


Abstract
In the constant evolutionary battle against mobile genetic elements (MGEs), bacteria have developed several defense mechanisms, some of which target the incoming, foreign nucleic acids e.g. restriction-modification (R-M) or CRISPR-Cas systems. Some of these MGEs, including bacteriophages, have in turn evolved different strategies to evade these hurdles. It was recently shown that the siphophage CAjan and 180 other viruses use 7-deazaguanine modifications in their DNA to evade bacterial R-M systems. Among others, phage CAjan genome contains a gene coding for a DNA-modifying homolog of a tRNA-deazapurine modification enzyme, together with four 7-cyano-7-deazaguanine synthesis genes. Using the CRISPR-Cas9 genome editing tool combined with the Nanopore Sequencing (ONT) we showed that the 7-deazaguanine modification in the CAjan genome is dependent on phage-encoded genes. The modification is also site-specific and is found mainly in two separate DNA sequence contexts: GA and GGC. Homology modeling of the modifying enzyme DpdA provides insight into its probable DNA binding surface and general mode of DNA recognition.
INTRODUCTION
Bacteriophages are the most abundant biological entities in the biosphere (1). Phages have an immense genetic diversity, different lifestyle strategies and have major influence on the composition of the bacterial communities. The constant struggle for persistence between phages and their hosts is often referred to as the oldest ‘arms race.’ On one side, bacteria have developed strategies to eliminate phages, such as the use of restriction endonucleases (REs) (2), whereas phages evolved counter measures to circumvent the bacterial antiphage defense systems, which include modifying their nucleotides (3). Phage genomes are very diverse, having either single or double stranded RNA or DNA as nucleic material and even some of the viral nucleotides can deviate from the canonical A, T, C and G (4,5).
Phage CAjan belongs to the Siphoviridae family (dsDNA genome, long non-contractile tail) and infects the well-characterized strain Escherichia coli K-12 MG1655 (6,7). The genome of CAjan is resistant to digestion by several restriction enzymes (REs) (8). REs have been known for decades as a part of the innate bacterial resistance arsenal against invading exogenous DNA, such as phage genomes (9). Moreover, it was recently shown that the genome of phage CAjan encodes for several proteins with a high level of similarity to enzymes of the 7-deazaguanine biosynthesis pathway which is reported to modify certain tRNAs as well as DNA (10,11). In fact, we showed that CAjan hypermodifies its genome with a 7-deazaguanine modification, specifically 7-cyano-7-deazaguanine (also known as preQ0) (8). Remarkably, the 7-deazaguanine modification gene cluster in phage CAjan consists of twelve genes (Figure 1). Six of them can be assigned a function: four preQ0 synthesis genes (folE, queD, queE, queC) as well as yhhQ gene which codes for a putative preQ0 transporter and dpdA gene for a paralog of the archaeal guanine-tRNA translgycosylase (arcTGT) enzyme. The latter removes the G base at position 15 in target tRNA and exchanges it with preQ0 (10). The rest of the genes in the 7-deazaguanine modification cluster remain without an assigned function (Figure 1). DNA modifications can be detected in genomes using various methodologies including restriction enzyme analysis, mass spectrometry or bisulphite sequencing (12). Recently, third generation sequencing technologies have been used to detect various modifications, mostly methylations, in DNA (13–15) and even in RNA (16). Nanopore-based sequencing applied by Oxford Nanopore Technologies (ONT) is a very promising method to detect a diverse set of modifications in DNA. Here, we investigate whether preQ0 modifications in DNA can be detected by ONT sequencing and whether these substitutions occur at specific sites within phage CAjan genome. To achieve this, an in vitro synthetic DNA template identical to phage CAjan DNA was generated using the canonical nucleotides in order to provide a modification-free DNA reference for Nanopore sequencing. Moreover, using the CRISPR-Cas9 technology, we generated a series of phage mutants deleted in specific genes of the 7-deazaguanine biosynthesis pathway to examine their involvement and effect on the modification of CAjan's DNA (17). The results provide insights into the preQ0 modifications in phage CAjan genome and offer a roadmap to detect additional 7-deazaguanine modifications in DNA and the sequence motifs in which they lie.
Figure 1.

(A) The 7-deazaguanine modification gene cluster in CAjan bacteriophage (NC_028776.1) located in the region 28 621–34 560 bp. Genes are colored by function: purple are genes involved in preQ0 synthesis, orange is the gene coding for the preQ0 transporter, blue is the dpdA gene and in white are genes coding for unknown functions. Numbers correspond to gene product numbers from NC_028776. On the right side, there is a 500 bp grey rectangle for size reference and rest of the Figure is drawn to this scale. (B) Biosynthesis pathway of preQ0. The biosynthesis starts with GTP which is transformed to preQ0 by enzymes FolE, QueD, QueE and QueC. Alternatively, YhhQ can transport preQ0 from the outside. The last step is DpdA enzyme that exchanges targeted G bases with the preQ0.
MATERIALS AND METHODS
Phage-host pair
The phage investigated is Enterobacteria phage CAjan (GenBank accession number NC_028776.1) and its host is E. coli K-12, MG1655 (U00096.3).
Unmodified whole genome amplification of DNA reference
An unmodified whole genome amplification (WGA) of phage CAjan was prepared using the illustra™ Ready-To-Go™ GenomiPhi™ V3 DNA amplification kit (GE Healthcare, Pittsburgh, US), which uses Phi-29 DNA polymerase. This was followed by debranching with S1 nuclease. Briefly, 10 μl of 5× nuclease buffer and 2 μl (200U) of S1 nuclease (ThermoFischer Scientific) were mixed with 38 μl of WGA product and incubated at 25°C for 30 min followed by inactivation at 70°C for 10 min. Product was then purified using the Clean-up AX kit (A&A Biotechnology) according to the manufacturer's instructions.
pL2Cas9 construction
sgRNA targeting the phage genes of interest were cloned into pL2Cas9 (Addgene.org, plasmid #9884) (17). Pairs of designed oligonucleotides (50 μm) were phosphorylated by mixing 2 μl of each with 10 μl of 5× T4 DNA ligase buffer (Thermo Scientific), 1 μl of T4 kinase (Thermo Scientific) and 32 μl of ddH2O followed by incubating at 37°C for 30 min, and then at 65°C for 20 min. The phosphorylated oligos were then ligated into Bsal (NEB) digested pL2Cas9 (molar ratio 5:1) using T4 DNA ligase (Thermo Scientific). The ligation products were then dialyzed on membranes and transformed into the bacterial host by electroporation. Successful transformation was verified by PCR and sequencing. Oligonucleotide pairs for each deletion mutant are listed in Supplementary Table S1.
pNZ123 construction
The repair templates were constructed using Gibson assembly (18). Regions flanking the targeted gene were amplified by PCR with the DreamTaq polymerase (Thermo Scientific) according to manufacturer's protocol. PCR products were cleaned with the Clean & Concentrator-5 PCR clean-up kit (Zymo Research). The primer pairs are listed in Supplementary Table S1. Then, a one-step isothermal assembly with Xbal digested pNZ123 was performed as described elsewhere (18). Again, the resulting product was dialyzed on membranes and transformed into the host by electroporation. PCR-confirmed transformants were purified and the repair template plasmids were isolated from overnight cultures using the Plasmid Mini kit (A&A Biotechnology). The purified plasmids were then transformed into the bacterial recombinant host harboring the modified pL2Cas9, as described previously (19). Bacterial cultures containing both plasmids were used to generate the phage mutants (17).
CRISPR–Cas9 mutagenesis
The phage mutants deleted in folE, queC, dpdA or yhhQ were generated using the CRISPR–Cas9 technology as previously described (17). In short, the selected phage genes were targeted by specifically designed sgRNA encoded into pL2Cas9-derivative plasmids. Repair templates of the target genes designed to remove 150–350 bp within each gene, including the protospacers, were supplied on pNZ123-derivative plasmids. Hosts harboring both plasmids were then used for multiple cycles of phage infection, resulting in the generation of deletion mutants. Deletions were verified by PCR and confirmed by whole genome sequencing to also verify the absence of off-target mutations.
Amplification of wild-type and deletion-mutant phages and isolation of viral DNA
Wild-type phage CAjan (CAjan WT) and its phage mutants deleted in either folE, queC, dpdA or yhhQ were amplified to high titers by separately growing them in 500 mL of host cultures (w/wo modification plasmids) to an early log-phase and infecting half of the volume (125 ml) with a low multiplicity of infection (∼0.01). The culture medium was Brain–Heart Infusion (BHI, Oxoid), and for the deletion mutants, BHI was supplemented with chloramphenicol (20 μg/ml) and erythromycin (150 μg/ml) to maintain the two plasmids. Both the infected and non-infected cultures were incubated at 37°C, with agitation (200 rpm). Phage lysates were treated essentially as described for lambda phage by Sambrook and Russell (20). Followed by the phenol–chloroform DNA extraction and ethanol precipitation. The DNA was dissolved in sterile deionized water.
Restriction endonuclease digestions
Restriction digestions were prepared in 10 μl volume reaction containing ∼500 ng of genomic DNA, 1 μL of restriction enzyme: AanI, BcuI, BstEII, EcoRV, HaeIII or NsiI (ThermoFisher) and 1 μl of a 10× corresponding reaction buffer. Reactions were performed at 37°C for 1 h. Afterwards, DNA fragments were visualized on a 1% agarose gel using GelRed as the DNA dye in a loading buffer.
3D protein modeling
A homology model of the CAjan dpdA gene product was generated using SWISS-MODEL (21). A template search in SWISS-MODEL returned the crystal structure of Pyrococcus horikoshii arcTGT as the top scoring template (PDB IDs 1IQ8 and 1IT8, (22)) with 15% sequence identity and 30% similarity to CAjan DpdA. The model quality is moderate as reflected by a mid-range GMQE (Global Model Quality Estimation) score of 0.43 and a QMEAN Z-score (an estimate of the degree of nativeness of the model) of –3.5 (21). A 10-base-pair dsDNA starting model containing an extrahelical G base was extracted from the crystal structure of the DNA repair enzyme AGT in complex with DNA (PDB ID 1T38, (23)), and its sequence changed to contain A, or GC 3′ of the flipped G nucleotide. The resulting model was then docked onto the protein model using the HADDOCK server (version 2.2, (24)). In the docking protocol, active site residues Asp105, Asp63, Asp206, His132 and Phe189, and the flipped G nucleotide of DNA were designated as active residues to apply distance restraints.
Mass spectrometry of DNA
Quantitative analysis of DNA modifications was performed as described previously but with several modifications (8). Purified DNA was hydrolyzed in 10 mM Tris–HCl (pH 7.9) with 1 mM MgCl2 with Benzonase (20 U), DNase I (4 U), calf intestine phosphatase (17 U) and phosphodiesterase (0.2 U) for 16 h at ambient temperature. Following passage through a 10 kDa filter to remove proteins, the filtrate was lyophilized and resuspended to a final concentration of 0.2 μg/μl (based on initial DNA quantity). Quantification of the modified 2′-deoxynucleosides (dADG, dQ, dPreQ0, dPreQ1 and dG+) and the four canonical 2′-deoxyribonucleosides (dA, dT, dG and dC) was achieved by liquid chromatography-coupled triple quadrupole mass spectrometry (LC–MS/MS) and in-line diode array detector (LC–DAD; for quantifying canonical nucleosides). Aliquots of hydrolyzed DNA were injected onto a Phenomenex Luna Omega Polar C18 column (2.1 × 100 mm, 1.6 μm particle size) equilibrated with 98% solvent A (0.1% v/v formic acid in water) and 2% solvent B (0.1% v/v formic acid in acetonitrile) at a flow rate of 0.25 mL/min and eluted with the following solvent gradient: 2–12% B in 10 min; 12–2% B in 1 min; hold at 2% B for 5 min. The HPLC column was coupled to an Agilent 1290 Infinity DAD and an Agilent 6490 triple quadruple mass spectrometer (Agilent). The column was kept at 40°C and the auto-sampler was cooled at 4°C. The UV wavelength of the DAD was set at 260 nm and the electrospray ionization of the mass spectrometer was performed in positive ion mode with the following source parameters: drying gas temperature 200°C with a flow of 14 l/min, nebulizer gas pressure 30 psi, sheath gas temperature 400°C with a flow of 11 l/min, capillary voltage 3000 V and nozzle voltage 800 V. Compounds were quantified in multiple reaction monitoring (MRM) mode with the following m/z transitions: 310.1 → 194.1, 310.1 → 177.1, 310.1 → 293.1 for dADG, 394.1 → 163.1, 394.1 → 146.1, 394.1 → 121.1 for dQ, 292.1 → 176.1, 176.1 → 159.1, 176.1 → 52.1 for dPreQ0, 296.1 → 163.1, 296.1 → 121.1, 296.1 → 279.1 for dPreQ1 and 309.1 → 193.1, 309.1 → 176.1, 309.1 → 159.1 for dG+. External calibration curves were used for the quantification of the modified canonical 2′-deoxynucleosides. The calibration curves were constructed from replicate measurements of eight concentrations of each standard. A linear regression with r2 > 0.995 was obtained in all relevant ranges. The limit of detection (LOD), defined by a signal-to-noise ratio (S/N) ≥ 3, ranged from 0.1 to 1 fmol for the modified 2′-deoxynucleosides. Data acquisition and processing were performed using MassHunter software (Agilent).
Nanopore sequencing
Barcoded Nanopore sequencing libraries were built from phage DNAs and the WGA DNA serving as negative control without modifications. Libraries were built using the Rapid Barcoding Sequencing kit RBK-004 (ONT) and sequenced on the MinION platform using a single R9.4 flowcell. Sequencing was performed using MinKNOW (25). Base calling was performed with Guppy v.2.1.3 (26) using default parameters. Only barcoded reads were used for downstream analysis.
Nanopore data analysis
Nanopore sequencing data were analyzed with the Tombo v.1.5 software (15) for detection of modified bases. Briefly, fast5 files for each barcoded library were compared against the reference CAjan phage genome, using the Tombo ‘resquiggle’ command. Modified bases were detected in the wild-type and mutants by comparing reads of the respective samples with the phi29-amplified negative control containing no DNA modifications, using the Tombo function ‘detect_modifications model_sample_compare’. Results were further parsed within the Tombo software suite and a DNA motif logo of the modified bases were made with the MEME software (27) using ‘Zero or One Occurrence Per Sequence (zoops) approach, as recommended in the Tombo manual. From Tombo, the fraction of modified reads (ModFrac) were used for WT and all mutants for plotting the CAjan genome with modified bases in Circos (28). ModFrac is defined as a value ranging from 0 to 1 that reflects how big a fraction of individual reads has a potential modification in the specific position of the genome. For example, when the total coverage of a certain nucleotide position is 100 and the signal of 50 of these reads deviates between the two samples then the ModFrac for this position is 0.5. The same data was imported into R for further analyses and plotting using the ggplot2 package. For generating ‘corrected ModFrac values’ in Table 2, a custom Python script was used. In short, it found Gs with a high ModFrac values (>0.883) and replaced the ModFrac of two upstream nucleotides (NN) with 0, provided they were not GA. In cases where upstream nucleotide was GA only the ModFrac of A was replaced by 0.
Table 2.
Percentage of modified bases using the identified ModFrac cut-off of 0.883
| Modified sites based on ModFrac | ||||
|---|---|---|---|---|
| Strain | A | C | G | T |
| WT | 9.2 | 9.4 | 34.9 | 6.7 |
| ΔfolE | 0.0 | 0.0 | 0.0 | 0.0 |
| ΔqueC | 0.0 | 0.0 | 0.0 | 0.0 |
| ΔdpdA | 0.0 | 0.0 | 0.0 | 0.0 |
| ΔyhhQ | 9.9 | 10.0 | 36.0 | 7.8 |
| Corrected ModFrac values | ||||
| WT | 1.91 | 0.1 | 23.11 | 0.20 |
RESULTS AND DISCUSSION
Validation of the presence of DNA modifications in CAjan phage
The quantitative landscape of 7-deazaguanine modifications in the CAjan phage genome was defined by LC–MS/MS analysis of the nucleosides dADG, dQ, dPreQ0, dPreQ1 and dG+ for WT and ΔqueC phage mutant. dPreQ0 was the only 7-deazaguanine detected and was present in both WT and ΔqueC phage CAjan DNA, though ΔqueC DNA contained 12-fold lower levels than WT (3.5 per 103 nt versus 44 per 103 nt, respectively). This was expected as, although the phage QueC does not contribute to the production of dPreQ0 in the ΔqueC mutant, the phage DpdA is still present and can insert a small amount of PreQ0 synthetized by the host into the phage. Interestingly, N6-methyl-dA was also detected in both ΔqueC and WT, but with 10-fold higher levels in ΔqueC (83 per 106 nt versus 865 per 106 nt, respectively). This is similar to what was observed for the phage T4, where mutants unable to produce glucosyl 5-hydroxymethylcytosine (glc-HMC) modification had a higher amount of methylation present in the 5′-GATC-3′ sequence (29) and suggests that the activity of adenine methylase is partially inhibited by glc-HMC or dPreQ0.
Nanopore sequencing can detect preQ0 modifications
Nanopore sequencing of the 59 670-bp genome (44.7 GC%) of the WT phage CAjan and its the deletion mutants, and the WGA DNA yielded more than adequate coverage for Tombo analyses (Table 1). In previous analysis, methylation sites were detected with a relatively low average coverage of ∼ 15× (U-test applied if at least five reads were present in both samples) (15). Here, we obtained much higher coverage (between 1207× and 42 970×), which significantly increases the statistical power, thus the relevance of this study (15). The control containing only canonical bases had the lowest average coverage of 1207×, likely due to the DNA pooling. On the other hand, the presence of preQ0 seems to partly impair the Nanopore sequencing process as manifested by a lower mean of Phred quality scores for samples with preQ0 modification (WT and ΔyhhQ) compared to the other samples (Table 1).
Table 1.
Basic characteristics of genomic DNA preparations used in this study
| DNA | Genome size | Coverage | Genomic position of deletion | Sensitivity to RE digestion | Number of reads | Total bases | Mean Phred score |
|---|---|---|---|---|---|---|---|
| WT | 59 670 | 31 007 | None | − | 149 692 | 1.3 Gbp | 6.3 |
| WGA | 59 670 | 1207 | None | n.a. | 77 522 | 70.6 Mbp | 8.0 |
| ΔfolE | 59 448 | 5 845 | 30 221 | + | 19 491 | 344.6 Mbp | 7.5 |
| ΔqueC | 59 380 | 1 953 | 32 786 | + | 12 816 | 113.9 Mbp | 7.5 |
| ΔdpdA | 59 353 | 42 970 | 29 212 | + | 140 692 | 1.2 Gbp | 7.6 |
| ΔyhhQ | 59 120 | 29 334 | 31 850 | − | 122 460 | 1.0 Gbp | 6.3 |
Comparison of the unmodified reads derived from the WGA genome to those of the wild-type CAjan phage and its mutants resulted in assigning the fraction of modified reads value (ModFrac) to each nucleotide position. The Tombo package does not suggest a fixed cut-off for ModFrac values to assess whether a base is modified or not. To find this cutoff, the density of ModFrac values for each of the four nucleotides were plotted for both strands (Figure 2).
Figure 2.

Density plot of the modified fraction for each nucleotide in the CAjan WT dataset. Continuous line shows plus strand, while dashed line shows minus strand. The vertical line indicates the ModFrac cutoff calculated as the mean of the rightmost peak for G (plus strand) subtracted by one standard deviation.
In the WT phage CAjan genome, the density distribution clearly shows two groups of peaks corresponding to unmodified and modified bases. As previously shown, phage CAjan modifications occur only on G nucleotides (8). Accordingly, the peak with high ModFrac (mean for G = 0.93) values was extracted for G nucleotides (ModFrac > 0.75) and a cut-off was defined as the mean minus one standard deviation (0.883). On the reverse strand these numbers were similar and were calculated to be 0.921 and 0.871, respectively, suggesting an asymmetric motif. While the G nucleotide had the most prominent peak in the high ModFrac range, the A, C and T bases also showed smaller peaks in the same range. As discussed below, this is likely due to a neighboring effect of modified bases (15). Nanopore sequencing of the DNA genome of phage mutant ΔyhhQ led to density plots comparable to the WT. Conversely, density plots of ModFrac values were similar for both DNA strands of phage mutants ΔfolE, ΔqueC and ΔdpdA (Figure S1), indicating that their deletions eliminated the modified peaks. Although ΔdpdA is completely lacking the modifications while ΔfolE and ΔqueC mutants have a secondary modification (ModFrac ∼ 0.3) peak which is likely caused by the 12-fold lower incorporation of preQ0 derived from the host metabolism. Because the ModFrac values for each base from the forward and reverse strands, are very similar (Figure 2), only the forward strand was used for further analysis. Plotting the fraction of modified read values (cut-off 0.883) of the phage variants against the CAjan WT genome confirmed that the WT and ΔyhhQ mutant have modified positions throughout the genome (Figure 3). This was an expected outcome because the products of folE, queC and dpdA are necessary for the synthesis of preQ0, whereas YhhQ is a preQ0 transporter dispensable for de novo formation of preQ0 in the host (30). Additionally, all phage mutants showed high ModFrac values in the genomic region corresponding to their respective deletion. This is a consequence of comparing the mutant reads to the WT genome, which is required for comparative analyses in Tombo (Figure 3). Because each deletion mutants lacks a part of its genome, the comparison artificially shows high ModFrac values at the deletion sites. The respective deleted regions for each mutant were removed from the datasets for subsequent analyses. According to LC–MS/MS data a small amount of dPreQ0 was incorporated to the ΔqueC phage mutant genome, however the signal derived from 12-fold lower level of dPreQ0 was below the set ModFrac cut-off (0.883).
Figure 3.

Circular representation of phage CAjan and the fraction of modified reads per nucleotide positions for WT and phage mutants. Starting from the inside, rings display, functional genetic modules (various colors), open reading frames (black: plus strand, grey: minus strand), genome track (yellow), GC-content. The five outermost rings show ModFrac in order from inside: mutants ΔyhhQ, ΔdpdA, ΔqueC, ΔfolE, and WT ModFrac. Each of the deletion mutants lacks a part of the WT genome and the comparison artificially shows high ModFrac values at the deletion sites.
Nanopore modification calling of preQ0 is convoluted by the neighboring effect
The identified cut-off (0.883, Figure 2) was used to calculate the percentage of modified positions for each of the four nucleotides on the positive strand (Table 2, top). The WT strain and ΔyhhQ mutant have similar percentages of modified positions for each of the four nucleotides, while the ΔfolE, ΔqueC and ΔdpdA mutants are below the set ModFrac cutoff (0.883).
While all other evidences indicated that only G’s are modified in phage CAjan genome, Table 2 suggests that other nucleotides are also modified in WT and ΔyhhQ. It is documented that nucleotides neighboring a modified base can also appear modified due to the nature of Nanopore sensing (15,31). This is because the nanopore signal comes from multiply contiguous bases occupying the pore (5–6 bases) with 3 bases mostly influencing the signal in newer pores (R9, R9.5) (32). In our dataset, the two bases immediately prior to (5’) and to a much lesser extent one subsequent base (3’) of the modified base had increased ModFrac values. This pattern is not fully consistent and is muddled if several modified bases are found in close proximity (Figure 4). This neighboring effect is responsible for false-positive results of bases other than G (Table 2) in the genomes of WT CAjan and its mutant ΔyhhQ. Using a custom Python script (see Materials and Methods), we corrected for this neighboring effect in the WT genome (Table 2, bottom part). After the correction, the percentage of modified Gs dropped to 23.11% (52 per 103 nt), which is lower than what we reported previously (8) but slightly higher compared to LC-MS/MS results obtained in this study (44 per 103 nt). Other factors could affect the percentage, as discussed below, secondary motifs (GGT, GGTT) may have somewhat elevated ModFrac values and could contribute to a higher fraction of modified Gs in the mass spectrometry data. On the other hand, LC–MS/MS data could be affected by residual, unmodified host DNA, while nanopore results are specific to the modified phage DNA.
Figure 4.

Neighbouring effect. In the top part random fragment of plus strain of CAjan phage genome is represented alongside ModFrac values of ΔqueC mutant (ΔqueC ModFrac +) and WT phage (WT ModFrac +). The bottom part shows the same region for minus strand (WT and ΔqueC ModFrac -, respectively). The modified Gs are highlighted in red. High ModFrac values of two preceding bases (the neighbouring effect) can be seen in orange. Gs that are not modified are marked in green. In the dashed square a position where G in GA motif is below the set threshold of modification.
DNA is modified at two separate sites with different lengths
The cut-off defined above was used to extract significantly modified regions of 15 bases centered on the base with highest ModFrac value, and then motifs of the aligned regions were generated as described in Materials and Methods. A single significant motif (E-value 10E–1220) was produced for both strands that are exactly complementary to each other (Figure 5). The motif 5’-GRH-3’ was identified, in which the initial G is always appearing as the most conserved base in modified regions, followed by an A or G (R) and then a less conserved A, C or T (H). To further investigate the neighboring effect, all 16 possible combinations of dinucleotides were extracted from the dataset together with their corresponding ModFrac values, and the average ModFrac value for the first of the two nucleotides was plotted (Figure 6A). In support of the 5’-GRH-3’ motif identified using MEME (Figure 5), the dinucleotides GA followed by GG showed the highest average ModFrac values of all the dinucleotide combinations (Figure 6A). The dinucleotides ending with a guanine (AG, TG and CG) had average ModFrac values elevated above the background level, which is likely due to the neighboring effect discussed above. The density plots of the ModFrac values of all dinucleotide combinations (Figure 7) clearly displayed two peaks for the NG motifs, corresponding to the unmodified (ModFrac ∼ 0.15) and modified (ModFrac ∼ 0.9) bases, whereas only one peak at ∼0.9 was present for the GA motif (Figure 7C). This is the expected outcome if the increased average ModFrac values are a result of the neighboring effect, as sequences ending with a G could be modified. The GG motif shows a considerably higher average ModFrac value and larger peak around the ModFrac value 0.9 than the other dinucleotides ending with a guanine (AG, TG and CG). To investigate this pattern, we looked at the average ModFrac value of all trinucleotides (Supplementary Table S2 and Figure 6B). All trinucleotide motifs containing GA (NGA and GAN) had high ModFrac averages (>0.8). Interestingly, the GGC motif also displayed a high ModFrac value (∼0.9) on par with the GA motifs, indicating that nearly all GGC motifs are modified. This is corroborated by the density plot of the GGC motif (Figure 8A). The ModFrac values of GG dinucleotides (unlike AG, TG and CG) are affected not only by the neighboring effect from modified GA motif but also secondary GGC motif.
Figure 5.
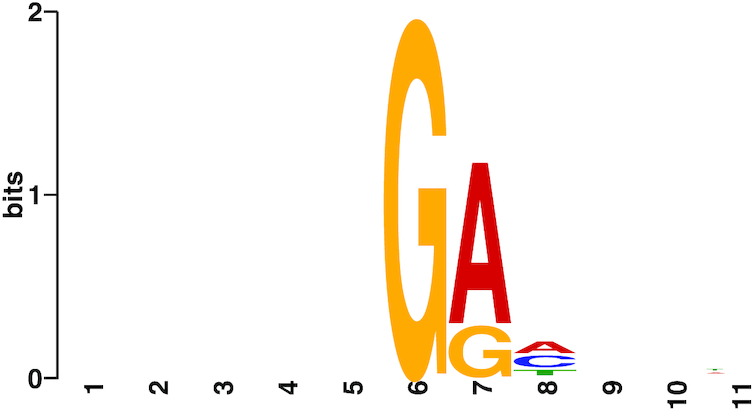
Motif predicted by MEME from significantly modified regions of the WT genome (ModFrac > 0.883). Five positions on either side of the most modified base per region were included for motif discovery.
Figure 6.

Mean ModFrac values of all combinations of dinucleotides (A) and selected trinucleotides (B) with standard errors. Only the ModFrac values of the first nucleotides are plotted.
Figure 7.

Density plots of fraction of modified dinucleotides for the WT phage CAjan dataset staring with A (panel A), C (panel B), G (panel C) and T (panel D).
Figure 8.

Density plots showing fraction of modified first bases for selected trinucleotide combinations. (A) All trinucleotide combinations starting in GG (GGN). (B) All trinucleotide combinations containing GA (NGA and GAN) with CTA included as a negative control, showing no modification nor neighbouring effect. (C) All trinucleotide combinations ending in guanine (NNG).
In the average ModFrac values of all possible 256 tetranucleotide combinations (Supplementary Table S2), all four base combinations where the first nucleotide has a ModFrac average above 0.45 can be explained by the presence of either a GGC or a GA motif.
Taken together, our data strongly indicate that G bases modified with preQ0 in the CAjan phage genome are located within GA and GGC motifs. The vast majority of these motifs were modified in the genome, however some of the sites do not cross the ModFrac threshold (Figure 4). The erroneous estimation of the motif GRH by the MEME software was caused by the fact that the modifications takes place at sites with two different motifs of different lengths. Identification of the correct motifs were further complicated by the large amounts of false positives resulting from the neighboring effect.
Restriction digestion confirms the prediction
We previously reported that preQ0 modification of phage DNA prevents some restriction enzymes from cutting viral DNA (8). In order to biologically confirm the double motif that was predicted, we selected several restriction enzymes and digested both the wild-type and the ΔqueC phage mutant genomic DNAs (Figure 9). Lower levels (12-fold lower) of dPreQ0 are insufficient to protect the majority of DNA from restriction digestion. As demonstrated in Figure 9, it is clear that an endonuclease (AanI), which does not have a G in its recognition site, is able to cut both DNA samples (Figure 9). When the recognition site contains a GA or GGC, the restriction enzymes (EcoRV and HaeIII) were able to digest the DNA from the ΔqueC phage mutant but not from the wild-type phage (Figure 9). The GC, GT and GGT containing-sites in both phage genomes were cut by restriction enzymes BcuI, NsiI and BstEII. The recognition site of BstEII contains a ubiquitous nucleotide (N) followed by an A, which could result in a GA motif in particular positions in the genome, thereby preventing the restriction enzyme activity, however confirmation of this requires further investigation.
Figure 9.

Restriction patterns for genomic DNA of WT CAjan and ΔqueC mutant. Six different restriction enzymes were used. Enzymes that recognize G in modified motif (GA and GGC) are indicated with red. Enzymes that recognize G in a motif not predicted to be modified are indicated with green (GT, GC and GGT). AanI enzyme does not have G in its recognition site. On each side of the gel is the 2-log ladder for size reference. On the right side there is a graphic representation of the expected restriction pattern. The number of predicted restriction sites is presented on the bottom.
Minor deviations from the discovered motifs
The TGA motif displayed both a slightly lower average ModFrac value than AGA, CGA and GGA (Supplementary Table S2) as well as a slightly distorted peak in the density plot (Figure 8B) compared to the other trinucleotide combinations containing the GA motif. This could indicate that this motif is slightly less likely to be modified than the other GA motifs, even though the difference is quite small. Another possibility is that not all bases are affected equally by the neighboring effect of a preQ0 modification and that the lower ModFrac values of TGA sites compared to AGA, GGA or CGA is a result of lower impacts of the neighboring effects on the thymine at the TGA sites.
The GGT motif has an average ModFrac value of 0.28 (Supplementary Table S2) however, if we exclude sites that are elevated as a result of the neighboring effects, the GGT motif showed a higher average ModFrac value than the background (Supplementary Table S2). To investigate whether the slightly higher ModFrac average was a result of a few GGT sites being completely modified or was due to a fraction of the reads for each site being modified, we inspected the density plot of the GGT motif (Figure 8A). The GGT motif showed no modified peak around the ModFrac value 0.9. Instead the GGT motif has the background peak around 0.15 shifted to the right, indicating that only a fraction of the reads for each GGT position was modified. However, when considering the 256 tetranucleotide combinations (Supplementary Table S2), not all possible GGT sites were equally modified. The GGTT site is responsible for a disproportionate amount of the increased ModFrac average of the GGT sites, particularly when the neighboring effect is considered. However, even GGTT sites did not appear to be completely modified, as none of the GGTT sites showing a ModFrac value >0.7 (Supplementary Table S2).
Structural insights into DNA recognition by CAjan DpdA
DpdA was initially identified as a paralog of the archaeal tRNA-guanine translgycosylase (arcTGT) enzyme, which removes the G base at position 15 in the D-loop of archaeal tRNA and exchanges it with preQ0 (11). ArcTGT utilizes a (β/α)8 TIM barrel catalytic domain to bind the D-loop of tRNA and places the G15 substrate in a deeply buried active site at the center of the barrel. The base exchange reaction starts with nucleophilic attack by a conserved active site Asp side chain on the C1′ of the substrate nucleotide to detach the guanine base, forming a covalent intermediate with tRNA. The incoming preQ0 replaces the detached guanine in the same pocket where its N9 atom, following deprotonation by another Asp residue, performs a second nucleophilic attack on C1′ to form the product (22,33).
Homology modeling of CAjan phage DpdA in SWISS-MODEL more closely reveals its structural similarity to the catalytic (N-terminal) domain of Pyrococcus horikoshii arcTGT (residues 1–350, PDB IDs 1IQ8 and 1IT8 (22), 1.14 Å over 276 Cα atoms, Figure 10), despites sequence identity and similarity of only 15% and 30%, respectively. 3D superposition and resulting structure-based sequence alignment show that the preQ0/guanine nucleotide binding pocket in arcTGT as well as the two catalytic aspartate residues are present in CAjan phage DpdA at the same positions (Figures 10A, B), suggesting a similar catalytic mechanism, even though the two enzymes act on different nucleic acid substrates. Based on this alignment, residues of the CAjan phage DpdA preQ0/guanine binding pocket would be Ser64, Phe67, Asp105, Gly153, Gly154, His132 and Phe189, the catalytic nucleophile for the first step of the transglycosylation reaction would be Asp206, and the second aspartate for deprotonation of incoming preQ0 is Asp63.
Figure 10.

Structural insights into DNA recognition by CAjan phage DpdA. (A) Homology model of CAjan phage DpdA (cyan) superposed on the catalytic domain of P. horikoshii arcTGT (brown, PDB ID 1IT8) viewed down the TIM barrel. (B) Close up of the superposed active sites showing preQ0 as seen bound to arcTGT. DpdA residues are labeled. (C) Docking model of dsDNA harboring a flipped G onto CAjan phage DpdA showing the putative DNA binding mode to the positively charged groove on the protein surface. The protein is shown in electrostatic surface potential representation. The active site in (A) and (C) is indicated with an asterisk.
Further, the model suggests that the active site is at the center of a positively charged groove that could accommodate double-stranded DNA (Figure 10C). Like the TGTs and DNA glycosylases involved in DNA repair (23,34), DpdA would be expected to access the substrate G by flipping the base out of the helix into the active site. However, docking of dsDNA harboring a flipped G onto that surface failed to place the extrahelical G deep enough into its binding pocket without significant clashes with the flanking nucleotides, suggesting that a conformational change in the protein and/or partial melting of the DNA duplex is needed to maximize surface complementarity with the enzyme. It is also possible that recognition of the nucleotides 3′ of the substrate G may occur via direct readout of flipped out bases as seen in DNA repair endonucleases (35). Overall, a detailed understanding of how DpdA enzymes recognize their target DNA to catalyze sequence-context-specific preQ0 insertion awaits further biochemical and structural characterization.
CONCLUSION
We have demonstrated that Nanopore sequencing can be used for detection of a novel hypermodifications (preQ0) in phage genomic DNA. Phages exhibit a large reservoir of DNA modifications and the possibility of detecting them can lead to the identification of novel phage defense and counter-defense systems. The identification of new enzymes involved in these pathways may also lead to new biotechnology applications. The workflow described in this study can also be applied to detection in other organisms, e.g. bacteria. Additionally, the Nanopore data provide information on the genomic localization of the nucleotide modification (motif), which is not the case when using only a mass spectrometry approach. Yet, our dataset also clearly demonstrated that extra care must be taken when using alignment-based software (e.g. MEME) for motif discovery. This is dictated by two main factors: (a) Nanopore sequencing is affected by the neighboring effect and (b) motifs can have different lengths, thereby complicating the analysis. Therefore, we recommend supplementing the detection process using alignment-based software with biological cross-verification, detection with an additional method (e.g. mass spectrometry), manual verification of the obtained results and in-depth evaluation of phage gene content.
DATA AVAILABILITY
Data were deposited in SRA under project PRJNA633876.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Marie-Laurence Lemay for her help with the genome editing of phage CAjan.
Contributor Information
Witold Kot, Department of Plant and Environmental Science, University of Copenhagen, Denmark.
Nikoline S Olsen, Department of Environmental Science, Aarhus University, Roskilde, Denmark.
Tue K Nielsen, Department of Plant and Environmental Science, University of Copenhagen, Denmark.
Geoffrey Hutinet, Department of Microbiology and Cell Science, University of Florida, Gainesville, FL USA.
Valérie de Crécy-Lagard, Department of Microbiology and Cell Science, University of Florida, Gainesville, FL USA; Genetics Institute, University of Florida, Gainesville, FL, USA.
Liang Cui, Antimicrobial Resistance Interdisciplinary Research Group, Singapore-MIT Alliance for Research and Technology, Singapore.
Peter C Dedon, Antimicrobial Resistance Interdisciplinary Research Group, Singapore-MIT Alliance for Research and Technology, Singapore; Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA.
Alexander B Carstens, Department of Plant and Environmental Science, University of Copenhagen, Denmark.
Sylvain Moineau, Département de biochimie, de microbiologie et de bio-informatique, Université Laval, Québec City, PQ, Canada; Groupe de recherche en écologie buccale, Faculté de médecine dentaire, Université Laval, Québec City, PQ, Canada; Félix d’Hérelle Reference Center for Bacterial Viruses, Université Laval, Québec City, PQ, Canada.
Manal A Swairjo, Department of Chemistry and Biochemistry and the Viral Information Institute, San Diego State University, San Diego, CA, USA.
Lars H Hansen, Department of Plant and Environmental Science, University of Copenhagen, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Villum Foundation [17595 to W.K.]; Human Frontier Science Program [RGP0024 to L.H., V.dC-L., S.M.]; National Research Foundation of Singapore through the Singapore-MIT Alliance for Research and Technology Antimicrobial Resistance IRG (to C.L. and P.C.D.); National Institute of General Medical Sciences [GM110588 to M.A.S.]; California Metabolic Research Foundation; S.M. holds the Tier 1 Canada Research Chair in Bacteriophages. Funding for open access charge: Villum Foundation [17595].
Conflict of interest statement. None declared.
REFERENCES
- 1. Edwards R.A., Rohwer F.. Opinion: viral metagenomics. Nat. Rev. Microbiol. 2005; 3:504–510. [DOI] [PubMed] [Google Scholar]
- 2. Tock M.R., Dryden D.T.F.. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005; 8:466–472. [DOI] [PubMed] [Google Scholar]
- 3. Samson J.E., Magadán A.H., Sabri M., Moineau S.. Revenge of the phages: defeating bacterial defences. Nat. Rev. Microbiol. 2013; 11:675–687. [DOI] [PubMed] [Google Scholar]
- 4. Wyatt G.R., Cohen S.S.. A new pyrimidine base from bacteriophage nucleic acids. Nature. 1952; 170:1072–1073. [DOI] [PubMed] [Google Scholar]
- 5. Warren R.A.J. Modified bases in bacteriophage DNAs. Annu. Rev. Microbiol. 1980; 34:137–158. [DOI] [PubMed] [Google Scholar]
- 6. Carstens A.B., Kot W., Lametsch R., Neve H., Hansen L.H.. Characterisation of a novel enterobacteria phage, CAjan, isolated from rat faeces. Arch. Virol. 2016; 161:2219–2226. [DOI] [PubMed] [Google Scholar]
- 7. Carstens A.B., Kot W., Hansen L.H.. Complete genome sequences of four novel Escherichia coli bacteriophages belonging to new phage groups. Genome Announc. 2015; 3:e00741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hutinet G., Kot W., Cui L., Hillebrand R., Balamkundu S., Gnanakalai S., Neelakandan R., Carstens A.B., Lui C.F., Tremblay D. et al.. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019; 10:5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Labrie S.J., Samson J.E., Moineau S.. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010; 8:317–327. [DOI] [PubMed] [Google Scholar]
- 10. Hutinet G., Swarjo M.A., de Crécy-Lagard V.. Deazaguanine derivatives, examples of crosstalk between RNA and DNA modification pathways. RNA Biol. 2017; 14:1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thiaville J.J., Kellner S.M., Yuan Y., Hutinet G., Thiaville P.C., Jumpathong W., Mohapatra S., Brochier-Armanet C., Letarov A.V., Hillebrand R. et al.. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E1452–E1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Plongthongkum N., Diep D.H., Zhang K.. Advances in the profiling of DNA modifications: Cytosine methylation and beyond. Nat. Rev. Genet. 2014; 15:647–661. [DOI] [PubMed] [Google Scholar]
- 13. Feng Z., Fang G., Korlach J., Clark T., Luong K., Zhang X., Wong W., Schadt E.. Detecting DNA modifications from SMRT sequencing data by modeling sequence context dependence of polymerase kinetic. PLoS Comput. Biol. 2013; 9:e1002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rand A.C., Jain M., Eizenga J.M., Musselman-Brown A., Olsen H.E., Akeson M., Paten B.. Mapping DNA methylation with high-throughput nanopore sequencing. Nat. Methods. 2017; 14:411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stoiber M.H., Quick J., Egan R., Lee J.E., Celniker S.E., Neely R., Loman N., Pennacchio L., Brown J.B.. De novo identification of DNA modifications enabled by genome-guided nanopore signal processing. 2017; bioRxiv doi:10 April 2017, preprint: not peer reviewed 10.1101/094672. [DOI]
- 16. Smith A.M., Jain M., Mulroney L., Garalde D.R., Akeson M., Jain M., Mulroney L., Garalde D.R., Akeson M.. Reading canonical and modified nucleotides in 16S ribosomal RNA using nanopore direct RNA sequencing. PLoS One. 2019; 14:e0216709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lemay M.L., Tremblay D.M., Moineau S.. Genome engineering of virulent lactococcal phages using CRISPR-Cas9. ACS Synth. Biol. 2017; 6:1351–1358. [DOI] [PubMed] [Google Scholar]
- 18. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 19. Chung C.T., Niemela S.L., Miller R.H.. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution. Biochemistry. 1989; 86:2172–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Russell D.W., Sambrook J.. Molecular Cloning: A Laboratory Manual. 1989; NY: Cold Spring Harbour. [Google Scholar]
- 21. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., De Beer T.A.P., Rempfer C., Bordoli L. et al.. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishitani R., Nureki O., Fukai S., Kijimoto T., Nameki N., Watanabe M., Kondo H., Sekine M., Okada N., Nishimura S. et al.. Crystal structure of archaeosine tRNA-guanine transglycosylase. J. Mol. Biol. 2002; 318:665–677. [DOI] [PubMed] [Google Scholar]
- 23. Daniels D.S., Woo T.T., Luu K.X., Noll D.M., Clarke N.D., Pegg A.E., Tainer J.A.. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat. Struct. Mol. Biol. 2004; 11:714–720. [DOI] [PubMed] [Google Scholar]
- 24. Van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., Van Dijk M., De Vries S.J., Bonvin A.M.J.J.. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 25. Ip C.L.C., Loose M., Tyson J.R., de Cesare M., Brown B.L., Jain M., Leggett R.M., Eccles D.A., Zalunin V., Urban J.M. et al.. MinION analysis and reference consortium: phase 1 data release and analysis. F1000Research. 2015; 4:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pomerantz A., Peñafiel N., Arteaga A., Bustamante L., Pichardo F., Coloma L.A., Barrio-Amorós C.L., Salazar-Valenzuela D., Prost S.. Real-time DNA barcoding in a rainforest using nanopore sequencing: opportunities for rapid biodiversity assessments and local capacity building. Gigascience. 2018; 7:giy033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bailey T.L., Williams N., Misleh C., Li W.W.. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006; 34:369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A.. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bryson A.L., Hwang Y., Sherrill-Mix S., Wu G.D., Lewis J.D., Black L., Clark T.A., Bushman F.D.. Covalent modification of bacteriophage T4 DNA inhibits CRISPR- Cas9. MBio. 2015; 6:e00648-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zallot R., Yuan Y., De Crécy-Lagard V.. The escherichia coli COG1738 member YhhQ is involved in 7-cyanodeazaguanine (preQ0) transport. Biomolecules. 2017; 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Q., Fang L., Yu G., Wang D., Xiao C.-L., Wang K.. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019; 10:2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rang F.J., Kloosterman W.P., de Ridder J.. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018; 19:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xie W., Liu X., Huang R.H.. Chemical trapping and crystal structure of a catalytic tRNA guanine transglycosylase covalent intermediate. Nat. Struct. Biol. 2003; 10:781–788. [DOI] [PubMed] [Google Scholar]
- 34. Lau A.Y., Schärer O.D., Samson L., Verdine G.L., Ellenberger T.. Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Cell. 1998; 95:249–258. [DOI] [PubMed] [Google Scholar]
- 35. Mol C.D., Hosfield D.J., Tainer J.A.. Abasic site recognition by two apurinic /apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat. Res. 2000; 460:211–229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were deposited in SRA under project PRJNA633876.