactions terminated by the addition of AMP-PNP and ddNTPs to 2 mM and 133 μM, respectively. DNA products were then digested with ScaI-HF (if not done earlier, see below), EcoRI-HF, and PvuI-HF for 10 min at 37°C, and analyzed by electrophoresis as described below.
Figure 4.
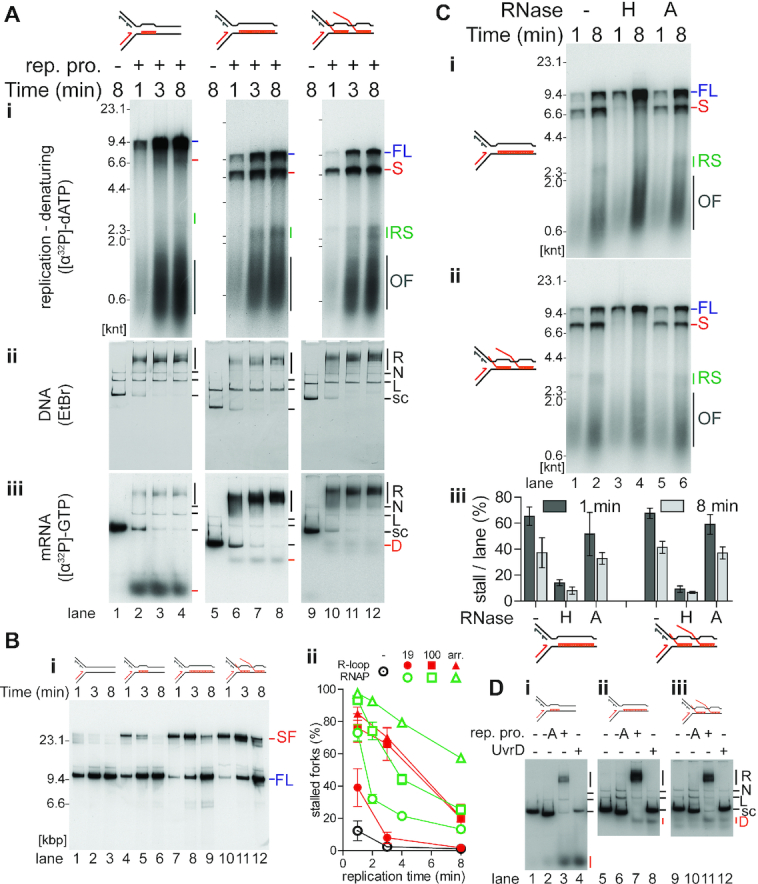
R-loops on the leading-strand template cause transient replisome stalling. (A) Time course of replisome collisions with three different R-loop species on the leading-strand template (n = 3). (i) Analysis of [α-32P]dATP-labeled replication reaction products on denaturing agarose gels. There is no DNA replication in the absence of the replication proteins as measured by the incorporation of [α-32P]dATP into acid-insoluble product. Thus, these samples, lanes 1, 5, and 9, were not included on the denaturing gel that is visualized by autoradiography. Native agarose gel of replication reaction products either stained with (ii) ethidium bromide or (iii) autoradiography of [α-32P]GTP-labeled RNA in the R-loops. Lanes 1–4, 19mer R-loop (on template CO19); lanes 5–8, 100mer R-loop (CO100); lanes 9–12, R-loop array (CO100). (B) Replication fork stalling increases with more complex R-loop species on the leading-strand template. (i) Native agarose gel of a replication time course of replisome collisions without or with leading-strand template R-loops. No R-loop (CO100), lanes 1–3; 19mer R-loop (CO19), lanes 4–6; 100mer R-loop (CO100), lanes 7–9; and R-loop array (CO100), lanes 10–12. Unlike the replication time courses in Figure 4A, these DNA templates had been linearized by ScaI digestion 40 s after replication initiation to allow for a more direct comparison between replication bypass of R-loops and CO replication-RNAP collisions. (ii) Quantification of stalled forks from native gels (n = 3, mean ± standard deviation) compared to the results from the quantification of stalled forks at CO replisome-RNAP collisions from Figure 2B lanes 5–16. R-loop collisions in red, RNAP collisions in open, green symbols. ⊙ no collision control, • 19mer, ▪ 100mer, ▴ array. (C) Replication stalling is relieved by treatment of the R-loop templates with RNase H, but not RNase A. Denaturing agarose gel of products in replication time courses of collisions with either the (i) CO 100mer R-loop template or (ii) CO R-loop array template subsequent to treatment with RNase H (lanes 3 and 4), RNase A (lanes 5 and 6), or no treatment (lanes 1 and 2). (iii) Quantification of the fraction of stall product (n = 3, mean ± standard deviation). (D) Either replication or UvrD treatment of R-loop templates can result in displacement of the RNA from the R-loop. Native agarose gel of [α-32P]GTP-labeled R-loop templates incubated (8 min) in replication buffer either without or with the indicated replication proteins and either with or without UvrD (100 nM), as indicated. (i) 19mer R-loop, (ii) 100mer R-loop, (iii) R-loop array. FL, full length leading strand; S, stalled leading strand product; RS, restarted leading strand products; OF, Okazaki fragments; R, replicated; N, nicked; L, linear; s.c., supercoiled; D, displaced RNA.
Figure 5.
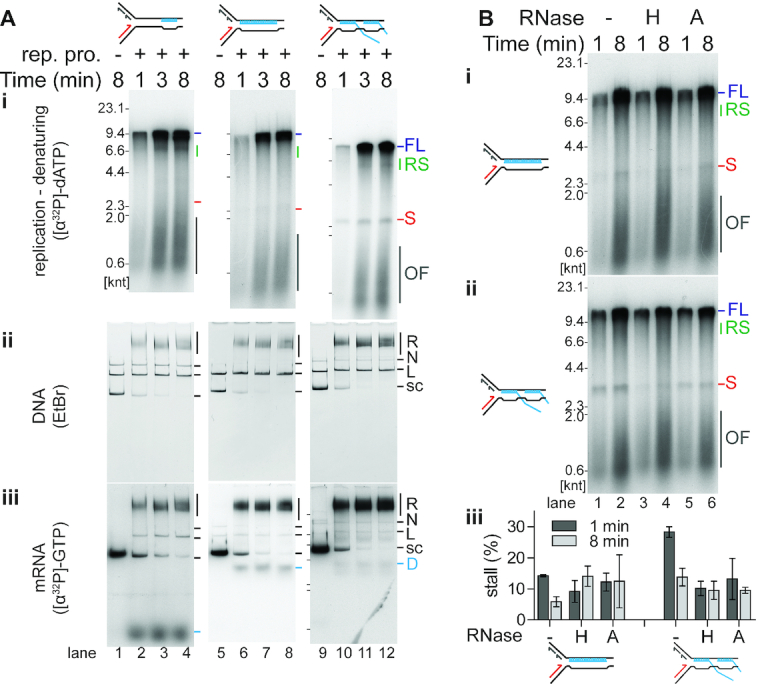
R-loops on the lagging-strand template are not a significant block to replication. (A) Time course of replisome collisions with three different R-loop species on the lagging-strand template (n = 3). (i) Analysis of [α-32P]dATP-labeled replication reaction products on denaturing agarose gels. There is no DNA replication in the absence of the replication proteins as measured by the incorporation of [α-32P]dATP into acid-insoluble product. Thus, these samples, lanes 1, 5, and 9, were not included on the denaturing gel that is visualized by autoradiography. Native agarose gel of replication reaction products either stained with (ii) ethidium bromide or (iii) autoradiography of [α-32P]GTP-labeled RNA in the R-loops. Lanes 1–4, 19mer R-loop (on template HO19); lanes 5–8, 100mer R-loop (HO100); lanes 9–12, R-loop array (HO100). (B) Both RNase H and RNase A relieve stalling on the HO R-loop array template. Denaturing gel analysis of the replication products of collisions with either the (i) HO 100mer R-loop template or (ii) HO R-loop array template subsequent to treatment with RNase H (lanes 3 and 4), RNase A (lanes 5 and 6), or no enzyme (lanes 1 and 2). (iii) Quantification of the fraction of stall product (n = 3, mean ± standard deviation).
Degradation of RNA in R-loops by RNases
RNases (20 μM RNase A (Roche) or 0.1 U/μl RNase H (NEB)) were added to reaction mixtures before the addition of replication proteins and incubated at 37°C for 10 min. The reactions were then either terminated by the addition of EDTA to 30 mM (Figure 3D) or replication was initiated by the addition of replication proteins (Figures 4C and 5B).
Figure 3.

Generation of R-loop templates. (A) Extension of the RNA in R-loops by either DnaG or Pol III* is blocked when the 3′-end of the RNA lacks a 3′-OH group. R-loops formed with [α-32P]GTP-labeled RNA as described in Materials and Methods on the CO19 template either without (G19) or with (3′dC20) incorporation of 3′-dCTP were incubated in replication buffer lacking either the complete complement of replication proteins (all) or either DNA Polymerase III* (III*) or DnaA (A). The products were analyzed by electrophoresis through a 20% 7M urea polyacrylamide gel (n = 2). (B) Schematic of the steps in preparing the R-loop templates as described in Results and Materials and Methods. (C) RNA transcripts present in the 19mer (lane 1), 100mer (lane 2), and R-loop array (lane 3) CO templates as analyzed by denaturing polyacrylamide gel electrophoresis. (D) RNase H, but not RNase A, digests RNA in the R loops. (i) 100mer R-loops and (ii) R-loop arrays formed on template CO100 were treated in replication buffer either without (–) or with the indicated RNases (RNase H, 0.1 U/μl; RNase A, 20 μM) for 10 min (n = 3). Native agarose gels of replication reaction products stained with ethidium bromide (DNA) or imaged by autoradiography of [α-32P]GTP-labeled RNA (mRNA) in the R-loops.
Visualizing replication intermediates
After incubation of the replication reaction mixtures for the indicated times, the reactions were terminated by the addition of EDTA to 30 mM. Replication products were not digested with any restriction enzyme to preserve replication intermediates. Linear DNA seen on the gels is the result of the inactivation of RNAP-free templates by PspOMI digestion prior to gel filtration.
Monitoring RNAP and R-loop bypass in pseudo-synchronous replication reactions
Pseudo-synchronous replication was achieved by the addition of ScaI-HF (0.33 U/μl) 40 s after the addition of replication proteins as the incubation continued. Template linearization by ScaI prevented any further replication initiation from oriC, so that the fate of either replication-RNAP (Figure 2B) or replication-R-loop (Figure 4B) collisions that formed in the first 40 s of the assay could be monitored over time. Reactions were performed with unlabeled R-loop templates when native gels were used for analysis because the signal of labeled R-loops co-migrated with full-length replication products.
Figure 2.

Analysis of collisions between RNAP-template complexes and replisomes. (A) Cartoon illustrating the potential products of a collision between a replisome and a CO-RNAP-template complex. (B) CO RNAP-replication collisions cause transient fork stalling, whereas HO RNAP-replication collisions are severe blockages to fork progression. Time courses of replisome collisions on templates carrying either no RNAP complexes (on template CO100, lanes 1–4); three different CO RNAP-templates: 19mer (CO100), lanes 5–8, 100mer (CO100), lanes 9–12, or RNAP array (CO100), lanes 13–16; or three different HO RNAP-templates: 19mer (HO100), lanes 17–20, 100mer (HO100), lanes 21–24, or RNAP array (HO100), lanes 25–28, analyzed by either (i) native agarose gel or (ii) denaturing agarose gel electrophoresis (n = 5). (C) Fork blockage becomes more severe as the complexity of the transcription complex increases. The fraction of stalled fork DNA products from native gels are plotted as a function of time for CO (i) and HO (ii) collisions. (iii) Reciprocal relationship between the fraction of stalled leading strand product and full-length material as a product of time for HO collisions on denaturing gels. (D) Relative efficiency of the DNA helicases Rep (blue) and UvrD (red) in resolving stalled forks in HO collisions with a 19mer RNAP (•, on template HO19) or an RNAP array (▴, HO100) (n = 3). SF, stalled fork; FL, full length product; RS, restart product; S, stalled leading-strand product; OF, Okazaki fragments.
Analysis of displacement of RNA from R-loops
Replication reaction mixtures containing [α-32P]GTP-labeled RNA and a complete set of replication proteins (+), all replication proteins, but no DnaA (-A), or 100 nM UvrD instead of replication proteins were incubated for 8 min. Reactions were terminated by the addition of EDTA to 30 mM without post-replicative digestion.
Gel electrophoresis
Native agarose gel electrophoresis was performed through vertical 0.8% agarose (SeaKem LE) gels using 50 mM Tris–HCl (pH 7.8), 40 mM NaOAc, and 1 mM EDTA as the electrophoresis buffer. A 5X loading buffer was added to samples to give final concentrations of 10 mM EDTA, 0.4% Sarcosyl, 2% Ficoll-400, and 0.015% Bromophenol blue. Gels were electrophoresed at 20–25 V for 380 V h.
Denaturing agarose gel electrophoresis was performed through horizontal 0.6% agarose (SeaKem LE) gels using 30 mM NaOH and 2 mM EDTA as the electrophoresis buffer. Samples were loaded containing 30 mM NaOH, 2% sucrose, and 1 μg/ml xylene cyanol. Gels were electrophoresed at 20–25 V for 380 Vh and then fixed by two 20 min incubations in 5% trichloroacetic acid.
Denaturing polyacrylamide gels were 1 mm thick and 20 cm long containing 7 M urea, 50 mM Tris-borate (pH 8), 1 mM EDTA and either 10% polyacrylamide-bisacrylamide (29:1) or 20% polyacrylamide-bisacrylamide (19:1). Reaction products were extracted with phenol-chloroform-isoamylalcohol, ethanol precipitated, and resuspended in 99.5% formamide, 20 mM EDTA, 0.05% Bromophenol blue, and 0.05% xylene cyanol. Electrophoresis was at 800 V for 75 min (10% polyacrylamide gels) or 1000 V for 120 min (20%) using 50 mM Tris-borate (pH 8), 1 mM EDTA as the electrophoresis buffer. Gels were fixed in 10% MeOH, 7% HOAc, and 5% glycerol for 15 min.
Gel imaging and presentation
Ethidium bromide-stained gels were imaged with a BioRad ChemiDoc XRS+ system.
Gels containing radioactive samples were dried onto chromatography paper (GE Healthcare, 3030-861) and imaged with a Typhoon FLA 7000 phosphorimager (GE Healthcare) for quantitative analyses. Dried gels were also exposed to Amersham Hyperfilm MP (GE Healthcare, 28906843) and scanned for data presentation.
RESULTS
Replisome collisions with transcription complexes
Our templates for examining CO collisions have a bacteriophage T7 A1 promoter approximately 6.9 kbp downstream of oriC. Clockwise-moving replication forks encounter transcription complexes in the CO orientation (Figure 1Ai). Templates CO19 and CO100 differ by their respective potential transcripts (Figure 1Aii). Transcription initiation using the dinucleotide ApC suppresses initiation at other promoters on the template. A 19mer transcript is formed on both templates in the presence of only ATP and GTP (Figure 1B, lanes 1 and 2). Gel filtration of the RNAP-19mer complex on CO100 removed unbound RNAP (Supplementary Figure S1B), allowing the subsequent extension of the 19mer to a 100mer in the presence of ATP, GTP and UTP (Figure 1B, lane 3). Alternatively, the addition of ATP, GTP and UTP with ApC from the start resulted in the formation of an RNAP array with up to three RNAPs occupying the same template (Figure 1B, lane 4). In order to generate HO replication-transcription collisions, the oriC-terB fragment of our template was inverted between the EcoRI and PvuI sites (Figure 1Ai). Template occupancy by RNAP blocks the PspOMI site (Figure 1Aii). Thus, any RNAP-free template was linearized using PspOMI, thereby inactivating those templates for replication. Templates were subsequently gel filtered to remove free RNAP and PspOMI before DNA replication.
Figure 1.
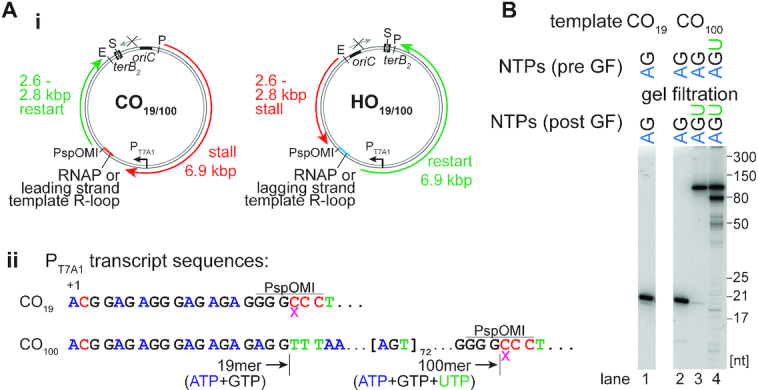
Templates and transcription products. (Ai) Replication templates with relevant features, restriction sites, and expected leading-strand lengths. (Aii) Bacteriophage T7 A1 promoter transcripts for template CO19 and CO100 with indicated transcript lengths for different nucleotide additions and relevant restriction sites. The pink x denotes the incorporation of the 3′-dCTP RNA chain terminator. (B) RNA transcripts formed on different templates by staged nucleotide omission as described in Results and Materials and Methods analyzed by electrophoresis through a 10% 7M urea-polyacrylamide gel. E, S, and P, EcoRI, ScaI and PvuI sites, respectively.
DNA replication reactions were initiated from oriC on either RNAP-free or RNAP-bound, column-isolated DNA templates. In the case of CO templates, counterclockwise-moving replication forks were blocked at the terB site bound by Tus, whereas clockwise-moving forks could replicate for ∼10 kb unless their progression was interrupted by RNAP–DNA complexes, generating a 6.9 kb stall fragment and a potential 2.6 kb restart fragment (Figure 1Ai). In the case of HO templates, clockwise-moving replication forks were blocked at the terB site. A stall of the counterclockwise-moving replication fork at an HO RNAP complex generates a 2.6 kbp fragment and a 6.9 kbp restart fragment (Figure 1Ai). Removal of the RNAP block and continuation of leading-strand synthesis generates a 9.6 kbp full-length fragment (Figure 1Ai). After 40 s of replication, the templates were linearized by ScaI digestion to prevent additional initiation at oriC, generating pseudo-synchronous progression of the replication forks that headed toward the potential blockage site that could be followed by gel electrophoresis after digestion with EcoRI and PvuI (Figure 2B). To maintain stalled RNAPs during replication reactions, CTP and UTP were omitted, which had no significant effect on replication (Supplementary Figure S1C).
The effects of replication-transcription collisions of both orientations were compared to replication fork progression in the absence of any bound RNAP using the three different gel-filtered RNAP-DNA complexes: a single RNAP and either (i) a 19mer transcript, (ii) a 100mer transcript or (iii) an array of up to three RNAPs with a maximum transcript length of 100 nt (Figures 2B, C, and D). On RNAP-free templates, replication generated a continuous leading strand of ∼9.6 knt and 1–3 knt long Okazaki fragments (Figure 2Bii, lanes 1–4). Native gel analysis of the replication products of the CO collisions showed the formation of stalled replication forks, with greater stalling paralleling increased complexity of the transcription complexes (Figure 2Bi, lanes 5–16). However, with time, the amounts of stalled forks decreased as full-length products increased. Even with an RNAP array, about half of all stalled forks were resolved after 8 min (Figure 2Ci), suggesting that the E. coli replisome can bypass CO RNAP complexes—even RNAP arrays—without additional factors (Figure 2A). Analysis of these products on denaturing gels revealed that replication generated a 6.9 knt nascent leading strand (Figure 2Bii, lanes 5–16), the expected length for a replisome-RNAP collision (Figures 1Ai and 2A, stall). With time, a ∼2.6 knt band—the expected length for leading-strand replication restart (Figures 1Ai and 2A, restart)—increased in intensity (Figure 2Bii, lanes 5–16). These data indicate that whereas the RNAP complexes created an obstacle to replication generating a stall, they were bypassed by initiation de novo (Figure 2A), as has been observed previously (11). The RNAP array had the most severe effect, generating a more prominent stall band than either the 100mer-RNAP template or the 19mer-RNAP template and exhibiting delayed replication restart (Figure 2Bii, lanes 5–16). Minor levels of full-length product were observed for the 19mer-RNAP template and the 100mer-RNAP template, possibly indicating dissociation of RNAP from some templates.
Native gel analysis showed the formation of a major species of stalled fork common to all three HO templates for RNAP complexes originating at the T7 promoter (Figure 2Bi, lanes 17–28, SFT7A1). Minor, slower-moving stalled fork species result from RNAP complexes with short transcripts originating at M13 promoters on the template, all of which are oriented HO with the counterclockwise-moving fork. Formation of full-length products was evident, although they accumulated slowly; even after 30 min, significant amounts of replication forks remained blocked (Figure 2Bi, lanes 17–28). As with the CO collisions, the severity of replication fork blockage increased with transcript length and RNAP number (Figures 2Bi, lanes 17–28, and 2Cii). Analysis of these reactions on denaturing agarose gels (Figure 2Bii, lanes 17–28) allowed us to compare the levels of nascent leading-strand stall and full-length products over time (Figure 2Ciii). This analysis confirmed that the severity of the stall increased with increasing complexity of the RNAP complex: lower complexity collisions exhibited a greater proportion of full-length product at all time points. Moreover, a reciprocal relationship was evident with time between the stall and full-length products, suggesting that the replisome remained idle at the HO collision for an extended period. Once the block was removed, presumably by dissociation of the RNAP, the 2.6 knt stall product was extended to a continuous full-length leading strand. Alternatively, the HO RNAP block can be removed by the action of accessory DNA helicases (24). Of the two DNA helicases known to associate with either RNAP (UvrD, (82)) or the replisome (Rep, with DnaB (23)), UvrD was more efficient at clearing the stalled RNAP (Figure 2D).
Thus, in either orientation, RNAP transcription complexes caused replisome stalling, as previously demonstrated, with replication progression resuming by restart in the CO orientation. HO collisions resulted in more potent blockages than CO collisions and increasing complexity of the transcription array in either orientation increased the severity of the stall.
Generation of templates carrying R-loops
We found that the most efficient way to produce templates for DNA replication carrying R-loops was to perform the transcription reactions as detailed above for the generation of RNAP-templates and then degrade the RNAP by digestion with proteinase K leaving R-loops behind. This method (Figure 3B) allowed us to generate three distinct R-loops that were directly relatable to the RNAP transcription complexes analyzed in the previous section: a 19mer, a 100mer, and an array where the longest R-loop could be 100 nt (Figure 3C). These R-loops were oriented to be either CO (RNA annealed to the leading-strand template) or HO (RNA annealed to the lagging-strand template) with respect to the oncoming replication fork (Figure 1Ai).
Because the R-loops are in the same position as the RNAP transcription complexes, the expected stall, restart, and full-length products during DNA replication are the same as for the RNAP-DNA templates (Figure 1Ai). In the case of the R-loop templates, however, in most replication reactions, except as noted below, templates were not linearized during replication and DNA gyrase was present to maintain superhelicity and R-loop stability. We found that the DNA Polymerase III Holoenzyme and DnaG, components of the reconstituted DNA replication system, could extend the RNA transcripts in the R-loop templates if they contained a free 3′-OH end (Figure 3A). Extension occurred in the absence of DnaA (Figure 3A, lane 3), where replication initiation from oriC was suppressed. Hence, R-loops with a free 3′-OH end can act as origins of replication as proposed for constitutive stable DNA replication (83,84). However, to suppress this reaction and monitor only replication fork collisions with R-loops, all RNAs in our experiments with R-loops were chain terminated (Figure 3B). To confirm that the isolated complexes were actual R-loops, we treated 100mer R-loop and R-loop array complexes made on the CO100 template with either RNase H or RNase A. After native gel electrophoresis, untreated DNA–RNA complexes showed superimposable patterns of ethidium bromide-stained DNA and radioactively-labeled RNA (Figure 3D), indicating that the 100mer and the RNA array remained stably bound to the DNA. Treatment with RNase H abrogated this interaction, whereas RNase A treatment did not (Figure 3D, compare lanes 5 and 6, and lanes 11 and 12).
R-loops on the leading-strand template cause transient replisome stalling
We tested the impact on DNA replication of a single 19mer R-loop, a single 100mer R-loop, or an R-loop array on the leading-strand template. [32P]R-loop templates were replicated in parallel in the presence or absence of [α-32P]dATP to monitor either DNA replication (denaturing agarose gels, Figure 4Ai) or the effect of replication on the R-loops themselves (native gels, Figures 4Aii and iii), respectively. RNAs were stable and not degraded during the replication reactions (Supplementary Figure S2A).
Full-length DNA products were formed during replication with all three leading-strand R-loop templates (Figure 4Ai). Some RNA dissociated from the DNA during gel filtration of the R-loop templates (∼20%), thus, a portion of full-length products resulted from replication of R-loop-free templates. A single 19mer R-loop on the leading-strand template did not pose an obstacle to DNA replication, as the full-length leading strand was essentially the only product (Figure 4Ai, lanes 2–4). In contrast, full-length leading-strand formation was reduced with the 100mer and R-loop array templates, most obviously visualized at early times in the reactions (Figure 4Ai, lanes 6 and 10). Significant amounts of the 6.9 knt leading-strand stall species was apparent. At later time points, a ∼2.6 knt restart product appeared, suggesting the replisome could bypass longer R-loops on the leading-strand template (Figure 4Ai, lanes 7, 8, 11 and 12). R-loop bypass was confirmed by analysis of replication reaction products by native gel electrophoresis. In this replication assay, the templates were linearized by ScaI digestion 40 s after replication initiation to prevent further initiation and to monitor the fate of the initial replication-R-loop collisions in a pseudo-synchronous manner. Stalled forks formed and were resolved into full-length products over time (Figure 4Bi). 100mer R-loops and R-loop arrays on the leading-strand template were equally as potent obstacles to fork progression (Figures 4Ai and B).
We compared the kinetics of stalled fork resolution using the pseudo-synchronous reactions for all the CO-RNAP templates and leading-strand R-loop templates (Figure 4Bii). This comparison showed that 100mer R-loops on the leading-strand template were nearly as potent obstacles as CO 100mer RNAP complexes, although a CO RNAP array was more potent than an R-loop array (Figure 4Bii).
To determine whether any of the RNA was displaced during the reaction, replication products formed with labeled RNA were analyzed by native gel electrophoresis without post-replicative restriction enzyme digestion to maintain the integrity of the replication intermediates. The majority of the RNA signal coincided with replication intermediates (compare ‘R’ in Figures 4Aii to Aiii). However, some of the RNA signal was displaced from the template DNA with all the R-loop templates (Figure 4Aiii, ‘D’; Supplementary Table S4). Displacement was confirmed by the formation of the same species upon incubation of R-loops with UvrD (Figure 4D, lanes 4, 8 and 12, respectively), which unwinds RNA–DNA hybrids (85). RNA displacement from R-loops was dependent on the presence of DnaA (Figure 4D, lanes 2, 6 and 10, respectively), demonstrating that replication forks that initiated at oriC were able to actively dissociate some R-loops. Dissociation was most efficient for the 19mer (Figure 4Aiii, lanes 2–4; Supplementary Table S4), which correlated with a lack of stall formation in the replication reactions (Figure 4Ai, lanes 2–4). Conversely, dissociation of longer R-loops was less efficient (Figure 4Aiii, lanes 6–8 and 10–12; Supplementary Table S4), which correlated with stronger replication fork stalling in replication reactions (Figure 4Ai, lanes 6–8 and 10–12). We further tested the nature of the obstacle by treatment with either RNase H or RNase A. RNase H treatment nearly completely ablated replication stalling, whereas RNase A treatment had little effect (Figure 4C). Hence, leading-strand synthesis can be stalled by R-loops on the leading-strand template unless the R-loops are short enough to be displaced by the replisome or removed, e.g. by RNase H.
R-loops on the lagging-strand template are not a significant block for replication
Templates HO19 or HO100 were used to form R-loops on the lagging-strand template. As with the leading-strand R-loops, lagging-strand R-loops were stable and not degraded during replication (Supplementary Figure S2B). Analysis of replication reactions on denaturing gels showed little stall product formation for the 19mer R-loop and the 100mer R-loop templates (Figure 5Ai, lanes 2–4 and 6–8). Only the R-loop array template showed low levels of the 2.6 knt stall product (Figure 5Ai, lanes 10–12). Replication resulted in similar levels of R-loop dissociation from the lagging-strand template as it did for replication collisions with R-loops on the leading-strand template (compare band D in Figures 5Aiii and 4Aiii; Supplementary Table S4). More efficient R-loop displacement was therefore not a likely mechanism to explain why R-loops on the lagging-strand template caused little fork stalling. Treatment of the 100mer R-loop with RNase H and RNase A yielded little in bypass improvement, as replication stalling was already low in the untreated samples (Figures 4Bi and 4Biii). RNase H treatment did however reduce stalling for the R-loop array. Additionally, RNase A treatment reduced the fraction of leading-strand stall products at early times in the reaction (Figure 5Bii, lane 5 and Figure 5Biii), suggesting that free single-stranded tails in the R-loop arrays might play a role in stalling the replisome. These results suggest that most R-loops on the lagging-strand template are not significant obstacles to DNA replication, although complex, multi-stranded R-loop arrays pose a slight blockage.
DISCUSSION
We found that the replisome was able to bypass R-loops on either template strand. We propose two different mechanisms for replisome bypass of leading-strand template R-loops (Figure 6A). Encounters with short R-loops could be resolved via the known strand-displacement activity of the DNA Polymerase III Holoenzyme (86), whereby Pol III could continue to extend the existing nascent leading strand and avoid the formation of a leading-strand gap (Figures 6Ai, ii, and iv). Longer leading-strand template R-loops are, however, too stable to be displaced in this fashion in a timely manner. We found that R-loop stability was determined mostly by the accumulated length of the RNA–DNA hybrid, not by transcript number, as 100mer R-loops and R-loop arrays blocked forks to similar levels (Figure 4B). Because the lagging-strand template should be free of obstacles, the replication fork helicase, DnaB, can continue to translocate and unwind the template downstream of the R-loops. DnaG, the primase, can then synthesize a new leading-strand primer downstream of the RNA–DNA hybrid, allowing the leading-strand polymerase to re-initiate synthesis. This process, like replisome skipping of a DNA template lesion (71), generates a ssDNA gap in the leading strand that must be filled post-replicatively (Figures 6Ai, ii, and iii). It is unclear how gap filling occurs after R-loops are removed, but if translesion polymerases are involved that could mean regions prone to making R-loops might have a higher mutation rate.
Figure 6.
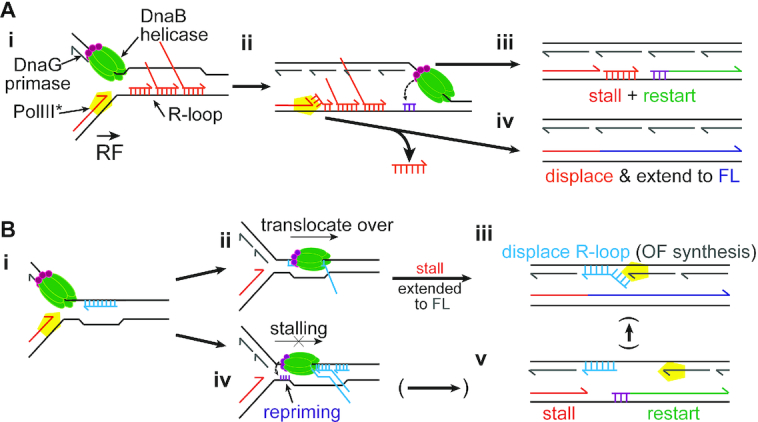
Models for replisome bypass of R-loops. Model for potential bypass mechanisms for replication fork encounters with either (A) R-loops on the leading-strand template, or (B) R-loops on the lagging-strand template. Details in the Discussion.
R-loops on the lagging-strand template barely caused fork stalling (Figure 5Ai). R-loops on the lagging-strand template are initially encountered by DnaB (Figure 6Bi). If the RNA is not hybridized at the 3′-end, DnaB can unwind short RNA–DNA duplexes (87). However, as the level of RNA displacement we observed was similar for replication-R-loop collisions on either template strand (Figures 4Aiii and 5Aiii, compare the amounts of band ‘D’; Supplementary Table S4), direct displacement of R-loops on the lagging-strand template by DnaB is rare. DnaB does not displace fully hybridized RNA–DNA hybrids (88), but can encircle and translocate over the RNA–DNA hybrid (Figure 6Bii) (89). RNA displacement likely resulted from Pol III strand displacement during Okazaki fragment synthesis (Figure 6Biii). Replication challenged with lagging-strand template R-loops generated mostly full-length products and little leading-strand stall (Figure 5Ai), suggesting that translocation over a single R-loop occurs fast, without the need for leading-strand re-priming or replisome skipping. DnaB can encircle up to three strands within its central channel (89), so single-stranded tails in complex R-loop arrays might interfere with DnaB translocation, causing delays or even a complete block to replication fork progression. DnaG could synthesize a new primer on the leading-strand template during extended fork pausing (Figure 6Biv). Overcoming such complex R-loop arrays would likely require further processing or degradation of the R-loop (tails) and could involve the formation of a leading-strand gap (Figure 6Bv).
Given the data that has accumulated in vivo attributing genome instability to replication collisions with R-loops (38,41–44), the ease by which the replisome bypassed R-loops on either template strand suggests that it is not the R-loops per se (at least not ones of the sizes examined herein) that are the etiological agent and that additional factors are required. Our data suggests that the likely additional factor is a protein bound to the R-loop, most generally RNAP.
Our data on collisions between replisomes and transcription complexes is in agreement with what has been published previously for collisions between the replisome and a single transcription complex (11,66). CO-RNAP-replication collisions caused a transient blockage to replication. Our data clearly indicate, with nearly 60% of forks stalled at a CO-RNAP array compared to 15–25% at a single RNAP after eight minutes of replication (Figure 2Ci), that increasing the number of RNAPs increases the severity of the replication obstacle. Nevertheless, replication restart picks up downstream, even when an array of up to three RNAPs is present (Figure 2B). Comparison of the kinetics of stalled fork resolution between CO-RNAP collisions and R-loop collisions showed the same general pattern, with the severity of the stall increasing as the complexity of either the transcription complexes or R-loops increased. Transcription complexes in general were a more severe obstacle, clearly seen when either single or arrays of transcription complexes and R-loops were compared (Figure 4Bii). However, both types of collisions are resolved, suggesting that neither a leading-strand R-loop nor a CO-RNAP transcription complex will be all that destabilizing to replication fork progression.
On the other hand, HO-RNAP collisions caused the most severe blocks to replication fork progression (Figure 2). Whereas transcript length and the number of RNAP complexes also increased blockage at HO collisions (Figure 2Cii), forks stalled for longer periods with each type of HO block compared to the equivalent CO block, with a majority of forks still blocked after 30 minutes (Figures 2Cii and 2Ciii). Unlike CO collisions, clearance of the RNAP by an accessory DNA helicase was required to relieve the blockage (Figure 2D). However, collisions between lagging-strand R-loops and the replisome barely caused any fork stalling (Figure 5). The length of R-loops does, however, affect replication-transcription collisions, as we observed an increase in replisome stalling for collisions with 100mers associated with a single RNAP compared to a single RNAP with a 19mer in either orientation (Figure 2Ci and Cii). These observations strongly suggest that the genome destabilizing effects of replisome-R-loop collisions are a result of proteins bound to the R-loops.
Our conclusions are consistent with observations in vivo. Increases in genome instability in vivo for replication collisions with R-loops on the lagging-strand template (HO), but not on the leading-strand template (CO), have been reported for B. subtilis (46) and human cells (47). In the former case, it is likely that effects attributed to R-loops were a result of the R-loops anchoring RNAPs to DNA, rendering the complexes more stable. A recent study observed replication fork blockage in yeast after collisions with R-loops on the leading-strand template, but only when the R-loop interacting protein Yra1 was overexpressed (90). Transcription of the R-loop forming region of mAIRN generated significantly longer R-loops (up to 600 nt) than those used in our study (47,91). It is certainly possible that long, complex R-loops are potent barriers to DNA replication even in the absence of any RNAP complexes. Increased stalling with the R-loop array on the lagging-strand template observed herein, compared to the single 100mer R-loop (Figures 5Ai and B), suggests that this may be the case.
In eukaryotes, the replicative helicase Mcm2–7 translocates along the leading-strand template and will be first to encounter R-loops. Mcm2–7 was capable of unwinding RNA-DNA hybrids in vitro (87), and leading-strand template R-loops were not potent replication obstacles in vivo (47,90). Thus, Mcm2–7 might remove RNA on the leading-strand template (47). Because eukaryotic replication and transcription proceed at similar speeds, CO encounters are unlikely to occur unless the RNAP is stalled, e.g., at DNA damage. In contrast, R-loops on the lagging-strand template would not be encountered and removed by Mcm2–7.
ATR activation was observed in a plasmid-based reporter assay in response to replisome collisions with HO, but not CO R-loops (47). Resolution of replication defects arising because of increased levels of R-loops in human cell lines also depended on ATR (60), suggesting that the eukaryotic replisome lacks an inherent mechanism to displace lagging-strand template R-loops. ATR mediated the resolution of replication-R-loop conflicts by stimulating cleavage of replication forks (59,60). The requirement for a cleaved fork intermediate might explain why HO R-loops are greater obstacles to replication in vivo (47,90). It was proposed that fork cleavage allows for transcript elongation of the R-loop-associated RNAP in the absence of torsional stress, so that the replication fork could be restored once the RNAP has passed (59). However, inactive RNAP complexes or binding of associated proteins to R-loops (92,93) that further stabilize R-loops could also exacerbate the need for fork processing, and consequently increase genome instability.
DATA AVAILABILITY
All data is held by the authors and is available on request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank John Petrini for a critical reading of the manuscript. Seth Darst (Rockefeller University) for the RpoZ expression plasmid, and Tim Lohman (Washington University in St. Louis) for purified UvrD.
Contributor Information
Jan-Gert Brüning, Molecular Biology Program, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10065, USA.
Kenneth J Marians, Molecular Biology Program, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10065, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [R35 GM126907 to K.J.M.]; Cancer Center Support Grant [NCI P30CA008748 to M.S.K.C.C.]. Funding for open access charge: NIH [R35 GM126907].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mirkin E.V., Mirkin S.M.. Mechanisms of transcription-replication collisions in bacteria. Mol. Cell. Biol. 2005; 25:888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vilette D., Ehrlich S.D., Michel B.. Transcription-induced deletions in Escherichia coli plasmids. Mol. Microbiol. 1995; 17:493–504. [DOI] [PubMed] [Google Scholar]
- 3. Deshpande A.M., Newlon C.S.. DNA replication fork pause sites dependent on transcription. Science. 1996; 272:1030–1033. [DOI] [PubMed] [Google Scholar]
- 4. Prado F., Aguilera A.. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005; 24:1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. French S. Consequences of replication fork movement through transcription units in vivo. Science. 1992; 258:1362–1365. [DOI] [PubMed] [Google Scholar]
- 6. Srivatsan A., Tehranchi A., MacAlpine D.M., Wang J.D.. Co-orientation of replication and transcription preserves genome integrity. PLoS Genet. 2010; 6:e1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang J.D., Berkmen M.B., Grossman A.D.. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:5608–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Petryk N., Kahli M., d’Aubenton-Carafa Y., Jaszczyszyn Y., Shen Y., Silvain M., Thermes C., Chen C.L., Hyrien O.. Replication landscape of the human genome. Nat. Commun. 2016; 7:10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brewer B.J., Fangman W.L.. A replication fork barrier at the 3' end of yeast ribosomal RNA genes. Cell. 1988; 55:637–643. [DOI] [PubMed] [Google Scholar]
- 10. Merrikh H., Machon C., Grainger W.H., Grossman A.D., Soultanas P.. Co-directional replication-transcription conflicts lead to replication restart. Nature. 2011; 470:554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pomerantz R.T., O’Donnell M.. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 2008; 456:762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu B., Wong M.L., Alberts B.. A transcribing RNA polymerase molecule survives DNA replication without aborting its growing RNA chain. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:10660–10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu B., Wong M.L., Tinker R.L., Geiduschek E.P., Alberts B.M.. The DNA replication fork can pass RNA polymerase without displacing the nascent transcript. Nature. 1993; 366:33–39. [DOI] [PubMed] [Google Scholar]
- 14. Condon C., French S., Squires C., Squires C.L.. Depletion of functional ribosomal RNA operons in Escherichia coli causes increased expression of the remaining intact copies. EMBO J. 1993; 12:4305–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trautinger B.W., Jaktaji R.P., Rusakova E., Lloyd R.G.. RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Mol. Cell. 2005; 19:247–258. [DOI] [PubMed] [Google Scholar]
- 16. Selby C.P., Sancar A.. Transcription preferentially inhibits nucleotide excision repair of the template DNA strand in vitro. J. Biol. Chem. 1990; 265:21330–21336. [PubMed] [Google Scholar]
- 17. Selby C.P., Drapkin R., Reinberg D., Sancar A.. RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res. 1997; 25:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tornaletti S., Maeda L.S., Hanawalt P.C.. Transcription arrest at an abasic site in the transcribed strand of template DNA. Chem. Res. Toxicol. 2006; 19:1215–1220. [DOI] [PubMed] [Google Scholar]
- 19. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herbert K.M., La Porta A., Wong B.J., Mooney R.A., Neuman K.C., Landick R., Block S.M.. Sequence-resolved detection of pausing by single RNA polymerase molecules. Cell. 2006; 125:1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Artsimovitch I., Landick R.. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dutta D., Shatalin K., Epshtein V., Gottesman M.E., Nudler E.. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011; 146:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guy C.P., Atkinson J., Gupta M.K., Mahdi A.A., Gwynn E.J., Rudolph C.J., Moon P.B., van Knippenberg I.C., Cadman C.J., Dillingham M.S. et al.. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol. Cell. 2009; 36:654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hawkins M., Dimude J.U., Howard J.A.L., Smith A.J., Dillingham M.S., Savery N.J., Rudolph C.J., McGlynn P.. Direct removal of RNA polymerase barriers to replication by accessory replicative helicases. Nucleic Acids Res. 2019; 47:5100–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boubakri H., de Septenville A.L., Viguera E., Michel B.. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010; 29:145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ivessa A.S., Lenzmeier B.A., Bessler J.B., Goudsouzian L.K., Schnakenberg S.L., Zakian V.A.. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell. 2003; 12:1525–1536. [DOI] [PubMed] [Google Scholar]
- 27. Merrikh C.N., Brewer B.J., Merrikh H.. The B. subtilis accessory helicase PcrA facilitates DNA replication through transcription units. PLos Genet. 2015; 11:e1005289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sabouri N., McDonald K.R., Webb C.J., Cristea I.M., Zakian V.A.. DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 2012; 26:581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park J.-S., Marr M.T., Roberts J.W.. E. coli Transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002; 109:757–767. [DOI] [PubMed] [Google Scholar]
- 30. Tehranchi A.K., Blankschien M.D., Zhang Y., Halliday J.A., Srivatsan A., Peng J., Herman C., Wang J.D.. The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell. 2010; 141:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sivaramakrishnan P., Sepulveda L.A., Halliday J.A., Liu J., Nunez M.A.B., Golding I., Rosenberg S.M., Herman C.. The transcription fidelity factor GreA impedes DNA break repair. Nature. 2017; 550:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Washburn R.S., Gottesman M.E.. Transcription termination maintains chromosome integrity. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:792–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu H.Y., Shyy S.H., Wang J.C., Liu L.F.. Transcription generates positively and negatively supercoiled domains in the template. Cell. 1988; 53:433–440. [DOI] [PubMed] [Google Scholar]
- 34. Thomas M., White R.L., Davis R.W.. Hybridization of RNA to double-stranded DNA: formation of R-loops. Proc. Natl. Acad. Sci. U.S.A. 1976; 73:2294–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wahba L., Amon J.D., Koshland D., Vuica-Ross M.. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell. 2011; 44:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanz L.A., Chedin F.. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2019; 14:1734–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leela J.K., Syeda A.H., Anupama K., Gowrishankar J.. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Costantino L., Koshland D.. Genome-wide map of R-loop-induced damage reveals how a subset of R-loops contributes to genomic instability. Mol. Cell. 2018; 71:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garcia-Muse T., Aguilera A.. R loops: from physiological to pathological roles. Cell. 2019; 179:604–618. [DOI] [PubMed] [Google Scholar]
- 40. Crossley M.P., Bocek M., Cimprich K.A.. R-loops as cellular regulators and genomic threats. Mol. Cell. 2019; 73:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Castellano-Pozo M., Garcia-Muse T., Aguilera A.. R-loops cause replication impairment and genome instability during meiosis. EMBO Rep. 2012; 13:923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gan W., Guan Z., Liu J., Gui T., Shen K., Manley J.L., Li X.. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011; 25:2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gomez-Gonzalez B., Garcia-Rubio M., Bermejo R., Gaillard H., Shirahige K., Marin A., Foiani M., Aguilera A.. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J. 2011; 30:3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wimberly H., Shee C., Thornton P.C., Sivaramakrishnan P., Rosenberg S.M., Hastings P.J.. R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat. Commun. 2013; 4:2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wellinger R.E., Prado F., Aguilera A.. Replication fork progression is impaired by transcription in hyperrecombinant yeast cells lacking a functional THO complex. Mol. Cell. Biol. 2006; 26:3327–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lang K.S., Hall A.N., Merrikh C.N., Ragheb M., Tabakh H., Pollock A.J., Woodward J.J., Dreifus J.E., Merrikh H.. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell. 2017; 170:787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hamperl S., Bocek M.J., Saldivar J.C., Swigut T., Cimprich K.A.. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 2017; 170:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dudas K.C., Kreuzer K.N.. UvsW protein regulates bacteriophage T4 origin-dependent replication by unwinding R-loops. Mol. Cell. Biol. 2001; 21:2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Drolet M., Phoenix P., Menzel R., Masse E., Liu L.F., Crouch R.J.. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:3526–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tuduri S., Crabbe L., Conti C., Tourriere H., Holtgreve-Grez H., Jauch A., Pantesco V., De Vos J., Thomas A., Theillet C. et al.. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009; 11:1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Masse E., Drolet M.. Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J. Biol. Chem. 1999; 274:16659–16664. [DOI] [PubMed] [Google Scholar]
- 52. Li X., Manley J.L.. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005; 122:365–378. [DOI] [PubMed] [Google Scholar]
- 53. Huertas P., Aguilera A.. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell. 2003; 12:711–721. [DOI] [PubMed] [Google Scholar]
- 54. Roy D., Zhang Z., Lu Z., Hsieh C.L., Lieber M.R.. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol. Cell. Biol. 2010; 30:146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Britton S., Dernoncourt E., Delteil C., Froment C., Schiltz O., Salles B., Frit P., Calsou P.. DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 2014; 42:9047–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li L., Germain D.R., Poon H.Y., Hildebrandt M.R., Monckton E.A., McDonald D., Hendzel M.J., Godbout R.. DEAD box 1 facilitates removal of RNA and homologous recombination at DNA Double-Strand breaks. Mol. Cell. Biol. 2016; 36:2794–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ohle C., Tesorero R., Schermann G., Dobrev N., Sinning I., Fischer T.. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell. 2016; 167:1001–1013. [DOI] [PubMed] [Google Scholar]
- 58. Sollier J., Stork C.T., Garcia-Rubio M.L., Paulsen R.D., Aguilera A., Cimprich K.A.. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell. 2014; 56:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chappidi N., Nascakova Z., Boleslavska B., Zellweger R., Isik E., Andrs M., Menon S., Dobrovolna J., Balbo Pogliano C., Matos J. et al.. Fork cleavage-religation cycle and active transcription mediate replication restart after fork stalling at Co-transcriptional R-Loops. Mol. Cell. 2020; 77:528–541. [DOI] [PubMed] [Google Scholar]
- 60. Matos D.A., Zhang J.M., Ouyang J., Nguyen H.D., Genois M.M., Zou L.. ATR protects the genome against R loops through a MUS81-triggered feedback loop. Mol. Cell. 2020; 77:514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen L., Chen J.Y., Zhang X., Gu Y., Xiao R., Shao C., Tang P., Qian H., Luo D., Li H. et al.. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Mol. Cell. 2017; 68:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sanz L.A., Hartono S.R., Lim Y.W., Steyaert S., Rajpurkar A., Ginno P.A., Xu X., Chedin F.. Prevalent, dynamic, and conserved R-Loop structures associate with specific epigenomic signatures in mammals. Mol. Cell. 2016; 63:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hamperl S., Cimprich K.A.. Conflict resolution in the genome: how transcription and replication make it work. Cell. 2016; 167:1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garcia-Pichardo D., Canas J.C., Garcia-Rubio M.L., Gomez-Gonzalez B., Rondon A.G., Aguilera A.. Histone mutants separate R loop formation from genome instability induction. Mol. Cell. 2017; 66:597–609. [DOI] [PubMed] [Google Scholar]
- 65. Zatreanu D., Han Z., Mitter R., Tumini E., Williams H., Gregersen L., Dirac-Svejstrup A.B., Roma S., Stewart A., Aguilera A. et al.. Elongation factor TFIIS prevents transcription stress and R-Loop accumulation to maintain genome stability. Mol. Cell. 2019; 76:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pomerantz R.T., O’Donnell M.. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010; 327:590–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu B., Alberts B.M.. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science. 1995; 267:1131–1137. [DOI] [PubMed] [Google Scholar]
- 68. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jiang Y., Chen B., Duan C., Sun B., Yang J., Yang S.. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015; 81:2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yeeles J.T., Marians K.J.. Dynamics of leading-strand lesion skipping by the replisome. Mol. Cell. 2013; 52:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yeeles J.T.P., Marians K.J.. The Escherichia coli replisome is inherently DNA damage tolerant. Science. 2011; 334:235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Twist K.A., Husnain S.I., Franke J.D., Jain D., Campbell E.A., Nickels B.E., Thomas M.S., Darst S.A., Westblade L.F.. A novel method for the production of in vivo-assembled, recombinant Escherichia coli RNA polymerase lacking the alpha C-terminal domain. Protein Sci. 2011; 20:986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bae B., Davis E., Brown D., Campbell E.A., Wigneshweraraj S., Darst S.A.. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of sigma70 domain 1.1. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:19772–19777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Parada C.A., Marians K.J.. Mechanism of DNA A protein-dependent pBR322 DNA replication. DNA A protein-mediated trans-strand loading of the DNA B protein at the origin of pBR322 DNA. J. Biol. Chem. 1991; 266:18895–18906. [PubMed] [Google Scholar]
- 75. Marians K.J. Phi X174-type primosomal proteins: purification and assay. Methods Enzymol. 1995; 262:507–521. [DOI] [PubMed] [Google Scholar]
- 76. Johanson K.O., Haynes T.E., McHenry C.S.. Chemical characterization and purification of the beta subunit of the DNA polymerase III holoenzyme from an overproducing strain. J. Biol. Chem. 1986; 261:11460–11465. [PubMed] [Google Scholar]
- 77. Marceau A.H., Bahng S., Massoni S.C., George N.P., Sandler S.J., Marians K.J., Keck J.L.. Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J. 2011; 30:4236–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lohman T.M., Green J.M., Beyer R.S.. Large-scale overproduction and rapid purification of the Escherichia coli ssb gene product. Expression of the ssb gene under lambda PL control. Biochemistry. 1986; 25:21–25. [DOI] [PubMed] [Google Scholar]
- 79. Hill T.M., Marians K.J.. Escherichia coli Tus protein acts to arrest the progression of DNA replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:2481–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maxwell A., Howells A.J.. Overexpression and purification of bacterial DNA gyrase. Methods Mol. Biol. 1999; 94:135–144. [DOI] [PubMed] [Google Scholar]
- 81. Kreuzer K.N., Alberts B.M.. Site-specific recognition of bacteriophage T4 DNA by T4 type II DNA topoisomerase and Escherichia coli DNA gyrase. J. Biol. Chem. 1984; 259:5339–5346. [PubMed] [Google Scholar]
- 82. Gwynn E.J., Smith A.J., Guy C.P., Savery N.J., McGlynn P., Dillingham M.S.. The conserved C-terminus of the PcrA/UvrD helicase interacts directly with RNA polymerase. PLoS One. 2013; 8:e78141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 1997; 61:212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Maduike N.Z., Tehranchi A.K., Wang J.D., Kreuzer K.N.. Replication of the Escherichia coli chromosome in RNase HI-deficient cells: multiple initiation regions and fork dynamics. Mol. Microbiol. 2014; 91:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Matson S.W. Escherichia coli DNA helicase II (uvrD gene product) catalyzes the unwinding of DNA.RNA hybrids in vitro. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:4430–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yuan Q., McHenry C.S.. Strand displacement by DNA polymerase III occurs through a tau-psi-chi link to single-stranded DNA-binding protein coating the lagging strand template. J. Biol. Chem. 2009; 284:31672–31679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shin J.H., Kelman Z.. The replicative helicases of bacteria, archaea, and eukarya can unwind RNA-DNA hybrid substrates. J. Biol. Chem. 2006; 281:26914–26921. [DOI] [PubMed] [Google Scholar]
- 88. Santamaria D., de la Cueva G., Martinez-Robles M.L., Krimer D.B., Hernandez P., Schvartzman J.B.. DnaB helicase is unable to dissociate RNA-DNA hybrids. Its implication in the polar pausing of replication forks at ColE1 origins. J. Biol. Chem. 1998; 273:33386–33396. [DOI] [PubMed] [Google Scholar]
- 89. Kaplan D.L., O’Donnell M.. Twin DNA pumps of a hexameric helicase provide power to simultaneously melt two duplexes. Mol. Cell. 2004; 15:453–465. [DOI] [PubMed] [Google Scholar]
- 90. Garcia-Rubio M., Aguilera P., Lafuente-Barquero J., Ruiz J.F., Simon M.N., Geli V., Rondon A.G., Aguilera A.. Yra1-bound RNA-DNA hybrids cause orientation-independent transcription-replication collisions and telomere instability. Genes Dev. 2018; 32:965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ginno P.A., Lott P.L., Christensen H.C., Korf I., Chedin F.. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012; 45:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wang I.X., Grunseich C., Fox J., Burdick J., Zhu Z., Ravazian N., Hafner M., Cheung V.G.. Human proteins that interact with RNA/DNA hybrids. Genome Res. 2018; 28:1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cristini A., Groh M., Kristiansen M.S., Gromak N.. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-Loop-Associated DNA damage. Cell Rep. 2018; 23:1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is held by the authors and is available on request.