

Abstract
Long-range electron transfer is coupled to proton transfer in a wide range of chemically and biologically important processes. Recently the proton-coupled electron transfer (PCET) rate constants for a series of biomimetic oligoproline peptides linking Ru(bpy)32+ to tyrosine were shown to exhibit a substantially shallower dependence on the number of proline spacers compared to the analogous electron transfer (ET) systems. The experiments implicated a concerted PCET mechanism involving intramolecular electron transfer from tyrosine to Ru(bpy)33+ and proton transfer from tyrosine to a hydrogen phosphate dianion. Herein these PCET systems, as well as the analogous ET systems, are studied with microsecond molecular dynamics, and the ET and PCET rate constants are calculated with the corresponding nonadiabatic theories. The molecular dynamics simulations illustrate that smaller ET distances are sampled by the PCET systems than by the analogous ET systems. The shallower dependence of the PCET rate constant on the ET donor-acceptor distance is explained in terms of an additional positive, distance-dependent electrostatic term in the PCET driving force, which attenuates the rate constant at smaller distances. This electrostatic term depends on the change in the electrostatic interaction between the charges on each end of the bridge and can be modified by altering these charges. On the basis of these insights, this theory predicted a less shallow distance dependence of the PCET rate constant when imidazole rather than hydrogen phosphate serves as the proton acceptor, even though their pKa values are similar. This theoretical prediction was subsequently validated experimentally, illustrating that long-range electron transfer processes can be tuned by modifying the nature of the proton acceptor in concerted PCET processes. This level of control has broad implications for the design of more effective charge-transfer systems.
Graphical Abstract

INTRODUCTION
Long-range electron transfer (ET) plays a significant role in a wide range of chemical and biological systems.1–4 Biomimetic model systems, in which the electron donor and acceptor are connected by molecular bridges, have been designed to investigate the fundamental physical principles underlying such long-range ET reactions. A variety of experimental and theoretical studies have been performed to elucidate the origin of the exponential dependence of ET rate constants on the length of the bridge between the electron donor and acceptor sites.1, 5–21 The nature of the bridges and their conformational flexibility can play significant roles in determining the distance dependence of the ET rate constants.17, 19, 22–26 Moreover, dependence of the driving force and reorganization energy on the distance may also influence the distance dependence of the ET rate constants.21, 27–30
Many long-range ET reactions in biological systems are also coupled to proton transfer (PT) reactions. These proton-coupled electron transfer (PCET) reactions play critical roles in biological processes such as nitrogen fixation,31 photosynthesis,32 respiration,33 and DNA synthesis.34 A significant amount of attention has been directed toward the dependence of PCET reactions on the proton donor-acceptor distance.35–43 However, only a relatively small number of studies have focused on the dependence of PCET reactions on the ET donor-acceptor distance.44–46 Recently, studies of PCET model systems with phenylene or p-xylene bridges44–46 indicated that PCET rate constants depend on the ET donor-acceptor distances in a similar manner as do the analogous ET systems with the same bridges.12, 47–49
Recently, a series of oligoproline peptides connecting Ru(bpy)32+ and tyrosine was designed and studied experimentally with a flash-quench transient absorption method.50 In these experiments, photogenerated Ru(bpy)33+ oxidized tyrosine by a concerted PCET process involving intramolecular ET from tyrosine to Ru(bpy)33+ and proton transfer from tyrosine to the hydrogen phosphate buffer in aqueous solution (Figure 1). The concerted PCET mechanism is supported by the phosphate buffer concentration dependence of the kinetics and the kinetic isotope effects. Moreover, the dependence of the PCET rate constant on the ET donor-acceptor distance was studied by varying the number of oligoproline linkers from n = 1 to 4. The experimentally measured ET distance dependence was found to be much shallower than the corresponding distance dependence measured for analogous bimetallic ET systems with oligoproline bridges 13, 51–53 (Figure 1). The shallow distance dependence for the PCET systems relative to the ET systems was proposed to be related to the necessity of employing stronger oxidants for tyrosine, thereby leading to lower tunneling barrier heights within the McConnell superexchange model.1, 54
Figure 1.

(A) ET and (B) PCET systems studied herein, where n = 1 – 4 for each system.
Herein we use a combination of microsecond molecular dynamics (MD) simulations and vibronically nonadiabatic PCET theory55–57 to further elucidate the ET distance dependence of these oligoproline metallopeptides. This PCET theory has been used to investigate the dependence of the PCET rate constant on the proton donor-acceptor distance for a wide range of systems,35–36, 40–41 but the dependence on the electron donor-acceptor distance has not been studied as extensively. We perform microsecond MD simulations of the solvated ET and PCET oligoproline systems with bridges of different lengths, namely one to four proline linkers. We also calculate the ET and PCET rate constants for these systems using nonadiabatic ET and PCET theories, respectively. Analysis of the results provides a physical explanation for the shallower dependence of the PCET rate constant on the ET distance in terms of the net charges on the redox species terminating the bridge, producing an attractive electrostatic interaction that is smaller for the product than for the reactant, thereby decreasing the driving force. This effect becomes more pronounced as the electron donor-acceptor distance decreases. Moreover, our theoretical model predicts that this distance dependence of the PCET rate constant will increase when imidazole rather than hydrogen phosphate is the proton acceptor, even though the relevant pKa values are similar. This theoretical prediction is subsequently validated experimentally, illustrating how the nature of the proton acceptor can significantly impact the dependence of the PCET rate constant on the ET distance.
METHODS
Molecular dynamics simulations
The ET and PCET systems studied in the present work are shown in Figure 1. The Cambridge Structural Database (CSD) entries TIRTIY58 and SOYRIJ59 were used to prepare the structures of the metal-containing terminal groups, and the CSD entry SOWJUL60 was used to build the proline linkage. For the MD simulations, the AMBER ff14SB force field61 was employed to model the proline linkage and the terminal Tyr group, the general AMBER force field (GAFF)62 was used to model the metal-containing terminal groups, and the TIP3P water force field63 was used to model the solvent. The partial charges of the terminal Tyr group were obtained through a two-stage RESP fitting procedure for two different configurations,64–65 and the parameters for the metal sites were generated with the MCPB.py program.66 Each of the four PCET systems was solvated in 0.1 M [HPO4]2− buffer solution and neutralized by the addition of K+ ions. Each of the four ET systems was solvated in a water box neutralized by the addition of Cl− ions. The long-range electrostatic interactions were treated with the particle mesh Ewald method.67 After a careful equilibration procedure, a 1 μs trajectory was propagated using the pmemd.cuda program68 for each of the eight systems, with configurations saved every 10 ps. These configurations were used for the analysis of the distributions of the ET donor-acceptor distance. Further details of the force field parameters and MD simulations are provided in the Supporting Informaton (SI).
Theoretical modeling of the ET and PCET reactions
For each ET system, the rate constant was calculated by thermal averaging of the standard nonadiabatic ET rate constant over the ET donor-acceptor distance:69
| (1) |
| (2) |
Here RET is the distance between the two Ru ions in the bimetallic ET system and P(RET) is the probability distribution obtained from the conformational sampling in the MD simulations. Moreover, kB is the Boltzmann constant, T is the temperature, and is the electronic coupling between the ET donor and acceptor sites with the ET distance corresponding to a single bridge unit (i.e., n = 1). The specific value of is not needed for this study because it simply corresponds to an additive constant to ln(kET). The coupling attenuation parameter βET was set to 1.4 Å−1 according to the ET study of the oligoproline bimetallic systems.13 The reorganization energy λ for each of the four ET systems was evaluated using a Marcus two-sphere model70 and averaging the resulting values according to the distribution P(RET) (see SI). The ET reaction free energy ΔGET was determined to be −26.9 kcal/mol on the basis of the relevant reduction potentials (see SI).
In the vibronically nonadiabatic PCET theory,55–57 the reaction is described in terms of nonadiabatic transitions between reactant and product electron-proton vibronic states. When fluctuations in the environment lead to the degeneracy between a pair of reactant and product vibronic states, the electron and proton tunnel simultaneously with a probability proportional to the square of the vibronic coupling. For each PCET system, the calculation of the rate constants required thermal averaging over both the ET donor-acceptor distance RET and the PT donor-acceptor distance RPT. The thermal averaging over RET was analogous to that described for the ET systems, where kET is replaced by kPCET in Eq. (1), and in this case the electron transfer distance is defined as the distance between the Ru3+ ion and the geometrical center of the six carbon atoms of the Tyr benzene ring. The probability distribution P(RET) was assumed to be the same for the PCET systems with either hydrogen phosphate or imidazole as the proton acceptor.
The nonadiabatic PCET rate constant kPCET(RET) at each fixed ET distance is given by
| (3) |
This PCET rate constant expression accounts for all pairs of reactant and product proton vibrational states μ and ν obtained by solving the one-dimensional Schrödinger equation for a proton moving in the reactant and product electronically diabatic proton potentials, respectively. The rate constant is calculated by averaging over the reactant proton vibrational states μ with Boltzmann populations Pμ and summing over the product proton vibrational states ν. The vibronic coupling is the product of the electronic coupling and the overlap integral Sμν(RPT) between the reactant and product proton vibrational wavefunctions. The angular brackets denote averaging over the proton donor-acceptor distance RPT with the Gaussian distribution function:
| (4) |
Note that this model neglects the dependence of the electronically diabatic proton potentials, as well as the reorganization energy, on RPT, thereby enabling the thermal averaging over RPT to be restricted to the overlap integrals.
In the present work, the electronically diabatic proton potentials were represented by Morse potentials corresponding to O-H or N-H bonds (see SI), and the overlap integrals were calculated analytically.71 Furthermore, , which is the equilibrium proton donor-acceptor distance in the reactant state, and the force constant fPT, which is related to the second derivative of the energy profile along RPT, were obtained from density functional theory (DFT) calculations (Table S4). As for the ET systems, the reorganization energy for each of the PCET systems was estimated by averaging the RET-dependent value obtained from the Marcus two-sphere model according to the distribution P(RET) (see SI). For the PCET systems, is defined analogously as the corresponding parameter for the ET system but is expected to assume a different value, which is not needed to compute the relevant slopes. For consistency, we used the same electronic coupling attenuation parameter βET for both the ET and PCET systems, although βET may be different for these two types of systems because of their different thermodynamic properties. To explore this possibility, we also investigated the impact of using different values of βET for the ET and PCET systems, but the qualitative conclusions did not change.
The free energy difference ΔGμν in the PCET rate constant is given by
| (5) |
where ΔGPCET is the reaction free energy of the PCET reaction, and εμ and εν are the proton vibrational energy levels for the states μ and ν relative to the lowest vibrational states of the corresponding diabatic proton potentials. ΔGPCET was determined to be −10.0 kcal/mol from the pKa values and redox potentials of the relevant species (see SI). The additional electrostatic term ΔEele, which depends on the distance between the metal center (Ru) and the proton acceptor (PA) group, accounts for the difference in electrostatic interactions between the charges localized on these sites in the reactant and product states in the PCET reaction and is given by
| (6) |
Here RRu-PA, which is the distance between the metal center and the proton acceptor group, is approximated by RET. In this expression, Δ(QRuQPA) is the difference between the product of the charges on the metal center and the proton acceptor group after and before the PCET reaction. For example, when [HPO4]2− serves as the proton acceptor, QRuQPA is (+3e)×(‒2e) before and (+2e)×(‒1e) after the PCET reaction, resulting in Δ(QRuQPA) = 4e2. In this case, because the attractive electrostatic interaction is smaller for the product than for the reactant, this difference is positive and therefore decreases the driving force, particularly at shorter distances. Finally, εeff is an empirical factor that accounts for the screening effects of the molecular and solvent environment as well as delocalization of the charges and thus depends on factors such as the solvent, ionic strength, ion pairing, and electronic structure of the donor-bridge-acceptor system. As discussed further below, this factor reflects an average over electrostatic interactions through the solution and/or the molecular bridge. In the present study, this factor was set to 5.1 to reproduce the slope of the experimentally measured ln(kPCET) versus the computed average ET distances for the system with hydrogen phosphate serving as the proton acceptor. We used the same value of this factor to model the system with imidazole as the proton acceptor and found good agreement with the experimentally measured slope for this system. We emphasize that ΔEele adjusts the driving force of the oligoproline peptides by including the electrostatic interactions between the electron donor and acceptor because ΔGPCET is computed from the thermodynamic properties of the isolated donor and acceptor species.
RESULTS AND DISCUSSION
Probability distributions of ET distances
To characterize the ET donor-acceptor distance fluctuations in the ET and PCET reactions, we analyzed the probability distributions of the ET distances obtained from microsecond MD trajectories. For the ET systems, RET was defined as the distance between the two Ru ions, and for the PCET systems, RET was defined as the distance between the Ru3+ ion and the geometrical center of the six carbon atoms of the Tyr benzene ring. As shown in Figure 2, the probability distribution for each ET system resembles a single Gaussian. In contrast, the probability distribution for each PCET system is broader with a shoulder at smaller distances, indicating that multiple conformations are sampled on the microsecond timescale. The average RET values, denoted , for the PCET systems are slightly smaller than those for the ET systems with the same number of proline residues and exhibit larger fluctuations (Table 1). These differences may be due to the flexibility of the neutral terminal Tyr electron donor group in the PCET systems, which enables sampling of conformations with smaller distances with respect to the positively charged Ru3+ center. In contrast, the metal centers in the ET systems are relatively bulky with the bound ligands and are both positively charged, preventing effective sampling of conformations with smaller distances. To understand the role of the counterions in the ET systems, we calculated the radial distribution function (RDF) between the Ru2+ and Ru3+ metal centers and the Cl− ions for each of the four systems (Figure S4). For all of these systems, the Cl− ions were found closer to the Ru3+ than to the Ru2+ because of its greater positive charge and smaller ligands.
Figure 2.
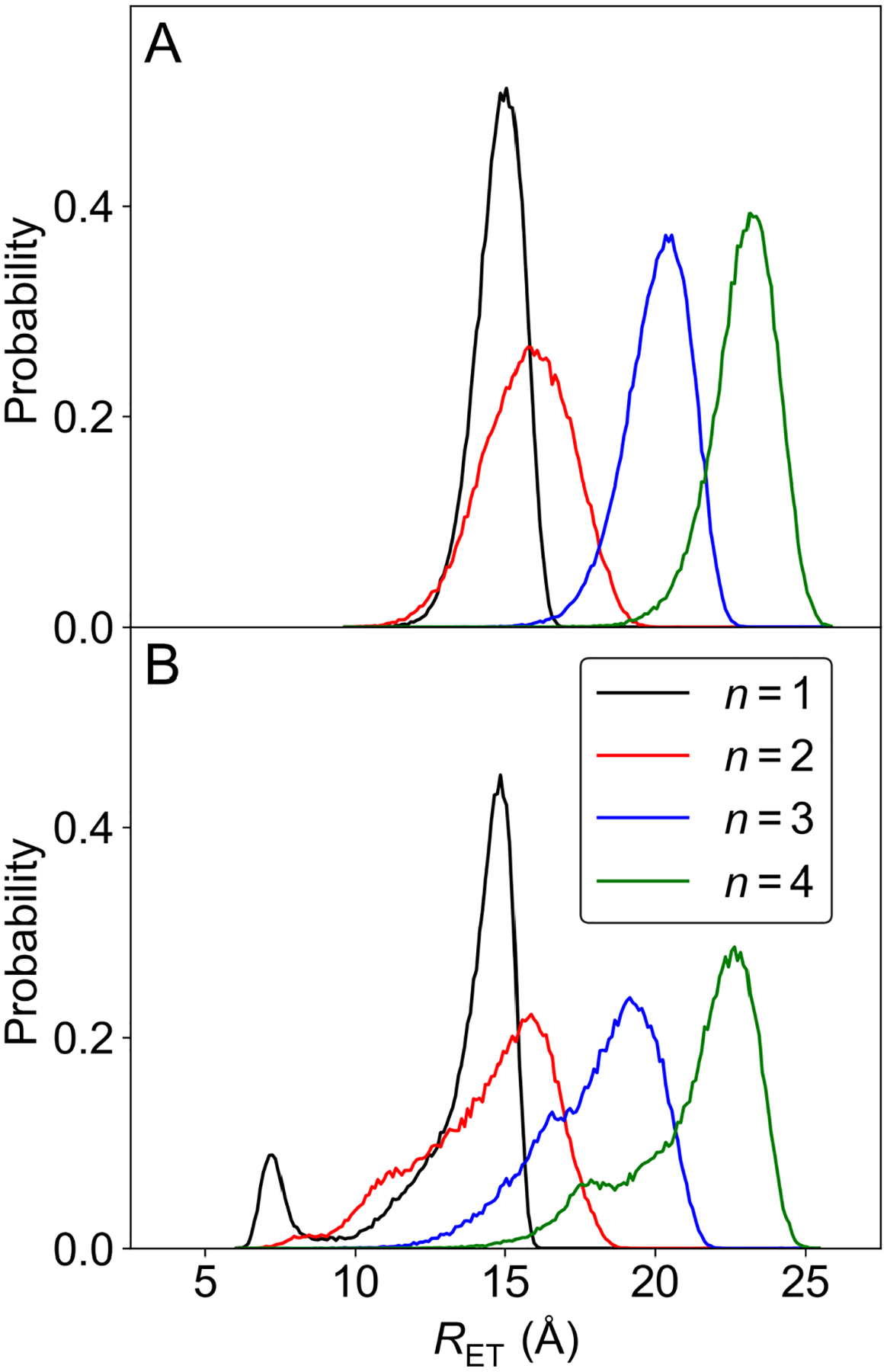
Probability distributions of the ET donor-acceptor distance in (A) the ET systems and (B) the PCET systems, where n is the number of proline residues in the bridge. These results are based on 1 μs MD trajectories for each of the eight systems.
Table 1.
Average ET Distances and Standard Deviations for the ET and PCET Systems.a
| n | ET systems | PCET systems |
|---|---|---|
| 1 | 14.8±0.8 | 13.4±2.3 |
| 2 | 15.7±1.5 | 14.3±2.2 |
| 3 | 20.1±1.1 | 18.1±1.9 |
| 4 | 22.9±1.1 | 21.3±2.1 |
These values were obtained from 1 μs MD trajectories for each system and are in units of Å.
To further analyze the probability distributions for the PCET systems, we fit the distribution for each system to a linear combination of three Gaussians (Table S6 and Figures S5 and S6). We also examined representative conformations corresponding to the distance associated with the maximum of the probability distribution function for the ET systems and the distance associated with each Gaussian component for the PCET systems, as depicted in Figures S7 and S8. The probability distribution function for the PCET system with n = 1 has an additional peak at short distances compared to the probability distribution functions for the longer peptide bridges. The conformation associated with this Gaussian component with RET ~7.2 Å (Figure S8) illustrates that the single proline linkage allows the Tyr to curl around in a manner that decreases its distance from the Ru3+ center, although it also samples more extended conformations. In contrast, the longer peptide bridges do not have the flexibility to curl in this manner, although they also sample a wide range of extended as well as bent conformations (Figure S8), leading to the shoulders exhibited in Figure 2B.
Experimental and calculated rate constants
The magnitudes of the slopes estimated from the dependence of the experimentally measured ln(k) values on the calculated values for the ET and PCET systems studied are provided in the last column of Table 2. The experimentally determined slope for the PCET reactions of the Ru/Tyr system, with hydrogen phosphate as the proton acceptor, is significantly smaller in magnitude than the slope for the ET reactions of the related Ru/Ru system. Note that the ET donor-acceptor distances can also be estimated on the basis of experimental data from related systems or using other strategies (see SI). We found that the slopes obtained with these different options for determining the ET distances do not vary significantly (Table S8 and Figures S9 and S10). For consistency, we present all of the experimental and calculated kinetic data in the main paper in terms of the calculated values.
Table 2.
Dependence of the Calculated (Calc.) and Experimentally Measured (Exptl.) ET and PCET Rate Constants on the Calculated .a
| Proton acceptor | Δ(QRuQPA) (e2) | pKa of proton acceptor | Calc. |slope| (Å−1) | Exptl. |slope| (Å−1) |
|---|---|---|---|---|
| ET reaction | ||||
| N/A | 0 | N/A | 1.37 | 1.34b |
| PCET reaction | ||||
| [HPO4]2− | 4 | 7.20c | 0.59 | 0.59d |
| Imidazole | 2 | 6.95e | 0.95 | 0.87f |
The calculated (experimental) magnitudes of the slopes are obtained from the calculated (experimental) ln(k) versus the calculated values from Table 1.
Value for ET in Ru/Ru system (Fig. 1A) determined by plotting the experimental rate constants from Ref. 13 as a function of the calculated values.
Value from Ref. 72.
Value for Ru/Tyr system (Fig. 1B) with phosphate as proton acceptor determined by plotting the experimental rate constants from Ref. 50 as a function of the calculated values.
Value from Ref. 73.
Value for Ru/Tyr system with imidazole as proton acceptor determined by plotting the experimental rate constants measured herein as a function of the calculated values.
To elucidate the factors that determine the shallow distance dependence of the PCET reaction rate constant with hydrogen phosphate as the proton acceptor, we calculated kET and kPCET for the eight systems using the nonadiabatic rate constant expressions and parameters given above. The slopes associated with the distance dependence of the rate constants were obtained from linear fits of the calculated values of ln(k) versus the corresponding average distances for the ET systems and the PCET systems with hydrogen phosphate as the proton acceptor (Figure 3). The calculated values of ln(k) versus were found to behave linearly with slopes that agree well with the corresponding slopes obtained experimentally (Table 2). Note that this agreement was obtained for the PCET systems by fitting the empirical parameter εeff to the experimentally determined slope (Figure 3B). As discussed below, however, the same value for εeff was used to successfully predict the increased magnitude of the slope for the PCET system with imidazole as the proton acceptor (Figure 3C).
Figure 3.

Comparison of the calculated (calc.) and experimentally measured (exptl.) ET and PCET rate constants as functions of the calculated values from Table 1: (A) ET systems, (B) PCET systems with hydrogen phosphate as the proton acceptor, and (C) PCET systems with imidazole as the proton acceptor. The experimentally measured PCET rate constants are second-order with units of M−1s−1, and the calculated rate constants are first-order with units of s−1, assuming the pre-equilibrium formation of a hydrogen-bonded complex. Given that these two rate constants are related by an unknown equilibrium constant, and the electronic coupling is unknown, the calculated ln(k) value for n = 1 was shifted to agree with the experimentally determined value for each system. Thus, the red and black data points are identical for n = 1 for each system and appear as red. For the PCET systems in part B, the experiments were performed in 0.05–0.60 M [HPO4]2− buffer solution at pH = 8.0. For the PCET systems in part C, the experiments were performed in 0.01–0.10 M imidazole at pH 7.6, with details given in the SI. For the ET systems, the experiments were performed in aqueous solutions of 0.13 M tert-butanol and NaClO4 at pH 6.53 The MD simulations used to compute the probability distributions of ET distances for the PCET systems with hydrogen phosphate as the proton acceptor were performed with 0.1 M [HPO4]2− buffer concentration, and the probability distributions were assumed to be the same for the PCET systems with imidazole as the proton acceptor. The MD simualtions used to compute the probability distributions of ET distances for the ET systems were performed through neutralization by Cl− counterions.
The parameter εeff should be viewed as an empirical factor that accounts for many different effects that are present in the experimental systems but are not included explicitly in our theoretical model. For example, this simple electrostatic model does not include the effects of charge delocalization, ion pairing, or the detailed electronic structure of the molecular system. To examine the fundamental basis for this electrostatic term, we performed constrained DFT calculations on three conformations of the n = 1 PCET system sampled during the MD simulations (Figure S8). The relative energies of the reactant and product states for the PCET reaction were computed by moving the proton from the donor to the acceptor and constraining the charge distribution using constrained DFT. As shown in Table S9, these relative reaction energies exhibit the trend that would be followed using the electrostatic term with εeff = 1, as expected for gas phase reactions. The conformations depicted in Figure S8 illustrate that the charges associated with the ends of the bridge interact through the solution or the molecular bridge or a combination thereof. Thus, the value of ~5 should be viewed as the result of averaging over all of these types of interactions, as well as accounting for the other effects mentioned above.
Impact of distance-dependent electrostatic term and validation of theoretical prediction
To understand the physical basis for the significantly different slopes observed for the ET systems and the PCET systems with hydrogen phosphate as the proton acceptor, we investigated the influence of the electrostatic term ΔEele on kPCET. We found that decreasing Δ(QRuQPA) with all other parameters, including εeff, held constant leads to a significant increase in the magnitude of the slope of ln(kPCET) versus . Specifically, this slope magnitude is 0.59 Å−1 for Δ(QRuQPA) = 4e2 and is 1.74 Å−1 when Δ(QRuQPA) = 0, illustrating that the slope becomes similar to that of the ET system when the ΔEele term in Eq. (5) is omitted. Moreover, the theoretical model predicts an intermediate slope magnitude of 0.96 Å−1 when Δ(QRuQPA) = 2e2 and all other parameters remain the same. This analysis also assumed that the electronic coupling attenuation parameter βET is the same for the ET and PCET systems. This assumption will be removed in additional calculations discussed below but does not change these qualitative trends.
To test this theoretical prediction, the PCET rate constants were experimentally measured for the same oligoproline metallopeptides with imidazole as the proton acceptor. The conjugate acid of imidazole has a pKa similar to the pKa of [H2PO4]− but is cationic rather than anionic, resulting in Δ(QRuQPA) = 2e2 in the numerator of the electrostatic contribution to the driving force. The magnitude of the slope of the experimental ln(kPCET) versus was found to be 0.87 Å−1, which is in qualitative agreement with our calculated value of 0.95 Å−1 for the system with imidazole as the proton acceptor. Note that using the pKa value of imidazole instead of phosphate and the Morse potential associated with an N-H rather than an O-H bond impacts the slope by only 0.01 Å−1. The agreement between the experimental measurement and our theoretical prediction provides validation for the theoretical model. It also supports our hypothesis that the electrostatic term ΔEele plays an important role in determining the shallow distance dependence of the PCET reaction rate constant with hydrogen phosphate as the proton acceptor.
To separate the effects of the electrostatic term and the probability distribution, we computed the slopes for both ET and PCET with two different electrostatic terms added to the driving force. If we set the electrostatic term ΔEele to 0 and 4e2/(εeffRET), we obtain different responses for the ET and PCET distance dependences. Specifically, the magnitude of the slope for the ET rate constant is 1.37 Å−1 and 1.14 Å−1, while the magnitude of the slope for the PCET rate constant with hydrogen phosphate as the proton acceptor is 1.74 Å−1 and 0.59 Å−1, respectively, for these two values of the electrostatic term. Thus, the electrostatic term exerts a much greater impact on the slope for the PCET reaction than for the ET reaction. As discussed below, our analysis suggests that this difference is due to the qualitative differences in the ET donor-acceptor distance probability distributions P(RET) for the ET and PCET systems shown in Figure 2.
Impact of probability distributions of ET distances
To further analyze the impact of P(RET), we fit the probability distributions obtained from the microsecond MD simulations to a linear combination of three Gaussians for each of the four PCET systems (Table S6 and Figures S5 and S6). To determine the contributions from these Gaussian components to the distance dependence of the PCET rate constants, we calculated the slopes for each of the four systems using only the third (major) Gaussian component, the sum of the second and third Gaussian components, or the sum of all three Gaussian components (Table 3). The magnitude of the slope calculated using only the third Gaussian component is larger than that calculated using the sum of the second and third Gaussian components, which in turn is larger than the magnitude of the slope calculated using the sum of all three Gaussian components. Thus, conformational sampling of the shorter ET distances effectively decreases the magnitude of the slope, leading to a shallower distance dependence. In contrast, the shorter distances are not as accessible for the ET systems, as indicated by the absence of the shoulders in the probability distributions (Figure 2). Note that when ΔEele = 0, this effect is not observed.
Table 3.
Influence of Electron Donor-Acceptor Distance Probability Distribution on Distance Dependence of PCET Rate Constant with Hydrogen Phosphate as Proton Acceptor.a
| P(RET) | Calc. |slope| (Å−1) | Calc. |slope| with ΔEele = 0 (Å−1) |
|---|---|---|
| Only third Gaussian | 0.93 | 1.65 |
| Only second and third Gaussians | 0.81 | 1.65 |
| All three Gaussians | 0.60 | 1.82 |
The first, second, and third Gaussian functions used to fit P(RET) are centered at increasing values of RET (Figures S5 and S6). The calculated magnitudes of the slopes were obtained from the calculated values of ln(kPCET) versus the calculated values using the fitted Gaussian distributions (Table S6).
Interplay between electrostatic term and probability distribution of ET distances
To investigate the interplay between the electrostatic term ΔEele and the probability distribution P(RET) in determining the distance dependence of the PCET rate constant, we analyzed the dependence of the various terms in the PCET rate constant kPCET(RET) on RET (Figure 4). This analysis includes only the term in Eq. (3) corresponding to the lowest reactant and product proton vibrational states, which was found to be the dominant contributor to the overall PCET rate constant (Table S10). This analysis used a constant reorganization energy corresponding to the value computed for the PCET system with n = 1 and hydrogen phosphate as the proton acceptor. The activation term increases as RET increases (Figure 4A) because ΔEele decreases as this distance increases. However, the squared electronic coupling, which is proportional to , exhibits the opposite trend (Figure 4B). The resulting ln[kPCET(RET)] exhibits a shallower distance dependence at RET distances in the range 12 – 16 Å and provides an explanation for the observation that these shorter distances are mainly responsible for the shallow distance dependence of the PCET rate constant (Figure 4C and Table 2). Thus, both the electrostatic term and the RET probability distribution play significant roles in determining the shallow distance dependence of the PCET rate constant. Changing ΔEele alters the slope in Figure 4A and therefore the curve shape in Figure 4C which, combined with the broadening of P(RET) toward shorter distances, leads to the shallow distance dependence of the overall PCET rate constant. This analysis indicates that the dependence of the rate constant on the ET distance can be influenced not only by the electronic coupling prefactor but also by the free energy barrier appearing in the exponential.
Figure 4.

Dependence of (A) negative activation free energy scaled by the thermal energy, −(ΔG00 + λ)2/4λkBT, (B) natural log of the square of the scaled electronic coupling, where , with βET taken to be 1.4 Å−1, and (C) ln[kPCET(RET)] on RET for the PCET system with hydrogen phosphate as the proton acceptor, including only the lowest reactant and product proton vibrational states.
The relatively flat curve for ln[kPCET(RET)] versus RET in Figure 4C at small RET predicts similar rate constants for the PCET systems with no proline linkage and with a single proline linkage, assuming the same value for . This behavior is consistent with experimental data indicating that the PCET system without a proline linkage has a rate constant of 3×106 M−1s−1,74 which is similar to the rate constant of 1.4×106 M−1s−1 measured experimentally50 for the PCET system with one proline linkage. As mentioned in the Introduction, only a few studies of the dependence of the PCET rate constant on the ET distance have been reported.12, 44, 47 In the study of PCET in iron carboxytetraphenylporphyrin complexes, a relatively small slope magnitude of 0.23 Å−1 for the overall distance dependence of the rate constant was measured,44 compared to the value of 0.4–0.46 Å−1 measured for ET reactions with the same bridge unit.12, 47 In this system, the PCET reaction has an electrostatic term with Δ(QRuQPA) = e2, while the ET reaction has an electrostatic term with Δ(QRuQPA) = −e2 or zero. These different electrostatic terms may account for the lower magnitude of the slope for PCET compared to ET in these systems. In general, these two factors, namely the broad distribution of ET distances and the distance-dependent electrostatic contributions to the driving force, could impact the distance dependence of the rate constants for ET systems as well as PCET systems.
Impact of coupling attenuation parameter
Previous work50 has suggested that the coupling attenuation parameter βET may be different for the ET and PCET systems, based on the different donor-bridge-acceptor energetics. To investigate this possibility, we repeated these calculations with βET = 1.2 Å−1 rather than 1.4 Å−1 for the PCET systems. We found that the experimentally determined slopes for the PCET systems with the two different proton acceptors, phosphate and imidazole, could also be reproduced after readjusting the empirical factor εeff in Eq. (6) (Table S11). However, a model in which the value of βET was different for the ET and PCET systems, without including the electrostatic term for PCET, cannot reproduce the larger magnitude of the slope with imidazole as the proton acceptor. Thus, the electrostatic term appears to be essential for describing the experimental data, but the different values of βET may also be a factor in describing the ET and PCET systems.
CONCLUSIONS
We investigated ET reactions in oligoproline bimetallic systems and PCET reactions in oligoproline metallopeptides using theoretical modeling. We performed microsecond MD simulations for the solvated ET and PCET systems and observed significant differences in the ET donor-acceptor distance probability distributions in these two systems. Specifically, the probability distributions for the ET systems are relatively narrow and symmetric, whereas those for the PCET systems are broader with shoulders at smaller distances, allowing the conformational sampling of these smaller distances on the microsecond timescale of the experiments. Another important distinction between the ET and PCET systems is the contribution to the driving force arising from the change in the electrostatic interaction between the charges at each end of the bridge. Although this electrostatic term is zero for the ET systems, it is positive and increases as the electron donor-acceptor distance decreases for the PCET systems. We emphasize that these effects are not related to fundamental differences between ET and PCET processes but rather are specific to these particular systems.
The ET and PCET rate constants computed with nonadiabatic theories reproduced the experimentally measured dependence of these rate constants on the average ET donor-acceptor distance for varying bridge lengths. The shallower distance dependence of the PCET rate constant with hydrogen phosphate as the proton acceptor was explained in terms of an additional positive, distance-dependent electrostatic term in the driving force, which attenuates the rate constant at smaller distances, in conjunction with the enhanced sampling of smaller distances observed in the MD simulations. The theoretical model predicted an increased magnitude of the slope for the PCET systems when imidazole is the proton acceptor because, although the conjugate acid of imidazole has a similar pKa value as dihydrogen phosphate, its cationic rather than anionic charge leads to a smaller positive electrostatic contribution to the driving force. This theoretical prediction of an increased magnitude of the slope was subsequently validated by experimental measurements.
This work highlights the impact of the nature of the proton acceptor, particularly its charge, on the ET distance dependence of the PCET rate constant. Because the ET distance dependence of the rate constant can be influenced by not only the conventional β parameter in the electronic coupling prefactor but also by a term in the free energy barrier, the slope associated with the distance dependence assumes a more complex interpretation. These insights into how short-range proton transfer reactions can be used to tune long-range electron transfer reactions have broad implications for designing charge-transfer systems relevant to solar cells, sensors, and other types of devices.
Supplementary Material
AKNOWLEDGMENT
We acknowledge the financial support from the National Institutes of Health Grant numbers GM056207 and GM50422. B.K. was supported by a Graduate Research Fellowship from the National Science Foundation. We thank the computational support from the Extreme Science and Engineering Discovery Environment (XSEDE),75 which is supported by the National Science Foundation (grant number ACI-1548562). This research used the GPU and CPU resources of Comet at San Diego Supercompueter Center (allocation number TG-MCB120097).
Funding Sources
Work by P.L., A.V.S., and S.H.-S. was supported by National Institutes of Health Grant GM056207. Work by B.K. and J.M.M. was supported by National Institutes of Health Grant 2R01GM50422. B.K. was supported by a Graduate Research Fellowship from the National Science Foundation.
Footnotes
Supporting Information
Details of the force field parameters, MD simulations, methods to calculate the reorganization energies and driving forces, parameters of the Morse potentials, experimentally determined rate constants and slopes, CDFT calculations, and supporting figures and tables.
The authors declare no competing financial interest.
REFERENCES
- (1).Gray HB; Winkler JR, Long-range electron transfer. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 3534–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mayo SL; Ellis WR; Crutchley RJ; Gray HB, Long-range electron transfer in heme proteins. Science 1986, 233, 948–952. [DOI] [PubMed] [Google Scholar]
- (3).Gray HB, Centenary Lecture. Long-range electron-transfer in blue copper proteins. Chem. Soc. Rev 1986, 15, 17–30. [Google Scholar]
- (4).Gray HB; Malmstroem BG, Long-range electron transfer in multisite metalloproteins. Biochemistry 1989, 28, 7499–7505. [DOI] [PubMed] [Google Scholar]
- (5).Miller JR; Calcaterra L; Closs G, Intramolecular long-distance electron transfer in radical anions. The effects of free energy and solvent on the reaction rates. J. Am. Chem. Soc 1984, 106, 3047–3049. [Google Scholar]
- (6).Warman JM; De Haas MP; Oevering H; Verhoeven J; Paddon-Row M; Oliver A; Hush N, Donor, acceptor, and self-quenching of the giant-dipole state of a rigid, σ-bond separated, donor-acceptor molecular assembly. Chem. Phys. Lett 1986, 128, 95–99. [Google Scholar]
- (7).Closs GL; Miller JR, Intramolecular long-distance electron transfer in organic molecules. Science 1988, 240, 440–447. [DOI] [PubMed] [Google Scholar]
- (8).Johnson MD; Miller JR; Green NS; Closs GL, Distance dependence of intramolecular hole and electron transfer in organic radical ions. J. Phys. Chem 1989, 93, 1173–1176. [Google Scholar]
- (9).Newton MD, Quantum chemical probes of electron-transfer kinetics: the nature of donor-acceptor interactions. Chem. Rev 1991, 91, 767–792. [Google Scholar]
- (10).Lambert C; Nöll G; Schelter J, Bridge-mediated hopping or superexchange electron-transfer processes in bis (triarylamine) systems. Nat. Mater 2002, 1, 69. [DOI] [PubMed] [Google Scholar]
- (11).Shin Y.-g. K.; Newton MD; Isied SS, Distance dependence of electron transfer across peptides with different secondary structures: the role of peptide energetics and electronic coupling. J. Am. Chem. Soc 2003, 125, 3722–3732. [DOI] [PubMed] [Google Scholar]
- (12).Weiss EA; Ahrens MJ; Sinks LE; Gusev AV; Ratner MA; Wasielewski MR, Making a Molecular Wire: Charge and Spin Transport through para-Phenylene Oligomers. J. Am. Chem. Soc 2004, 126, 5577–5584. [DOI] [PubMed] [Google Scholar]
- (13).Malak RA; Gao Z; Wishart JF; Isied SS, Long-range electron transfer across peptide bridges: The transition from electron superexchange to hopping. J. Am. Chem. Soc 2004, 126, 13888–13889. [DOI] [PubMed] [Google Scholar]
- (14).Eng MP; Albinsson B, Non-Exponential Distance Dependence of Bridge-Mediated Electronic Coupling. Angew. Chem 2006, 118, 5754–5757. [DOI] [PubMed] [Google Scholar]
- (15).Cordes M; Köttgen A; Jasper C; Jacques O; Boudebous H; Giese B, Influence of Amino Acid Side Chains on Long-Distance Electron Transfer in Peptides: Electron Hopping via “Stepping Stones”. Angew. Chem. Int. Ed 2008, 47, 3461–3463. [DOI] [PubMed] [Google Scholar]
- (16).Gray HB; Winkler JR, Electron flow through proteins. Chem. Phys. Lett 2009, 483, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hanss D; Walther ME; Wenger OS, Importance of covalence, conformational effects and tunneling-barrier heights for long-range electron transfer: insights from dyads with oligo-p-phenylene, oligo-p-xylene and oligo-p-dimethoxybenzene bridges. Coord. Chem. Rev 2010, 254, 2584–2592. [Google Scholar]
- (18).Ding F; Wang H; Wu Q; Van Voorhis T; Chen S; Konopelski JP, Computational study of bridge-assisted intervalence electron transfer. J. Phys. Chem. A 2010, 114, 6039–6046. [DOI] [PubMed] [Google Scholar]
- (19).Wenger OS, How donor-bridge-acceptor energetics influence electron tunneling dynamics and their distance dependences. Acc. Chem. Res 2010, 44, 25–35. [DOI] [PubMed] [Google Scholar]
- (20).Wenger OS, Photoinduced electron and energy transfer in phenylene oligomers. Chem. Soc. Rev 2011, 40, 3538–3550. [DOI] [PubMed] [Google Scholar]
- (21).Kuss-Petermann M; Wenger OS, Electron transfer rate maxima at large donor-acceptor distances. J. Am. Chem. Soc 2016, 138, 1349–1358. [DOI] [PubMed] [Google Scholar]
- (22).Beratan DN; Onuchic JN; Hopfield J, Limiting forms of the tunneling matrix element in the long distance bridge mediated electron transfer problem. J. Chem. Phys 1985, 83, 5325–5329. [Google Scholar]
- (23).Cave RJ; Newton MD; Kumar K; Zimmt MB, Theoretical study of solvent effects on the electronic coupling matrix element in rigidly linked donor-acceptor systems. J. Phys. Chem 1995, 99, 17501–17504. [Google Scholar]
- (24).Sachs SB; Dudek SP; Hsung RP; Sita LR; Smalley JF; Newton MD; Feldberg SW; Chidsey CE, Rates of interfacial electron transfer through π-conjugated spacers. J. Am. Chem. Soc 1997, 119, 10563–10564. [Google Scholar]
- (25).Lewis FD; Liu J; Weigel W; Rettig W; Kurnikov IV; Beratan DN, Donor-bridge-acceptor energetics determine the distance dependence of electron tunneling in DNA. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 12536–12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cave RJ; Newton MD, Multistate Treatments of the Electronic Coupling in Donor-Bridge-Acceptor Systems: Insights and Caveats from a Simple Model. J. Phys. Chem. A 2014, 118, 7221–7234. [DOI] [PubMed] [Google Scholar]
- (27).Isied SS; Vassilian A; Wishart JF; Creutz C; Schwarz HA; Sutin N, The distance dependence of intramolecular electron-transfer rates: Importance of the nuclear factor. J. Am. Chem. Soc 1988, 110, 635–637. [Google Scholar]
- (28).Tachiya M; Murata S, New explanation for the lack of the inverted region in charge separation reactions. J. Phys. Chem 1992, 96, 8441–8444. [Google Scholar]
- (29).Tachiya M; Murata S, Non-Marcus energy gap dependence of back electron transfer in contact ion pairs. J. Am. Chem. Soc 1994, 116, 2434–2436. [Google Scholar]
- (30).Georgievskii Y; Burshtein AI; Chernobrod BM, Electron transfer in the inverted region: Adiabatic suppression and relaxation hindrance of the reaction rate. J. Chem. Phys 1996, 105, 3108–3120. [Google Scholar]
- (31).Lanzilotta WN; Christiansen J; Dean DR; Seefeldt LC, Evidence for coupled electron and proton transfer in the [8Fe-7S] cluster of nitrogenase. Biochemistry 1998, 37, 11376–11384. [DOI] [PubMed] [Google Scholar]
- (32).Alexov E; Gunner M, Calculated protein and proton motions coupled to electron transfer: electron transfer from QA- to QB in bacterial photosynthetic reaction centers. Biochemistry 1999, 38, 8253–8270. [DOI] [PubMed] [Google Scholar]
- (33).Belevich I; Verkhovsky MI; Wikström M, Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature 2006, 440, 829. [DOI] [PubMed] [Google Scholar]
- (34).Wörsdörfer B; Conner DA; Yokoyama K; Livada J; Seyedsayamdost M; Jiang W; Silakov A; Stubbe J; Bollinger JM Jr; Krebs C, Function of the diiron cluster of Escherichia coli class Ia ribonucleotide reductase in proton-coupled electron transfer. J. Am. Chem. Soc 2013, 135, 8585–8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Soudackov A; Hatcher E; Hammes-Schiffer S, Quantum and dynamical effects of proton donor-acceptor vibrational motion in nonadiabatic proton-coupled electron transfer reactions. J. Chem. Phys 2005, 122, 014505. [DOI] [PubMed] [Google Scholar]
- (36).Hammes-Schiffer S; Hatcher E; Ishikita H; Skone JH; Soudackov AV, Theoretical studies of proton-coupled electron transfer: models and concepts relevant to bioenergetics. Coord. Chem. Rev 2008, 252, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhang M-T; Irebo T; Johansson O; Hammarström L, Proton-coupled electron transfer from tyrosine: A strong rate dependence on intramolecular proton transfer distance. J. Am. Chem. Soc 2011, 133, 13224–13227. [DOI] [PubMed] [Google Scholar]
- (38).Markle TF; Rhile IJ; Mayer JM, Kinetic effects of increased proton transfer distance on proton-coupled oxidations of phenol-amines. J. Am. Chem. Soc 2011, 133, 17341–17352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Markle TF; Tenderholt AL; Mayer JM, Probing quantum and dynamic effects in concerted proton-electron transfer reactions of phenol-base compounds. J. Phys. Chem. B 2012, 116, 571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Soudackov AV; Hammes-Schiffer S, Nonadiabatic rate constants for proton transfer and proton-coupled electron transfer reactions in solution: Effects of quadratic term in the vibronic coupling expansion. J. Chem. Phys 2015, 143, 194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Soudackov AV; Hammes-Schiffer S, Proton-coupled electron transfer reactions: analytical rate constants and case study of kinetic isotope effects in lipoxygenase. Faraday Discuss 2016, 195, 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Markle TF; Zhang M-T; Santoni M-P; Johannissen LO; Hammarström L, Proton-Coupled Electron Transfer in a Series of Ruthenium-Linked Tyrosines with Internal Bases: Evaluation of a Tunneling Model for Experimental Temperature-Dependent Kinetics. J. Phys. Chem. B 2016, 120, 9308–9321. [DOI] [PubMed] [Google Scholar]
- (43).Glover SD; Parada GA; Markle TF; Ott S; Hammarström L, Isolating the effects of the proton tunneling distance on proton-coupled electron transfer in a series of homologous tyrosine-base model compounds. J. Am. Chem. Soc 2017, 139, 2090–2101. [DOI] [PubMed] [Google Scholar]
- (44).Warren JJ; Menzeleev AR; Kretchmer JS; Miller III TF; Gray HB; Mayer JM, Long-range proton-coupled electron-transfer reactions of bis (imidazole) iron tetraphenylporphyrins linked to benzoates. J. Phys. Chem. Lett 2013, 4, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Chen J; Kuss-Petermann M; Wenger OS, Distance Dependence of Bidirectional Concerted Proton-Electron Transfer in Phenol-Ru (2, 2′-bipyridine) 32+ Dyads. Chem. Eur. J 2014, 20, 4098–4104. [DOI] [PubMed] [Google Scholar]
- (46).Chen J; Kuss-Petermann M; Wenger OS, Dependence of Reaction Rates for Bidirectional PCET on the Electron Donor-Electron Acceptor Distance in Phenol-Ru (2, 2′-Bipyridine) 32+ Dyads. J. Phys. Chem. B 2015, 119, 2263–2273. [DOI] [PubMed] [Google Scholar]
- (47).Helms A; Heiler D; McLendon G, Electron transfer in bis-porphyrin donor-acceptor compounds with polyphenylene spacers shows a weak distance dependence. J. Am. Chem. Soc 1992, 114, 6227–6238. [Google Scholar]
- (48).Hanss D; Wenger OS, Electron tunneling through oligo-p-xylene bridges. Inorg. Chem 2008, 47, 9081–9084. [DOI] [PubMed] [Google Scholar]
- (49).Hanss D; Wenger OS, Tunneling barrier effects on photoinduced charge transfer through covalent rigid rod-like bridges. Inorg. Chem 2009, 48, 671–680. [DOI] [PubMed] [Google Scholar]
- (50).Koronkiewicz B; Swierk J; Regan K; Mayer JM, Shallow Distance Dependence for Proton-Coupled Tyrosine Oxidation in Oligoproline Peptides. J. Am. Chem. Soc 2020, Accepted, DOI: 10.1021/jacs.0c01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Isied SS; Vassilian A; Magnuson RH; Schwarz HA, Electron transfer across polypeptides. 5. Rapid rates of electron transfer between osmium (II) and cobalt (III) in complexes with bridging oligoprolines and other polypeptides. J. Am. Chem. Soc 1985, 107, 7432–7438. [Google Scholar]
- (52).Vassilian A; Wishart JF; Van Hemelryck B; Schwarz H; Isied SS, Electron transfer across polypeptides. 6. Long-range electron transfer in osmium-ruthenium binuclear complexes bridged with oligoproline peptides. J. Am. Chem. Soc 1990, 112, 7278–7286. [Google Scholar]
- (53).Ogawa MY; Wishart JF; Young Z; Miller JR; Isied SS, Distance dependence of intramolecular electron transfer across oligoprolines in [(bpy) 2RuIIL. bul.-(Pro) n-CoIII (NH3) 5] 3+, n= 1–6: different effects for helical and nonhelical polyproline II structure. J. Phys. Chem 1993, 97, 11456–11463. [Google Scholar]
- (54).McConnell HM, Intramolecular charge transfer in aromatic free radicals. J. Chem. Phys 1961, 35, 508–515. [Google Scholar]
- (55).Soudackov A; Hammes-Schiffer S, Multistate continuum theory for multiple charge transfer reactions in solution. J. Chem. Phys 1999, 111, 4672–4687. [Google Scholar]
- (56).Soudackov A; Hammes-Schiffer S, Derivation of rate expressions for nonadiabatic proton-coupled electron transfer reactions in solution. J. Chem. Phys 2000, 113, 2385–2396. [Google Scholar]
- (57).Hammes-Schiffer S; Soudackov AV, Proton-coupled electron transfer in solution, proteins, and electrochemistry. J. Phys. Chem. B 2008, 112, 14108–14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Nickita N; Belousoff MJ; Bhatt AI; Bond AM; Deacon GB; Gasser G; Spiccia L, Synthesis, structure, spectroscopic properties, and electrochemical oxidation of ruthenium (II) complexes Incorporating monocarboxylate bipyridine ligands. Inorg. Chem 2007, 46, 8638–8651. [DOI] [PubMed] [Google Scholar]
- (59).Souza ML; Castellano EE; Telser J; Franco DW, Secondary Coordination Sphere Effects in Ruthenium (III) Tetraammine Complexes: Role of the Coordinated Water Molecule. Inorg. Chem 2015, 54, 2067–2080. [DOI] [PubMed] [Google Scholar]
- (60).Wilhelm P; Lewandowski B; Trapp N; Wennemers H, A crystal structure of an oligoproline PPII-helix, at last. J. Am. Chem. Soc 2014, 136, 15829–15832. [DOI] [PubMed] [Google Scholar]
- (61).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA, Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (63).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (64).Bayly CI; Cieplak P; Cornell W; Kollman PA, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem 1993, 97, 10269–10280. [Google Scholar]
- (65).Cieplak P; Cornell WD; Bayly C; Kollman PA, Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem 1995, 16, 1357–1377. [Google Scholar]
- (66).Li P; Merz KM Jr, MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model 2016, 56, 599–604. [DOI] [PubMed] [Google Scholar]
- (67).Darden T; York D; Pedersen L, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (68).Salomon-Ferrer R; Götz AW; Poole D; Le Grand S; Walker RC, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 2013, 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- (69).Marcus RA; Sutin N, Electron transfers in chemistry and biology. Biochim. Biophys. Acta, Rev. Bioenerg 1985, 811, 265–322. [Google Scholar]
- (70).Marcus RA, On the theory of oxidation-reduction reactions involving electron transfer. I. J. Chem. Phys 1956, 24, 966–978. [Google Scholar]
- (71).López V; Rivera A; Smirnov YF; Frank A, Simple evaluation of Franck-Condon factors and non-Condon effects in the Morse potential. Int. J. Quantum Chem 2002, 88, 280–295. [Google Scholar]
- (72).Powell KJ; Brown PL; Byrne RH; Gajda T; Hefter G; Sjöberg S; Wanner H, Chemical speciation of environmentally significant heavy metals with inorganic ligands. Part 1: The Hg2+-Cl−, OH−, CO32−, SO42−, and PO43-aqueous systems (IUPAC Technical Report). Pure Appl. Chem 2005, 77, 739–800. [Google Scholar]
- (73).Walba H; Isensee RW, Acidity constants of some arylimidazoles and their cations. J. Org. Chem 1961, 26, 2789–2791. [Google Scholar]
- (74).Irebo T; Reece SY; Sjödin M; Nocera DG; Hammarström L, Proton-coupled electron transfer of tyrosine oxidation: Buffer dependence and parallel mechanisms. J. Am. Chem. Soc 2007, 129, 15462–15464. [DOI] [PubMed] [Google Scholar]
- (75).Towns J; Cockerill T; Dahan M; Foster I; Gaither K; Grimshaw A; Hazlewood V; Lathrop S; Lifka D; Peterson GD, XSEDE: accelerating scientific discovery. Comput. Sci. Eng 2014, 16, 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.