


Abstract
G-quadruplexes (G4s) are nucleic acid structure motifs that are of significance in chemistry and biology. The function of G4s is often governed by their interaction with G4-binding proteins. Few categories of G4-specific tools have been developed to inhibit G4–protein interactions; however, until now there is no aptamer tool being developed to do so. Herein, we present a novel L-RNA aptamer that can generally bind to D-RNA G-quadruplex (rG4) structure, and interfere with rG4–protein interaction. Using hTERC rG4 as the target for in vitro selection, we report the shortest L-aptamer being developed so far, with only 25 nucleotides. Notably, this new aptamer, L-Apt.4-1c, adopts a stem–loop structure with the loop folding into an rG4 motif with two G-quartet, demonstrates preferential binding toward rG4s over non-G4s and DNA G-quadruplexes (dG4s), and suppresses hTERC rG4–nucleolin interactions. We also show that inhibition of rG4–protein interaction using L-RNA aptamer L-Apt.4-1c is comparable to or better than G4-specific ligands such as carboxypyridostatin and QUMA-1 respectively, highlighting that our approach and findings expand the current G4 toolbox, and open a new avenue for diverse applications.
INTRODUCTION
RNA has significant biological roles in cells, including but not limited to genetic information storage, RNA catalysis and gene regulation (1,2). The function of RNA is often governed by its own structure, as well as its interactions with other biomolecules such as RNA-binding proteins (3–6). To better understand RNA biology and function, one can target the RNA of interest to reveal its regulatory role in gene activity. Over the past decades, targeting RNAs were mostly achieved via antisense oligonucleotide-based or RNA interference-based approach (7); however, this approach is limited to accessible or unstructured region of the RNA of interest. Recently, new chemical tools were being developed to target structured regions of RNA, including classical and non-classical structural motifs (8–13).
One non-classical RNA structure motif of general importance is referred to as RNA G-quadruplex (rG4). rG4s are secondary structures assembled by two or more layers of guanine (G)-quartets, in which each G-quartet is formed by four Gs via hydrogen bonds (Figure 1A) (14). G-quartets in rG4 are further stabilized by monovalent ion (Figure 1A), preferably potassium ion (K+) and sodium ion (Na+), but not in lithium ion (Li+) (15). One rG4 of particular interest is human telomerase RNA component (hTERC) rG4. Early reports have found that the 5′ end of hTERC RNA sequence is G-rich, and can fold into a parallel rG4 structure that is important for its recognition with protein partners and its cellular function (16–18). In addition, high-resolution structural study has revealed the hTERC rG4 is non-canonical, which contains 3-layered G-quartet with a single C bulge at the seventh nucleotide position (19) (Figure 1B), also commonly known as bulged rG4.
Figure 1.

Comparative sequence analysis of aptamer candidates identifies a conserved RNA secondary structure that binds to hTERC rG4. (A) Chemical structure of a G-quartet. (B) Schematic representation of hTERC non-canonical rG4 structure with a C-bulge at nucleotide position 7. (C) RNA secondary structure prediction of full-length aptamer 4 (Apt. 4) by Mfold. Three stem regions (Stem 1, 2, and 3), 1 internal loop (Loop 2), 1 hairpin loop (Loop 1), and 2 bulges (Bulge 1 and 2) are presented in this predicted structure of full-length Apt.4. (D) The sequences of the N40 region (after removing the fixed primer region) for the representative aptamer candidates are shown. Apt. 2 has one less nucleotide as identified by Sanger sequencing result. From the sequences and Mfold predicted structures of the aptamers (see Supplementary Figure S2), a conserved nucleotide region (red) is identified (also marked by asterisk below), and a stem region (green) with base pair co-variations is observed (Stem1 5′ and Stem1 3′).
Aptamers are single-stranded oligonucleotides that fold into unique structure scaffold to interact with specific region of target of interest with high affinity and selectivity (20). Among all classes, aptamers that are composed of L-RNA nucleotides, mirror image of naturally occurring D-RNA nucleotides, are referred to as L-RNA aptamer or spiegelmer (20,21). Since L-RNA nucleotide is unnatural, L-RNA aptamer is less susceptible to nuclease digestion and have a better biostability with much longer half-life than its D-counterparts (22–24). Over the years, studies have demonstrated that L-RNA aptamers can be used to detect small molecules, peptides and proteins (23–29), and several of them are currently under clinical trials (30). More recently, L-RNA aptamers that target RNA structure were also reported (26,31–33); however, they are limited to stem–loop/hairpin structured RNA region.
So far, only one L-RNA aptamer, L-Ap3-7, was developed to target rG4 structure (34). Our data demonstrated that the binding is highly selective to TERRA rG4 over other rG4s, dG4s and non-G4s, and L-Ap3-7 can inhibit TERRA rG4–RHAU53 peptide interaction (34). Motivated by these earlier findings, we wonder (i) whether this simple and robust L-RNA-based strategy can be applied to other biologically important rG4 subtypes such as bulged rG4, (ii) whether novel L-RNA aptamer can be developed to provide a more general selectivity toward rG4s (in contrast to individual rG4 selectivity) over non-G4s and dG4s; and (iii) whether the newly developed L-RNA aptamer can be used to suppress rG4-protein interactions as effectively as current rG4 ligands. In this work, we employ hTERC rG4 as the selection target and develop the first L-RNA aptamer that binds hTERC rG4 structure with strong affinity. It folds into an interesting RNA secondary structure and is the shortest L-aptamer ever reported. Application of our L-RNA aptamer showed that it preferentially recognizes rG4 over non-G4 or dG4 motifs, and can specifically interferes with newly identified hTERC rG4–nucleolin interactions, with inhibition efficiency comparable to or better than the state-of-the-art rG4 targeting small molecules.
MATERIALS AND METHODS
Materials
All oligonucleotides, including primers used in selection cycles, M13 primers (for colony polymerase chain reaction; pcr), aptamer candidate templates, as well as DNA N40 library template, D-RNA oligos and 5′ FAM-labeled G4s used in this study were chemically synthesized from either Beijing Genomics Institute (BGI), Integrated DNA Technologies (IDT) or Genewiz. While 5′ biotinylated L-hTERC rG4, 5′ FAM-labeled L-hTERC rG4 and 5′ FAM – L-Apt.4-1c were synthesized from Bio-synthesis Inc., while unlabeled L-RNA aptamers used were synthesized either from Bio-synthesis Inc. or ChemGene. Dynabeads™ MyOne™ Streptavidin C1 magnetic beads (Invitrogen) were used in capturing bound RNAs. T7 in vitro transcription, reverse transcription, plasmid preparation and plasmid extraction were performed using the New England BioLabs (NEB)’s HiScribe™ ‘T7 High Yield RNA Synthesis Kit’, SuperScript™ III (SSIII) First-Strand Synthesis System (Invitrogen), TOPO-TA Cloning Kit (Invitrogen) and Tiangen Biotech (Beijing)’s TIANprep Mini Plasmid Kit, respectively. Trans5α chemically competent cells used for cloning were purchased from Transgen Biotech (Beijing). The RHAU53 peptide and nucleolin protein used for the binding and inhibition study were ordered from OriGene. With the exception of last mining cycle, all PCRs amplifications were carried out with NEB’s Q5 Hot Start High-Fidelity Master Mix, while Thermo Scientific's DreamTaq Polymerase was used in the last mining cycle to introduce A-tail at the 3′ end of the double-stranded DNA (dsDNA) library for TOPO-TA cloning purpose.
Methods
In vitro selection (SELEX)
RNA library preparation
The library reaction of 100 μl was prepared consisting of 10 U/μl SSIII reverse transcriptase, 1 mM dNTP mixture and reverse transcription buffer of 1× concentration containing; [1 mM DTT, 4 mM MgCl2, 150 mM LiCl and 20 mM Tris-HCl (pH 7.5)] and 5 μM DNA N40 library template with 5 μM reverse selection primer (Supplementary Table S1) were mixed to generate dsDNA (see step 1 Supplementary Figure S1). The extension products obtained were column purified (Zymoclean Gel DNA Recovery kit, NEB) and employed as the template dsDNA in a 40 μl T7 in vitro transcription in accordance with the manufacturer’s instruction the NEB’s HiScribeTM ‘T7 High Yield RNA Synthesis Kit’. The reaction mixture was well mixed and incubated for 3.5 h at 37°C, followed by 15 min after the addition of 2 U/μl Turbo DNase. 2× RNA stopping dye (NEB) was added, the samples were heated at 95°C for 3 min and resolved by 12% denaturing polyacrylamide gel electrophoresis (PAGE). Bands of correct size were cut using new razor blade, crushed using sterile pipette tip and incubated at 4°C overnight in 80 μl/well extraction buffer containing 1× Tris-EDTA and 0.8 M LiCl while shaking at 1300 rpm. Zymo RNA clean and concentrator column purification were then followed according to manufacturer’s instructions to obtain the purified D-RNA pool for next step (see step 2 Supplementary Figure S1).
Negative selection
As depicted in Supplementary Figure S1, this step was conducted before the positive selection to reduce the amount of non-specific RNAs. First, 3 mg streptavidin dynabeads were activated and washed as per manufacturer’s instructions, then incubated at room temperature for an hour with 0.1 mg/ml yeast tRNA (to reduce beads non-specific binding sites) as shown in step 3 Supplementary Figure S1. While in a different reaction, a 300 μl reaction mixture containing the RNA library pool, KCl, MgCl2 and Tris-HCl with concentrations according to corresponding selection round condition as shown in Supplementary Table S2 was prepared. This condition was maintained throughout the negative and positive selections of each selection rounds. Then, the reaction mixture was heated at 70°C for 3 min and followed by cooling for 15 min at the selection temperature (see Supplementary Table S2). Negative selection begins with the addition of 1 mg of prepared dynabeads to the mixture. The mixture was then incubated at 300 rpm according to the corresponding temperature and negative selection time (refer to Supplementary Table S2 for details). The supernatant was extracted (by placing the reaction tube to a magnet) and used directly for positive selection, while beads binding RNA were discarded (step 5 Supplementary Figure S1).
Positive selection
5′biotinylated L-hTERC rG4 was added to the reaction mixture (as highlighted in Supplementary Table S2 and step 6 Supplementary Figure S1) and incubated at 300 rpm for 30 min (note: this changes to 5 min in the last selection round). Then, the reaction was added to a 2 mg of the prepared dynabeads (step 7 Supplementary Figure S1) and incubated for additional half an hour. The supernatant was discarded (step 8 Supplementary Figure S1), and 600 μl washing buffer with the same concentrations of KCl, MgCl2 and Tris-HCl was added to the dynabeads, then pipette mixed and incubated for the stated washing time (see Supplementary Table S2). Washing was repeated five consecutive times before the captured RNAs were eluted with 250 μl of elution buffer containing 25 mM NaOH and 1 mM EDTA. Five microliters of (of 1 M) Tris-HCl (pH 7.5) was added to neutralize the mixture, and then followed by column purification (Zymo RNA clean and concentrator), step 9 Supplementary Figure S1. The purified RNAs were then reversed transcribed (as shown in step 10 Supplementary Figure S1) in 60 μl reaction mixture, consisting of 10 U/μl Superscript III reverse transcriptase, 1 mM dNTP mixture and 1× concentration of reverse transcription (RT) buffer containing; [1mM DTT, 4 mM MgCl2, 150 mM LiCl and 20 mM Tris-HCl (pH 7.5)] for 15 min. After the reverse transcription, 3 μl of 2 M NaOH was added and incubated at 95°C for 10 min to denature the SSIII and degrade the unwanted RNA template. Then, the mixture was neutralized by the addition of 15 μl of 1 M Tris-HCl (pH 7.5) before column purification (zymo RNA clean and concentrator, NEB). The corresponding single-stranded DNA (ssDNA/cDNA) products were PCR amplified using the forward and reverse selection primers as shown in Supplementary Table S1, also see step 11 Supplementary Figure S1. The number of PCR cycles chosen is shown in Supplementary Table S2, and subsequently the dsDNA products were used for T7 in vitro transcription for the next selection round. Overall, seven selection rounds were performed in this study and their corresponding conditions are listed in Supplementary Table S2.
TA cloning
The ssDNA from the last SELEX round (seventh round) were PCR amplified using DreamTaq DNA polymerase (Thermo Scientific) to introduce an A-tail to the PCR products for effective ligation and vector transformation. As depicted in step 12 Supplementary Figure S1, the dsDNAs were ligated with TOPO vector and cloned into Trans5α chemically competent cell (Transgen Biotech) using Invitrogen’s TOPO-TA Cloning Kit. The bacteria were incubated at 37°C for 15 h on a 50 μg/ml ampicillin containing LB agar plate. Individual colonies (with good shape, see Supplementary Figure S1) were picked using pipette tip and added to a premixed polymerase reaction mixture containing Q5® Hot Start High-Fidelity Master Mix, and M13 forward and reverse primers. The mixture was PCR amplified (step 13 Supplementary Figure S1) and 2 μl of 6× orange loading dye (NEB) was added to the products and electrophoresed by 2% agarose gel at 120 V for 40 min. Colonies with correct size were picked and dropped (with sterile pipette tips) into 5 ml of 50 μg/ml ampicillin-containing LB medium for replication and growth. Samples are then incubated for 16 h at 250 rpm and at 37°C, TIANprep Mini Plasmid Kit (Tiangen) was used for the plasmid extraction and sent for Sanger Sequencing (Genewiz service was used), see steps 14 and 15 Supplementary Figure S1.
Sequence analysis and RNA secondary structure prediction
Sequencing data are analyzed using SnapGene software. The linkers/primers regions were identified by the add primer function. Then, the N40 random region was identified [middle of the two primers, as in the oligos design (see Supplementary Table S1)].
Structure of the sequences were predicted using Mfold web server (RNA folding form at http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form). For Supplementary Figure S3, bases in the conserved region (I,i+1,…,i+k-1) were forced to be single stranded by inputting P I 0 k function on 1 line in the constraint box. This was to better understand the similarities between the candidates, as the co-variation in the stem loops of some of the candidates interrupt with the loop folding prediction.
Electrophoretic mobility shift assay (EMSA)
A 20 μl reaction mixtures consisting of 1 nM 5′ FAM labeled D/L-hTERC rG4 (wild-type or mutants rG4s), 25 mM Tris-HCl of pH 7.5, 150 mM KCl (except for Li+-dependent experiment where 150 mM LiCl was used instead of KCl), 1 mM MgCl2 (except for Mg2+-dependent experiment, where no Mg2+ was added in reaction mixture, reaction buffer and gel), 8% sucrose with varying aptamer concentrations were prepared and heated at 75°C for 3 min, then slowly cool down to 4°C, samples are then kept at 4°C overnight. Fifteen microliters of samples are loaded onto a 12% native non-denaturing polyacrylamide gel (acrylamide:bis-acrylamide,19:1) consisting of 150 mM potassium acetate, 1 mM magnesium chloride and 25 mM Tris-HCl (pH 7.5) and electrophoresed at 70 mA for 120 min on ice. At the end of 120 min, gel scanning was done using FLA-9000 FujiFilm Gel Imager at 650 V and quantified by ImageJ software. The curve fitting and Kd evaluations was done using Graphpad Prism software using the one site-specific binding model.
Circular dichroism spectroscopy
CD experiment was conducted as we previous reported (35,36). Briefly, the reaction mixture consisted of 5 μM L-Apt.4-1c, 10 mM LiCac (pH 7.0), 150 mM KCl or 150 mM LiCl in a 2 ml total reaction. Then, the reaction mixtures were thoroughly mixed, followed by heating for 5 min at 95°C (to denature the RNAs) and cooled down for 15 min at 25°C (for RNAs renaturation). Samples were transferred into a quartz cuvette (Hellma Analytics) with 1-cm length path and CD spectra were recorded every 1 nm by scanning the aptamer at 220–310 nm range at room temperature. Data shown were average of two scans at interval of 2 s/nm (37,38). They were then standardized to ‘molar residue ellipticity and smoothed over 5 nm’ (39). The measurement was conducted using a Jasco CD spectrophotometer (J-1500).
UV-melting spectroscopy
Cary 100 UV-Vis spectrophotometer was employed for the UV-melting experiment. The reaction mixture was prepared exactly with same conditions as mentioned for the CD experiment. Data were collected over 0.5°C while heating over the temperature range of 20–95°C and monitoring the aptamer transitions at 295 nm. Results are smoothed over 5 nm.
Ligand enhanced fluorescence assay
Reaction mixture of 100 μl containing 1 μM L-Apt.4-1c, 10 mM LiCac buffer (pH 7.0), 150 mM KCl or 150 mM LiCl and 1 μM ligand; N-methyl mesophorphyrin IX (NMM) or Thioflavin T (ThT) ligands were prepared. The reaction was mixed well (without ligand) then denatured for 3 min at 95°C then allowed to cooled down for 15 min at 25°C. Then, transferred into a quartz cuvette (obtained from Wuxi Jinghe Optical Instrument Co.) of 1-cm length path. Samples were excited at 394 and 425 nm for NMM and ThT, respectively. Using an entrance and exit slit of 5 nm, samples were scanned from 550 to 750 nm and 440 to 700 nm for NMM and ThT, respectively. Data were recorded every 2 nm at room temperature. This experiment was conducted using HORBIA FluoroMax – 4 instruments. All data analysis was performed using Microsoft Excel.
EMSA for binding specificity
Reaction mixtures consisting of 1 nM 5′ FAM labeled target, 25 mM Tris-HCl (pH 7.5), 150 mM KCl, 1 mM MgCl2 and 8% sucrose were prepared. In positive reaction samples, 543 nM of L-Apt. 4-1c was added. The samples were denatured for 3 min at 75°C and slowly cool down to 4°C for half an hour. Then, 15 μl was loaded onto 12% non-denaturing PAGE containing 150 mM potassium acetate, 1 mM magnesium chloride and 25 mM Tris-HCl (pH 7.5) and electrophoresed at 70 mA (current) for 120 min followed by gel scanning using FLA-9000 FujiFilm Gel Imager at 650 V and quantified by ImageJ software. The curve fitting and Kd evaluations were done using Graphpad Prism software by using the one site-specific binding model.
EMSA for RHAU53/nucleolin interaction with D-hTERC rG4 or L-Apt.4-1c
A 20 μl reaction mixtures consisting of 5 nM 5′ FAM labeled D-hTERC rG4 or L-Apt.4-1c, 25 mM Tris-HCl of pH 7.5, 150 mM KCl, 1 mM MgCl2, 8% sucrose and varying protein (RHAU53 or Nucleolin) concentrations were prepared and denatured for 3 min at 75°C, then slowly cool down to 4°C for half an hour. Fifteen microliters of samples are loaded to a 6% native non-denaturing polyacrylamide gel (acrylamide:bis-acrylamide,37.5:1) containing 40 mM potassium acetate, 1 mM magnesium chloride and 0.5× TBE buffer of pH 8.3 (Biochemical). Then, electrophoresed at 25 mA (current) for 75 min on ice. At the end of 75 min, gel scanning was done as mentioned earlier.
EMSA for inhibition assay
Two sets of 120 μl reactions consisting of 25 mM Tris-HCl (pH 7.5), 150 mM KCl, 1 mM MgCl2 and 8% sucrose were prepared. Each set of reactions was splinted into 12 pcr tubes. To one set an increasing concentration of L-Apt., 4-1c was added (aptamer reactions), while 5 nM 5′ FAM labeled hTERC rG4 was added to the other reactions (hTERC reactions). Both reactions were then denatured for 3 min at 75°C and slowly cool down to 4°C. Then, 80 nM protein/peptide (Nucleolin/RHAU53) was added to the hTERC reactions (hTERC–protein reactions) except control tubes and incubated for 30 min at 37°C. Then, the aptamer reactions were added to the hTERC–protein reactions and incubated for additional 30 min at 37°C, then slowly cool down to 4°C for half an hour. Then, 15 μl of samples are loaded to a 6% native non-denaturing polyacrylamide gel (acrylamide:bis-acrylamide,37.5:1) containing 40 mM potassium acetate, 1 mM magnesium chloride and 0.5× Tris Borate EDTA (TBE) buffer of pH 8.3 (Biorad). Then, electrophoresed at 25 mA (current) for 75 min on ice. The gel was scanned and quantified as mentioned above. The curve fitting and IC50 evaluations were done using the log[inhibitor] versus normalized response – variable slope model of Graphpad Prism software.
Microscale thermophoresis (MST) binding assays in standard reaction buffer
Sixteen sets of 10 μl reaction consisting of 25 mM Tris-HCl (pH 7.5), 150 mM KCl, 1 mM MgCl2, 40 nM FAM hTERC rG4 and increasing concentrations of L-Apt.4-1c or carboxypyridostatin (cPDS) or QUMA-1 ligand or GAR peptide were prepared and heated at 75°C for 3 min and slowly cool down to 4°C. Note, in the case of cPDS, QUMA-1 ligand and GAR peptide assays, the hTERC rG4 was denatured alone and cool down to 4°C before cPDS or QUMA-1 or GAR peptide are added to the reaction, then incubated at 37°C for 30 min and slowly cool down to 4°C for half an hour. Samples were then loaded to MST (Nano-Temper Monolith NT.115) capillary tubes, then measurement were conducted at 25°C using blue light mode from the binding software. Data were analyzed with MST nano temper analysis (nta) analysis software using the Kd mode analysis.
Microscale thermophoresis (MST) inhibition assays in standard reaction buffer
Sixteen sets of 10 μl reaction consisting of 25 mM Tris-HCl (pH 7.5), 150 mM KCl, 1 mM MgCl2, 40 nM FAM hTERC rG4 or VEGF dG4 were prepared and heated at 75°C for 3 min and slowly cool down to 4°C. While increasing concentrations of L-Apt.4-1c were denatured and cooled down separately, 80 nM of nucleolin was then added into each of the tubes and incubated at 37°C for 30 min. Then, the varying L-Apt.4-1c or cPDS or QUMA-1 was added and incubated for additional 30 min at 37°C and slowly cool down to 4°C for half an hour. Samples were then loaded to MST capillary tubes, then measurement was conducted at 25°C using blue light mode from the binding software. Data were analyzed with MST nta analysis software using the Kd mode analysis.
Bio-stability test on L/D Apt.4-1c
About 10 nM initial concentration of either L or D-Apt.4-1c was dissolved in ultra-pure water and denatured for 3 min at 75°C and slowly cool down to 37°C. Then, 5% fetal bovine serum (FBS) final concentration was added to L-Apt.4-1c reaction and 0.2% FBS added to D-Apt.4-1c reaction except for the negative control reaction (where no FBS is added), reactions are then mixed well, vortex and incubated at 37°C immediately. About 5 μl of samples are withdrawn at varying time intervals (2–120 min), and 1× denaturing formamide gel loading orange dye is added immediately to quench the reaction and stored at −20°C pending analysis. To monitor the stability of the aptamers, 15% denaturing PAGE was prepared and samples were first defrosted and heated at 95°C for 3 min before loading to the gel. Gel was pre-heated at 300 V for 15 min before samples are loaded and run at 300 V for 25 min after samples are loaded. Gel was stained in 1× SYBR Gold solution for 5 min and visualized using Bio-Rad ChemiDoc™ touch imaging system.
Microscale thermophoresis (MST) binding assays in fetal bovine serum (FBS)
Sixteen sets of 10 μl reaction consisting of 25 mM Tris-HCl (pH 7.5), 150 mM KCl, 1 mM MgCl2 and 40 nM FAM hTERC rG4 were heated at 75°C for 3 min and slowly cool down to 4°C, then 1× ribonuclease inhibitor final concentration was added to each tubes followed by 0.2% FBS final concentration. Separately, an increasing concentration of L-Apt.4-1c (0.15 – 5000 nM) were prepared and heated at 75°C for 3 min and slowly cool down to 4°C. Then, the varying L-Apt.4-1c were added (to the hTERC rG4 plus FBS tubes) and incubated for additional 30 min at 37°C and slowly cool down to 4°C for half an hour. Samples were then loaded to MST capillary tubes, then measurement was conducted at 25°C using blue light mode from the binding software. Data were analyzed with MST nano temper analysis (nta) analysis software using the Kd mode analysis.
Microscale thermophoresis (MST) inhibition assays in fetal bovine serum (FBS)
Sixteen sets of 10 μl reaction consisting of 25 mM Tris-HCl (pH 7.5), 150 mM KCl, 1 mM MgCl2 and 40 nM FAM hTERC rG4 were prepared and heated at 75°C for 3 min and slowly cool down to 4°C, then 1× final concentrations of ribonuclease and protease inhibitors were added to each tubes and followed by 0.2% FBS final concentration. While increasing concentrations of L-Apt.4-1c were denatured and cooled down separately. About 80 nM of nucleolin was then added into each of the tubes and incubated at 37°C for 30 min. Then, the varying L-Apt.4-1c were added and incubated for additional 30 min at 37°C and slowly cool down to 4°C for half an hour. Samples were then loaded to MST capillary tubes, then measurement was conducted at 25°C using blue light mode from the binding software. Data were analyzed with MST nta analysis software using the Kd mode analysis.
RESULTS
Development of D-RNA aptamer to bind with L-rG4 in hTERC RNA
To develop L-RNA aptamer for D-rG4 in hTERC RNA, we initially designed the hTERC rG4 in L-RNA form and attached a biotin group to its 5′ end for streptavidin bead pull down purpose in the Systematic Evolution of Ligands by EXponential enrichment (SELEX) experiment (Supplementary Table S1 and Supplementary Figure S1). Then, we performed SELEX using D-RNA library pool with a 40-nucleotide randomized aptamer region (N40) (Supplementary Table S1). The D-RNA library underwent a negative selection and beads-specific RNAs were removed. Then, the beads non-specific RNAs were incubated with L-hTERC rG4 (positive selection) under defined conditions as shown in Supplementary Table S2 (see ‘Materials and Methods’ section and Supplementary Figure S1 for details). After seven rounds of negative and positive selections (Supplementary Table S2), enriched D-RNA aptamer candidates were cloned and selected colonies were sent for Sanger sequencing to obtain the sequences of the D-aptamer candidates for downstream analysis (Supplementary Figure S1).
From the Sanger sequencing results, five representative sequences were identified (Supplementary Table S3). A representative of the candidates (Apt.4) was presented in Figure 1C. Using RNA secondary structure prediction program Mfold (40), the full-length Apt.4, which consisted of 78 nucleotides (N40 aptamer region plus 38-nucleotide fixed primer region), was predicted to contain 3 stems, 2 loops and 2 bulges (Figure 1C). It is of note that Apt.2 has one less nucleotide as revealed from the Sanger sequencing data (Supplementary Table S3), which may be due to deletion occurred either in transcription, reverse transcription or PCR step in SELEX (Supplementary Figure S1), which is not uncommon. To verify the selected D-RNA aptamer candidates binding to L-hTERC rG4, we have performed direct binding assays between full-length D-Apt.4 and D-Apt.5 to L-hTERC rG4, monitored by Electrophoretic Mobility Shift Assay (EMSA) at 4°C, and found both D-aptamers to interact with its L-target (Supplementary Figure S2), illustrating the validity of our approach.
Comparative sequence analysis was further performed by first extracting the N40 region of the five aptamer candidates, and then aligned their sequences to reveal any similarity and difference between the candidates. Interestingly, we found a conserved region (Figure 1D, highlighted in red, and asterisk beneath). For the rest of the sequences in the N40 region, no apparent sequence conservation can be detected in the five candidates. Since the universally conserved region is located in the predicted loop I of the aptamer (Figure 1C), we constrained these nucleotides to be single-stranded in the RNA secondary structure prediction in Mfold, and found that a similar stem–loop structure was obtained for all five candidates (Supplementary Figure S3). By mapping the base pairing patterns of these predicted secondary structures to their corresponding sequences in Figure 1D, we identified variable stem lengths and more importantly base pair co-variations in the stem region (Figure 1D), which strongly supports the presence of a stem region in the aptamer. For this, we focused on Apt.4 and its derivatives in this work (Supplementary Table S4).
Mutagenesis and spectroscopic analysis of selected D-RNA aptamer
The comparative sequence analysis performed above has motivated us to examine the predicted secondary structure of D-Apt.4 further and revealed its importance in binding to L-hTERC rG4. Given the variable stem length observed in the aptamer candidates (Figure 1D), we designed a truncated version of Apt.4 aptamer of only 25 nucleotides long (Apt.4-1), which was predicted by Mfold to fold into a stem region with five base pairs and a loop region with 15 nucleotides (Figure 2A). Using EMSA, we found that D-Apt.4-1 interacted with L-hTERC rG4 with a dissociation constant (Kd) value of 153 ± 24 nM (Supplementary Figure S4), suggesting that this construct with shorten stem region also binds strongly with its target.
Figure 2.
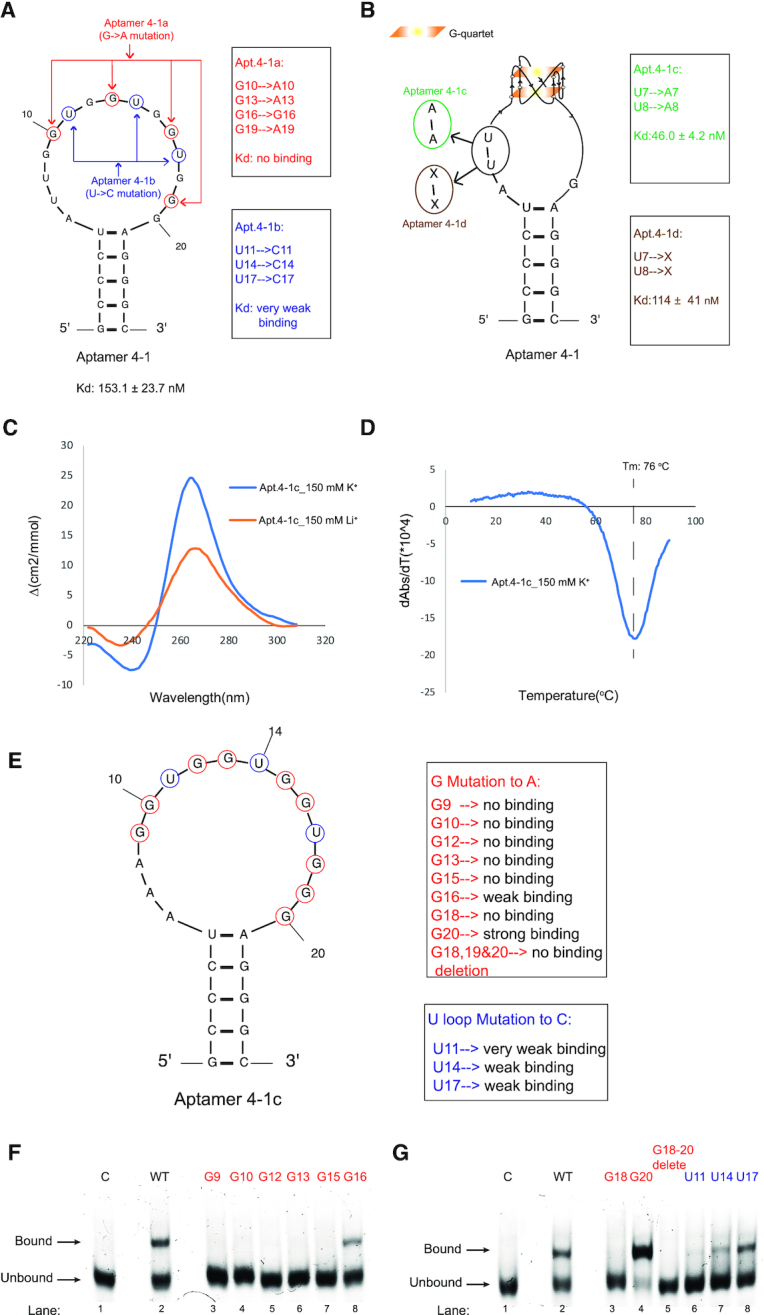
Mutagenesis and biophysical analysis of Apt. 4-1 uncovers the formation of rG4 in the conserved region and is required for binding to hTERC rG4. (A) Mutation of Apt. 4-1 to Apt. 4-1a and Apt. 4-1b: Apt. 4-1a replaces 4 Gs potentially involved in G-quartet of rG4 to As (G10, G13, G16, G19 to A10, A13, A16, A19) and this mutant abolishes the binding (see Supplementary Figure S5A). Apt. 4-1b replaces 3 Us potentially involved in the loops of rG4 to Cs (U11, U14, U17 to C11, C14, C17) and this mutant shows very weak binding (see Supplementary Figure S5B). (B) Mutation of Apt. 4-1 to Apt. 4-1c and Apt. 4-1d: Apt. 4-1c replaces U7U8 to A7A8 and this mutant strengthens the binding, with a Kd of 46.0 ± 4.2 nM (see Supplementary Figure S6). Apt.4-1d deletes U7U8 and this mutant shows very weak binding (see Supplementary Figure S7). (C) CD profile of D-Apt. 4-1c. The CD signal is dependent on monovalent ion (150 mM K+ versus 150 mM Li+), and the CD spectra shows a positive peak at 264 nm and negative peak at 240 nm under K+ condition, suggestive of parallel rG4 formation. (D) UV-melting of D-Apt. 4-1c monitored at 295 nm. A distinctive negative peak (hypochromic shift) is observed in the derivative plot shown, supporting the formation of rG4, with a melting temperature (Tm) of 76°C under 150 mM K+ condition. (E) Single point mutations of Apt. 4-1c where G9, G10, G12, G13, G15, G16, G18 and G20 are individually mutated to A9, A10, A12, A13, A15, A16, A18 and A20 respectively, and G18-20 deletion. Also, U11, U14 and U17 were individually mutated to C11, C14 and C17 respectively. (F–G) Except G20 to A20 that showed stronger binding to hTERC rG4 as compared to wild-type aptamer, G16 to A16, U14 to C14 and U17 to C17 and all remaining mutants showed weaker or no binding to hTERC rG4 as compared to wild-type aptamer.
The 15 nucleotides long loop predicted in Apt. 4-1 is somewhat unexpected (Figure 2A) as RNA generally has loop size of 4–8 nucleotides long (41,42). A closer inspection of the conserved sequence region (Figure 1D) within the long loop identified a putative rG4 sequence, with potential to form two-layered G-quartets. To test this possibility, mutagenesis experiments were conducted and analysed by EMSA. We mutated the Gs potentially involved in G-quartet of rG4 to As (G10, G13, G16, G19 to A10, A13, A16, A19) and referred this mutant as Apt.4-1a (Figure 2A, red), and found no binding (Supplementary Figure S5A). Moreover, we mutated the 3 Us potentially involved in the loops of rG4 to Cs (U11, U14, U17 to C11, C14, C17) and referred this mutant as Apt.4-1b (Figure 2A, blue), and found very weak binding with Kd > 1 μM (Supplementary Figure S5B). These results suggested that these nucleotides are critical for the binding to L-hTERC rG4, and we tentatively assigned rG4 formation in the loop region of the aptamer (Figure 2B). Furthermore, we attempted to examine the binding affinity by mutating the U7U8 to A7A8 in Apt.4-1, as an attempt to minimize the Gs in the putative rG4 to potentially base pair with U7U8 in the loops, and referred this mutant as Apt.4-1c (Figure 2B, green), and revealed a significant enhanced binding to hTERC rG4 and the Kd was found to be 46.0 ± 4.2 nM (Supplementary Figure S6). We also designed an additional construct by deleting U7U8 and referred this mutant as Apt.4-1d (Figure 2B, brown), and found it to bind very weakly with Kd > 1 μM (Supplementary Figure S7). As such, we used the stronger binding Apt.4-1c for downstream experiments.
To further assess the formation of rG4 in the aptamer, we performed a number of spectroscopic assays that can provide distinctive signals for rG4, if present (43). First, circular dichroism (CD) was carried out on Apt.4-1c under 150 mM K+ or Li+-containing conditions at room temperature (Figure 2C). The CD signal was found to be dependent on monovalent cation (K+ versus Li+), suggestive of rG4 formation (Figure 2C). In addition, the CD profile showed the presence of a positive peak at 264 nm and negative peak at 240 nm under K+ condition (Figure 2C), indicating the formation of an rG4 with parallel topology in Apt.4-1c. Second, UV-melting monitored at 295 nm was performed on Apt.4-1c under 150 mM K+, and a hyperchromic shift UV profile was detected (Figure 2D), supporting rG4 formation. The melting temperature (Tm) was determined to be 76°C (Figure 2D), highlighting that the rG4 in Apt.4-1c is thermostable. Last, we also conducted ligand-enhanced fluorescence assays at room temperature using G4-specific ligands N-methyl mesophorphyrin IX (NMM) and Thioflavin T (ThT), and the data showed that enhanced fluorescence signals were observed for both ligands (2.5- and 3-fold increase for NMM and ThT, respectively) under 150 mM K+ as compared to 150 mM Li+ (Supplementary Figure S8).
To investigate the critical nucleotides in aptamer that contribute to the binding to hTERC rG4, we designed 12 additional Apt.4-1c mutants (Supplementary Table S4) and conducted binding analysis using EMSA (Figure 2E–G). We found that mutating individual Gs that may be involved in rG4 formation either disrupts or weaken the binding, except for G20 to A20 mutation (Figure 2G). This is possibly because G18, instead of G20, is involved in rG4 formation in the fourth G-tract (together with G19), and mutating G20 to A20 likely led to more defined rG4 (with two Gs in each tract), and thus enhanced binding affinity. Interestingly, U14 to C14, G16 to A16, U17 to C17 mutations showed weaker binding (but not complete disruption) when compared to Apt.4-1c wild-type (Figure 2F–G), suggesting some flexibility in these three nucleotides in recognition and binding to hTERC rG4. Collectively, these results revealed the important nucleotides of aptamer that are involved in binding, and provided substantial evidence that Apt.4-1c contains a thermostable rG4 motif with parallel topology.
Binding affinity and enantiomeric specificity of L-Apt.4-1c with D-hTERC rG4
To evaluate the effect of aptamer and target chirality on binding, the mirror images of the D-Apt.4-1c and L-hTERC rG4 were first designed, and binding assay was performed on them (L-Apt.4-1c and D-hTERC rG4). EMSA showed that L-Apt.4-1c interacted with D-hTERC rG4 (Figure 3A). We further investigated and compared the binding affinities between D-Apt.4-1c with L-hTERC rG4 versus L-Apt.4-1c with D-hTERC rG4, and found them to be similar to each other, with the D-Apt.4-1c with L-hTERC rG4 pair to be 46.0 ± 4.2 nM and L-Apt.4-1c with D-hTERC rG4 pair to be 74.9 ± 7.5 nM (Figure 3B). The difference in KdS observed was not uncommon and have been reported by us and others (33,34), and we reasoned this may be caused by slight difference in qualities of oligonucleotides obtained. To study whether the binding is enantiomeric-specific, we tested combinations of aptamer and target in the same or opposite chirality, and found that bindings were only observed when the aptamer and target were in opposite chirality (Figure 3C), whereas no binding was detected when aptamer and target were in same chirality, supporting the interaction between Apt.4-1c and hTERC rG4 is enantiomeric-specific. Overall, we demonstrated that L-RNA aptamer can be developed to bind with D-rG4 (i.e. D-hTERC rG4 in this work), and it is worth highlighting that our aptamer L-Apt.4-1c is the shortest L-aptamer developed so far (only 25 nt) for any target of interest.
Figure 3.
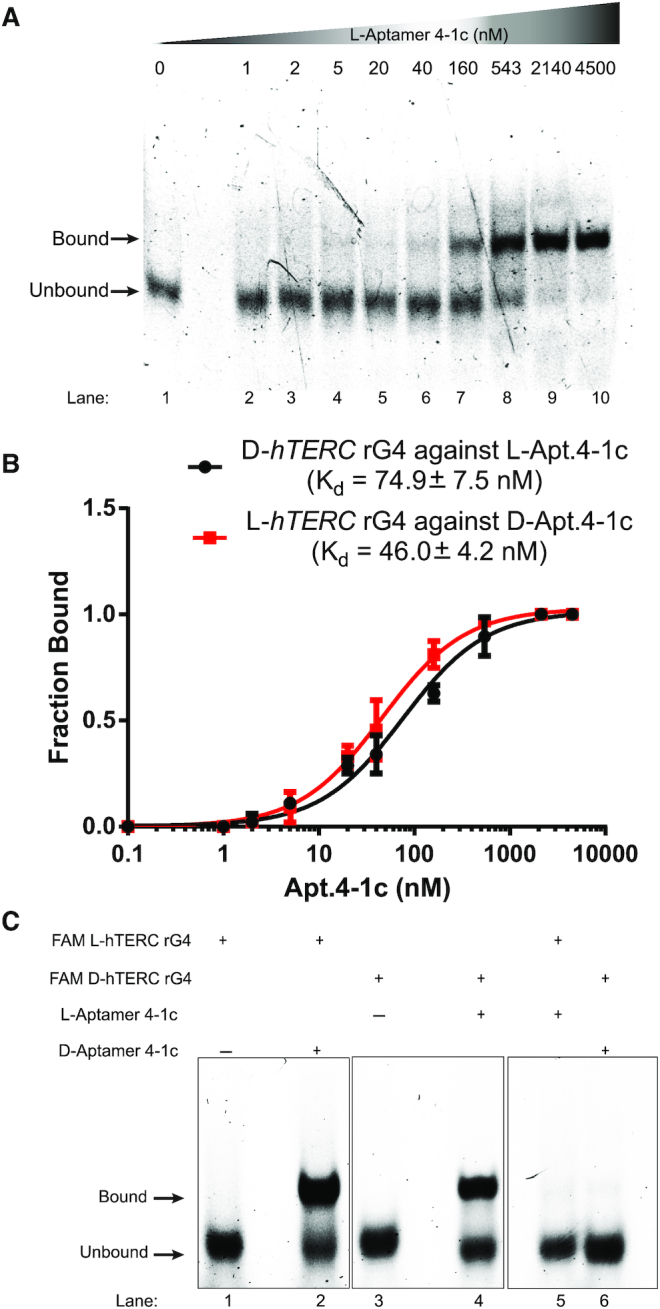
Binding analysis reports the enantiomeric specificity of hTERC rG4 with Apt. 4-1c. (A) The binding between FAM-D-hTERC rG4 and L-Apt. 4-1c was monitored by EMSA. About 12% native gel was used to resolve the bound and unbound fraction. Each lane contained 1 nM FAM-D-hTERC, and L-Apt. 4-1c was added from 0 to 4500 nM. With increasing concentration of L-Apt. 4-1c (from left to right on the gel), the bound band (upper band) became darker, whereas the unbound band (lower band) became weaker. This suggests direct interaction between D-hTERC rG4 and L-Apt. 4-1c. (B) Binding curves of FAM-D-hTERC rG4 against L-Apt. 4-1c (black), and FAM-L-hTERC rG4 against D-Apt. 4-1c (red). The dissociation constants (Kd) were found to be 74.9 ± 7.5 nM and 46.0 ± 4.2 nM, respectively. The error bars represent the standard deviation of three independent replicates. (C) Binding test of FAM-hTERC rG4 with L-Apt. 4-1c of same or opposite chirality, monitored by EMSA. 1 nM FAM-D/L-hTERC rG4 and 543 nM of D/L-Apt. 4-1c were used. In lanes 1–2, an upper band (bound) was observed only when the D-apt. 4-1c was added to the FAM-L-hTERC rG4. In lanes 3–4, an upper band (bound) was observed only when the L-Apt.4-1c was added to the FAM-D-hTERC rG4. In lanes 5–6, only a lower band (unbound) was observed when FAM-hTERC rG4 and Apt. 4-1c of same chirality was mixed.
Metal ion requirements for binding and target specificity of L-Apt.4-1c
To understand the aptamer–target interactions better, we tested the dependence of metal ions for binding. We repeated the binding experiments in the absence of magnesium ion (Mg2+), and observed that the Kd increased from 74.9 ± 7.5 to 586 ± 95 nM (Supplementary Figure S9), suggesting that although Mg2+ is not absolutely required for the binding, the presence of magnesium (Mg2+) enhances the binding significantly. We also repeated the binding experiments by replacing the K+ to Li+, and found that no binding was detected (Supplementary Figure S10), supporting that K+ is essential for the binding to occur, and likely also stabilize the folding of the rG4 in both the aptamer and target.
We also explored the specificity of L-Apt.4-1c toward other targets besides D-hTERC rG4 (Figure 4). First, we designed five non-G4 targets (Supplementary Table S4), including RNA and DNA hairpin, as well as poly rA, rC, rU RNAs, and no binding was observed with any of them, suggesting that L-Apt.4-1c can distinguish hTERC rG4 over non-G4 structural motifs. Second, we designed 4 D-hTERC rG4 mutants (Figure 4B and Supplementary Table S4) to study the importance of loop and bulge nucleotide of hTERC rG4 in binding, and found that a much weaker binding with mutants 1, 3 and 4 (Figure 4C, E and F) compared to the wild-type hTERC rG4 (Figure 3A), thus indicating the significance of these mutated nucleotides (loop nucleotides in hTERC rG4) in the hTERC rG4 recognition by L-Apt.4-1c. Deletion of the C-bulge nucleotide in hTERC rG4 has similar binding with L-4-1c (Figure 4D) compared with the wild-type hTERC rG4 (Figure 3A), suggesting that this C bulge may not be involved in specific interactions with the aptamer. Last, we also designed a panel of dG4s and rG4s, and found that 4 out of 5 dG4s did not bind with L-Apt.4-1c, whereas 5 out of 7 of rG4s interacted with L-Apt.4-1c, highlighting that L-Apt.4-1c has a binding preference toward rG4s over dG4s. As rG4s generally fold into parallel topology (14,15), we tested whether the observed binding difference may be related to topology of dG4s used. From our CD results, we found that with exception of hTELO dG4 that showed mixed topology in 150 mM KCl (Supplementary Figure S11), all the other dG4s are of parallel topologies in 150 mM KCl (Supplementary Figure S11 and Supplementary Figure S12A). Therefore, this excludes the possibility that G4 topology plays a key role in the binding to L-Apt.4.1c. Interestingly, the only tested dG4 that showed interaction with L-Apt.4-1c is the DNA version of hTERC rG4, hTERC dG4, which folds into parallel topology and binds to L-Apt.4-1c weakly, with a Kd value of 315 ± 81 nM (Supplementary Figure S12B-C) as compared to hTERC rG4 with a Kd value of 74.9 ± 7.5 nM (Figure 3A). Given the relatively simplistic secondary structure of L-Apt.4-1c (Figure 2B), it is not unexpected that G4 of closely related structure may be recognized as well. We also cannot rule out the possibility that the aptamer may change its conformation upon binding. In sum, L-Apt.4-1c offers great distinction power between G4 versus non-G4 targets, and has a preference for rG4s versus dG4s tested.
Figure 4.

Binding analysis reveals L-Apt. 4-1c interacts with hTERC rG4 wild-type, variants, and a few other rG4s. The binding between FAM-D-targets and L-Apt. 4-1c was monitored by EMSA. About 12% native gel was used to resolve the bound and unbound fraction. Each lane contained 1 nM FAM-D-targets with or without 543 nM L-Apt. 4-1c (for panels A and G). For panels C–F, all lanes contained 1 nM FAM-D-hTERC rG4 mutant and increasing concentration of L-Apt.4-1c 0–4500 nM. (A) Binding test of L-Apt.4-1c to hTERC rG4 and non-G4s including RNA hairpin, DNA hairpin, poly rA, poly rC and poly rU (B). An illustration of hTERC rG4 mutant’s positions. Mutant 1 replaces U4-U5 with A4-A5, mutant 2 is a deletion of C7 bulge, while mutant3 replaces A10 to U10 and mutant4 replaces U14 to A14. (C–F) Binding between L-Apt.4-1c and hTERC rG4 mutants 1–4. Mutants 1,3,4 (panels C, E, F) showed much weaker binding compared with wild-type (Figure 3A), whereas mutant 2 (panel D) showed similar binding with wildtype (Figure 3A). (G) Binding test of L-Apt.4-1c to G4s. From the gel, TERRA, NRAS, MT3, Bcl2 and TRF2 rG4s and hTERC dG4 showed binding (upper bound band) with L-Apt. 4-1c, whereas the L-Apt. 4-1c showed no binding (no upper bound band) to ZIC1 and ADAM10 rG4s, hTELO dG4, VEGF dG4, Bcl2Mid dG4, c-Kit dG4s. The differences in the fluorescence intensity of the unbound form in the rG4 and dG4 constructs were likely due to the varying degree of quenching caused by the 5′ G and G4 structure.
Suppression of D-hTERC rG4–RHAU53 peptide interaction with L-Apt.4-1c
To demonstrate the utility of L-Apt.4-1c, we attempted to explore the possibility for it to inhibit interactions involving D-hTERC rG4 and its binding partners (Figure 5A). RNA Helicase associated with AU-rich elements (RHAU) is a family member of the ATP-dependent RNA helicase that specifically bind and unwind G4 structures (16–18,44). The high-resolution structure of RHAU in complex with G4 was resolved recently (45), and together with other prior studies (46,47), the key protein region involving in G4 interactions were determined to be near the N-terminus of RHAU. As a truncated version of RHAU full-length protein, the amino acid residues 53–105 of the RHAU, commonly referred as RHAU53, has been shown to bind preferentially to parallel DNA and RNA G4 (46,48).
Figure 5.

L-Apt. 4.1c interferes with hTERC rG4–RHAU53 peptide interactions. (A) Schematic representation of L-Apt. 4-1c interference with hTERC rG4–RHAU53 peptide interactions. (B) The binding of RHAU53 and FAM-D-hTERC was monitored by EMSA. About 6% native gel was used to resolve the bound and unbound fraction (see ‘Materials and Methods’ section). Each lane contained 5 nM FAM hTERC rG4 and 80 nM RHAU53 (except lanes 1 and 2 from left). In presence of increasing concentration of L-Apt.4-1c (lanes 4–12 from left), the rG4–RHAU53 complex (upper bands) diminishes while the rG4–L-Apt.4-1c complex becomes darker (lower bands). This suggests the interaction of D-hTERC and RHAU53 (see also Supplementary Figure S11), and its subsequent inhibition by L-Apt.4-1c. (C) IC50 value of L-Apt.4-1c for its inhibition of hTERC rG4–RHAU53 interaction is 1.69 ± 0.01 μM. Error bars represent standard deviation from three independent replicates.
We first test whether D-hTERC rG4 interact with RHAU53 peptide using EMSA, and found that at around 40 nM RHAU53, a complex shift was observed that indicated the formation of D-hTERC rG4–RHAU53 peptide complex, with a Kd value of 24.5 ± 3.9 nM (Supplementary Figure S13). Since L-Apt.4-1c also contains an rG4 motif (in L-form), we also examined its potential to bind to RHAU53 (Supplementary Figure S14). Our data showed that they interacted very weakly (>1 μM), suggesting L-Apt.4-1c functions by targeting the hTERC rG4 motif in the hTERC rG4–RHAU53 peptide complex. To evaluate the ability of L-Apt.4-1c to suppress D-hTERC rG4–RHAU53 peptide complex formation, we initially assembled D-hTERC rG4–RHAU53 complex, and then titrated L-Apt.4-1c at increasing concentrations. Using EMSA, we monitored the disruption of D-hTERC rG4–RHAU53 interaction and the appearance of D-hTERC rG4–L-Apt.4-1c bands (Figure 5B). We found that as the L-Apt.4-1c concentrations increased from 0 to 20 μM, the D-hTERC rG4–RHAU53 complexes diminished while the D-hTERC rG4-L-Apt.4-1c complexes increased, with a half maximal inhibitory concentration (IC50) value of 1.69 ± 0.1 μM (Figure 5B and C), suggesting that our aptamer L-Apt.4-1c can suppress D-hTERC rG4–RHAU53 peptide interaction. RHAU53 peptide was previously reported to interact with the 5′-end G-quartet, and to a lesser extent to the 3′ end G-quartet in parallel G4s (46,49), and given our finding that L-Apt.4-1c can successfully disrupt D-hTERC–RHAU53 peptide interactions (Figure 5), it is possible that L-Apt.4-1c binds to D-hTERC rG4 at the 5′ and/or 3′-end G-quartet of hTERC rG4. To evaluate this, we have designed two D-hTERC rG4 constructs with 3As overhanging nucleotides either on 5′ end or 3′ end, and performed the binding with L-Apt.4-1c. Our results showed that both the 5′ or 3′ A3 extension construct caused a significant disruption in binding, with Kd >1 μM (Supplementary Figure S15), underlining that the 5′ end and 3′ end G-quartet interface is also important for L-Apt.4-1c recognition, and this result provides support that the D-hTERC rG4–RHAU53 complex is likely disrupted by L-Apt.4-1c via competitive inhibition mechanism.
Comparison between L-Apt.4-1c and G4-targeting ligand on the inhibition of D-hTERC rG4–nucleolin protein interaction
Currently, G4-specific small molecules are generated and used for G4-targeting and for interfering with G4–protein interactions (50); however, no aptamer has been developed and reported to do so. To demonstrate the broad application of our newly developed L-RNA aptamer, L-Apt.4-1c, we aimed to further examine its ability to interfere with D-hTERC rG4–protein interaction (Figure 6A). For this application, nucleolin protein was chosen, as it is a well-studied G4 binding protein. Example of both dG4s and rG4s that interacted with nucleolin were reported, and its complex with G4 was proposed to be important for biology (43,50–53). One early report suggests that nucleolin interacted with the human telomerase reverse transcriptase subunit (hTERT) through binding with its carboxyl-terminal GAR domain and RNA-binding domain 4, and this interaction also involved the hTERC RNA (54); however, the direct binding of D-hTERC rG4 with nucleolin was never explored. To address this, we first studied the binding of D-hTERC rG4 with nucleolin, and found that the D-hTERC–nucleolin complex band appeared at 20 nM and saturated at 160 nM nucleolin (Supplementary Figure S16), indicating that they indeed directly interacted with each other. Motivated by this novel finding, we further investigated the potential of hTERC rG4 binding with the GAR domain of nucleolin, as GAR domain was commonly identified in G4 binding proteins (55). Our results suggested that the binding of GAR domain with D-hTERC rG4 to be strong, with Kd value of 53.0 ± 13.7 nM (using two-state binding mode). Also, it appeared that the D-hTERC rG4–GAR domain binding maybe three-state, as the binding curve fit better to a three-state model, with a Kd1 value of 900 ± 600 pM and Kd2 value of 299 ± 116 nM (Supplementary Figure S17). Overall, we have revealed for the first time the molecular recognition of hTERC rG4 with nucleolin, likely via the GAR domain.
Figure 6.

L-Apt. 4.1c suppresses hTERC rG4–nucleolin interactions. (A) Schematic representation of L-Apt. 4-1c interference with hTERC rG4–nucleolin interactions. (B) The binding of nucleolin and FAM-D–hTERC was monitored by EMSA. Each lane contained 5 nM FAM hTERC rG4 and 80 nM nucleolin (except lanes 1 and 2 from left). In the presence of increasing concentration of L-Apt.4-1c (lanes 4–11 from left), the rG4–nucleolin complex (upper bands) diminishes while the rG4–L-Apt.4-1c complex becomes darker (lower bands). (C) IC50 value of L-Apt.4-1c for its inhibition of hTERC rG4–nucleolin interaction is 279 ± 1 nM.
To assess the ability of L-Apt.4-1c to inhibit D-hTERC rG4–nucleolin interaction, we constructed the D-hTERC rG4–nucleolin complex, and titrated with increasing concentration of L-Apt.4-1c. Our EMSA data showed that as the L-Apt.4-1c concentrations increased from 0 to 10 μM, the D-hTERC rG4–nucleolin complexes diminished while the D-hTERC rG4-L–Apt.4-1c complexes increased, with an IC50 value of 279 ± 1 nM (Figure 6B and C). To support the EMSA results above, we performed an independent assay to examine the interference of hTERC rG4–nucleolin interaction using L-Apt.4-1c. By employing microscale thermophoresis (MST) at room temperature, we first tested the binding between L-Apt.4-1c and hTERC rG4, and found the Kd to be 59.1 ± 11.9 nM (Figure 7B), which is similar to Kd value obtained by EMSA (74.9 ± 7.5 nM) in Figure 3B, suggesting EMSA gel-based and MST-based assays report consistent results. To determine the IC50 value in MST assay, we titrated increasing concentration of L-Apt.4-1c into the assembled D-hTERC rG4–nucleolin complex, and found the IC50 value to be 219 ± 30 nM (Figure 7C), also similar to EMSA results of 279 ± 1 nM in Figure 6C. We also carried out a negative control using VEGF dG4, and found that the IC50 is about five times weaker, with a value of 1190 ± 425.2 nM (Supplementary Figure S18). Altogether, the results from EMSA and MST strongly supported that L-Apt.4-1c can effectively inhibit D-hTERC rG4–nucleolin interaction.
Figure 7.

L-Apt. 4.1c is comparable to cPDS and better than QUMA-1 in inhibiting hTERC rG4-nucleolin interactions. (A) Schematic representation of L-Apt. 4-1c or cPDS interference with hTERC rG4–nucleolin interactions. (B) The binding saturation of L-Apt.4-1c–hTERC rG4 interaction monitored by MST. Reaction mixture contained 40 nM FAM hTERC rG4 and varying concentrations of L-Apt.4-1c (0.15–5000 nM). The Kd was found to be 59.1 ± 11.9 nM. (C) Saturation plot of L-Apt.4-1c for its inhibition of hTERC rG4–nucleolin interaction. Reaction mixture contained 40 nM FAM hTERC rG4, 80 nM nucleolin and increasing concentrations of L-Apt.4-1c (0.15–5000 nM). The IC50 was found to 219 ± 30 nM. (D) Molecular structure of carboxypyridostatin (cPDS) rG4 specific ligand. (E) Similar set up as used in (B) except cPDS ligand was used. The Kd for the interaction was found to be 19.5 ± 11.5 nM. (F) Similar set up as (C) except cPDS rG4 ligand was used. The IC50 value was found to be 122 ± 18 nM. (G) Chemical structure of QUMA-1 rG4 specific ligand. (H) Similar set up as used in (B) except QUMA-1 ligand was used. The Kd for the interaction was found to be 171 ± 43 nM. (I) Similar set up as (C) except QUMA-1 rG4 ligand was used. The IC50 value was found to be 1.05 ± 0.31 μM. All error bars represent standard deviation from three independent replicates.
Next, we compared our L-RNA aptamer results with G4-specific ligand, which is currently the major class of G4-targeting tool. For this comparison, we used rG4-specific ligands referred to as carboxypyridostatin (cPDS) and QUMA-1. cPDS is a synthetic small molecule derived from parent molecule PDS, with demonstrated molecular specificity toward rG4 compared to dG4 (56). QUMA-1 is developed initially to track the folding of rG4s in live human cells, and has been recently applied to rG4s in other systems (57,58). As the cPDS and QUMA-1 ligands are small in size (Figure 7D and G) and thus unobservable gel-shift in EMSA, we have carried out the comparison using MST assay (Figure 7E,F, H–I). From the MST data, we identified tight binding between hTERC rG4 and cPDS, with Kd value of 19.5 ± 11.5 nM (Figure 7E). We then assembled nucleolin–hTERC rG4 followed by increasing concentrations of cPDS and monitored the IC50 to be 122 ± 18 nM (Figure 7F), which is in same order of magnitude with our L-Apt.4-1c (only ∼1.8 fold difference), highlighting that the L-Apt.4-1c inhibits D-hTERC rG4–nucleolin interactions comparably with cPDS. On the other hand, the Kd value of D-hTERC rG4 and QUMA-1 was 171 ± 43 nM (Figure 7H), and the IC50 was 1.05 ± 0.31 μM (Figure 7I), which is ∼4.8 times weaker than L-Apt.4-1c. Last, we also demonstrated that the L-Apt.4-1c is biotstable compared to its D-counterpart (Supplementary Figure S19), and the L-Apt.4-1c can bind to D-hTERC rG4 and inhibit D-hTERC rG4–nucleolin in serum-related condition, although with eight times weaker in Kd and 3.5 times less effective in IC50 under 0.2% FBS condition (Supplementary Figure S20), suggesting that the L-Apt.4-1c needs to be further developed and optimized to be applicable to more complex conditions. In sum, we have demonstrated for the first time an L-RNA aptamer to interfere rG4–protein interaction, and its inhibitory effect is promising.
DISCUSSION
In the present work, we show that L-Apt.4-1c fold into a unique RNA secondary structure to preferentially target rG4s over dG4s and non-G4s, and demonstrate it can interfere with D-hTERC rG4–RHAU53 peptide and D-hTERC rG4–nucleolin protein interactions. There are three key insights from this proof-of-concept study.
First, the Apt.4-1c is the shortest L-aptamer developed so far (Figure 2B). Based on our data above, it is very likely that the D-hTERC rG4 interact with the rG4 in L-Apt.4-1c in the formation of the target–aptamer complex. We wish to note that such D-rG4 to L-rG4 binding was similarly reported in our recent study with another target–aptamer pair on L-Ap3-7–D-TERRA rG4, supporting that this structural feature may be general and should be valuable for future design of L-aptamer tools for G4-targeting. Notably, there are also clear distinctions between these two works. Interestingly, in this study we found strong sequence conservation and base pair co-variation in our aptamer candidates from our cloning results (Figure 1D) to allow us to propose the aptamer secondary structure and refine our aptamer constructs (Figures 1D; 2A and B), and these sequence conservation and base pair co-variation features were not observed in the cloning results in our previous study (34). More importantly, although both L-Ap3-7 and L-Apt.4-1c aptamers contain rG4 motif, there are also substantial structural differences observed, in which the secondary structure of L-Ap3-7 (36 nt in length) is more complex, with a four base pair RNA stem, a long internal loop (10 nt total) and a 3-layered rG4 motif (34), whereas the secondary structure of L-Apt4-1c (25 nt in length) is simpler, with a five base pair RNA stem, a short internal loop (4 nt total), and a two-layered rG4 motif (Figure 2B). This difference in aptamer structure complexity has turned out to contribute a significant effect on target specificity achieved. Unlike L-Ap3-7 that is highly selective to its selection target, TERRA rG4, among all other rG4s, dG4s, and non-G4s tested (34), L-Apt.4-1c exhibited properties that behaves more like a general rG4 binder, in which it has a strong preference to bind to rG4s (five out of seven tested) over non-G4s (zero out of five tested) and dG4s (one out of five tested) (Figure 4A and G). The lack of interaction with ADAM rG4 and ZIC1 rG4 (Figure 4G) is potentially related to the loop composition and structure, as we found mutation in the loops of hTERC rG4 also has some impact on the binding (Figure 4C–F). Overall, the development of both individual rG4-specific (e.g. L-Ap3-7) and general rG4-specific (e.g. L-Apt.4-1c) tool is both essential and complementary for understanding rG4 structure, dissecting G4 biology and developing new applications. Besides binding to G4 targets (this study and our previous work (34)), other L-aptamers bearing G4 structure were also reported to bind to protein (59) and stem–loop RNA (31), highlighting that G4 motif in L-aptamer is potentially a versatile structural scaffold that enables strong and specific recognition of its D-target.
Second, extensive mutagenesis was carried out on aptamer (Apt.4-1c) and target (hTERC rG4) to understand critical nucleotides that are required for the binding (Figures 2 and 4; Supplementary Figure S15). While most single point mutation on the aptamer completely abolished the binding (Figure 2), we found that G20 to A20 mutation, but not G18 to A18, led to stronger binding, and we reasoned that G18 and G19, instead of G19 and G20 were participated in the formation of rG4 in the aptamer. The G20 to A20 mutation likely reduced the potential G4 polymorphism in the last G-tracts and led to more defined aptamer secondary structure for binding. Previous G4 studies have shown shorter loops to have higher G4 thermostability (60), thus involving G18 over G20 in rG4 formation in Apt.4-1c is not unusual, as the use of G20 instead of G18 will lead to lengthening of the third loop by one nucleotide in rG4, which may affect the loop structure and thermostability of the rG4, and therefore its binding. Another interesting observation was that mutations in U14, G16 and U17 only caused partial disruption on binding, suggesting these three nucleotides that are on/near the third G-tracts can tolerate some degree of mutations. Regarding the target, mutations on the loops of rG4, but not the C-bulge of rG4, weaken the binding, indicating the RNA base identity of these nucleotides in the loops of rG4 have some roles in the recognition (Figure 4). Likewise, adding 5′ and 3′ extension to the target disrupted the binding significantly, highlighting that the 5′ and 3′ G-quartet interface of target was indeed critical for binding (Supplementary Figure S15). With our current data, we propose the D-hTERC rG4–L-Apt.4-1c complex to involve rG4–rG4 stacking interaction and additional recognition from the loop nucleotides, as a few of the aptamer loop mutants disrupted or weaken the binding (Figure 2E–F). Based on the hTERC rG4 5′ and 3′ extension data, it is possible that an unobstructed end G-quartet interface is needed for maximal stacking and interaction. Future high-resolution 3D structure of D-hTERC rG4 (with and without L-Apt.4-1c bound) will provide the structural basis of the target–aptamer complex formation.
Last, besides using hTERC rG4–RHAU53 peptide complex to showcase the utility of our developed aptamer, we also explored and reported nucleolin binding to D-hTERC rG4 directly for the first time (Supplementary Figures S16 and 17). A previous study has shown that nucleolin associates with the hTERT through binding with its GAR domain and RNA-binding domain 4, and suggested hTERC RNA was likely involved (54), yet the direct interaction, as well as the critical region involved in the potential hTERC RNA–nucleolin interaction was never investigated. In this study, we have verified that the rG4 motif of hTERC rG4 was involved in the binding to the nucleolin (Supplementary Figure S16), and we further confirmed the GAR domain of nucleolin can directly and strongly interact with the hTERC rG4 (Supplementary Figure S17), providing a potential binding mechanism of how nucleolin can interact with telomerase (hTERC RNA and hTERT protein). Based on the binding curve (Supplementary Figure S17), we speculate that multiple binding events occurred as our data fits better to three-state model than two-state model, and the GAR domain may bind to both ends of the rG4 with different binding affinities. This novel discovery of hTERC rG4–nucleolin GAR interaction is consistent with recent findings in literatures, in which several dG4s and more recently a few rG4s have been reported to bind to nucleolin directly (43,50–53), and a number of G4-binding proteins contains the GAR domain (55). Notably, we showcased that L-Apt.4-1c can suppress D-hTERC rG4–nucleolin interaction effectively, with IC50 value highly comparable to or better than the state-of-the-art G4-specific ligands such as cPDS and QUMA-1 (Figure 7), underscoring the fact that our research findings have expanded the current G4 targeting toolkit to include L-aptamer as well, which open a new door for investigating G4 biology and developing G4-related applications in the near-future. In addition, we have demonstrated the feasibility of performing the binding and inhibition assays in serum-related condition, although the Kd and IC50 were found to be much eight times weaker and 3.5 times less effective, respectively (Supplementary Figures S19 and 20), which is likely due to the introduction of FBS in the reaction. Further investigation and refinement of the properties of L-Apt.4-1c will be needed be make it more applicable for working as an inhibitor for rG4–protein interaction in native conditions.
CONCLUSION
In sum, we have developed a novel L-RNA aptamer, L-Apt.4-1c, and demonstrated it to bind with hTERC rG4 structure strongly and interfere with hTERC rG4–RHAU53 and hTERC rG4–nucleolin interactions effectively, with potential to be applied to more complex conditions. Contrary to a recent individual rG4-specific binder developed in our lab, we found that our newly developed aptamer in this work has a preference to bind to rG4 motif over non-G4s and dG4 motif, and can be utilized as a general rG4-specific binder, enabling different applications in the near future. Interestingly, through our extensive spectroscopy and mutagenesis analysis, we show that this aptamer contains an rG4 motif with two G-quartets and is the shortest L-aptamer being reported so far, with 25 nt in length. Together, these findings provide the general framework for the simple and robust development of L-RNA aptamer for binding rG4 and suppressing rG4-protein interactions of interest. We expect that further development of this exciting approach will make L-aptamers practical for diverse chemical and biological applications.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mr Chun-Yin Chan for the discussion.
Contributor Information
Mubarak I Umar, Department of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon Tong, Hong Kong SAR, China.
Chun Kit Kwok, Department of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon Tong, Hong Kong SAR, China; Shenzhen Research Institute of City University of Hong Kong, Shenzhen, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Shenzhen Basic Technology Research Project [JCYJ20180507181642811]; Research Grants Council of the Hong Kong SAR, China Projects; [CityU 11101519, CityU 11100218, N_CityU110/17, CityU 21302317]; Croucher Foundation [9500030, 9509003, 9500039]; City University of Hong Kong Projects [6000711, 9680261] to C.K.K.. M.I.U received support from the Nigerian government through the Petroleum Technology Development Fund. Funding for open access charge: Research Grants Council of the Hong Kong SAR CityU [11100218].
Conflict of interest statement. None declared.
REFERENCES
- 1. Cech T.R. The RNA worlds in context. Cold Spring Harb. Perspect. 2012; 4:a006742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cech T., Steitz J.A., Atkins J.F.. RNA Worlds: New Tools for Deep Exploration. 2019; Cold Spring Harbor Laboratory Press. [Google Scholar]
- 3. Wan Y., Kertesz M., Spitale R.C., Segal E., Chang H.Y.. Understanding the transcriptome through RNA structure. Nat. Rev. Genet. 2011; 12:641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kwok C.K., Tang Y., Assmann S.M., Bevilacqua P.C.. The RNA structurome: transcriptome-wide structure probing with next-generation sequencing. Trends Biochem. Sci. 2015; 40:221–232. [DOI] [PubMed] [Google Scholar]
- 5. Qian X., Zhao J., Yeung P.Y., Zhang Q.C., Kwok C.K.. Revealing lncRNA structures and interactions by sequencing-based approaches. Trends Biochem. Sci. 2019; 44:33–52. [DOI] [PubMed] [Google Scholar]
- 6. Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell. Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
- 7. Kole R., Krainer A.R., Altman S.. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012; 11:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Disney M.D., Winkelsas A.M., Velagapudi S.P., Southern M., Fallahi M., Childs-Disney J.L.. Inforna 2.0: A platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol. 2016; 11:1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Disney M.D. Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc. 2019; 141:6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eubanks C.S., Zhao B., Patwardhan N.N., Thompson R.D., Zhang Q., Hargrove A.E.. Visualizing RNA conformational changes via pattern recognition of RNA by small molecules. J. Am. Chem. Soc. 2019; 141:5692–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morgan B.S., Sanaba B.G., Donlic A., Karloff D.B., Forte J.E., Zhang Y., Hargrove A.E.. R-BIND: An interactive database for exploring and developing RNA-targeted chemical probes. ACS Chem. Biol. 2019; 14:2691–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donlic A., Hargrove A.E.. Targeting RNA in mammalian systems with small molecules. Wiley Interdiscip. Rev. RNA. 2018; 9:e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raguseo F., Chowdhury S., Minard A., Di Antonio M.. Chemical-biology approaches to probe DNA and RNA G-quadruplex structures in the genome. Chem. Commun. (Camb.). 2020; 56:1317–1324. [DOI] [PubMed] [Google Scholar]
- 14. Kwok C.K., Marsico G., Balasubramanian S.. Detecting RNA G-Quadruplexes (rG4s) in the Transcriptome. Cold Spring Harb. Perspect. 2018; 10:a032284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neidle S., Balasubramanian S.. Quadruplex Nucleic Acids. 2006; Cambridge: Royal Society of Chemistry. [Google Scholar]
- 16. Booy E.P., Meier M., Okun N., Novakowski S.K., Xiong S., Stetefeld J., McKenna S.A.. The RNA helicase RHAU (DHX36) unwinds a G4-quadruplex in human telomerase RNA and promotes the formation of the P1 helix template boundary. Nucleic Acids Res. 2012; 40:4110–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sexton A.N., Collins K.. The 5′ guanosine tracts of human telomerase RNA are recognized by the G-quadruplex binding domain of the RNA helicase DHX36 and function to increase RNA accumulation. Mol. Cell. Biol. 2011; 31:736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lattmann S., Stadler M.B., Vaughn J.P., Akman S.A., Nagamine Y.. The DEAH-box RNA helicase RHAU binds an intramolecular RNA G-quadruplex in TERC and associates with telomerase holoenzyme. Nucleic Acids Res. 2011; 39:9390–9404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martadinata H., Phan A.T.. Formation of a stacked dimeric G-quadruplex containing bulges by the 5′-terminal region of human telomerase RNA (hTERC). Biochemistry. 2014; 53:1595–1600. [DOI] [PubMed] [Google Scholar]
- 20. Ozer A., Pagano J.M., Lis J.T.. New technologies provide quantum changes in the scale, speed, and success of SELEX methods and aptamer characterization. Mol. Ther. Nucleic Acids. 2014; 3:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Young B.E., Kundu N., Sczepanski J.T.. Mirror-image oligonucleotides: history and emerging applications. Chemistry. 2019; 25:7981–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhong W., Sczepanski J.T.. A mirror image fluorogenic aptamer sensor for live-cell imaging of MicroRNAs. ACS Sens. 2019; 4:566–570. [DOI] [PubMed] [Google Scholar]
- 23. Kluszmann S., Nolte A., Bald R., Erdmann V.A., Furste J.P.. Mirror-image RNA that binds D-adenosine. Nat. Biotechnol. 1996; 14:1112–1115. [DOI] [PubMed] [Google Scholar]
- 24. Nolte A., Klussmann S., Bald R., Erdmann V.A., Furste J.P.. Mirror-design of L-oligonucleotide ligands binding to L-arginine. Nat. Biotechnol. 1996; 14:1116–1119. [DOI] [PubMed] [Google Scholar]
- 25. Williams K.P., Liu X.H., Schumacher T.N., Lin H.Y., Ausiello D.A., Kim P.S., Bartel D.P.. Bioactive and nuclease-resistant L-DNA ligand of vasopressin. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:11285–11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olea C. Jr, Weidmann J., Dawson P.E., Joyce G.F.. An L-RNA aptamer that binds and inhibits RNase. Chem. Biol. 2015; 22:1437–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oberthur D., Achenbach J., Gabdulkhakov A., Buchner K., Maasch C., Falke S., Rehders D., Klussmann S., Betzel C.. Crystal structure of a mirror-image L-RNA aptamer (Spiegelmer) in complex with the natural L-protein target CCL2. Nat. Commun. 2015; 6:6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schwoebel F., van Eijk L.T., Zboralski D., Sell S., Buchner K., Maasch C., Purschke W.G., Humphrey M., Zollner S., Eulberg D. et al.. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood. 2013; 121:2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helmling S., Maasch C., Eulberg D., Buchner K., Schröder W., Lange C., Vonhoff S., Wlotzka B., Tschöp M.H., Rosewicz S. et al.. Inhibition of ghrelin action in vitro and in vivo by an RNA-Spiegelmer. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:13174–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vater A., Klussmann S.. Turning mirror-image oligonucleotides into drugs: the evolution of Spiegelmer therapeutics. Drug Discov. Today. 2015; 20:147–155. [DOI] [PubMed] [Google Scholar]
- 31. Dey S., Sczepanski J.T.. In vitro selection of l-DNA aptamers that bind a structured d-RNA molecule. Nucleic Acids Res. 2020; 48:1669–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kabza A.M., Sczepanski J.T.. An l-RNA aptamer with expanded chemical functionality that inhibits MicroRNA biogenesis. ChemBioChem. 2017; 18:1824–1827. [DOI] [PubMed] [Google Scholar]
- 33. Sczepanski J.T., Joyce G.F.. Binding of a structured D-RNA molecule by an L-RNA aptamer. J. Am. Chem. Soc. 2013; 135:13290–13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan C.Y., Kwok C.K.. Specific Binding of a d-RNA G-Quadruplex Structure with an l-RNA Aptamer. Angew. Chem. Int. Ed. 2020; 59:5293–5297. [DOI] [PubMed] [Google Scholar]
- 35. Chan K.L., Peng B., Umar M.I., Chan C.Y., Sahakyan A.B., Le M.T.N., Kwok C.K.. Structural analysis reveals the formation and role of RNA G-quadruplex structures in human mature microRNAs. Chem. Commun. (Camb.). 2018; 54:10878–10881. [DOI] [PubMed] [Google Scholar]
- 36. Chan C.Y., Umar M.I., Kwok C.K.. Spectroscopic analysis reveals the effect of a single nucleotide bulge on G-quadruplex structures. Chem. Commun. (Camb.). 2019; 55:2616–2619. [DOI] [PubMed] [Google Scholar]
- 37. Kwok C.K., Sherlock M.E., Bevilacqua P.C.. Effect of loop sequence and loop length on the intrinsic fluorescence of G-quadruplexes. Biochemistry. 2013; 52:3019–3021. [DOI] [PubMed] [Google Scholar]
- 38. Kwok C.K., Ding Y., Shahid S., Assmann S.M., Bevilacqua P.C.. A stable RNA G-quadruplex within the 5′-UTR of Arabidopsis thaliana ATR mRNA inhibits translation. Biochem. J. 2015; 467:91–102. [DOI] [PubMed] [Google Scholar]
- 39. Sosnick T.R. Characterization of tertiary folding of RNA by circular dichroism and urea. Curr. Protoc. Nucleic Acid Chem. 2001; doi:10.1002/0471142700.nc1105s04. [DOI] [PubMed] [Google Scholar]
- 40. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Woese C.R., Winker S., Gutell R.R.. Architecture of ribosomal RNA: constraints on the sequence of “tetra-loops”. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:8467–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schudoma C., May P., Nikiforova V., Walther D.. Sequence-structure relationships in RNA loops: establishing the basis for loop homology modeling. Nucleic Acids Res. 2010; 38:970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kwok C.K., Merrick C.J.. G-Quadruplexes: Prediction, characterization, and biological application. Trends Biotechnol. 2017; 35:997–1013. [DOI] [PubMed] [Google Scholar]
- 44. Mendoza O., Bourdoncle A., Boule J.B., Brosh R.M. Jr, Mergny J.L.. G-quadruplexes and helicases. Nucleic Acids Res. 2016; 44:1989–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen M.C., Tippana R., Demeshkina N.A., Murat P., Balasubramanian S., Myong S., Ferre-D’Amare A.R.. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature. 2018; 558:465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Heddi B., Cheong V.V., Martadinata H., Phan A.T.. Insights into G-quadruplex specific recognition by the DEAH-box helicase RHAU: Solution structure of a peptide-quadruplex complex. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:9608–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lattmann S., Giri B., Vaughn J.P., Akman S.A., Nagamine Y.. Role of the amino terminal RHAU-specific motif in the recognition and resolution of guanine quadruplex-RNA by the DEAH-box RNA helicase RHAU. Nucleic Acids Res. 2010; 38:6219–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen M.C., Murat P., Abecassis K., Ferre-D’Amare A.R., Balasubramanian S.. Insights into the mechanism of a G-quadruplex-unwinding DEAH-box helicase. Nucleic Acids Res. 2015; 43:2223–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heddi B., Cheong V.V., Schmitt E., Mechulam Y., Phan A.T.. Recognition of different base tetrads by RHAU (DHX36): X-ray crystal structure of the G4 recognition motif bound to the 3′-end tetrad of a DNA G-quadruplex. J. Struct. Biol. 2020; 209:107399. [DOI] [PubMed] [Google Scholar]
- 50. Sun Z.-Y., Wang X.-N., Cheng S.-Q., Su X.-X., Ou T.-M.. Developing novel G-Quadruplex Ligands: from interaction with nucleic acids to interfering with nucleic Acid-Protein interaction. Molecules. 2019; 24:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fay M.M., Lyons S.M., Ivanov P.. RNA G-quadruplexes in biology: principles and molecular mechanisms. J. Mol. Biol. 2017; 429:2127–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bian W.X., Xie Y., Wang X.N., Xu G.H., Fu B.S., Li S., Long G., Zhou X., Zhang X.L.. Binding of cellular nucleolin with the viral core RNA G-quadruplex structure suppresses HCV replication. Nucleic Acids Res. 2019; 47:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lago S., Tosoni E., Nadai M., Palumbo M., Richter S.N.. The cellular protein nucleolin preferentially binds long-looped G-quadruplex nucleic acids. Biochim. Biophys. Acta. Gen. Subj. 2017; 1861:1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khurts S., Masutomi K., Delgermaa L., Arai K., Oishi N., Mizuno H., Hayashi N., Hahn W.C., Murakami S.. Nucleolin interacts with telomerase. J. Biol. Chem. 2004; 279:51508–51515. [DOI] [PubMed] [Google Scholar]
- 55. Huang Z.L., Dai J., Luo W.H., Wang X.G., Tan J.H., Chen S.B., Huang Z.S.. Identification of G-Quadruplex-binding protein from the exploration of RGG Motif/G-Quadruplex interactions. J. Am. Chem. Soc. 2018; 140:17945–17955. [DOI] [PubMed] [Google Scholar]
- 56. Rocca R., Talarico C., Moraca F., Costa G., Romeo I., Ortuso F., Alcaro S., Artese A.. Molecular recognition of a carboxy pyridostatin toward G-quadruplex structures: Why does it prefer RNA. Chem. Biol. Drug Des. 2017; 90:919–925. [DOI] [PubMed] [Google Scholar]
- 57. Chen X.C., Chen S.B., Dai J., Yuan J.H., Ou T.M., Huang Z.S., Tan J.H.. Tracking the dynamic folding and unfolding of RNA G-quadruplexes in live cells. Angew. Chem. Int. Ed. 2018; 57:4702–4706. [DOI] [PubMed] [Google Scholar]
- 58. Shao X., Zhang W., Umar M.I., Wong H.Y., Seng Z., Xie Y., Zhang Y., Yang L., Kwok C.K., Deng X.. RNA G-Quadruplex structures mediate gene regulation in bacteria. mBio. 2020; 11:e02926-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yatime L., Maasch C., Hoehlig K., Klussmann S., Andersen G.R., Vater A.. Structural basis for the targeting of complement anaphylatoxin C5a using a mixed L-RNA/L-DNA aptamer. Nat. Commun. 2015; 6:6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pandey S., Agarwala P., Maiti S.. Effect of loops and G-quartets on the stability of RNA G-quadruplexes. J. Phys. Chem. B. 2013; 117:6896–6905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.