


Abstract
The RNA helicase RIG-I plays a key role in sensing pathogen-derived RNA. Double-stranded RNA structures bearing 5′-tri- or diphosphates are commonly referred to as activating RIG-I ligands. However, endogenous RNA fragments generated during viral infection via RNase L also activate RIG-I. Of note, RNase-digested RNA fragments bear a 5′-hydroxyl group and a 2′,3′-cyclic phosphate. How endogenous RNA fragments activate RIG-I despite the lack of 5′-phosphorylation has not been elucidated. Here we describe an endogenous RIG-I ligand (eRL) that is derived from the internal transcribed spacer 2 region (ITS2) of the 45S ribosomal RNA after partial RNase A digestion in vitro, RNase A protein transfection or RNase L activation. The immunostimulatory property of the eRL is dependent on 2′,3′-cyclic phosphate and its sequence is characterized by a G-quadruplex containing sequence motif mediating guanosine-5′-triphosphate (GTP) binding. In summary, RNase generated self-RNA fragments with 2′,3′-cyclic phosphate function as nucleotide-5′-triphosphate binding aptamers activating RIG-I.
INTRODUCTION
The recognition of pathogen-derived RNA by the innate immune system is mediated by pattern recognition receptors (PRRs) like endosomal/lysosomal Toll-like receptors (1) or by cytoplasmic RNA helicases (also called RIG-like receptors, RLR) such as RIG-I and MDA5 (2,3). PRR activation leads to cytokine induction and the establishment of an antiviral state via type I interferon (IFN). The different RLRs show similarities such as the association with AU-rich viral mRNAs but also differ in their ligand specificity (4). MDA5 recognizes long double-stranded RNA and single-stranded positive-sense RNA viruses such as picornaviruses (3,5). In particular, it was shown that the encephalomyocarditis virus leader protein encoding-region of antisense RNA is recognized by MDA5 when bound to LGP2 (6). In the case of the measles virus, MDA5 is activated by the nucleoprotein-coding region (7). In contrast, RIG-I senses double-stranded (ds) RNAs or panhandle structures bearing 5′-tri- or diphosphates (3,8–11). For example, copy-back defective interfering genomes of influenza virus and measles virus as well as 3′ untranslated viral genomic regions were shown to be RIG-I activators (7,12). In addition to triggering PRR-mediated cytokine release, dsRNA also binds and activates interferon-stimulated oligoadenylate-synthetases (OAS1–3), which subsequently synthesize the second messenger 2′-5′-linked oligoadenylate (2–5A). 2–5A activates RNase L (13,14), which degrades viral genomes and cellular RNA to constrain viral gene expression. Strikingly, RNase L-generated cellular RNA fragments also activate RIG-I amplifying the dsRNA mediated danger signal in a positive feedback loop (15). In addition, cellular stress and the unfolded protein response (UPR) initiated by the kinase/endoribonuclease IRE1 also generates self-RNA fragments that stimulate RIG-I (16). Both RNase L and IRE1 as well as RNase A belong to the family of metal-ion-independent RNases which cleave single-stranded RNA generating fragments with 5′-hydroxyl and 2′,3′-cyclic phosphate termini (17–19). The question why these RNA degradation products activate RIG-I although their phosphorylation pattern does not correspond to the classical 5′-triphosphorylated RIG-I stimulating RNA has not yet been elucidated. We hypothesized that RNase digestion of endogenous RNA may release RNA fragments that function as aptamer and bind triphosphorylated nucleotides leading to RIG-I activation. Interestingly, RNA sequences with G-quadruplex have been identified as GTP-binding RNAs that could fulfill this function (20). Indeed, we identified a G-quadruplex containing 72 nt sequence released after single-strand specific RNase digestion that is derived from the internal transcribed spacer 2 region (ITS2) of the 45S ribosomal RNA. In general, the ITS2 region encompasses 1167 bp in humans and gets excised and degraded upon 45S rRNA processing (21). Since 45S rRNA transcription and content (and therefore ITS2 amount) is enhanced in tumor cells compared to non-transformed cells (22), digested self-RNA from strongly proliferating cells shows enhanced immunostimulatory potential.
In summary, RNase generated self-RNA fragments from proliferating cells function as nucleotide 5′-triphosphate binding aptamers with a new way of RNA-mediated RIG-I activation.
MATERIALS AND METHODS
Ethics statement
The local Ethics committees of Justus-Liebig-University Giessen and Philipps-University Marburg approved the use of human blood samples (buffy coats) for this study. Informed consent was obtained from all blood donors and the experiments performed conformed to the principles of the WMA Declaration of Helsinki. For experiments with murine immune cells, male and female C57BL/6 WT or IPS-1-deficient mice (23) at the age of 8 to 16 weeks were sacrificed and tissue/organs removed. These experiments were performed in accordance with the National German welfare law §4 (3) TierSchG and §2 and Annex 2 (TierSchVerV) of the National Order for the use of animals in research and do not need the approval by a local ethics committee. According to the regulations, the number of mice used was reported to the animal welfare officer of the Philipps-University Marburg.
Reagents
Transfection reagent DOTAP was obtained from Roche (Basel, Switzerland) and Lipofectamine 2000 was obtained from ThermoFisher (Waltham, MA, USA). RNA40 (5′-GCCCGUCUGUUGUGUGACUC-3′) was synthesized by IBA, Göttingen. Poly(I:C) and 5′ppp-dsRNA were obtained from InvivoGen (Toulouse, France).
Cells
HEK293, HEK293 ΔRIG-I, Huh7.5 and A549 cells were cultivated in DMEM (Pan-Biotech, Aidenbach, Germany) supplied with 10% FCS, 2 mM l-glutamine, 100 units/ml penicillin and 10 μg/ml streptomycin. HeLa, MDCK and Vero cells were grown in RPMI (Pan-Biotech, Aidenbach, Germany) with 5% FCS and 2 mM l-glutamine, 100 units/ml penicillin and 10 μg/ml streptomycin. The medium for HEK293-RIG-I-IFN-ß reporter cells (HEK-RIG-I) and MDA5 or RIG-I transfected Huh7.5 cells (24) contained in addition 0.7 mg/ml Geneticin and 10 μg/ml blasticidin as selection markers, respectively. All cell lines were tested with a TLR2 luciferase reporter assay for mycoplasma contamination and tested negative. For cell stimulation, cell numbers were seeded in 96-well plates as follows: A549 cells 2.8 × 104 cells/well, HEK293-RIG-I-IFN-ß reporter cells and human monocytes 105 cells/well and WT or IPS-1- (MAVS, CARDIF) deficient murine macrophages 2 × 105 cells/well. In 12-well plates, Huh7.5 cells were seeded at 4 × 105 cells/well and murine macrophages at 2.4 × 106 cells/well. PBMCs were purified from buffy coats using Ficoll gradient centrifugation and seeded at 3 × 105 cells/well for cytokine induction in RPMI supplemented with 2% serum of AB positive donors, 2 mM l-Glutamine, 100 units/ml penicillin and 10 μg/ml streptomycin, amino acids and sodium pyruvate. To induce proliferation, PBMCs were additionally supplemented with 10% FCS and 1 μg/ml phytohemagglutinin (PHA, InvivoGen, Toulouse, France) and grown for 4–5 days. Monocytes were purified from PBMCs using elutriation and grown in PBMC medium. Bone marrow-derived M-CSF induced murine macrophages were grown as described (25).
Cell stimulation
For cytokine induction or luciferase assay, primary cells, A549 and HEK-RIG-I cells were incubated for 16–20 h with various stimuli as indicated. For RNA extraction of stimulated primary murine macrophages and Huh7.5 cells were incubated for 6 h with respective stimuli. Nucleic acids used for stimulation were 10 μM CpG-ODN 2216, 0.6 μg/ml RNA40 + DOTAP, 1 μg/ml poly(I:C), 2 μg/ml 5′ppp-dsRNA (InvivoGen, Toulouse, France) and different digested and undigested RNA samples at concentrations indicated in the corresponding figure legends. RNA was complexed with Lipofectamine 2000 Reagent (ThermoFisher, Waltham, Massachusetts) or DOTAP (Roche, Basel, Switzerland) according to the manufacturer's instructions. Per 96-well, RNA in 25 μl Opti-MEM was added to 2.5 μl Lipofectamine or 1.5 μl DOTAP diluted in 25 μl Opti-MEM (ThermoFisher, Waltham, MA, USA). After 5 min incubation at room temperature, 50 μl growth medium was added. The solution was mixed and 100 μl were used for stimulation of cells in a 96-well seeded in 100 μl growth medium. In some experiments, the cells were incubated prior to RNA stimulation with 10 μM mycophenolic acid (MA, Sigma-Aldrich, St. Louis, MO, USA) for 24 h. Huh7.5 cells expressing RLRs were seeded in 12-well plates at 70% confluency and stimulated 6 h after stimulation, RNA was extracted using Trizol and subjected to qRT-PCR.
Cytokine measurement
Concentrations of murine IFN-α and human RANTES were determined by ELISA according to the manufacturer's instructions (PBL Interferon Source, Piscataway, New Jersey for murine IFN-α, R&D Biosystems, Wiesbaden, Germany, for human RANTES). Concentrations of human IFN-α were determined using mouse monoclonal anti-human IFN-α (PBL Interferon Source, Piscataway, NJ, USA) as capture antibody and anti-human IFN-α HRP-Conjugate (eBioscience, San Diego, USA) as detection antibody. The IFN-α standard used was recombinant human IFN-α from PeproTech (Rocky Hill, NJ, USA).
Limited protease digestion assay
To monitor RIG-I activation, A549 cells were transfected with 200 ng/ml of tRNA (negative control), 5'-triphosphorylated double-stranded RNA (positive control; InvivoGen, Toulouse, France) or the endogenous RNase fragment (eRL) using Endofectin (Biocat, Heidelberg, Germany). After 2 h, cells were subjected to RIG-I activation assay as previously described (26). Briefly, one part of the lysate was left untreated (input control) and the other part was subjected to limited protease digestion. Therefore, the sample was digested for 10 min with 0.2 μg/μl TPCK-treated trypsin (Sigma-Aldrich, St. Louis, MO, USA) at 37°C. The digestion was stopped by adding 4-fold sample buffer (200 mM Tris–HCl pH 6.8, 8% SDS, 40% glycerol, 25% β-mercaptoethanol, 0.4% Bromphenol Blue) and boiling for 5 min at 100°C. Samples were subjected to 15% SDS-PAGE and western blot analysis using mouse monoclonal anti-RIG-I antibody (clone ALME-1, Adipogen, San Diego, California). Band intensity was quantified using Image Lab 5.2.1 (BioRad, Hercules, California) and activated RIG-I represents the ratio of trypsin-resistant RIG-I to undigested RIG-I. Means of three independent experiments are shown. P values were determined using a paired two-tailed Student's t-test.
Luciferase assay
HEK293-RIG-I-IFN-ß reporter cells (HEK-RIG-I cells) were seeded in 96-well plates at 5 × 104 cells/well and stimulated in triplicates. 16–20 h post stimulation, cells were lysed in 1× Lysis-Juice 2 buffer (PJK, Kleinblittersdorf, Germany) and firefly luciferase activity was measured using Berthold Detection Systems (Pforzheim, Germany).
Next generation sequencing (NGS)
Construction of small RNA Transcription library, Ion Torrent sequencing and initial bioinformatical analysis were performed by CeGaT (Tübingen, Germany). Sequences were displayed in sense direction. Sequences longer than 29 bp were mapped to human 45S rRNA (NCBI Reference Sequence: NR_046235.1) accepting maximal three mismatches. To detect RNA fragments shorter than 29 bp, unique Ion Torrent sequences were searched for identical hits to the expected sequence using USEARCH version 8.1.1861 (27) with the comprehensive local search option ‘search_local’. In order to find all identical hits of at least 13 bp length, the settings -hspw 5 -mincols 13 -id 1 -maxaccepts 0 -maxrejects 0 -strand both -blast6out were used. The sequences producing local hits were further examined to identify those identical to parts of the human 45S rRNA. In order to exclude contamination from constituents of the cell culture medium extracted from other organisms, hit sequences were further tested for their occurrence in sense direction in Bos taurus, Sus scrofa, Penicillium chrysogenum and Streptomyces griseus.
NGS sequencing of RNase A-digested and gel purified RNA fragments from an in vitro transcribed ITS2 fragment (45S rRNA, nt 7143-7500) was performed by Vertis Biotechnologie AG (Weihenstephan, Germany). In brief, the RNA was first treated with T4 Polynucleotide Kinase (PNK). Then, oligonucleotide adapters were ligated to the 5′- and 3′-ends of the RNA sample. First-strand cDNA synthesis was performed using M-MLV reverse transcriptase and the 3′-adapter as primer. The resulting cDNA was amplified with PCR using a high fidelity DNA polymerase (12 cycles). The cDNA was purified using the Agencourt AMPure XP Kit (Beckman Coulter Genomics, Brea, CA, USA) and was analysed by capillary electrophoresis. The cDNA pool was single-end sequenced with a NextSeq 500 system using 150 bp read-length. Sequences were analysed with the Galaxy analysis interface using standard settings (https://usegalaxy.org/).
RNA isolation and qRT-PCR
Total RNA was isolated using Trizol according to the manufacturer's protocol (Invitrogen, Carlsbad, CA, USA) or by the method of Chomczynski and Sacchi (28). Contaminating DNA were eliminated using DNase I (Roche, Basel, Switzerland). Human ISG56 was determined using QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) and corresponding QuantiTect Primer Assays (hISG56/IFIT1 QT00201012). cDNA was amplified with Brilliant III Ultra-Fast SYBR Green qPCR Master Mix (Agilent Technologies, Ratingen, Germany). All gene expression data are presented as relative expression to murine tubulin (murine cells) or human GAPDH (human cells). The sequences were as follows: GAPDH-sense 5′-AAGGTGAAGGTCGGAGTCAACG-3′, GAPDH-antisense 5′-GGCAGAGATGATGACCCTTTTGGC-3′, murine tubulin sense 5′-CACATCCAGGCCGGACA-3′, tubulin-antisense 5′-TGTTCATCGCTTATGACCTCCC-3′.
Quantification of 45S rRNA was performed with primers located in the 5.8S and ITS2 region of rRNA. The sequences were: 5.8S rRNA sense 5′-CCGGGGCTACGCCTGTCTGA-3′ and ITS2 antisense 5′-GCCGCCGGGTCTGCGCTTAG-3′.
Following immunoprecipitation, RNA samples (input and IP) were processed using the RNeasy Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. The isolated RNA was used for cDNA synthesis using PrimeScript RT Master Mix (Takara Bio, Kusatsu, Japan) followed by RT-PCR using SYBR Premix Ex Taq (Tli RNase H Plus) (Takara, Kusatsu, Japan) with detection of 18S ribosomal RNA (Cat. No. QT00199367, Qiagen, Hilden, Germany), VSV RNA (fwd primer 5′ GATAGTACCGGAGGATTGACGACTA 3′ and rev primer 5′ TCAAACCATCCGAGCCATTC 3′) (29) and ITS2 with primers ITS2-short 5′ GAAGGTCCCGTGCCCGTCG-3′ and ITS2-rev 5′-GCGCTCCCCCGACCCTCTC-3′. PCR conditions for the latter PCR were: initial denaturation at 95°C for 2 min, 30 s and hot start followed by 30 cycles including denaturation at 95°C for 30 s, annealing at 65°C for 30 s and elongation at 72°C for 15 s. Results of immunoprecipitation are displayed as direct CT-value comparison between ΔMx and RIG-I.
For the detection of the ratio of cut ITS2 to uncut ITS2 fragment a three-primer-PCR using the primers ITS2-long 5′-GGGAGGAGGAAGGCGGGTC, ITS2-short 5′ GAAGGTCCCGTGCCCGTCG-3′ and ITS2-rev 5′-GCGCTCCCCCGACCCTCTC-3′ was performed. PCR conditions were: initial denaturation at 95°C for 2 min, 30 s and hot start followed by 21 cycles including denaturation at 95°C for 30 s, annealing at 65°C for 30 s and elongation at 72°C for 15 s. Fragments were separated on a 12% PAGE and visualized with SYBR™ Gold Nucleic Acid Gel Stain. Band intensity was quantified using GelQuantNET (omicX, Le Petit-Quevilly, France). The measured values were adjusted by subtracting the background measured in an area without stained bands.
Immunoprecipitation
HEK293 ΔRIG-I (7,5 × 105 cells/well) were transfected with 500 ng of either pI.18 3xFLAG-ΔMx or pI.18 3xFlag RIG-I, using TransIT (Mirus Bio, Madison, Wisconsin) according to manufacturer's instruction. After 24 h, the cells were stimulated by transfection with either VSV RNA (8) or eRL using EndoFectin (GeneCopeia, Rockville, MD, USA) according to manufacturer's instruction, or left untreated. After 6 h of RNA stimulation, the cells were scraped off in PBS, and 25% of the cells were transferred to a separate tube as RNA input. All cells were then pelleted by centrifugation (800 x g, 5 min). The pellet for immunoprecipitation (IP) was lysed in 500 μl/IP RIPA buffer (50 mM Tris–HCl (pH 7,5), 150 mM NaCl, 1% NP-40, 1× protease inhibitor cocktail (cOmplete) (Roche, Basel, Switzerland) and cell debris were removed by centrifugation (10 000 × g, 10 min, 4°C). As protein input, 5% of the lysates were saved in a separate tube. The remaining lysates (480 μl) were added to Dynabeads Protein G (Invitrogen, Carlsbad, CA, USA) coupled with FLAG M2 antibody (clone F3165, Sigma-Aldrich, St. Louis, MO, USA). To prevent unspecific binding of RNA to the beads, tRNA (Merck Millipore, Burlington, MA, USA) was added to the lysates with a final concentration of 1 mg/ml. The IP was performed at 4°C for 16 h with head-over-tail rotation followed by three washes with TBS-T. After the last wash, the bead-suspension was divided in half for protein and RNA elusion, respectively. The protein samples (input and IP) were analysed by SDS-PAGE and western blot with primary antibodies against Flag (Sigma-Aldrich, St. Louis, Missouri) and β-tubulin (Abcam, Cambridge, UK) using Mouse TrueBlot ULTRA (clone eB144, BioMol, Hamburg, Germany) and Rabbit TrueBlot (clone eB182, BioMol, Hamburg, Germany) as secondary antibodies, respectively. RNA was reversed transcribed using QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) with RT Primer Mix according to the manufacturers' protocol.
RNA manipulation
For digestion of total RNA, RNase A (Qiagen, Hilden, Germany), RNase T1 and RNase T2 (both from Sigma-Aldrich, St. Louis, MO, USA) were used at 2.5 × 10−5 , 3.3 or 0.85 U/μg RNA, respectively. For ultrasonic treatment, an UW 2070 (Bandelin, Berlin, Germany) was utilized and RNA was treated for 60 s at power setting of 75%. Digestion of double-stranded RNA was achieved by incubation with RNase III (Epicentre, Madison, WI, USA) at 2.5 × 10−2 U/μg RNA. For hydrolyzation of 2′,3′-cyclic phosphate, RNA was incubated with 10 mM HCl for 30 min at 37°C and subsequently neutralized with 10 mM NaOH. eRL was treated with FastSAP (Shrimp Alkaline phosphatase) (ThermoFisher, Waltham, MA, USA) and PNK according to the manufactures protocol in buffer A supplied with PNK.
3′-phosphorylated RNA40 (5′-GCCCGUCUGUUGUGUGACUC-3′) was obtained from Microsynth AG (Balgach, Switzerland). Recombinant rtcA was prepared as GST fusion protein and the RNA40 with a 2′-3′-cyclic phosphate was generated in T4 RNA ligase buffer (ThermoFisher, Waltham, MA, USA) with incubation at 16°C for at least 16 h.
In vitro RNA transcription
For generation of in vitro transcribed (IVT) RNAs the AmpliScrib High Yield Transcription Kits with T7 RNA polymerase (Epicentre, Madison, USA) was used according to the manufacturer's protocol. In a typical 40 μL reaction, 1 μg template was incubated in the presence of 7.5 mM each NTP, 5 mM DTT, and 100 units T7 RNA polymerase in 1x transcription buffer at 37°C for 2 to 4 h. Various synthetic gene fragments with flanking EcoRI sites and a 5′ T7 promotor were synthesized by GeneART (Thermo Fisher Scientific, Regensburg, Germany) and served as DNA template after EcoRI digestion and fragment purification. The sequences or nucleotide references are as follows: IVT_ITS2 (T7 promotor and DNA sequence position 7143–7500 of 45S rRNA, NR_046235.1) and IVT_ITS2_ΔG (G-quadruplex replaced within region 7143–7500) are depicted in Supplementary Figure S4. In general, in vitro transcribed RNA was dephosphorylated two times with excess of calf intestine phosphatase (CIP) according to the manufacturer's protocol. IVT RNA was digested with RNase A (Qiagen, Hilden, Germany at 2.5 × 10–5 U/μg RNA for 20 min. Digestion was stopped by adding phenol/chloroform and RNA fragments were precipitated with isopropanol.
RNA fractionation, visualization and purification
RNase digestion fragments were fractionated via anionic exchange chromatography using NucleoBond-columns DNA/RNA400 or DNA/RNA80 (MACHEREY-NAGEL, Düren, Germany) and stepwise salt elution (ranging from 400–850 mM KCl) according to the manufacturer's protocol. Alternatively, RNA fragments from RNase A-digested in vitro transcribed ITS2 RNA (IVT_ITS2 or IVT_ITS2_ΔG) were separated via 12% PAA gel electrophoresis, excised from gel and extracted by incubation with 0.3 mM Na-acetate, 0.01% SDS for 2 h at 42°C on a thermo shaker or in HEPES-buffered saline for 16 h at 4°C and precipitated after phenol/chloroform purification. For sequencing of RNase A digests, RNA fragments were 3′-dephosphorylated by calf intestine phosphatase (CIP) and 5′-phosphorylated by T4 polynucleotide kinase (PNK) (NEB, Ipswich, USA). In general, RNA was visualized by PAA gel electrophoresis and stained with SYBR™ Gold Nucleic Acid Gel Stain (Invitrogen/ThermoFisher, Waltham, Massachusetts, USA, # S11494) or analysed on an Agilent 2100 Bioanalyser System utilizing RNA Pico Chips according to the manufacturers' protocol.
Northern blotting
Gel loading buffer II (ThermoFisher, Waltham, MA, USA) was added to 60 μg total RNA (or 10 μg RNase A treated RNA, 0.1 μg synthesized RNA). Samples were denatured at 95°C for 5 min and then separated on a 15% PAA gel containing 8 M urea. The RNA was transferred to a Nylon membrane, UV-crosslinked (120 mJ/cm²) and hybridized with a DIG-labeled DNA probe overnight. at 42°C in DIG EasyHyb Buffer (Roche, Basel, Switzerland). The probe (5′-CGGACCCGCCTTCCTCCTC-3′) was labelled using the second Generation DIG Tailing Kit (Roche, Basel, Switzerland) according to the manufacturer's protocol. Detection was performed using washing/blocking reagents, anti-DIG antibody and chemiluminescence reagent CDP STAR from Roche according to the manufacturer's protocol. The 72-mer ITS2 RNA fragment 7219–7290 was in vitro transcribed using corresponding ODNs with T7 promotor sequence and served as positive control.
Generation of RNase L-deficient HEK293-RIG-I-IFN-ß reporter cells and RNase L-reconstituted cells
HEK293-RIG-I-IFN-ß reporter cells were transfected with CRISPR/Cas9 vector px459 (30) expressing the RNase L-specific guide RNA GAGGACGUGGGUCCCUCCUG which was selected with E-CRISP (http://www.e-crisp.org/E-CRISP/) (31). After transfection, cells were selected with 1 μg/ml puromycin for 4 days and cloned. Resistant clones were sequenced and tested by western blotting for RNase L expression. RNase L-deficient clones were transfected with human RNase L expression plasmid EX-U1047-M68 (Genecopoeia, Rockville, USA) selected with 2 μg/ml puromycin and cloned. Resistant clones were tested for RNase L expression by western blotting (see below).
Western blotting
1.2 × 106 cells were seeded in 6 well plates and harvested after 16 h. Cells were lysed in 150 μl PBS with 1% Triton-X100 and protease inhibitors. After 30 min of incubation at 4°C, lysate was centrifuged at 14 000 rpm for 20 min. Supernatant was analysed for protein content and 30 μg were loaded and run on a 4–20% SDS-Page gradient gel (Expedeon, Biozol, Eching, Germany, #NXG42012) and blotted according to the manufacturer's protocol. RNase L was detected with mouse anti-human RNase L-specific antibody 2E9 (Novusbiologicals, Bio-Techne GmbH, Wiesbaden, Germany #NB100–351SS) at a 1:1000 dilution at 4°C overnight and goat anti-mouse-peroxidase labelled antibody (1:5000) for 1 h at room temperature.
RNase L-mediated RNA digestion within cell lysates
2 × 106 cells were lysed in 350 μl buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 2.5 mM MgCl2, 1 mM ATP, 4 mM Fructose-1–6-bisphosphate, 2 mM DTT, 0,1% Triton-X100, 1× protease-inhibitor-cocktail (Pierce/ThermoFisher, Waltham, Massachusetts, USA, #1862209), 1× phosphatase-inhibitor-cocktail (Pierce/ThermoFisher, Waltham, MA, USA, #1862495), 400 U/ml RiboLock RNase inhibitor (ThermoFisher, Waltham, MA, USA, #EO0381) and 50 μg/ml purified total HEK293 RNA. Lysate was incubated 5 min at 4°C and centrifuged at 12 000 rpm for 5 min. Supernatant was transferred to new tubes and 100 μl incubated for 1 h at 37°C under various conditions such as mock treatment or addition of 2′-5′-oligoadenylate (2–5A) or a 2–5A negative control (see below). After the incubation, RNA was purified by solvent extraction using phenol/chloroform/isoamylalcohol mixture. 60 μl of the upper phase were purified by size exclusion using Micro Bio-Spin columns P-30 (Bio-Rad, Feldkirchen, Germany, #732–6250) according to the manufacturer's protocol. RNA was further used for visualization, RT-PCR or immune stimulation.
Generation of 2′-5′-oligoadenylate
2′-5′-oligoadenylate (2–5A) containing solution was generated according to Chitrakar A et al (32). Briefly, HeLa cells were plated in 10 cm plates at 70% confluency, incubated for 4 h with 100 U/ml universal type I interferon alpha (PBL, Piscataway, USA, #11200-2) and transfected with 1 μg/ml poly(I:C) complexed to Lipofectamine 2000. After further 6 h, cells were lysed with 1 ml RIPA buffer (ThermoFisher Scientific) containing 1× protease-inhibitor-cocktail (Pierce/ThermoFisher, Waltham, MA, USA, #18622091). The lysate was spun down at 12 000 × g for 10 min at 4°C. The supernatant was centrifuged through a 3 kDa Amicon Ultra-4 centrifugal filter (Amicon, Merck Millipore, Darmstadt, Germany, #UFC800324) and the eluate collected containing the fraction of small nucleic acids. As a negative control 1 μg/mL poly(I:C) was added to the cells directly before washing and lysis of the cells and processed the same way as above. Equivalent amounts (10 μl each) of small RNA fraction (2–5A and 2–5Aneg) were used for RNA digestion in RNase L containing cellular lysates.
Protein transfection
HEK293-RIG-I-IFN-ß reporter cells were transfected with 3.5 μg/ml RNase A or 3.5 μg/ml murine IgG using SAINT-Protein (Synvolux, Leiden, Netherlands) in 96-well plates according to the manufacturer's protocol. Briefly, 2 μg protein were diluted in 50 μl PBS. 20 μl SAINT-Protein was added and solution gently resuspended. After incubation of 5 minutes, 14 μl were added to a single 96-well containing 30 000 cells in 100 μl serum-free Opti-MEM medium (ThermoFisher, Waltham, MA, USA). Cells were lysed 24 hours after transfection and analyzed for luciferase activity (see above).
RNA nucleotide competition assay
2 μM RNA (eRL or eRL ΔG) was incubated in 3 mM MgCl2, 200 mM KCl, 20 mM HEPES pH 7.1 with 0.2 μl γ-32P GTP (3,3 μM, (3000 Ci/mmol) (Hartmann Analytic, Braunschweig, Germany) and 500 μM of following competitor nucleotides / nucleosides: GTP, ATP, UTP, CTP and dGTP (ThermoFisher, Waltham, MA, USA), GMP, GDP, guanosine (Sigma-Aldrich, St. Louis, MO, USA) and ITP, 6-thio-GTP, 7-methyl-GTP (Jena Bioscience, Jena, Germany). After 1 h incubation at 37°C samples were gel filtrated over Micro Bio-Spin columns P-30 (Bio-Rad, Feldkirchen, Germany, #732-6250) according to the manufacturer's protocol. Radioactivity in the eluate was measured in a ß-counter (Beckmann Coulter, Brea, CA, USA). Percentage GTP binding was determined by the (amount of radiolabelled GTP bound in the presence of indicated unlabelled competitor/amount of radiolabelled GTP bound in the absence of unlabelled competitor) × 100.
Statistical analysis
Experiments that were assessed for statistical significance were performed three to six times (luciferase assays, binding studies, qPCR studies, stimulation). Data were analysed for normality using D’Agostino–Pearson and Kolmogorov–Smirnov tests. If not stated otherwise, data are presented as arithmetic means + SD and statistical analyses of the generally normally distributed data were based on paired or unpaired two-tailed t-tests.
RESULTS
RNA digestion by single-strand specific RNases generates immunostimulatory fragments
To characterize immunostimulatory self-RNA fragments, we digested total RNA from HEK293 cells with metal-ion-independent single-strand (ss)-specific RNases such as RNase A, T1 or T2 (Figure 1A) and tested for type I interferon production in human peripheral blood mononuclear cells (PBMCs) (Figure 1B). Limited RNase digestion by all three RNases generated RNA fragments <200 nt in size as visualized by Agilent bioanalyzer RNA analysis (Figure 1A). Importantly, transfection of RNA fragments induced IFN-α from PBMCs, whereas in contrast to a previous report (15) RNA-mediated cytokine induction and ISG56 upregulation were IPS-1 and RIG-I but not MDA-5-dependent (Supplementary Figure S1). Of note, RNA fragments induced by ultrasonic wave (UW) treatment were not immunostimulatory (Figure 1B). Therefore, the immunostimulatory property of endogenous RNA fragments does not depend on small fragment size but on certain RNA sequences and properties released upon RNase digestion.
Figure 1.

RNA digestion by single-strand specific RNases generates immune-stimulatory fragments. (A) Agilent bioanalyzer mediated RNA analysis of undigested HEK293 RNA (–), RNase-digested HEK293 RNA (A, T1 and T2) and ultrasonic waves (UW) fragmented HEK293 RNA. (B) RNAs visualized in panel A were complexed to DOTAP or Lipofectamine 2000 and incubated with human PBMCs at 2 μg/ml. After a 20 h incubation IFN-α levels were detected by ELISA. Left panel shows one individual experiment, right panel combines three independent experiments each in biological duplicates (six measurements per data point ± S.D.), *** P < 0.001. (C) Polyacrylamide gel electrophoresis of RNase A (A) and RNase A / RNase III (A/III) treated HEK293 RNA (upper panel). RNase A-digested RNA was size-fractionated by anionic exchange chromatography into fraction with lower (A') and higher (A'') molecular RNA size (lower panel). (D) PBMCs or human monocytes were stimulated with undigested, RNase A- or RNase A / RNase III treated RNA at 0.4 μg/ml and IFN-α levels were detected by ELISA. Graph combines six independent experiments each in biological duplicates (twelve measurements per data point ± S.D.), *** P < 0.001. (E and F) Human monocytes (E) or A549 cells (F) were stimulated with size fractionated RNA fragments (A', A'') for 20 h at 2 or 0.4 μg/ml, respectively. IFN-α or RANTES levels were determined by ELISA. Graph combines three independent experiments each in biological duplicates (six measurements per data point ± S.D.), *** P < 0.001, * P < 0.05.
Analysis of RNase-digested fragments of HEK293 RNA by polyacrylamide gel electrophoresis identified a distinct RNA band pattern, which was characterized by a prominent band of ∼50 nt length and additional four to five bands in the range up to 80 nt (Figure 1C, upper panel). Importantly, subsequent digestion of RNase A-treated RNA samples with ds-specific RNase III removed higher molecular bands leading to the loss of immunostimulatory capacity on human PBMCs or monocytes (Figure 1D). To further verify that these bands contained the immunostimulatory component, we size-fractionated the RNase A digest and analyzed two fractions, A' and A'', which contained RNA fragments of different size (Figure 1C, lower panel).
Of note, only the fraction enriched for the higher molecular bands (A'') elicited IFN-α release from human PBMCs (Figure 1E) or RANTES production from A549 cells, respectively (Figure 1F).
Identification of an ITS2-derived immunostimulatory RNA fragment
In order to determine the sequence of the endogenous RIG-I-stimulating RNA ligand (eRL), we subjected size-fractionated RNase A and RNase A/RNase III digests (Figure 1C, upper panel sample A and A/III) to next generation sequencing (NGS). Further analysis uncovered three major groups of sequences locating to the 18S, 28S and internal transcribed spacer 2 (ITS2) region of ribosomal 45S RNA (Figure 2A). Sequences corresponding to helix 44 of 18S rRNA (position 5376–5419 ± 5nt on 5′- or 3′-ends on 45S rRNA, NR_046235) were found in both samples with 2784 and 2543 reads for RNase A and RNase A/III treated probes, respectively. In contrast, sequences corresponding to the ITS2 or 28S region were enriched over 100-fold in the RNase A treated sample. Accordingly, sequences corresponding to the 28S rRNA expansion segment ES7L (position 8690–8751 + ≤ 8 nt on 5′-end of 45S rRNA) were identified by 160 reads in the RNase A treated sample only (Table 1). The other group of sequences corresponded to the ITS2 region (position 7219–7290 ± 3nt on 5′- or 3′-ends of 45S rRNA) and was identified by 1369 reads in the RNase A treated sample and only by 11 reads in the RNase A/III digested sample (Table 1). We could verify the generation of the ITS2 derived fragment by RNase A digestion using northern blot detection (Figure 2C). Of note, combined RNase A and RNase III digestion led to loss of the specific signal for the ITS2 fragment supporting the data derived from NGS sequencing. Due to higher representation of the ITS2 sequence (Figure 2B) in the RNase digest, we focused on this fragment for detailed characterization.
Figure 2.

Identification of an ITS2-derived immunostimulatory RNA fragment. (A) Schematic illustration of 45S rRNA, consisting of 5.8S, 18S and 28S as well as internal and external transcribed spacers (ETS, ITS). Positions and number of sequence reads in RNase A and RNase A / RNase III digested and size-fractionated samples A' and A'' (see Figure 1C) are depicted. (B) Sequence of most common read specifically found in the RNase A-digested but not in the RNase A / RNase III-digested sample. (C) Northern blot analysis of undigested, RNase A, RNase III or RNase A/RNase III digested HEK293 RNA with a specific probe for 45S rRNA 7219–7290. IVT-transcribed fragment 7219–7290 served as positive control. (D) Human PBMCs were stimulated for 20 h with 2 μg/ml mock-treated (grey symbols) or RNase A-digested RNA (black symbols) from primary tissue such as human PBMCs (five samples, circle), murine liver (one sample, square) or tumor cell lines such as HeLa (one sample, triangle up), Vero (two samples, hexagonal), MDCK (two samples, triangle down) and HEK293 (five samples, diamond). IFN-α was measured by ELISA. *** P < 0.001. (E) Human PBMCs and monocytes were stimulated with 2 μg/ml mock-treated (grey bars) or RNase A-digested RNA (black bars) from PBMCs, or with RNA from PBMCs activated with PHA for 4 days. IFN-α was measured by ELISA. Graph combines three independent experiments each in biological duplicates (six measurements per data point ± S.D.), ** P < 0.01. (F) 45S rRNA levels of unstimulated or PHA stimulated PBMCs were determined by RT-PCR targeting the 5.8S/ITS2 region. Three combined independent experiments are shown each in biological duplicates (six measurements per data point ± S.D.), * P < 0.05.
Table 1.
NGS analysis of RNase A and RNase A / RNase III digested HEK293 RNA. HEK293 RNA was subjected to RNase A and RNase A / RNase III digestion and size-fractionated (samples A and A/III from Figure 1C, upper panel). RNA isolations were used for cDNA library preparation and sequence analysis. Identified sequences were mapped to the 45S rRNA (NR_046235.1) and number of identified reads in each sample as well as their ratio are depicted
| Number of reads | ||||||
|---|---|---|---|---|---|---|
| Start | End | length | Sequence | RNase A | RNase A/III | RNase A / RNase A/III |
| 28S rRNA | ||||||
| 8698 | 8751 | 54 | GUCCGUCCGUCCUCCUCCUCCCCCGUCUCCGCCCCCCGGCCCCGCGUCCUCCCU | 12 | 0 | - |
| 8694 | 8751 | 58 | GUCCGUCCGUCCGUCCUCCUCCUCCCCCGUCUCCGCCCCCCGGCCCCGCGUCCUCCCU | 47 | 0 | - |
| 8690 | 8751 | 62 | GUCCGUCCGUCCGUCCGUCCUCCUCCUCCCCCGUCUCCGCCCCCCGGCCCGCCGUCCUCCCU | 101 | 0 | - |
| internal transcribed spacer 2 (ITS2) | ||||||
| 7222 | 7290 | 69 | GGUGGGGGAGGAGGAAGGCGGGUCCGGAAGGGGAAGGGUGCCGGCGGGGAGAGAGGGUCGGGGGAGCGC | 104 | 1 | 104.0 |
| 7221 | 7290 | 70 | UGGUGGGGGAGGAGGAAGGCGGGUCCGGAAGGGGAAGGGUGCCGGCGGGGAGAGAGGGUCGGGGGAGCGC | 16 | 0 | - |
| 7220 | 7291 | 72 | GUGGUGGGGGAGGAGGAAGGCGGGUCCGGAAGGGGAAGGGUGCCGGCGGGGAGAGAGGGUCGGGGGAGCGCA | 32 | 0 | - |
| 7219 | 7290 | 72 | GGUGGUGGGGGAGGAGGAAGGCGGGUCCGGAAGGGGAAGGGUGCCGGCGGGGAGAGAGGGUCGGGGGAGCGC | 652 | 7 | 93.1 |
| 7219 | 7288 | 70 | GGUGGUGGGGGAGGAGGAAGGCGGGUCCGGAAGGGGAAGGGUGCCGGCGGGGAGAGAGGGUCGGGGGAGC | 10 | 1 | 10.0 |
| 7218 | 7291 | 74 | UGGUGGUGGGGGAGGAGGAAGGCGGGUCCGGAAGGGGAAGGGUGCCGGCGGGGAGAGAGGGUCGGGGGAGCGCA | 21 | 0 | - |
| 7217 | 7290 | 74 | GUGGUGGUGGGGGAGGAGGAAGGCGGGUCCGGAAGGGGAAGGGUGCCGGCGGGGAGAGAGGGUCGGGGGAGCGC | 534 | 2 | 267.0 |
| 18S rRNA | ||||||
| 5380 | 5426 | 47 | GGUUUAGUGAGGCCCUCGGAUCGGCCCCGCCGGGGUCGGCGGAGGGC | 46 | 46 | 1.0 |
| 5376 | 5427 | 51 | GGAUGGUUUAGUGAGGCCCUCGGAUCGGCCCCGCCGGGGUCGGCGGAGGGC | 398 | 284 | 1.4 |
| 5376 | 5419 | 44 | GGAUGGUUUAGUGAGGCCCUCGGAUCGGCCCCGCCGGGGUCGGC | 2340 | 2213 | 1.1 |
The ITS2 is part of the ribosomal RNA precursors and gets excised and degraded upon processing (21). Therefore, we postulated that RNA from proliferating cells shows enhanced stimulatory potential highlighting the importance of the ITS2-derived eRL. Supporting this view, RNase A-digested RNA from proliferating tumor cell lines (e.g. HeLa, Vero, MDCK and HEK293) induced higher levels of IFN-α when compared to RNase A-digested RNA from primary tissue samples (human PBMCs or murine liver) (Figure 2D). Of note, induction of PBMC proliferation with the T-cell mitogen phytohemagglutinin (PHA) strongly increased the immunostimulatory potential of RNase A-digested RNA (Figure 2E) as well as 45S rRNA levels (Figure 2F) linking cellular proliferation to ITS2-mediated immune stimulation.
To determine the exact nature of the eRL released by RNase digestion of the ITS2 region, we generated by in vitro transcription a 358 nucleotide ITS2 RNA fragment (ITS2-IVT) flanking the eRL (position 7143–7500 of 45S rRNA) (Supplementary Figure S2A). Subsequently, ITS2-IVT was treated with calf intestine phosphatase to remove the triphosphate at the 5′-end, digested with RNase A (ITS2-RA) and the released internal eRL gel-purified under non-denaturing conditions (Figure 3A). NGS-analysis of the eRL identified two major groups of sequences at position 7217–7290/7219–7290 and 7369–7400/7369–7401 (Supplementary Figure S2). The 72/74-mer fragments were identical to the sequences identified after RNase A digestion of total RNA (Figure 2B).
Figure 3.
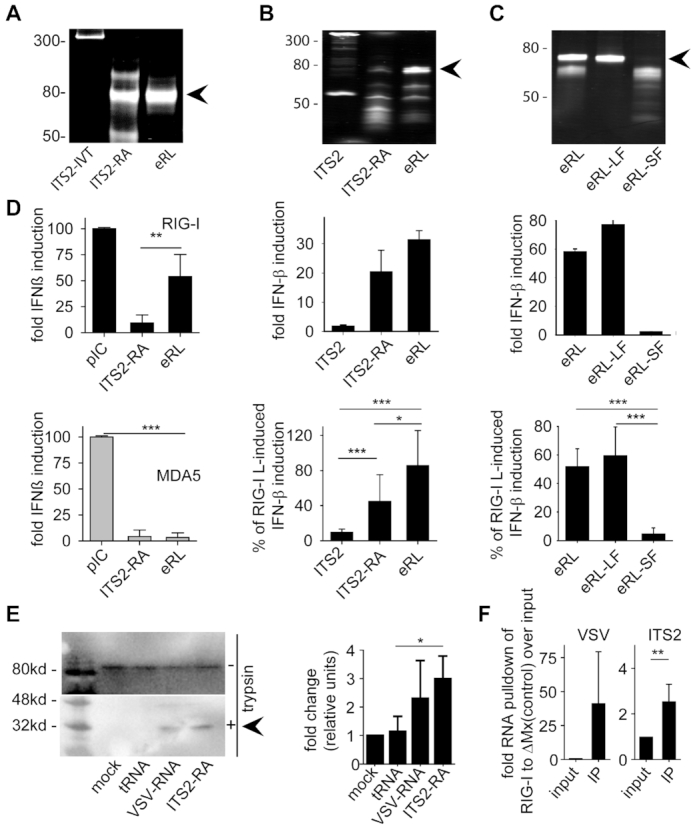
The eRL is single stranded and located in the ITS2 region. (A) Non-denaturing PAGE analysis of various RNA samples: a 358 nucleotide ITS2 RNA fragment (ITS2-IVT), RNase A-digested ITS2-IVT (ITS2-RA) and gel purified band (eRL, endogenous RIG-I ligand). (B) HEK293-RIG-I-IFN-ß reporter cells were stimulated with 0.5 μg/ml ITS2-IVT, ITS2-RA and eRL and fold IFN-ß induction was determined after 12 h. (C) HEK293-RIG-I-IFN-ß reporter cells were stimulated with 0.5 μg/ml eRL, large fragment (eRL-LF) and small fragment (eRL-SF) and fold IFN-ß induction was determined after 12 h. Integrity and composition of RNA samples used for stimulation in (B) and (C) were monitored by denaturing PAGE analysis with subsequent SYBR-Gold staining (upper row). Fold IFN-ß induction is shown for one individual experiment (middle row) and a diagram combining three independent experiments each in biological triplicates with data adjusted to % of pIC induced IFN-β activation (lower row; nine measurements per data point ± S.D.). * P < 0.05, *** P < 0.001. (D) Huh7.5 cells expressing RIG-I or MDA5 were stimulated with 1 μg/ml pIC and 0.4 μg/ml ITS2, ITS2-RA or eRL for 6 h and IFN-β upregulation was determined by RT-PCR. Graph combines three independent experiments each in biological duplicates (six measurements per data point ± S.D.), *** P < 0.001, ** P < 0.01, * P < 0.05. (E) A549 cells were transfected with controls and eRL at 200 ng/ml for 1 h. RIG-I activation was marked by a remaining 30 kDa fragment after limited trypsin digestion in conformational switch assay. Western blot shows one representative result out of three independent experiments. Diagram combines three independent experiments with data adjusted to % of mock-induced conformational switch (three measurements per data point ± S.D.), * P < 0.05. (F) HEK293 ΔRIG-I were transfected with flag-tagged RIG-I or ΔMx and stimulated with 250 ng VSV-RNA or eRL 24 h later. Immunoprecipitation of flag-tagged proteins was performed 6 h after stimulation and RNA binding was determined in qRT-PCR. First panel shows pulldown of VSV-RNA as a positive control, last panel shows pulldown of eRL. Diagram combines three independent experiments each in biological duplicates (six measurements per data point ± S.D), * P < 0.05.
The eRL is single stranded
To further address the immunostimulatory potential of the ITS2-derived eRL, we stimulated RIG-I transfected HEK293 cells carrying an IFN-ß luciferase reporter. The 358 nucleotide fragment (ITS2-IVT) and RNase A-treated 358 nucleotide fragment (ITS2-RA) served as control (Figure 3B). Of note, ITS2-RA and the eRL strongly activated the IFN-ß reporter, whereas the ITS2-IVT was not stimulatory (Figure 3B). Denaturing PAGE analysis revealed the existence of a band at just below the 80 nt marker and additional smaller bands possibly reflecting short fragments of the opposite strand. Therefore, we PAGE purified and separated the eRL into a large fragment (eRL-LF) and small fragments (eRL-SF). Subsequent stimulation of HEK293-RIG-I-IFN-ß reporter cells revealed that the eRL-LF retained immunostimulatory activity, whereas the smaller fragments were inactive (Figure 3C). In addition, Huh-7.5 cells responded to eRL with IFN-ß upregulation that was strictly RIG-I but not MDA5 dependent (Figure 3D). Direct activation of RIG-I by eRL was demonstrated by conformational switch assay (29) (Figure 3E). In addition, eRL-RIG-I interaction could be monitored by immunoprecipitation using 5′-3xFlag-tagged human RIG-I or as a control 5′-3xFlag-tagged ΔMx1 (33) (containing nucleotide +4 to +318 of the Mx1 ORF (XM_005260978.2, not binding to RNA) (Figure 3F and Supplementary Figure S3A). In summary, this observation strongly supports the view that the 72-nt component of the eRL identified by NGS is solely responsible for immunostimulation of RIG-I.
The eRL is generated by intracellular RNase activity
To ensure that eRL is accessible under natural conditions and not protected from RNase digestion by ribosomal proteins or ribosome biogenesis factors, RNase A protein was directly transfected into HEK293-RIG-I-IFN-ß reporter cells using SAINT-Protein delivery system. Importantly, RNase A transfection induced IFN-β reporter activation whereas a control protein (murine IgG) did not (Figure 4A). Since detection of the ITS2 fragment by northern blotting was not sensitive enough we used a three primer PCR strategy to determine the ratio of short eRL fragments (≙64 bp, cut ITS2 at or in the vicinity of nt 7219) to fragments that were not cut at this position (≙143 bp) (Figure 4B). cDNA preparations from untreated, RNase A- or IgG-transfected cells were PCR-amplified and fragments quantified. This analysis demonstrated a weak increase in the ratio of cut and undigested ITS2 fragments (Figure 4C) suggesting a release of eRL. To extend the generation of eRL to other RNases such as RNase L, we first generated RNase L-deficient HEK293-RIG-I-IFN-ß reporter cells (ΔRNase L) utilizing CRISPR/Cas9 technology and created clones with reconstituted RNase L (RNase L K1, K2) thereof. Absence or expression of RNase L was detected by Western blotting (Figure 4D). RNase L-mediated RNA degradation was then analyzed with cell lysates from both cell types. Purified HEK293 RNA was added to lysates from RNase L-deficient (ΔRNase L) or RNase L-reconstituted cells (RNase L K2) and lysates were mock-treated or incubated with 2′-5′ oligoadenylate (2–5A) or a negative control (2–5A neg). The 2′-5′ oligoadenylate containing or negative control solution was obtained from HeLa cells by poly(I:C) or mock stimulation as described (32). Of note, after incubation of 1 h at 37°C and RNA purification, the analysis of RNA integrity by bioanalyzer demonstrated that RNA degradation was dependent on 2–5A and presence of RNase L (Figure 4E). In addition, immune stimulation of HEK293-RIG-I-IFN-ß reporter cells with the same RNA preparations indicated that only the RNase L-degraded RNA was immunostimulatory (Figure 4F). Utilizing the eRL fragment specific RT-PCR (Figure 4B) we could further demonstrate that RNase L-mediated digestion of RNA releases eRL from ribosomal RNA similar to RNase A (Figure 4G). In summary, eRL is generated by RNase A and RNase L and intracellular generated eRL levels are sufficient to induce immune activation.
Figure 4.
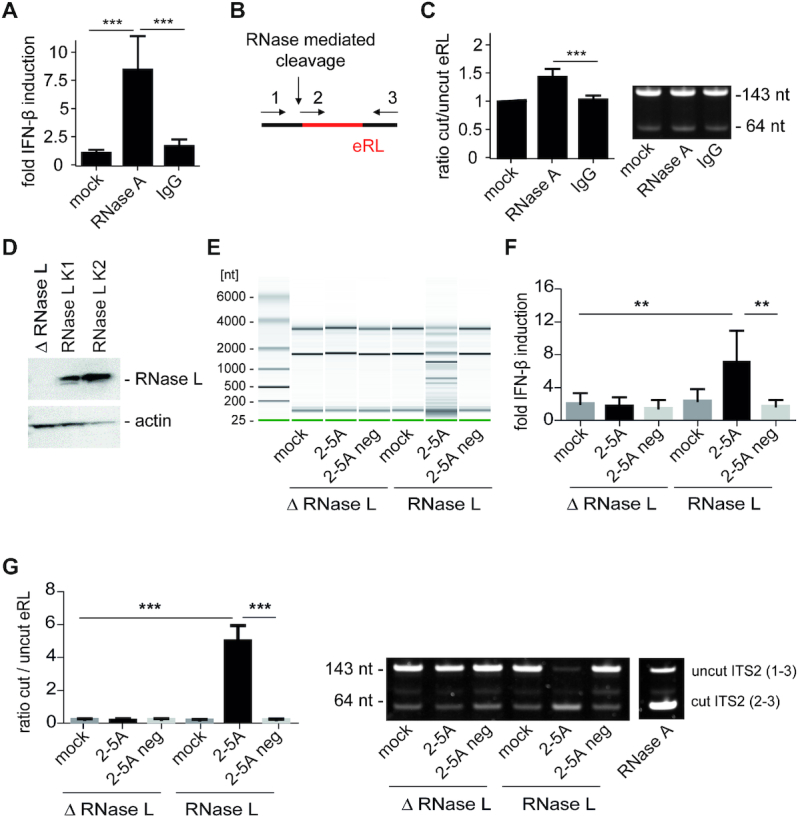
The eRL is generated intracellularly and induces immune activation. (A) HEK293-RIG-I-IFN-ß reporter cells were mock-treated or transfected with 3.5 μg/ml RNase A and 3.5 μg/ml murine IgG using SAINT-Protein transfection reagent and interferon reporter activity was measured 24 h after transfection. Graph combines three independent experiments each in biological quadruplicates (12 measurements per data point ± S.D.), *** P < 0.001. (B) Schematic illustration of the RT-PCR strategy using three primers (depicted as 1, 2 and 3, see Materials and Methods) in one reaction to analyze uncut or cut ITS2 fragments (eRL). (C) Cleavage of eRL was determined in qRT-PCR after 24 h. Left panel combines three independent experiments each in technical duplicates (six measurements per data point ± S.D). *** P < 0.001. Right panel depicts a representative fragment analysis in a 12% PAA gel stained with SYBRGold. One individual experiment of three independent experiments is shown. (D) Western blot of HEK293-RIG-I-IFN-ß reporter cells devoid of or reconstituted with RNase L. ß-actin served as loading control. (E) RNase L-deficient (△ RNase L) and RNase L-competent (RNase L K2) HEK cell lysates were untreated (mock) or incubated with 2′-5′-oligoadenylate (2–5A) containing lysate or control lysate (2–5A neg). After incubation of 1 h at 37°C, RNA was purified and RNA integrity investigated utilizing Pico RNA chips on an Agilent bioanalyzer. One individual representative experiment of three independent experiment is shown. (F) RNAs depicted in (E) were transfected into HEK293-RIG-I-IFN-β reporter cells at 5 μg/ml RNA each. Fold IFN-ß induction was determined after 12–16 h. Graph combines three independent experiments each in biological duplicates (6 measurements per data point ± S.D.), ** P < 0.01. (G) RNAs depicted in (E) were analyzed for generation of eRL by RT-PCR. Left panel combines three independent experiments each in technical duplicates (six measurements per data point ± S.D), *** P < 0.001. Right panel depicts a representative fragment analysis in a 12% PAA gel stained with SYBRGold. One individual experiment of three independent experiments is shown. In vitro RNase A-digested HEK293 RNA served as positive control.
The eRL depends on a 2′,3′-cyclic phosphate
Since the hydrolytic activity of RNase A leads to the release of the eRL with a 5′-hydroxyl and a 2′,3′-cyclic phosphate end, the immunostimulatory potential is independent of a 5′-triphosphate (5′ppp). Accordingly, treatment with calf intestine phosphatase or shrimp alkaline phosphatase did not influence its immunostimulatory potential (Supplementary Figure S3B).
To further characterize the role of the 3′-end for immunostimulation, we hydrolyzed the 2′,3′-cyclic phosphate to monophosphate by treatment with 10 mM HCl for 30 min (34–36). Importantly, both the eRL and RNase A-digested total HEK293 RNA lost their immunostimulatory capacity, whereas 5′ppp-RL and poly(I:C) (pIC) were unaffected (Figure 5A and B). The integrity of the eRL after cyclic phosphate hydrolysation was confirmed via denaturing PAGE analysis (Supplementary Figure S3C). To demonstrate the hydrolyzing effect of HCL on the 2′,3′-cyclic phosphate (>P) we utilized the synthetic oligoribonucleotide (ORN) RNA40 (37) carrying a 3′-P (RNA40–3P) or a 2′,3′-cyclic phosphate (RNA40>P). RNA40>P was created from RNA40–3P by treatment with RNA 3′-terminal phosphate cyclase (rtcA). Alteration in RNA phosphorylation status by incubation with HCL and/or Shrimp Alkaline Phosphatase (SAP) was analysed via differences in electrophoretic mobility of the ORN (Figure 5C). SAP hydrolyzed RNA40–3P to RNA40–3′OH leading to a shift in electrophoretic mobility (Figure 5C, upper panel, compare lane ‘mock’ to lane ’SAP’). In contrast, SAP treatment of RNA40>P did not alter the electrophoretic mobility since SAP cannot hydrolyze 2′,3′-cyclic phosphate (38) (Figure 5C, lower panel, compare lane ‘mock’ to lane ’SAP’). However, when RNA40>P was treated with HCL which hydrolyzes a 2′,3′-cyclic phosphate to a mixture of 2′- and 3′ phosphates (34–36) subsequent SAP incubation lead to the formation of RNA40–3′OH (compare Figure 5C, lower panel, compare lane ‘HCL/SAP’ to lane ‘mock’). Since a 2′P-end cannot be efficiently hydrolyzed by SAP, two bands with differential electrophoretic mobility are visible. An additional method to remove the 2′,3′-cyclic phosphate is the use of T4 polynucleotide kinase (PNK). This enzyme harbors a 2′,3′-cyclic phosphodiesterase activity that generates a terminal 3′-phosphate that is removed by its phosphatase activity (39). We combined PNK and SAP treatment since PNK alone was not efficiently dephosphorylating RNA40–3P suggesting a weak phosphatase activity (Figure 5C, upper panel, lane ‘PNK’). PNK/SAP treatment of RNA40>P removed the cyclic phosphate and generated RNA40–3′OH, whereas SAP again had no effect. Importantly, eRL treated with SAP and PNK (eRL + P + S) showed reduced IFN-ß induction (Figure 5D) similar to HCL-treated eRL. In summary, the immunostimulatory capacity of the eRL is strictly dependent on a 2′,3′-cyclic phosphate.
Figure 5.

The activity of eRL depends on a 2′,3′-cyclic phosphate. (A and B) HEK293-RIG-I-IFN-ß reporter cells were stimulated with 2 μg/ml 5'-triphosphate RNA (5′ppp-RL), RNase A-digested HEK293 RNA (dig. RNA), 1 μg/ml pIC, and eRL-LF (untreated or treated with 10 mM HCl for 30 min) and luciferase activity was determined 12 to 20 h later. One individual experiment (A) and a diagram combining three independent experiments each in biological duplicates or triplicates with data adjusted to the ratio of IFN-β activation with and without HCl treatment (B) (6 to 8 measurements per data point ± S.D.) are shown, *** P < 0.001, ** P < 0.01. (C) 3′-phosphorylated RNA40 (RNA40–3′-P) and RNA40 with a 2′-3′-cyclic phosphate (generated with rtcA) (RNA40>P) were incubated with 10 mM HCL, SAP, PNK or in combinations as indicated. RNAs were analyzed at 50°C on a 12% polyacrylamide gel containing 8 M urea and stained with SyBRGold. (D) HEK293-RIG-I-IFN-ß reporter cells were stimulated with 0.0625 μg/ml eRL that was mock treated or incubated with SAP and PNK. The commercially available RIG-I activator 3p-hpRNA served as control. Graph combines six independent experiments each in biological duplicates (twelve measurements per data point ± S.D.), *** P < 0.001.
The eRL functions as GTP-binding aptamer
We further hypothesized that the eRL noncovalenty binds intracellular GTP due to a GTP-binding aptamer function substituting for a covalently linked 5′-triphosphate. Importantly, the eRL contains a sequence motif that is reminiscent of GTP-binding sequences (20). Indeed, competitor studies using IVT eRL, 32P-labeled gamma GTP and various nucleotides as competitors revealed that the eRL bound GTP but not UTP, CTP or ATP (Figure 6A). Extending the studies to guanosine and 5′-di- or monophosphate demonstrated that the base guanosine is efficiently bound. In this context, binding also depends on position 6 and 7 of the guanosine ring since 6-thio-GTP and 7-methyl-GTP interfere with nucleotide binding to the RNA fragment (Figure 6B). To show the impact of GTP-binding aptamers on eRL activity, a mutant construct (eRL ΔG) was designed in which G-quadruplexes were disrupted without changing secondary structure (Supplementary Figure S4). RIG-I activating activity of eRL ΔG was significantly reduced (Figure 6C, D) and GTP-binding activity was diminished (Figure 6E). Supporting a role of the eRL in presenting GTP, incubation of HEK293-RIG-I-IFN-ß reporter cells with mycophenolic acid (MA), a IMP-dehydrogenase inhibitor that leads to reduction in GTP levels within cells (40), strongly reduced the immunostimulatory potential of transfected RNase A digested ITS2 (ITS2-RA) (Figure 6F). In contrast, 5′-triphosphate-RNA mediated immune activation was not negatively affected. Therefore, RNase generated self-RNA fragments have the potential to function as GTP binding aptamers activating RIG-I despite the lack of a covalently bound 5′-triphosphate.
Figure 6.

The eRL functions as an aptamer and binds GTP. (A) Competitive binding studies with IVT-eRL, γ-32P GTP and indicated unlabeled competitors. Percentage γ-32P GTP binding = [(amount of radiolabeled GTP bound in the presence of unlabeled competitor) / (amount of radiolabeled GTP bound in the absence of indicated competitor)] × 100. Graph combines two independent experiments each in duplicates (four measurements per data point ± S.D.), * P < 0.05. (B) Data from competitive binding experiments mapped to the structure of GTP. (C, D) HEK293-RIG-I-IFN-ß reporter cells were stimulated with eRL, eRL ΔG or 3p-hpRNA at 0.25; 0.063, 0.016, 0.0039 or 0.00098 μg/ml. Luciferase activity was measured 12 h after ligand stimulation. (C) One representative experiment is shown and (D) a statistical analysis at the concentration of 0.063 μg/ml combining three independent experiments each in biological duplicates (6 measurements per data point ± S.D.) is depicted, *** P < 0.001. (E) GTP binding studies utilizing 0,1 nmol eRL, eRL ΔG or poly A and γ-32P GTP. GTP binding by eRL is set to 100. Graph combines three independent experiments each in duplicates (six measurements per data point ± S.D.), * P < 0.05. (F) Stimulation of HEK293-RIG-I-IFN-ß reporter cells that where mock-treated or pre-incubated with 10 μM mycophenolic acid (MA) for 24 h and subsequently incubated with 0.5 μg/ml 5′ppp-RL or 1 μg/ml ITS2-RA. Luciferase activity was measured 12 h after ligand stimulation. One individual experiment is shown (first panel) and a diagram combining three independent experiments each in triplicates with data adjusted to the ratio of IFN-β activation without and with MA treatment (last panel; nine measurements per data point ± S.D.), *** P < 0.001.
DISCUSSION
The cytoplasmic RNA sensor RIG-I senses viral double-stranded (ds) RNAs, poly(I:C) or RNA panhandle structures bearing 5′-tri- or diphosphates (3,8–10). Additionally, endogenous RNA fragments generated by RNase digestion during viral infection (via RNase L) (15) or the unfolded protein response (via IRE1) (16) are recognized by RIG-I. Importantly, metal ion-independent RNases (such as RNase L, RNase A, IRE1) generate RNA fragments with a 5′-hydroxyl group and a 2′,3′-cyclic phosphate (17–19) and therefore create a RIG-I ligand that differs structurally from known 5′-tri- or diphosphorylated RIG-I ligands. Here we show that the endogenous RIG-I ligand (eRL) is derived from the internal transcribed spacer 2 region (ITS2) of the 45S ribosomal RNA (Figure 7). The eRL is specifically generated both by intracellular RNase A and RNase L and induces immunostimulation under physiological conditions. Its immunostimulatory property depends on 2′,3′-cyclic phosphate and is characterized by a G-quadruplex containing sequence motif mediating guanosine-5′-triphosphate (GTP) binding (20).
Figure 7.
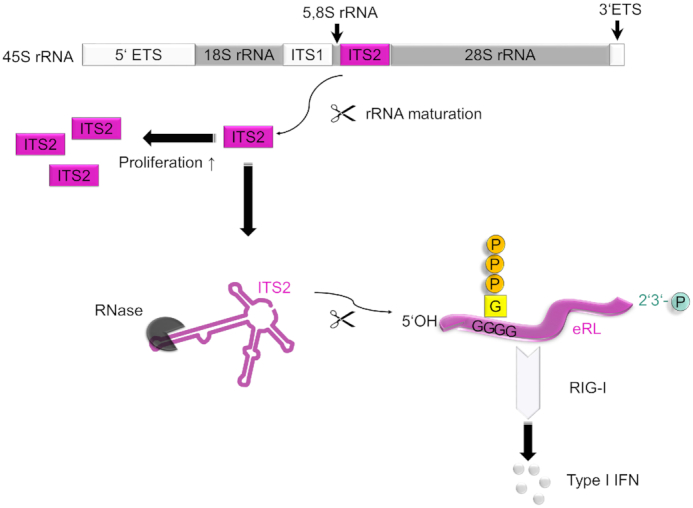
Mechanism of RIG-I signaling activation by endogenous RNA. The endogenous RIG-I ligand (eRL) is derived from the internal transcribed spacer 2 (ITS2) region of polycistronic 45S ribosomal RNA. ITS2 is excised by processing of 45S precursor rRNA to mature ribosomal RNA species and more abundant in proliferating than in resting cells. RNase treatment exposes eRL as a specific sequence derived from ITS2 region. This stimulatory RNA fragment binds GTP by internal G-quadruplexes and bears a 2′,3′-cyclic phosphate. Both these features are crucial for eRL dependent RIG-I activation.
The ITS2 part of the 45S ribosomal RNA precursor is excised and degraded upon rRNA processing. It is conserved among eukaryotes, suggesting a widespread source of potential stimulatory RIG-I activating self-RNA after RNase digestion. Further characteristics of the ITS2 region are a high GC-content (83%) and an extensive formation of double-stranded regions which protect against digestion by single-strand specific RNases (41). Interestingly, the 45S ribosomal precursor RNA transcription is downregulated upon growth arrest (21), whereas tumor cells show an enhanced 45S rRNA content compared to non-transformed cells (22). Therefore, RNA from proliferating cells showed a higher potential to generate stimulatory fragments after RNase digestion. Since rRNA synthesis is mostly silenced in resting cells (21) and pre-rRNA species harboring ITS2 are completely exhausted in less than 4h post transcription (41), the proliferation status of cells influences the availability of stimulatory RNA fragments. Since RNase L and RIG-I have been defined as tumor suppressor genes in addition to their role in innate immune sensing (42,43), the ITS2-derived stimulatory self-RNA might function as a tumor suppressor by linking the proapoptotic and anticancer properties of RIG-I and RNase L.
The functional role of the 2′,3′-cyclic phosphate for the immunostimulatory capacity is less clear. However, it is a molecular feature necessary for RIG-I-mediated type I IFN induction by self-RNA fragments, similar to its activity in the enhancement of RNase L-digested RNA-mediated IL-1β production by NLRP3 (44). Recently, also activation of TLR8 by RNase T2-degraded RNA has been shown to depend on 2′,3′-cyclic phosphate (45). 2′,3′-cyclic phosphate may protect RNA fragments from exonuclease activity, as it has been reported for U6 RNA (46). It could also lead to self-circularization of the RNA, again protecting it from nuclease digestion by the lack of free ends. This way, the circular eRL might exhibit similarities to viral RNAs like the HDV genome (47) which is also immunostimulatory via IPS-1 (and presumably RIG-I) (48). Of note, it has been reported recently that RIG-I senses circular RNA and that discrimination of foreign and self-circular RNA depends on its accessibility, since the endogenous RNA splicing machinery possibly shields the intron lariat from RIG-I recognition (49). In contrast, Wesselhoeft et al. demonstrated that circular RNA is able to bypass immune sensing via RIG-I (50). Although the ATPase activity of RIG-I impedes self-RNA recognition by RIG-I (51,52), the importance of masking self-RNA for avoiding recognition was also demonstrated recently by showing that viral unmasking of a cellular 5S rRNA pseudogene transcript induces RIG-I activation (53). In a similar manner an inducible lncRNA acts as a molecular decoy that restricts endogenous RNA recognition by RIG-I (54).
However, the latter observations did not explain how circular RNA and RNase-digested RNA devoid of a 5′-triphosphate strongly activate RIG-I. Screening the ITS2 derived RIG-I stimulatory sequence, we observed that it contains a G-quadruplex containing sequence motif described to mediate guanosine-5′-triphosphate binding (20). We therefore hypothesized that the eRL noncovalenty acts as an aptamer, binding intracellular GTP that substitutes for a covalently linked 5′-triphosphate. Recently RNA aptamers that specifically target RIG-I have been identified, but these sequences contained polyU motifs and were devoid of G-quadruplexes (55). However, eRL sequence competitor studies demonstrated that the ITS2 fragment of interest binds guanosine and guanosine-mono-, di- and triphosphate where binding involves position 6 and 7 of the guanosine ring since 6-thio GTP and 7-methyl GTP interfere with binding. Of note, GTP is the major constituent of the cellular guanosine-nucleotide pool exceeding the concentration of GDP, GMP and other deoxyguanosine nucleotides by 3- to 100-fold (56,57). Therefore, the eRL acts as aptamer and mainly presents GTP (and to a minor extend GDP) to RIG-I mimicking covalently bound 5′-triphosphate RNA.
In summary, we describe that RNase generated self-RNA fragments with a 2′,3′-cyclic phosphate and a G-quadruplex sequence motif supporting guanosine 5′-triphosphate binding activate RIG-I. Moreover, we hypothesize that recognition of self-RNA fragments generated by IRE1, RNase L and RNase A may function as DAMP signals in tumor cell recognition and surveillance. The fact that eRL could also be generated by intracellular application of RNase A opens up the possibility of new clinical developments. In particular, this could lead to the generation of new RNA-based adjuvants. In addition, low intracellular RNase A levels may also be used for the treatment of cancer, as the antitumor activity of RIG-I ligands has already been reported (58,59). Thus, these results not only open up mechanistic insights but also prospects for possible clinical applications.
DATA AVAILABILITY
GenBank® is an open genetic sequence database provided by the National Institutes of Health (NIH) (https://www.ncbi.nlm.nih.gov/genbank/).
Basic Local Alignment Search Tool (BLAST) compares regions of similarity between nucleotide sequences and is provided by the NIH (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Clustal Omega was used for pairwise sequence alignment to identify regions of similarity between RNA sequences (https://www.ebi.ac.uk/Tools/psa/).
Galaxy analysis is an open source platform for data intensive sequence analysis (https://usegalaxy.org/).
RNAfold web server predicts secondary structures of RNA sequences (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi).
NGS data is publicly available under BioProject ID: PRJNA643769.
Supplementary Material
ACKNOWLEDGEMENTS
We thank H. Hackstein and G. Bein, Institute for Clinical Immunology and Transfusion Medicine, Justus-Liebig-University Giessen, for providing human buffy coats, Marco Binder for providing RIG-I and MDA5-expressing Huh7.5 cells. We thank Ulrike Protzer for extensive support and Hannah Busen, Core Facility Helmholtz Center Munich, for statistical consulting.
Notes
Present address: Stephanie Jung, Institut für Virologie, Helmholtz Zentrum München / Technische Universität München, Medizinische Fakultät, Trogerstraße 30, 81675 München, Germany.
Present address: Ines Yang, NIFE – Niedersächsisches Zentrum für Biomedizintechnik, Implantatforschung und Entwicklung, Klinik für Zahnärztliche Prothetik und Biomedizinische Werkstoffkunde, Medizinische Hochschule Hannover, Stadtfelddamm 34, 30625 Hannover, Germany.
Contributor Information
Stephanie Jung, Institut für Immunologie, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
Tina von Thülen, Institut für Immunologie, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
Ines Yang, Institut für Medizinische Mikrobiologie und Krankenhaushygiene, Medizinische Hochschule Hannover, Carl Neuberg Straße 1, 30625 Hannover, Germany.
Viktoria Laukemper, Institut für Immunologie, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
Benjamin Rupf, Institut für Immunologie, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
Harshavardhan Janga, Institut für Lungenforschung/iLung, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
Georgios-Dimitrios Panagiotidis, Institut für Virologie, Fachbereich Veterinärmedizin (FB10), Justus-Liebig-Universität Gießen, Schubertstr. 81, 35392 Gießen, Germany.
Andreas Schoen, Institut für Virologie, Fachbereich Veterinärmedizin (FB10), Justus-Liebig-Universität Gießen, Schubertstr. 81, 35392 Gießen, Germany.
Marina Nicolai, Institut für Immunologie, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
Leon N Schulte, Institut für Lungenforschung/iLung, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany; Deutsches Zentrum für Lungenforschung (DZL), 35392 Gießen, Germany.
Hannah-Lena Obermann, Institut für Immunologie, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
Friedemann Weber, Institut für Virologie, Fachbereich Veterinärmedizin (FB10), Justus-Liebig-Universität Gießen, Schubertstr. 81, 35392 Gießen, Germany.
Andreas Kaufmann, Institut für Immunologie, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
Stefan Bauer, Institut für Immunologie, Philipps-Universität Marburg, BMFZ, Hans-Meerwein-Straße 2, 35043 Marburg, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Center for Infection Research [TTU 09.806 to S.B.]; Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [369799452 – TRR237-A02, 114933180 – TR84-C10, 284237345 – KFO 309 (BA 1618/8-1), BA 1618/5-1 to S.B. and 284237345 – KFO 309 (We 2616/8-1) to F.W.]; German Academic Scholarship Foundation (to S.J.). Funding for open access charge: DFG
Conflict of interest statement. None declared.
REFERENCES
- 1. Blasius A.L., Beutler B.. Intracellular toll-like receptors. Immunity. 2010; 32:305–315. [DOI] [PubMed] [Google Scholar]
- 2. Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T.. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004; 5:730–737. [DOI] [PubMed] [Google Scholar]
- 3. Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J. et al.. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006; 441:101–105. [DOI] [PubMed] [Google Scholar]
- 4. Runge S., Sparrer K.M.J., Lässig C., Hembach K., Baum A., García-Sastre A., Söding J., Conzelmann K.-K., Hopfner K.-P.. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog. 2014; 10:e1004081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kato H., Takeuchi O., Mikamo-Satoh E., Hirai R., Kawai T., Matsushita K., Hiiragi A., Dermody T.S., Fujita T., Akira S.. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008; 205:1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deddouche S., Goubau D., Rehwinkel J., Chakravarty P., Begum S., Maillard P.V., Borg A., Matthews N., Feng Q., van Kuppeveld F.J. et al.. Identification of an LGP2-associated MDA5 agonist in picornavirus-infected cells. Elife. 2014; 3:e01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sanchez David R.Y., Combredet C., Sismeiro O., Dillies M.A., Jagla B., Coppée J.Y., Mura M., Guerbois Galla M., Despres P., Tangy F. et al.. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. Elife. 2016; 5:e11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C.. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science (New York, N.Y.). 2006; 314:997–1001. [DOI] [PubMed] [Google Scholar]
- 9. Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M. et al.. 5′-Triphosphate RNA is the ligand for RIG-I. Science (New York, N.Y.). 2006; 314:994–997. [DOI] [PubMed] [Google Scholar]
- 10. Schlee M., Roth A., Hornung V., Hagmann C.A., Wimmenauer V., Barchet W., Coch C., Janke M., Mihailovic A., Wardle G. et al.. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009; 31:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goubau D., Schlee M., Deddouche S., Pruijssers A.J., Zillinger T., Goldeck M., Schuberth C., Van der Veen A.G., Fujimura T., Rehwinkel J. et al.. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014; 514:372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baum A., Sachidanandam R., García-Sastre A.. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. PNAS. 2010; 107:16303–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dong B., Silverman R.H.. A bipartite model of 2–5A-dependent RNase L. J. Biol. Chem. 1997; 272:22236–22242. [DOI] [PubMed] [Google Scholar]
- 14. Clemens M.J., Williams B.R.. Inhibition of cell-free protein synthesis by pppA2′p5′A2′p5′A: a novel oligonucleotide synthesized by interferon-treated L cell extracts. Cell. 1978; 13:565–572. [DOI] [PubMed] [Google Scholar]
- 15. Malathi K., Dong B., Gale M. Jr., Silverman R.H.. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007; 448:816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eckard S.C., Rice G.I., Fabre A., Badens C., Gray E.E., Hartley J.L., Crow Y.J., Stetson D.B.. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol. 2014; 15:839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011; 44:1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuchillo C.M., Nogues M.V., Raines R.T.. Bovine pancreatic ribonuclease: fifty years of the first enzymatic reaction mechanism. Biochemistry. 2011; 50:7835–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cooper D.A., Jha B.K., Silverman R.H., Hesselberth J.R., Barton D.J.. Ribonuclease L and metal-ion-independent endoribonuclease cleavage sites in host and viral RNAs. Nucleic Acids Res. 2014; 42:5202–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Curtis E.A., Liu D.R.. Discovery of widespread GTP-binding motifs in genomic DNA and RNA. Chem. Biol. 2013; 20:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hannan K.M., Kennedy B.K., Cavanaugh A.H., Hannan R.D., Hirschler-Laszkiewicz I., Jefferson L.S., Rothblum L.I.. RNA polymerase I transcription in confluent cells: Rb downregulates rDNA transcription during confluence-induced cell cycle arrest. Oncogene. 2000; 19:3487–3497. [DOI] [PubMed] [Google Scholar]
- 22. Uemura M., Zheng Q., Koh C.M., Nelson W.G., Yegnasubramanian S., De Marzo A.M.. Overexpression of ribosomal RNA in prostate cancer is common but not linked to rDNA promoter hypomethylation. Oncogene. 2012; 31:1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J.. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005; 437:1167–1172. [DOI] [PubMed] [Google Scholar]
- 24. Binder M., Eberle F., Seitz S., Mucke N., Huber C.M., Kiani N., Kaderali L., Lohmann V., Dalpke A., Bartenschlager R.. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J. Biol. Chem. 2011; 286:27278–27287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stanley E.R., Chen D.M., Lin H.S.. Induction of macrophage production and proliferation by a purified colony stimulating factor. Nature. 1978; 274:168–170. [DOI] [PubMed] [Google Scholar]
- 26. Weber M., Weber F.. Monitoring activation of the antiviral pattern recognition receptors RIG-I and PKR by limited protease digestion and native PAGE. J. Vis. Exp. 2014; 89:e51415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010; 26:2460–2461. [DOI] [PubMed] [Google Scholar]
- 28. Chomczynski P., Sacchi N.. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat. Protoc. 2006; 1:581–585. [DOI] [PubMed] [Google Scholar]
- 29. Weber M., Gawanbacht A., Habjan M., Rang A., Borner C., Schmidt A.M., Veitinger S., Jacob R., Devignot S., Kochs G. et al.. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe. 2013; 13:336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heigwer F., Kerr G., Boutros M.. E-CRISP: fast CRISPR target site identification. Nat. Methods. 2014; 11:122–123. [DOI] [PubMed] [Google Scholar]
- 32. Chitrakar A., Rath S., Donovan J., Demarest K., Li Y., Sridhar R.R., Weiss S.R., Kotenko S.V., Wingreen N.S., Korennykh A.. Real-time 2–5A kinetics suggest that interferons β and λ evade global arrest of translation by RNase L. PNAS. 2019; 116:2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kainulainen M., Habjan M., Hubel P., Busch L., Lau S., Colinge J., Superti-Furga G., Pichlmair A., Weber F.. Virulence factor NSs of rift valley fever virus recruits the F-box protein FBXO3 to degrade subunit p62 of general transcription factor TFIIH. J. Virol. 2014; 88:3464–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Usher D.A., McHale A.H.. Hydrolytic stability of helical RNA: a selective advantage for the natural 3′,5′-bond. PNAS. 1976; 73:1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lutay A.V., Chernolovskaya E.L., Zenkova M.A., Vlassov V.V.. The nonenzymatic template-directed ligation of oligonucleotides. Biogeosciences. 2006; 3:243–249. [Google Scholar]
- 36. Zenkova M.A. Artificial Nucleases. 2004; Springer Berlin Heidelberg. [Google Scholar]
- 37. Heil F., Hemmi H., Hochrein H., Ampenberger F., Kirschning C., Akira S., Lipford G., Wagner H., Bauer S.. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (New York, N.Y.). 2004; 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- 38. Zhelkovsky A.M., McReynolds L.A.. Polynucleotide 3′-terminal phosphate modifications by RNA and DNA ligases. J. Biol. Chem. 2014; 289:33608–33616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das U., Shuman S.. Mechanism of RNA 2′,3′-cyclic phosphate end healing by T4 polynucleotide kinase-phosphatase. Nucleic Acids Res. 2013; 41:355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cohen M.B., Sadee W.. Contributions of the depletions of guanine and adenine nucleotides to the toxicity of purine starvation in the mouse T lymphoma cell line. Cancer Res. 1983; 43:1587–1591. [PubMed] [Google Scholar]
- 41. Joseph N., Krauskopf E., Vera M.I., Michot B.. Ribosomal internal transcribed spacer 2 (ITS2) exhibits a common core of secondary structure in vertebrates and yeast. Nucleic Acids Res. 1999; 27:4533–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carpten J., Nupponen N., Isaacs S., Sood R., Robbins C., Xu J., Faruque M., Moses T., Ewing C., Gillanders E. et al.. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat. Genet. 2002; 30:181–184. [DOI] [PubMed] [Google Scholar]
- 43. Li X.Y., Guo H.Z., Zhu J.. Tumor suppressor activity of RIG-I. Mol Cell Oncol. 2014; 1:e968016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chakrabarti A., Banerjee S., Franchi L., Loo Y.M., Gale M. Jr., Nunez G., Silverman R.H.. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe. 2015; 17:466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Greulich W., Wagner M., Gaidt M.M., Stafford C., Cheng Y., Linder A., Carell T., Hornung V.. TLR8 is a sensor of RNase T2 degradation products. Cell. 2019; 179:1264–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shchepachev V., Wischnewski H., Soneson C., Arnold A.W., Azzalin C.M.. Human Mpn1 promotes post-transcriptional processing and stability of U6atac. FEBS Lett. 2015; 589:2417–2423. [DOI] [PubMed] [Google Scholar]
- 47. Puttaraju M., Perrotta A.T., Been M.D.. A circular trans-acting hepatitis delta virus ribozyme. Nucleic Acids Res. 1993; 21:4253–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suarez-Amaran L., Usai C., Di Scala M., Godoy C., Ni Y., Hommel M., Palomo L., Segura V., Olague C., Vales A. et al.. A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN-beta induction. J. Hepatol. 2017; 67:669–679. [DOI] [PubMed] [Google Scholar]
- 49. Chen Y.G., Kim M.V., Chen X., Batista P.J., Aoyama S., Wilusz J.E., Iwasaki A., Chang H.Y.. Sensing self and foreign circular RNAs by intron identity. Mol. Cell. 2017; 67:228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wesselhoeft R.A., Kowalski P.S., Parker-Hale F.C., Huang Y., Bisaria N., Anderson D.G.. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell. 2019; 74:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jang M.A., Kim E.K., Now H., Nguyen N.T., Kim W.J., Yoo J.Y., Lee J., Jeong Y.M., Kim C.H., Kim O.H. et al.. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 2015; 96:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lassig C., Matheisl S., Sparrer K.M., de Oliveira Mann C.C., Moldt M., Patel J.R., Goldeck M., Hartmann G., Garcia-Sastre A., Hornung V. et al.. ATP hydrolysis by the viral RNA sensor RIG-I prevents unintentional recognition of self-RNA. Elife. 2015; 4:e10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chiang J.J., Sparrer K.M.J., van Gent M., Lassig C., Huang T., Osterrieder N., Hopfner K.P., Gack M.U.. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018; 19:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jiang M., Zhang S., Yang Z., Lin H., Zhu J., Liu L., Wang W., Liu S., Liu W., Ma Y. et al.. Self-recognition of an inducible host lncRNA by RIG-I feedback restricts innate immune response. Cell. 2018; 173:906–919. [DOI] [PubMed] [Google Scholar]
- 55. Hwang S.Y., Sun H.Y., Lee K.H., Oh B.H., Cha Y.J., Kim B.H., Yoo J.Y.. 5′-Triphosphate-RNA-independent activation of RIG-I via RNA aptamer with enhanced antiviral activity. Nucleic Acids Res. 2012; 40:2724–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Traut T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994; 140:1–22. [DOI] [PubMed] [Google Scholar]
- 57. Zhao H., Chiaro C.R., Zhang L., Smith P.B., Chan C.Y., Pedley A.M., Pugh R.J., French J.B., Patterson A.D., Benkovic S.J.. Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J. Biol. Chem. 2015; 290:6705–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Poeck H., Besch R., Maihoefer C., Renn M., Tormo D., Morskaya S.S., Kirschnek S., Gaffal E., Landsberg J., Hellmuth J. et al.. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat. Med. 2008; 14:1256–1263. [DOI] [PubMed] [Google Scholar]
- 59. Kübler K., Gehrke N., Riemann S., Böhnert V., Zillinger T., Hartmann E., Pölcher M., Rudlowski C., Kuhn W., Hartmann G. et al.. Targeted activation of RNA helicase retinoic acid-inducible gene-I induces proimmunogenic apoptosis of human ovarian cancer cells. Cancer Res. 2010; 70:5293–5304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GenBank® is an open genetic sequence database provided by the National Institutes of Health (NIH) (https://www.ncbi.nlm.nih.gov/genbank/).
Basic Local Alignment Search Tool (BLAST) compares regions of similarity between nucleotide sequences and is provided by the NIH (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Clustal Omega was used for pairwise sequence alignment to identify regions of similarity between RNA sequences (https://www.ebi.ac.uk/Tools/psa/).
Galaxy analysis is an open source platform for data intensive sequence analysis (https://usegalaxy.org/).
RNAfold web server predicts secondary structures of RNA sequences (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi).
NGS data is publicly available under BioProject ID: PRJNA643769.