

Abstract
Aims
Haemodynamic determinants of blood pressure (BP) include cardiac output (CO), systemic vascular resistance (SVR), and arterial stiffness. We investigated the heritability of these phenotypes, their association with BP-related single-nucleotide polymorphisms (SNPs), and the causal association between BP and arterial stiffness.
Methods and results
We assessed BP, central BP components, and haemodynamic properties (during a single visit) including CO, SVR, and pulse wave velocity (PWV, measure of arterial stiffness) in 3531 (1934 monozygotic, 1586 dizygotic) female TwinsUK participants. Heritability was estimated using structural equation modelling. Association with 984 BP-associated SNP was examined using least absolute shrinkage and selection operator (LASSO) and generalized estimating equation regression. One and two-sample Mendelian randomization (MR) was used to estimate the causal direction between BP and arterial stiffness including data on 436 419 UK Biobank participants. We found high heritability for systolic and pulsatile components of BP (>50%) and PWV (65%) with overlapping genes accounting for >50% of their observed correlation. Environmental factors explained most of the variability of CO and SVR (>80%). Regression identified SNPs (n = 5) known to be associated with BP to also be associated with PWV. One-sample MR showed evidence of bi-directional causal association between BP and PWV in TwinsUK participants. Two-sample MR, confirmed a bi-directional causal effect of PWV on BP (inverse variance weighted (IVW) beta = 0.11, P < 0.02) and BP on arterial stiffness (IVW beta = 0.004, P < 0.0001).
Conclusion
The genetic basis of BP is mediated not only by genes regulating BP but also by genes that influence arterial stiffness. Mendelian randomization indicates a bi-directional causal association between BP and arterial stiffness.

Keywords: Blood pressure, Genes, Haemodynamics, Arterial stiffness, Mendelian randomization
See page 3323 for the editorial comment on this article (doi: 10.1093/eurheartj/ehaa434)
Introduction
Hypertension is one of the most important risk factors for cardiac, cerebrovascular, and renal-associated morbidity and mortality and the largest contributor to the global burden of disease.1 Twin and family studies have identified a substantial (49–54%) heritable component to blood pressure (BP)2 and genetic association studies have now identified a large number of individual single-nucleotide polymorphisms that associate with BP.3 Haemodynamic determinants of BP include cardiac output (CO), systemic vascular resistance (SVR, determined by the microvasculature), and stiffness of large arteries (Table 1). Cardiac output and SVR determine steady state or mean arterial pressure (MAP). Stroke volume (SV) and large artery stiffness (and other properties), determine pulse pressure (PP). We investigated the heritability and shared heritability of these underlying haemodynamic properties with those of conventional peripheral BP and components of central aortic BP (Figure 1) in the TwinsUK cohort. To understand the mechanism by which genetic polymorphisms influence BP, we examined the association of BP phenotypes and cardiovascular properties with genetic variants previously associated with BP and examined the direction of causality between BP and heritable haemodynamic properties using Mendelian randomization (MR). This was performed in the TwinsUK and UK Biobank cohorts as these two cohorts have complimentary properties. Arterial stiffness [measured using the ‘gold standard’ carotid-femoral pulse wave velocity (PWV)] and BP were available within TwinsUK. In the much larger UK Biobank cohort, BP was available and arterial stiffness was estimated from a pulse wave-derived index.
Table 1.
Glossary of key definitions relating to heritability and cardiovascular measures
| Heritability definitions | |
|---|---|
| ACE model | This model assumes that the source of phenotype variance can be attributed to genetic influences (A), shared environmental factors (C), and unique environmental factors (E). Environmental factors are all those that are not inherited irrespective of whether they are explicitly measured. |
| CE model | Assumes that the source of phenotype variance can be attributed to shared environmental factors (C) and unique environmental factors (E) |
| Heritability | Proportion of population variance of a phenotype attributed to genetic factors at a particular time point. |
| Cardiovascular definitions | |
| Augmentation pressure | Augmentation pressure (AP) is the difference between central systolic blood pressure and P1 (see Figure 1). |
| Cardiac output | Volume of blood ejected by the left ventricle per minute. |
| Pressure at P1 | Pressure at the first systolic shoulder of the central pressure waveform. P1 represents the pressure at the first systolic shoulder or peak and corresponds to the time of peak myocardial wall stress (see Figure 1). |
| Pulse pressure | Pulse pressure is the pressure difference between systolic and diastolic pressure. |
| Pulse wave velocity | Pulse wave velocity is the velocity at which the pressure pulse propagates along the arterial tree and is regarded as the gold-standard measure of arterial stiffness. |
| Stiffness index | An index of arterial stiffness derived from the finger photoplethysmography that both theoretically and empirically relates to PWV. |
| Systemic vascular resistance | Resistance to blood flow offered by the systemic vasculature. |
Figure 1.

Example of a central blood pressure waveform separated into its components P1 (pressure at the first systolic shoulder) and augmentation pressure (AP), systolic blood pressure (SBP), and diastolic blood pressure (DBP).
Methods
Participants
Study participants were 3531 monozygotic (MZ) and dizygotic (DZ) female twins (2442 with genotyping) enrolled in the TwinsUK national registry of adult twins without regard to phenotype status.4 TwinsUK began in 1992 and initially only recruited middle-aged women to investigate osteoarthritis and osteoporosis in women. As a result, the cohort is predominantly female and only women were included in the present study.4 Peripheral BP, central BP (including height of the first systolic shoulder, P1 and augmentation pressure, AP, Figure 1), and carotid-femoral PWV were measured in all participants. In addition, 1625 participants underwent echocardiography to measure left ventricular outflow track (LVOT) diameter, SV, and CO. Systemic vascular resistance was calculated from MAP and CO. The study was approved by St Thomas’ Hospital research ethics committee and written informed consent was obtained from all participants. Details of the genotyping, BP, and cardiovascular measurements are provided in the Supplementary material online.
Heritability
Influence of genetic factors (A) and environmental factors was modelled in twins using the ACE twin model. Environmental influences were partitioned into those that are shared between twins (C) and therefore make them more similar (e.g. raised in same household); and those that are unique to individuals (E) and result in differences between twins (and which includes measurement error). Shared environment was assumed to correlate perfectly for both MZ and DZ twins whereas unique environment was assumed to be uncorrelated in twins. Environmental factors represent the totality of all such factors whether measured or unmeasured. Details of the heritability modelling are provided in the Supplementary material online.
Blood pressure associated gene variants
To determine to what extent genes that influence BP associate with specific haemodynamic determinants of BP, we selected 984 single-nucleotide polymorphisms (SNPs) shown to be robustly associated with BP in the most recent genome-wide association studies (GWAS).3 Of these, data for 896 SNPs were available from genotyping in the TwinsUK. It was not expected that all known SNPs would contribute to BP in our cohort. Therefore, in order to identify the most informative SNPs associated with BP and to protect against weak instrument bias5 in MR, we performed least absolute shrinkage and selection operator (LASSO) regression using Stata version 14 and the cvlasso function on 799 SNPs (number of variables is limited to 800 in the cvlasso function, so we selected polymorphisms with an allele frequency >0.10). Least absolute shrinkage and selection operator regression performs variable selection and shrinkage at the same time by penalizing parameters that contribute little to the fit of the model. This analysis is based on a type of machine learning where data is split into training (30%) and validation datasets (70%) and results are based on 10-fold cross-validation analysis.6 Selecting SNPs that associated with BP in LASSO regression, we then examined the association of those SNPs to BP components and heritable haemodynamic properties using generalized estimating equations (GEE) which account for the relationship structure of twins. All SNPs were included in the model at the same time. In addition, we repeated the analysis using LASSO regression. Results are shown for SNPs with both a P-value <0.05 in GEE and that were selected in LASSO regression. Augmentation pressure was transformed (square root) for the analysis.
Mendelian randomization—TwinsUK
To determine the direction of causality between BP and arterial stiffness, we first performed one-sample bi-directional MR using a two-stage least squares regression analysis with STATA software and the command ivregress using a multiple instruments model7 in TwinsUK. For this analysis, the exposure is estimated by the genotypes (instrumental variables, IV) by calculating predictive values from the regression of the exposure on the genotypes and then regressing the outcome variable (PWV) on the predicted exposure to obtain a causal effect estimate.8 The IV was all SNPs identified from LASSO regression analysis to associate with BP in the TwinsUK cohort (n = 56). Genotypes were coded 0, 1, and 2 and an additive genetic model was assumed (we also performed the analysis with all SNPs irrespective of whether they were selected by LASSO). Secondly, the causal effect of PWV on BP was investigated. In this case, the IVs were two SNPs previously identified to robustly associate with PWV from GWAS (P < 5 × 10−8),9 the exposure was PWV and the outcome was BP. Sensitivity analysis was performed including only one twin in the analysis to ensure the twin family structure did not influence the results.
Mendelian randomization—Biobank UK
Since one-sample MR may provide biased estimates of effect size,10 a two-sample MR was also performed using summary-level GWAS data available from Biobank UK and the MR-Base platform (http://www.mrbase.org). UK Biobank comprises 502 000 genotyped adults aged between 40 and 69 years of age of whom 436 419 have BP data. To determine whether PWV-associated SNPs are associated with BP, the IV were built considering GWAS significant SNPs (P < 5 × 10−8) and suggestive SNPs (P < 1 × 10−5) from separate loci defined by linkage disequilibrium (LD) structure (r 2 < 0.80). The more liberal P-value threshold of P < 1 × 10−5 was adopted because only one SNP was available for the more conservative P-value analysis. Statistical associations between individual SNPs and PWV were taken from Mitchel et al. 9 If a SNP was absent in the summary GWAS statistics, a proxy SNP in high LD with r 2 ≥ 0.80 was used where available. However, if this was not successful, the SNP was excluded and thus not all 18 SNPs were included in the final analysis. The association between IV and outcome was assessed using inverse-variance weighted (IVW) regression models. We also assessed the association using the weighted median method which is less sensitive to outliers.11 We performed an MR-Egger test to look for directional pleiotropy.12 Leave-one-out sensitivity was performed to exclude the possibility of one SNP having a large effect on the overall results. To determine whether BP-associated SNPs were associated with arterial stiffness, IV were built considering the 984 SNPs previously identified from GWAS. In this case, the outcome was arterial stiffness index (SI), an estimate of arterial stiffness obtained using the PulseTrace (PCA2, CareFusion, USA) device, which is correlated to carotid-femoral PWV.13
Results
Participant characteristics (n = 3531, 1934 MZ and 1586 DZ) in the TwinsUK cohort by zygosity are shown in Table 2. Mean (±SD) age for women was 57.7 ± 12.9 years, with average peripheral systolic BP (SBP) and diastolic BP (DBP) of 126 ± 17 and 74 ± 9 mmHg, respectively. Twenty-two per cent were on antihypertensive treatment and 14% were on lipid-lowering therapy. Three per cent were treated for diabetes mellitus and 9% were current smokers. Compared with MZ twins, DZ twins were older, had higher SBP and DBP, and a higher percentage were current smokers and on treatment for hypertension and hypercholesterolaemia.
Table 2.
Table of participant characteristics for the TwinsUK cohort and by zygosity
| Variable | N | TwinsUK Cohort | MZ twins (N = 1934) | DZ Twins (N = 1586) | P-value |
|---|---|---|---|---|---|
| Age (years) | 3531 | 56.7 ± 12.9 | 55.2 ± 13.8 | 58.6 ± 11.1 | <0.001 |
| SBP (mmHg) | 3416 | 125.6 ± 17.3 | 124.5 ± 17.3 | 127.0 ± 12.4 | <0.001 |
| DBP (mmHg) | 3416 | 73.7 ± 8.9 | 73.2 ± 8.8 | 74.2 ± 9.0 | <0.001 |
| PP (mmHg) | 3416 | 52.0 ± 12.7 | 51.3 ± 12.9 | 52.8 ± 12.3 | <0.001 |
| Antihypertensive treatment (%) | 3502 | 21.5 | 20.3 | 23.2 | =0.040 |
| Lipid-lowering treatment (%) | 3503 | 14.2 | 12.7 | 16.1 | =0.004 |
| Diabetes mellitus treatment (%) | 3531 | 2.5 | 2.1 | 3 | =0.070 |
| Current smoker (%) | 3528 | 9.4 | 8.0 | 11.0 | =0.002 |
| AP (mmHg) | 3371 | 13.8 ± 8.0 | 13.2 ± 8.1 | 14.6 ± 7.6 | <0.001 |
| P1 (mmHg) | 3371 | 28.7 ± 6.8 | 28.4 ± 6.9 | 29.1 ± 6.6 | =0.001 |
| PWV (m/s) | 3309 | 9.2 ± 2.1 | 9.1 ± 2.1 | 9.4 ± 2.1 | <0.001 |
| LVOT diameter (mm) | 1625 | 20.0 ± 1.9 | 19.9 ± 1.9 | 20.1 ± 1.8 | =0.008 |
| Cardiac output (l/min) | 1582 | 4.49 ± 1.2 | 4.46 ± 1.2 | 4.52 ± 1.3 | =0.290 |
| SVR (dyn□s□cm−5) | 1540 | 1761 ± 563 | 1747 ± 543 | 1779 ± 588 | =0.280 |
Subject characteristics are summarized as means and standard deviation unless otherwise stated. Comparison between groups were made using Students’ t-test and χ2 test.
AP, augmentation pressure; DBP, diastolic blood pressure; DZ, dizygotic; LVOT, left ventricular outflow tract; MAP, mean arterial pressure; MZ, monozygotic; PP, pulse pressure; PWV, pulse wave velocity; SBP, systolic blood pressure; SVR, systemic vascular resistance.
Heritability of blood pressure and haemodynamic parameters
Unadjusted intra-class correlation coefficients for all BP components were higher for MZ compared with DZ twin pairs suggesting a genetic influence on these measures (Supplementary material online, Table S1). Compared with BP, differences in intra-class correlations between MZ and DZ twins for SV, CO, LVOT diameter, and SVR were smaller suggesting a comparatively smaller genetic influence on these measures. After adjusting for age, univariable model fitting confirmed a substantial additive genetic component for peripheral SBP and DBP (63% and 58%, respectively) and for other BP components including PP, AP, and P1: the additive genetic component was >55% for these components in the ACE model (Figure 2). Out of all the cardiovascular determinants of BP, a substantial additive genetic component was observed only for PWV (67%) after age adjustment. Estimates of shared environment were close to zero and the most parsimonious model for BP components and PWV was the AE model (Supplementary material online, Table S2).
Figure 2.

Bar graph of ACE modelling estimates. AP, augmentation pressure; CO, cardiac output; DBP, diastolic blood pressure; LVOT, left ventricular outflow tract; PP, pulse pressure; PWV, pulse wave velocity; SBP, systolic blood pressure; SV, stroke volume; SVR, systemic vascular resistance. Bars represent 95% confidence intervals.
Heritability estimates for SV (8%), CO (15%), LVOT diameter (17%), and SVR (5%) were much lower compared with those for BP components and PWV in the ACE model (Figure 2). Further adjusting the model for height or BMI did not appreciably change the estimates. For these phenotypes, the CE model was the most parsimonious model suggesting a non-significant genetic effect (Supplementary material online, Table S2). Shared environment accounted for 28%, 29%, 52%, and 32% of the variability for SV, CO, LVOT diameter, and SVR in the CE model, respectively. Sensitivity analysis excluding individuals on antihypertensive therapy produced comparable results for all phenotypes (Supplementary material online, Table S3).
Phenotypic correlation and shared genetic heritability between blood pressure components and pulse wave velocity
We next performed bivariate heritability analysis to determine to what extent the correlation between BP components and PWV (which were highly heritable) can be explained by overlapping genetic factors (Supplementary material online, Table S4). The phenotypic correlation between SBP and other BP components (PP, AP, P1), apart from DBP was moderately high (r ≥ 0.49). Similarly, the phenotypic correlation between BP components, apart from DBP and AP, with PWV was moderately high (r ≥ 0.55). Bivariate heritability analysis of the association between BP components and PWV suggested a large genetic overlap (>50% of the co-variance explained by additive genetic factors, Supplementary material online,Table S4). Common genetic factors explained a large percentage of the correlation between P1 and PWV (49%) but only a modest proportion between AP and PWV (20%).
Association between blood pressure-associated single-nucleotide polymorphisms with components of blood pressure and haemodynamic parameters
LASSO regression identified 56 SNPs associated with SBP, DBP, or PP in the current cohort (Supplementary material online, Table S5). We next tested the association between these 56 SNPs with BP components and PWV (Table 3). From LASSO regression analysis, we observed an association between six SNPs (rs10923038, rs3184504, rs3745318, rs10842991, rs3742182, and rs1055144) and AP (Table 3). Five SNPs (rs2390258, rs9888615, rs9860290, rs4810332, and rs11909120) associated with P1. Five SNPs (rs9888615, rs2390258, rs4553000, and rs4980515) associated with PWV. There was little overlap between association of SNPs with different BP components and PWV except for P1 and PWV for which two SNPs associated with both P1 and PWV (rs2390258 and rs9888615, Table 3).
Table 3.
Association between blood pressure single-nucleotide polymorphisms with components of blood pressure components and pulse wave velocity
| Augmentation pressure |
Blood pressure at P1 |
Pulse wave velocity |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SBP ID | Gene | BP trait | beta | P | OLS | beta | P | OLS | beta | P | OLS |
| rs10923038 | DBP | −0.09 | <0.01 | −0.09 | — | — | — | — | — | — | |
| rs3742182 | DBP | 0.11 | <0.01 | 0.09 | — | — | — | — | — | — | |
| rs3184504 | SHB3 | DBP/SBP/PP | 0.08 | <0.05 | 0.06 | — | — | — | — | — | — |
| rs3745318 | KLF2 | DBP | 0.07 | <0.05 | 0.07 | — | — | — | — | — | — |
| rs10842991 | DBP | 0.09 | <0.05 | 0.08 | — | — | — | — | — | — | |
| rs1055144 | LOC100506236 | SBP | −0.1 | <0.05 | −0.11 | — | — | — | — | — | — |
| rs11909120 | N6AMT1 | DBP | — | — | — | −0.6 | <0.05 | −0.67 | — | — | — |
| rs4810332 | SBP | — | — | — | 0.55 | <0.05 | 0.55 | — | — | — | |
| rs9860290 | CMSS1 | PP | — | — | — | −0.52 | 0.05 | −0.58 | — | — | — |
| rs9888615 | FERMT2 | SBP | — | — | — | −0.59 | <0.05 | −0.54 | −0.17 | <0.05 | −0.16 |
| rs2390258 | DBP/SBPPP | — | — | — | 0.45 | 0.05 | 0.47 | 0.23 | 0.001 | 0.26 | |
| rs4553000 | UBAP1 | DBP/SBP/PP | — | — | — | — | — | — | −0.22 | 0.001 | −0.19 |
| rs4980515 | DBP | — | — | — | — | — | — | −0.21 | 0.001 | −0.23 | |
DBP, diastolic blood pressure; OLS, ordinary least squares; P1, pressure at the first systolic shoulder; PP, pulse pressure; SBP, systolic blood pressure.
One-sample Mendelian randomization between blood pressure and arterial stiffness
Using all SNPs associated with SBP in LASSO regression as an IV, a 1 SD increase in IV-predicted SBP associated with 0.08 m/s increase in PWV (P < 0.0001, Table 4). This was similar to the association estimated from the observational data (Table 4). Using SNPs associated with DBP as an IV, an 1-SD increase in predicted DBP was associated with a 0.07 m/s increase in PWV (P < 0.0001), and a 1-SD increase in PP predicted by PP-associated alleles was associated with a 0.12 m/s increase in PWV (P < 0.0001). When using all SNPs as IV rather than the sub-sample identified by LASSO, point estimates of the beta coefficients did not differ appreciably (data not shown). Previous GWAS have identified SNPs associated with PWV which are independent of those associated with BP and vice versa. Using alleles previously associated with PWV (rs3742207 of gene COL4A1 and rs7152623of gene 3ʹ-BCL11B) as instruments, a 1-SD increase in predicted PWV associated with a 4.84 mmHg increase in SBP (P = 0.011) and a 3.34 mmHg increase in PP (P < 0.01) but not with DBP or MAP (Table 4). Sensitivity analysis including only one twin produced comparable beta coefficients for all phenotypes (data not shown).
Table 4.
One-sample bi-directional Mendelian randomization between blood pressure components and pulse wave velocity in the TwinsUK cohort
| Association exposure-outcome |
MR (IV-exposure-PWV) |
|||||
|---|---|---|---|---|---|---|
| Exposure variable | Outcome variable | N | beta | P | beta | P |
| SBP | PWV | 2088 | 0.07 | <0.0001 | 0.08 | <0.0001 |
| DBP | PWV | 2088 | 0.09 | <0.0001 | 0.07 | <0.0001 |
| PP | PWV | 2088 | 0.10 | <0.0001 | 0.12 | <0.0001 |
| PWV | SBP | 1758 | 4.84 | <0.0001 | 7.42 | 0.011 |
| PWV | DBP | 1758 | 1.5 | <0.0001 | 0.83 | 0.613 |
| PWV | PP | 1758 | 3.34 | <0.0001 | 6.59 | 0.006 |
| PWV | MAP | 1758 | 2.87 | <0.0001 | 3.17 | 0.207 |
DBP, diastolic blood pressure; MAP, mean arterial pressure; MR, Mendelian randomization; PP, pulse pressure; PWV. pulse wave velocity; SBP, systolic blood pressure.
Two-sample Mendelian randomization using MR-base
Of the 18 alleles included in the two-sample MR analysis with PWV as the exposure, six were available in summary-level outcome data. The main MR results are shown in Figure 3. Based on MR analysis with an inverse weighted method, we found evidence for a causal effect of PWV on SBP (Figure 3A, inverse-variance weighted analysis beta = 0.11, P < 0.02) and DBP (Figure 3B, beta = 0.09, P < 0.0001) suggesting a causal effect of PWV on both SBP and DBP. The weighted median regression estimates were consistent with these findings. Positive effects of similar magnitude and significance were found for leave-one-out sensitivity analysis. The MR-Egger regression intercept did not suggest any evidence of horizontal pleiotropy (beta = −0.017, P = 0.291 for SBP and beta = −0.006, P = 0.489 for DBP).
Figure 3.
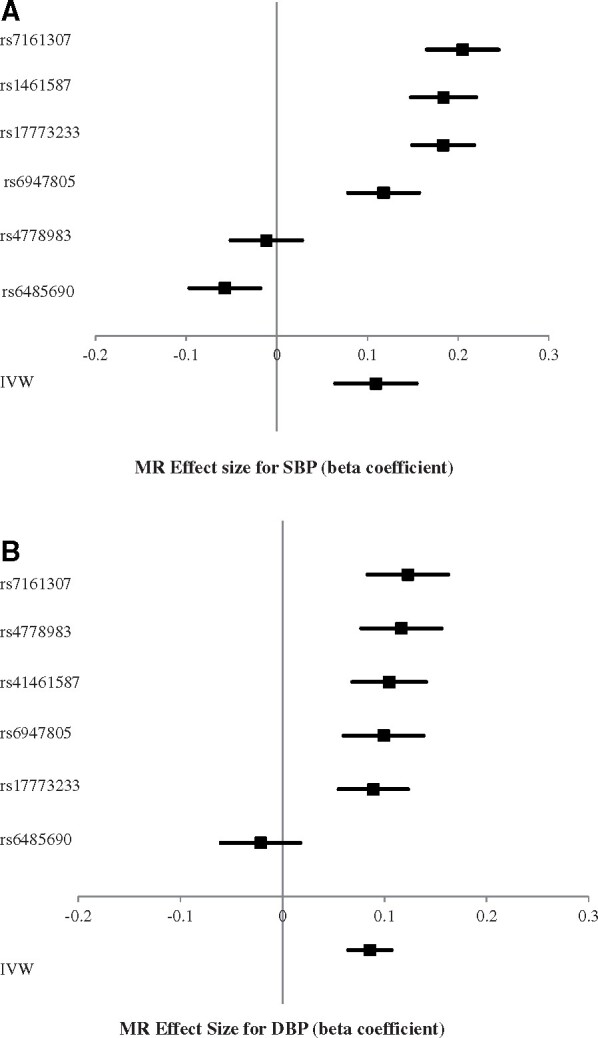
(A) Forest plot of two-sample Mendelian randomization (MR) with systolic blood pressure (SBP) as the outcome (single-nucleotide polymorphisms are ordered according to strength of association). (B) Forest plot of two-sample Mendelian randomization (MR) with diastolic blood pressure (DBP) as the outcome (single-nucleotide polymorphisms are ordered according to strength of association).
Of the 984 alleles included in the 2-sample MR analysis with BP as the exposure, 575 were available in summary-level outcome data. The main MR results are shown in Figure 4. Based on MR with inverse weighted method, we found evidence for a causal effect of BP on arterial stiffness (Figure 4, inverse-variance weighted analysis beta = 0.004, P < 0.0001). The weighted median regression estimates were consistent with these findings. The MR-Egger regression intercept did not suggest any evidence of horizontal pleiotropy (beta = 0.00, P = 0.871). A sensitivity analysis as recommended by Burgess et al.14 using fewer but stronger genetic variants to investigate bias resulting from an overlap in participants in the discover sets for the BP-associated SNPs and participants in the MR analysis did not influence our conclusions (data not shown).
Figure 4.

The association between the effect of blood pressure associated single-nucleotide polymorphisms (SNPs) on the arterial stiffness index (y-axis) plotted against the effect of blood pressure associated SNPs on blood pressure (x-axis). The slope of the regression line represents the causal association estimated using different regression methods.
Take home figure.
Figure summarizing heritability of cardiovascular phenotypes, pleiotropic effects of blood pressure single nucleotide polymorphisms and bi-directional causal association between blood pressure and arterial stiffness.
Discussion
Understanding the mechanism by which genetic polymorphisms influence BP is key to identifying novel pathways underlying hypertension. To date, GWAS investigating the genetic cause of hypertension have mostly focused on SBP and DBP. However, these values provide limited information on the BP phenotype. Mean and DBP are determined mainly by CO and SVR whereas SBP and pulsatile components of BP are more closely related to SV and arterial stiffness.
To the best of our knowledge, this is the first study to examine the heritability and shared heritability of BP components and their haemodynamic determinants. The main finding is that, of the cardiovascular properties that determine BP, arterial stiffness is the one with the highest heritability and that shared genes account for a large proportion of the correlation between SBP and arterial stiffness. When examining gene variants known to relate to BP, to determine to which BP components and haemodynamic determinants of BP they relate most strongly, our finding of a number being related to PWV is consistent with high shared heritability of BP with PWV. Such genes could influence PWV through BP or through a direct influence on the arterial wall. In this regard, it is notable that we identified a SNP that is likely to act through a direct effect on the arterial wall. rs9888615 is located on chromosome 14 within gene FERMT2 which has been implicated in cell–extracellular matrix interactions15 that could affect arterial stiffness. Of the components of central SBP, it is notable that AP shows high heritability. Augmentation pressure refers to the portion of central systolic PP arising after myocardial wall stress has peaked early in systole but left ventricular pressure and central BP continues to rise. It is thought to depend less on aortic stiffness than the other components of PP and more on cardiac dynamics and wave reflection. That shared genes account for only a small proportion of the phenotypic correlation between AP and PWV is consistent with AP being only weakly linked to PWV and relating more closely to other aspects of ventricular–vascular coupling.16 Relatively high heritability of DBP but low heritability of CO and SVR, the main determinants of DBP (which is close to MAP), would appear a paradox at first sight but might be explained by a genetic regulation of BP itself rather than genetic regulation of CO and SVR; CO and SVR, despite being influenced by environmental factors, may be balanced through feedback mechanisms to achieve a genetically regulated ‘set point’ of MAP or DBP. Such a set point could occur through renal (pressure-natriuretic)17 or neural (long-term effects of baroreceptor setting or other neural set point) mechanisms.18 The finding of no statistically significant effect of shared environment on BP is consistent with a recent family study19 that found a greater effect of genetic and unique environmental factors compared with shared environmental factors on BP.
Shared heritability of BP and PWV could be due to a bi-directional relationship between BP and PWV. Although PWV is a haemodynamic determinant of the pulsatile components of BP, it is influenced by BP via the non-linear elastic properties of wall of the artery which result in a functional stiffening of the artery when distended by a higher BP. Long-term effect of steady state or pulsatile BP components may also lead to stiffening of the arterial wall through growth or remodelling processes. Thus, whether arterial stiffening is the cause or consequence of hypertension has been debated, with previous epidemiological studies differing in their conclusions.20 The Framingham Heart study found that higher aortic stiffness was associated with a higher incidence of hypertension but not vice versa for progression of PWV.21 However, in a younger cohort Chen et al.22 used cross-lagged path coefficients to investigate the temporal association between BP and PWV in 584 adults aged between 32 and 51 years in the Bogalusa Heart study. Over a 7-year follow-up they concluded that a BP rise preceded large artery stiffening. Our finding of shared heritability of BP and PWV and of an association between BP-associated SNPs and PWV suggests a causal relationship between BP and PWV but does not identify the direction of causality. Using MR, which may be less susceptible to bias from confounding than studies using phenotypic correlations and which provides evidence of longer-term influences of potential determinants of outcomes, we found evidence of a causal role of BP in increasing aortic stiffness using 56 BP-associated SNPs as instrumental variables but also a causal role of PWV to increase SBP and PP but not MAP or DBP. Two-sample MR in the Biobank cohort confirmed the causal role of BP to increase arterial stiffness, as measured by arterial SI, using 575 GWAS significant BP SNPs. The analysis in Biobank also identified a role of PWV to increase BP when using GWAS significant PWV SNPs (P < 5 × 10−8) and suggestive PWV SNPs (P < 1 × 10−5) as instruments. These results were supported by several sensitivity analyses including leave-one-out analysis, MR Egger and weighted median MR. An important assumption of MR is that genotype is related to the outcome only via its association with its risk factors (exclusion restriction assumption) i.e. that gene variants influence BP or PWV via specific mechanisms on one or other of these properties. For the majority of gene variants used in the present analysis the mechanism underlying their association with BP or PWV is unknown and could potentially be linked to one or both of these properties (i.e. exhibit horizontal pleiotropy). However, a lack of horizontal pleiotropy is supported by the low P-value in MR Egger analysis. Furthermore, use of multiple genetic variants as instrumental variables that are located on separate chromosomes and with independent effects on the risk factor is likely to minimize the effect of pleiotropy and strengthens the evidence for a bi-directional causal association between aortic stiffness and BP.7 Gottsäter et al.,23 investigated the causal association between SBP and PWV using SBP-associated SNPs as instrumental variables and found no causal association. However, this study used 29 SBP-associated SNPs as instrumental variables which is likely to have accounted for a smaller percentage of BP variance and thus be more susceptible to weak instrument bias which in MR biases the results towards the null.24 High-shared heritability of PWV and BP together with a bi-directional causal relationship between these two phenotypes suggest that PWV GWAS with similar power to that recently achieved for BP is an important objective for future studies to identify genetic determinants of BP regulation that are mediated through arterial stiffness.
Strengths and limitations
The present study has several strengths. We used detailed cardiovascular phenotyping in a relatively large Twin cohort to determine the heritability of haemodynamic properties that determine BP and to explore haemodynamic mechanisms through which BP-associated polymorphisms may influence BP. While we are not able to infer the contribution from individual environmental factors, a major advantage of the twin design is that we can quantify the contribution of the totality of environmental factors on phenotypes, since by definition the environmental factors incorporate all those that are not inherited. Mendelian randomization techniques have the advantage of overcoming confounding by unmeasured/unknown factors due to the independent assortment of the instrumental variable risk alleles with confounding factors. Using both one- and two-sample, MR design allowed us to use a large sample size maximizing our statistical power and providing evidence of causality. In addition, we used multiple SNPs as instrumental variables instead of creating a weighted allele score. This has higher statistical power7 and protects against bias arising from horizontal pleiotropy.12
The study also has several limitations. We cannot rule out that our measures of heritability include effects on DNA methylation which may play a role in regulating BP independently of known genetic variants. Most of our analysis is limited to female twins and cannot be generalized to men. However, this cohort has been shown to be comparable with the general female population in the UK.25 Although there were some significant differences between MZ and DZ twins (BP, medication use, and haemodynamic properties), most of these differences were explained by DZ twins being older compared with MZ. We accounted for this by adjusting for age in the heritability analysis. Our study did not identify pathways lying upstream of the intermediate phenotypes (BP and stiffness). Replication studies are required to confirm the link between gene variants associated with BP and PWV that we identified together with functional studies to determine the specific biological pathways through which these may act. Many pathways are likely to be involved and may including those related to telomere length, glucose, and inflammation.26–28
Limitations of MR have been reviewed elsewhere29 and include failure to establish associations between genotype and intermediate phenotype, confounding of these associations, pleiotropy and canalization and developmental compensation. In the present study, these were mitigated by selection of gene variants that were robustly associated with phenotypes, the use of multiple gene variants located on different chromosomes and consistency of results in two populations. It should, however, be noted that, in the Biobank MR analysis, arterial stiffness was estimated using arterial SI which is an indirect measure of arterial stiffness that may be influenced by other haemodynamic properties.30 Instrumental variables for arterial stiffness in the Biobank analysis were also less robust than for BP. There was an overlap in participants for the discovery sets that led to identification of the 984 BP SNPs and the participants included in MR analysis (but a sensitivity analysis to guard against bias introduced by this,14 did not influence our main conclusions). Further work in other cohorts and/or direct measurement of arterial stiffness in Biobank would therefore be valuable.
We used MR to examine the association between intermediate phenotypes (BP and stiffness) but there are other important categories of inference that can be derived from MR such as propensity to exposure to a risk factor, the category of exposure of importance, characterizing ‘difficult to measure’ environmental exposures and modifiers of environmental exposure. Future work using MR to explore the specific environmental determinants of BP phenotypes, particularly those with a large environmental component, is likely to be productive.
Conclusion
We provide evidence of significant heritability of BP components and of large artery stiffness but not CO or SVR, which appear to be influenced more by environmental rather than genetic factors. Bivariate heritability analysis identified a high proportion of shared genes underlying the association of pulsatile components of BP other than AP with arterial stiffness and several of the gene variants known to be associated with BP are associated with arterial stiffness. Mendelian randomization suggests a bi-directional causal relationship between BP and arterial stiffness. The genetic basis of BP may be mediated at a haemodynamic level by genes that influence arterial stiffness and in part by genes that act directly to regulate BP. The finding of a bi-directional relationship between BP and PWV is key to tackling the epidemic of predominantly systolic hypertension in our ageing societies characterized by elevated PWV. It suggests that the most effective treatments will be a combination of conventional antihypertensive agents to lower BP and specific agents to lower PWV.
Funding
This work was supported by a British Heart Foundation Centre for Research Excellence Career Development Fellowship and a British Heart Foundation Special Project grant SP/12/4/29573. TwinsUK is funded by the Wellcome Trust; European Community’s Seventh Framework Programme (FP7/2007–13). The study also received support from the National Institute for Health Research BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St. Thomas’ NHS Foundation Trust and King’s College London.
Conflict of interest: none declared.
Supplementary Material
Contributor Information
Marina Cecelja, Cardiovascular Division, Department of Clinical Pharmacology, King’s College London British Heart Foundation Centre, St Thomas’ Hospital, Westminster Bridge Road, London SE1 7EH, UK.
Louise Keehn, Cardiovascular Division, Department of Clinical Pharmacology, King’s College London British Heart Foundation Centre, St Thomas’ Hospital, Westminster Bridge Road, London SE1 7EH, UK.
Li Ye, Cardiovascular Division, Department of Clinical Pharmacology, King’s College London British Heart Foundation Centre, St Thomas’ Hospital, Westminster Bridge Road, London SE1 7EH, UK.
Tim D Spector, Department of Twin Research and Genetic Epidemiology, King’s College London, St Thomas’ Hospital, Westminster Bridge Road, London SE1 7EH, UK.
Alun D Hughes, Department of Population Science and Experimental Medicine, Institute of Cardiovascular Sciences, University College London, 69 Chenies Mews, London W1T 7HA, UK.
Phil Chowienczyk, Cardiovascular Division, Department of Clinical Pharmacology, King’s College London British Heart Foundation Centre, St Thomas’ Hospital, Westminster Bridge Road, London SE1 7EH, UK.
References
- 1.GBD 2017 Risk Factor Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392:1923–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang B, Liao C, Zhou B, Cao W, Lv J, Yu C, Gao W, Li L. Genetic contribution to the variance of blood pressure and heart rate: a systematic review and meta-regression of twin studies. Twin Res Hum Genet 2015;18:158–170. [DOI] [PubMed] [Google Scholar]
- 3. Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, Ntritsos G, Dimou N, Cabrera CP, Karaman I, Ng FL, Evangelou M, Witkowska K, Tzanis E, Hellwege JN, Giri A, Velez Edwards DR, Sun YV, Cho K, Gaziano JM, Wilson PWF, Tsao PS, Kovesdy CP, Esko T, Mägi R, Milani L, Almgren P, Boutin T, Debette S, Ding J, Giulianini F, Holliday EG, Jackson AU, Li-Gao R, Lin W-Y, Luan J, Mangino M, Oldmeadow C, Prins BP, Qian Y, Sargurupremraj M, Shah N, Surendran P, Thériault S, Verweij N, Willems SM, Zhao J-H, Amouyel P, Connell J, de Mutsert R, Doney ASF, Farrall M, Menni C, Morris AD, Noordam R, Paré G, Poulter NR, Shields DC, Stanton A, Thom S, Abecasis G, Amin N, Arking DE, Ayers KL, Barbieri CM, Batini C, Bis JC, Blake T, Bochud M, Boehnke M, Boerwinkle E, Boomsma DI, Bottinger EP, Braund PS, Brumat M, Campbell A, Campbell H, Chakravarti A, Chambers JC, Chauhan G, Ciullo M, Cocca M, Collins F, Cordell HJ, Davies G, de Borst MH, de Geus EJ, Deary IJ, Deelen J, Del Greco M F, Demirkale CY, Dörr M, Ehret GB, Elosua R, Enroth S, Erzurumluoglu AM, Ferreira T, Frånberg M, Franco OH, Gandin I, Gasparini P, Giedraitis V, Gieger C, Girotto G, Goel A, Gow AJ, Gudnason V, Guo X, Gyllensten U, Hamsten A, Harris TB, Harris SE, Hartman CA, Havulinna AS, Hicks AA, Hofer E, Hofman A, Hottenga J-J, Huffman JE, Hwang S-J, Ingelsson E, James A, Jansen R, Jarvelin M-R, Joehanes R, Johansson Å, Johnson AD, Joshi PK, Jousilahti P, Jukema JW, Jula A, Kähönen M, Kathiresan S, Keavney BD, Khaw K-T, Knekt P, Knight J, Kolcic I, Kooner JS, Koskinen S, Kristiansson K, Kutalik Z, Laan M, Larson M, Launer LJ, Lehne B, Lehtimäki T, Liewald DCM, Lin L, Lind L, Lindgren CM, Liu Y, Loos RJF, Lopez LM, Lu Y, Lyytikäinen L-P, Mahajan A, Mamasoula C, Marrugat J, Marten J, Milaneschi Y, Morgan A, Morris AP, Morrison AC, Munson PJ, Nalls MA, Nandakumar P, Nelson CP, Niiranen T, Nolte IM, Nutile T, Oldehinkel AJ, Oostra BA, O’Reilly PF, Org E, Padmanabhan S, Palmas W, Palotie A, Pattie A, Penninx BWJH, Perola M, Peters A, Polasek O, Pramstaller PP, Nguyen QT, Raitakari OT, Ren M, Rettig R, Rice K, Ridker PM, Ried JS, Riese H, Ripatti S, Robino A, Rose LM, Rotter JI, Rudan I, Ruggiero D, Saba Y, Sala CF, Salomaa V, Samani NJ, Sarin A-P, Schmidt R, Schmidt H, Shrine N, Siscovick D, Smith AV, Snieder H, Sõber S, Sorice R, Starr JM, Stott DJ, Strachan DP, Strawbridge RJ, Sundström J, Swertz MA, Taylor KD, Teumer A, Tobin MD, Tomaszewski M, Toniolo D, Traglia M, Trompet S, Tuomilehto J, Tzourio C, Uitterlinden AG, Vaez A, van der Most PJ, van Duijn CM, Vergnaud A-C, Verwoert GC, Vitart V, Völker U, Vollenweider P, Vuckovic D, Watkins H, Wild SH, Willemsen G, Wilson JF, Wright AF, Yao J, Zemunik T, Zhang W, Attia JR, Butterworth AS, Chasman DI, Conen D, Cucca F, Danesh J, Hayward C, Howson JMM, Laakso M, Lakatta EG, Langenberg C, Melander O, Mook-Kanamori DO, Palmer CNA, Risch L, Scott RA, Scott RJ, Sever P, Spector TD, van der Harst P, Wareham NJ, Zeggini E, Levy D, Munroe PB, Newton-Cheh C, Brown MJ, Metspalu A, Hung AM, O’Donnell CJ, Edwards TL, Psaty BM, Tzoulaki I, Barnes MR, Wain LV, Elliott P, Caulfield MJ; the Million Veteran Program. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 2018;50:1412–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moayyeri A, Hammond CJ, Hart DJ, Spector TD. The UK Adult Twin Registry (TwinsUK Resource). Twin Res Hum Genet 2013;16:144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vaarhorst AA, Lu Y, Heijmans BT, Dolle ME, Bohringer S, Putter H, Imholz S, Merry AH, van Greevenbroek MM, Jukema JW, Gorgels AP, van den Brandt PA, Muller M, Schouten LJ, Feskens EJ, Boer JM, Slagboom PE. Literature-based genetic risk scores for coronary heart disease: the Cardiovascular Registry Maastricht (CAREMA) prospective cohort study. Circ Cardiovasc Genet 2012;5:202–209. [DOI] [PubMed] [Google Scholar]
- 7. Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, Davey Smith G, Sterne JA. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res 2012;21:223–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Teumer A. Common methods for performing Mendelian randomization. Front Cardiovasc Med 2018;5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mitchell GF, Verwoert GC, Tarasov KV, Isaacs A, Smith AV, Yasmin Rietzschel ER, Tanaka T, Liu Y, Parsa A, Najjar SS, O’Shaughnessy KM, Sigurdsson S, De Buyzere ML, Larson MG, Sie MP, Andrews JS, Post WS, Mattace-Raso FU, McEniery CM, Eiriksdottir G, Segers P, Vasan RS, van Rijn MJ, Howard TD, McArdle PF, Dehghan A, Jewell ES, Newhouse SJ, Bekaert S, Hamburg NM, Newman AB, Hofman A, Scuteri A, De Bacquer D, Ikram MA, Psaty BM, Fuchsberger C, Olden M, Wain LV, Elliott P, Smith NL, Felix JF, Erdmann J, Vita JA, Sutton-Tyrrell K, Sijbrands EJ, Sanna S, Launer LJ, De Meyer T, Johnson AD, Schut AF, Herrington DM, Rivadeneira F, Uda M, Wilkinson IB, Aspelund T, Gillebert TC, Van Bortel L, Benjamin EJ, Oostra BA, Ding J, Gibson Q, Uitterlinden AG, Abecasis GR, Cockcroft JR, Gudnason V, De Backer GG, Ferrucci L, Harris TB, Shuldiner AR, van Duijn CM, Levy D, Lakatta EG, Witteman JC. Common genetic variation in the 3’-BCL11B gene desert is associated with carotid-femoral pulse wave velocity and excess cardiovascular disease risk: the AortaGen Consortium. Circ Cardiovasc Genet 2012;5:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taylor AE, Davies NM, Ware JJ, VanderWeele T, Smith GD, Munafo MR. Mendelian randomization in health research: using appropriate genetic variants and avoiding biased estimates. Econ Hum Biol 2014;13:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bell JA, Carslake D, Wade KH, Richmond RC, Langdon RJ, Vincent EE, Holmes MV, Timpson NJ, Davey Smith G. Influence of puberty timing on adiposity and cardiometabolic traits: a Mendelian randomisation study. PLoS Med 2018;15:e1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Millasseau SC, Kelly RP, Ritter JM, Chowienczyk PJ. Determination of age-related increases in large artery stiffness by digital pulse contour analysis. Clin Sci (Lond) 2002;103:371–377. [DOI] [PubMed] [Google Scholar]
- 14. Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol 2016;40:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell 2003;113:37–47. [DOI] [PubMed] [Google Scholar]
- 16. Li Y, Jiang B, Keehn L, Gu H, Boguslavskyi A, Cecelja M, Vennin S, Spector T, Alastruey J, Chowienczyk P. Hemodynamic mechanism of the age-related increase in pulse pressure in women. Hypertension 2019;73:1018–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guyton AC. Dominant role of the kidneys and accessory role of whole-body autoregulation in the pathogenesis of hypertension. Am J Hypertens 1989;2:575–585. [DOI] [PubMed] [Google Scholar]
- 18. Floras JS, Hassan MO, Jones JV, Osikowska BA, Sever PS, Sleight P. Consequences of impaired arterial baroreflexes in essential hypertension: effects on pressor responses, plasma noradrenaline and blood pressure variability. J Hypertens 1988;6:525–535. [DOI] [PubMed] [Google Scholar]
- 19. Tan Q, Duan H, Wang A, Zhu D, Li S. Longitudinal analysis of sibling correlation on blood pressure using mixed modeling. Ann Epidemiol 2019;33:49–53. [DOI] [PubMed] [Google Scholar]
- 20. Mitchell GF. Arterial stiffness and hypertension: chicken or egg? Hypertension 2014;64:210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA 2012;308:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen W, Li S, Fernandez C, Sun D, Lai CC, Zhang T, Bazzano L, Urbina EM, Deng HW. Temporal relationship between elevated blood pressure and arterial stiffening among middle-aged black and white adults: the Bogalusa Heart Study. Am J Epidemiol 2016;183:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gottsäter M, Hindy G, Orho-Melander M, Nilsson PM, Melander O. A genetic risk score for fasting plasma glucose is independently associated with arterial stiffness: a Mendelian randomization study. J Hypertens 2018;36:809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burgess S, Thompson SG; CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 2011;40:755–764. [DOI] [PubMed] [Google Scholar]
- 25. Andrew T, Hart DJ, Snieder H, de Lange M, Spector TD, MacGregor AJ. Are twins and singletons comparable? A study of disease-related and lifestyle characteristics in adult women. Twin Res 2001;4:464–477. [DOI] [PubMed] [Google Scholar]
- 26. Strazhesko I, Tkacheva O, Boytsov S, Akasheva D, Dudinskaya E, Vygodin V, Skvortsov D, Nilsson P. Association of insulin resistance, arterial stiffness and telomere length in adults free of cardiovascular diseases. PLoS One 2015;10:e0136676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yasmin, McEniery CM, Wallace S, Mackenzie IS, Cockcroft JR, Wilkinson IB. C-reactive protein is associated with arterial stiffness in apparently healthy individuals. Arterioscler Thromb Vasc Biol 2004;24:969–974. [DOI] [PubMed] [Google Scholar]
- 28. McNulty M, Mahmud A, Feely J. Advanced glycation end-products and arterial stiffness in hypertension. Am J Hypertens 2007;20:242–247. [DOI] [PubMed] [Google Scholar]
- 29. Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol 2004;33:30–42. [DOI] [PubMed] [Google Scholar]
- 30. Millasseau SC, Ritter JM, Takazawa K, Chowienczyk PJ. Contour analysis of the photoplethysmographic pulse measured at the finger. J Hypertens 2006;24:1449–1456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.