

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
RGS10 and RGS18 cooperate to restrain unnecessary platelet activation, while RGS18 promotes platelet production.
Deleting RGS10 and RGS18 results in platelet preactivation, reduced survival, thrombocytopenia, and occlusive hemostatic plugs.
Abstract
G protein–coupled receptors are critical mediators of platelet activation whose signaling can be modulated by members of the regulator of G protein signaling (RGS) family. The 2 most abundant RGS proteins in human and mouse platelets are RGS10 and RGS18. While each has been studied individually, critical questions remain about the overall impact of this mode of regulation in platelets. Here, we report that mice missing both proteins show reduced platelet survival and a 40% decrease in platelet count that can be partially reversed with aspirin and a P2Y12 antagonist. Their platelets have increased basal (TREM)-like transcript-1 expression, a leftward shift in the dose/response for a thrombin receptor–activating peptide, an increased maximum response to adenosine 5′-diphosphate and TxA2, and a greatly exaggerated response to penetrating injuries in vivo. Neither of the individual knockouts displays this constellation of findings. RGS10−/− platelets have an enhanced response to agonists in vitro, but platelet count and survival are normal. RGS18−/− mice have a 15% reduction in platelet count that is not affected by antiplatelet agents, nearly normal responses to platelet agonists, and normal platelet survival. Megakaryocyte number and ploidy are normal in all 3 mouse lines, but platelet recovery from severe acute thrombocytopenia is slower in RGS18−/− and RGS10−/−18−/− mice. Collectively, these results show that RGS10 and RGS18 have complementary roles in platelets. Removing both at the same time discloses the extent to which this regulatory mechanism normally controls platelet reactivity in vivo, modulates the hemostatic response to injury, promotes platelet production, and prolongs platelet survival.
Visual Abstract

Introduction
Rapid and robust platelet activation is a critical step in the hemostatic response to injury, but can be problematic in the setting of thrombosis or vessel wall disease. Most platelet agonists exert their effects via G protein–coupled receptors (GPCRs). In the presence of a platelet agonist, exchange of guanosine diphosphate for guanosine triphosphate (GTP) on receptor-associated G proteins leads to dissociation of the G protein α subunit from the βγ heterodimer. The duration of subsequent signaling is limited in part by the intrinsic GTPase activity of the α subunit, which can be enhanced by regulators of G protein signaling (RGSs), which stabilize the GTP to guanosine diphosphate transition state.1-4 The predominant members of the RGS family expressed in human and mouse platelets are RGS10 and RGS18, neither of which is platelet specific.5-7 The expression of RGS18 has been shown to be limited to hematopoietic cell lineages.8-11 RGS10 is expressed more widely.12-16
We and others have previously shown that deletion of either RGS10 or RGS18 alone results in a gain of function with respect to GPCR-mediated platelet activation.17-20 We have also shown that a substitution in Gi2α (G184S) that renders Gi2 resistant to interactions with RGS proteins, also causes a gain of function in platelets.21 Gi2 is the major Gi family member expressed in platelets and a critical mediator of platelet activation via P2Y12 adenosine 5′-diphosphate (ADP) receptors.22 While these observations are consistent with a role for RGS proteins as regulators of the platelet signaling network, critical questions remain unanswered. These include their contribution, if any, to platelet biology in the absence of injury, the extent to which RGS10 and RGS18 modify the hemostatic response when injury occurs, and whether their roles are complementary or redundant. Here, we have attempted to address all 3 of these questions, deleting both RGS10 and RGS18 to permit us to compare the double knockout, the individual knockouts, and matched controls, all in the same background.
The results show that the full impact of RGS10 and RGS18 can only be appreciated when both are deleted at the same time. Mice missing both proteins show reduced platelet lifespan and a 40% decrease in platelet counts that can be partially rescued by aspirin and a P2Y12 antagonist. Their platelets show evidence of preactivation in the circulation, and, when tested in vitro, show a leftward shift in the dose/response for a thrombin receptor–activating peptide, an increased maximum response to ADP and TxA2, and an exaggerated hemostatic response to injuries in the cremaster muscle microcirculation. Neither of the individual knockouts displays this same constellation of findings. Platelets from RGS10−/− mice have an enhanced response to agonists in vitro, but their platelet count is normal, and so is their platelet lifespan. Platelets from RGS18−/− mice have a normal lifespan and a nearly normal response to agonists, but the mice have a 15% reduction in platelet count that appears to reflect a production defect and is not rescued with antiplatelet agents. Collectively, these results show that RGS10 and RGS18 have complementary rather than identical roles, jointly regulating the hemostatic response to injury, improving platelet production, prolonging platelet survival, and preventing unwarranted platelet activation in the absence of injury.
Methods
Mice
Generation of RGS10/18 double-knockout mice using the CRISPR-Cas9 genome-editing system was performed essentially as described.23 See supplemental Methods (available on the Blood Web site) for additional details. All mouse protocols were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Platelet flow cytometric analysis
Diluted whole blood was prepared as outlined in supplemental Methods and incubated with 1 mM aspirin and 1 U/mL apyrase for 30 minutes at 37°C to eliminate secondary signaling (except for ADP measurements, incubated only with 1 mM aspirin). Following inhibitor treatment, diluted blood was treated with agonist for 15 minutes at 37°C in the presence of saturating concentrations of the fluorescently labeled monoclonal antibody against P-selectin and activated αIIbβ3 integrin (Jon/A) and F(ab′)2 fragments against CD41 (αIIb) and analyzed on a FACSCanto II cell analyzer (BD Biosciences, San Jose, CA). The platelet population was gated based on forward scatter/side scatter and CD41 positivity. For thiazole orange (TO) studies, platelets were incubated with 1 μg/mL TO for 20 minutes at 37°C prior to staining with CD41 (αIIb). For (TREM)-like transcript-1 (TLT-1) studies, platelets were coincubated with Alexa Fluor 488–labeled anti-TLT-1 in place of anti-P-selectin.
Vascular injury: platelet and fibrin accumulation
Hemostatic thrombus formation was observed in the cremaster muscle microcirculation of male mice aged 8 to 12 weeks, as previously described24 and detailed in supplemental Methods.
Platelet clearance
To assess the rate of platelet clearance, mice were injected via the retro-orbital plexus with nonsaturating concentrations (1 μg/g body weight) of rat anti-GPIbβ-Dylight488 (Emfret Analytics, Eibelstadt, Germany) to label the existing platelet pool while avoiding excess free antibody in circulation. Twenty minutes after injection, ∼90% of CD41+ platelets were GPIbβ+ for each genotype as assessed by flow cytometry, and the percentage of GPIbβ+ CD41+ platelets was then tracked every 24 hours for 4 days to assess the rate of platelet clearance.
Antiplatelet agents
Mice were given aspirin (50 mg/kg) and prasugrel (1.875 mg/kg) in 0.5% methylcellulose via oral gavage daily for 10 days. Prior to the first dose and every 5 days thereafter, blood was obtained via the retro-orbital plexus, and platelet counts were determined with a Procyte Hematological Analyzer (Idexx Laboratories, Westbrook, ME). Five days after treatment ceased, platelet counts were determined once more as described.
Statistics
Results are reported as mean ± standard error of the mean (SEM). For group comparisons, 1- or 2-way analysis of variance was used for global hypothesis testing, followed by Tukey’s honestly significant difference test for pairwise multiple comparisons when appropriate. For paired comparisons, Student t test was used for hypothesis testing. P ≤ .05 was considered statistically significant.
Additional methods and materials are described in supplemental Methods.
Results
Generation and characterization of transgenic mice
To generate RGS10−/−18−/− mice via CRISPR-Cas9, 2 single guide RNAs were designed for each gene, each targeting Exon 4 and Intron 4 (Figure 1A). Exon 4 encodes part of the RGS domain for both proteins. Complete regional deletion within the Rgs10 gene resulted in loss of RGS10 expression (Figure 1B, left). The same founder possessed a 5-bp deletion in exon 4 of Rgs18, producing a premature stop codon and loss of RGS18 expression (Figure 1B, right).
Figure 1.
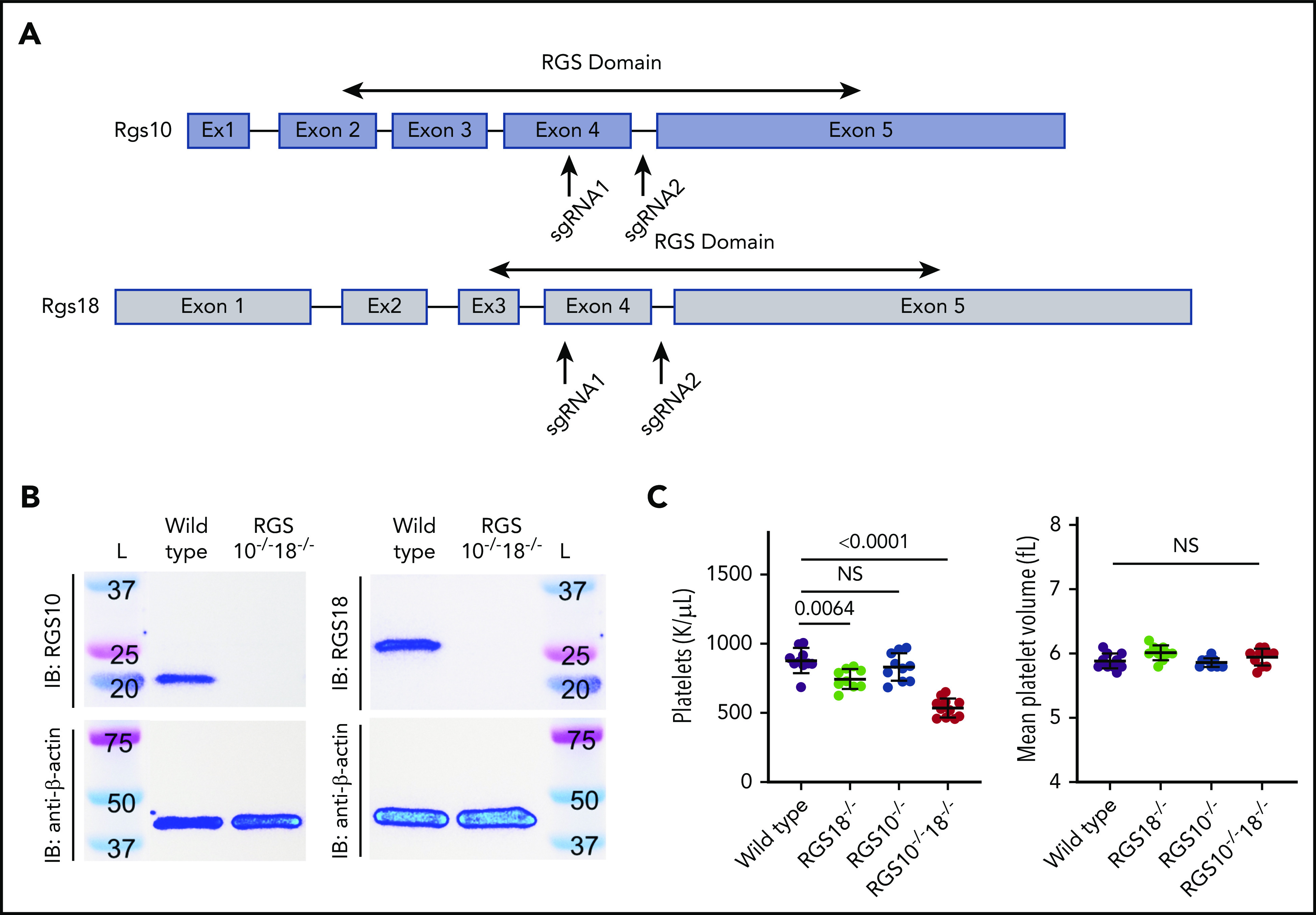
Generation and characterization of Rgs deletion mice. (A) Graphical depiction of Rgs10 and Rgs18 genes. Arrows indicate approximate locations targeted by single guide RNAs during CRISPR-Cas9. In both cases, regions within the sequence that encode the RGS domain were targeted. (B) Representative RGS10 and RGS18 immunoblots (IB) (top) of platelet lysates from RGS10+/+18+/+ (denoted “Wild type”) and RGS10−/−18−/− mice with β-actin (bottom) as the loading control. (C) Platelet counts and mean platelet volume of 8-week-old WT, RGS18−/−, RGS10−/−, and RGS10−/−18−/− mice. At least 9 measurements were collected per genotype. NS indicates P > .05; mean ± SEM.
RGS10−/−18−/− mice were viable, grossly normal in appearance, and gained weight normally (supplemental Figure 1A). Their blood counts were normal, except for the platelet count, which was reduced by ∼40% (Figure 1C; supplemental Figure 1B). In agreement with previous reports, RGS10−/− and RGS18−/− mice were also grossly normal in appearance and did not differ in their initial weight gains (supplemental Figure 1A).17-19 Consistent with previous work, RGS10−/− mice had normal blood counts, while RGS18−/− mice had a 15% reduction in platelet count (Figure 1C).18-20 Once the lines were established, subsequent pups were produced by breeding homozygous parents of each individual genotype.
Agonist-mediated platelet activation in vitro was detected using 2 independent markers: P-selectin, which is exposed on the platelet surface during α granule exocytosis,25,26 and the activated conformation of αIIbβ3 integrin, detected using Jon/A antibody.27 Platelets were stimulated with either a PAR4 agonist peptide (PAR4P; AYPGKF), ADP, or the TxA2 mimetic, U46619. Resting P-selectin expression was the same as controls for all 3 knockouts, as was the maximal response to PAR4P. However, there was a pronounced leftward shift in the PAR4P dose/response curve for RGS10−/−18−/− platelets (50% effective concentration, 74 ± 2 µM, mean ± SEM) and RGS10−/− platelets (79 ± 3 µM) relative to the wild-type (WT) control (126 ± 7 μM) and a smaller shift for RGS18−/− platelets (100 ± 4 μM) (Figure 2A; supplemental Figure 2A). The same pattern was observed using Jon/A (50% effective concentration, 83 ± 4, 89 ± 7, 104 ± 4, and 129 ± 12 µM for RGS10−/−18−/−, RGS10−/−, RGS18−/−, and WT platelets, respectively) (Figure 2B; supplemental Figure 2B).
Figure 2.
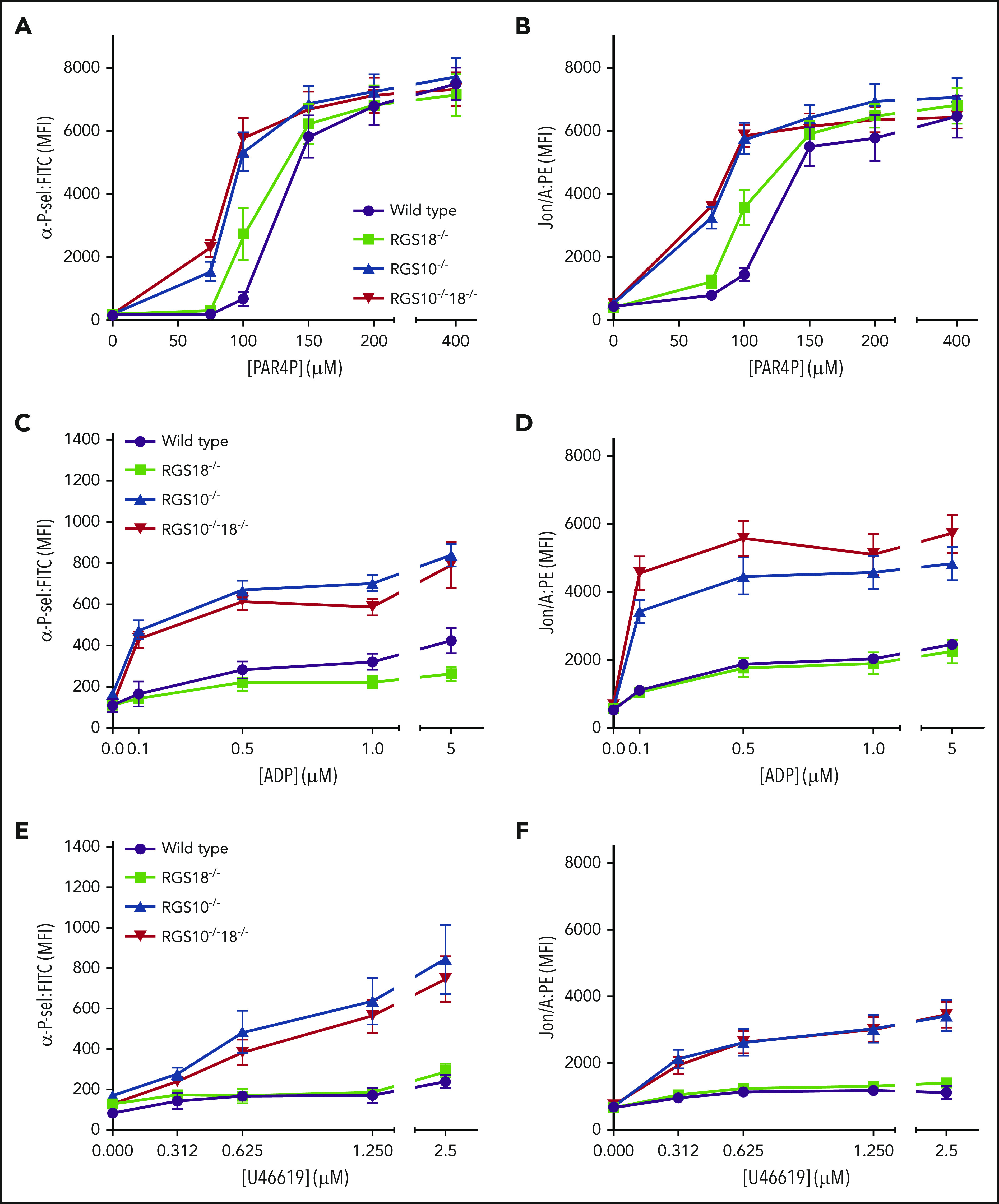
In vitro platelet activation dose/response curves. Flow cytometric analysis of (A,C,E) P-selectin expression and (B,D,F) integrin αIIbβ3 activation of platelets from matched WT, RGS18−/−, RGS10−/−, and RGS10−/−18−/− mice. Platelets were stimulated with increasing doses of PAR4P (AYPGKF) (A-B), ADP (C-D), and TxA2 analog (U46619) (E-F) and gated by forward scatter/side scatter and CD41 positivity. At least 4 measurements were collected per genotype per condition (mean ± SEM). See supplemental Figure 2 for statistical comparisons between genotypes. FITC, fluorescein isothiocyanate; MFI, mean fluorescence intensity; PE, phycoerythrin.
In contrast to these results, RGS10−/−18−/− and RGS10−/− platelets stimulated with ADP or U46619 showed an increase in their maximal P-selectin and Jon/A response over resting values that was substantially greater than in the WT mice (Figure 2C-F; supplemental Figure 3). RGS18−/− platelets were indistinguishable from the controls. Thus, combined loss of RGS10 and RGS18, and solitary loss of RGS10 enhances platelet responses to PAR4AP, ADP, and TxA2. Loss of RGS18 has a much smaller effect.
Hemostatic response to injury in vivo
Platelet function in vivo was assessed with real-time confocal fluorescence microscopy following a laser-inflicted penetrating injury in cremaster muscle arterioles.24 Because the vessel wall is penetrated and blood escapes, we view this as a model of hemostasis rather than thrombosis. The hemostatic thrombi formed in this setting have a characteristic architecture in which a densely packed core of fully activated, P-selectin+ platelets is overlaid by a shell of loosely packed, minimally activated P-selectin− platelets.24 We have shown that the core is driven primarily by thrombin, while the shell is driven mainly by released ADP and TxA2.24,28
Representative end-point images of hemostatic plugs formed in WT and RGS10−/−18−/− mice 3 minutes after injury are shown in Figure 3A. Mean total platelet accumulation was 77% greater in RGS10−/−18−/− mice than in controls (Figure 3B,E, left; supplemental Video 1). The P-selectin+ core region increased by 111% (Figure 3C,E, middle). By subtracting the core area from the total platelet area, we estimated the area of the P-selectin− shell region. It was on average 69% larger in RGS10−/−18−/− mice than in controls. Thus, both the core and the shell are bigger after injury in RGS10−/−18−/− mice. The net result is a larger hemostatic thrombus in which the proportion that becomes P-selectin+ is essentially unchanged (18% in WT mice and 22% in RGS10−/−18−/− mice). Fibrin accumulation was unaffected (Figure 3D,E, right).
Figure 3.
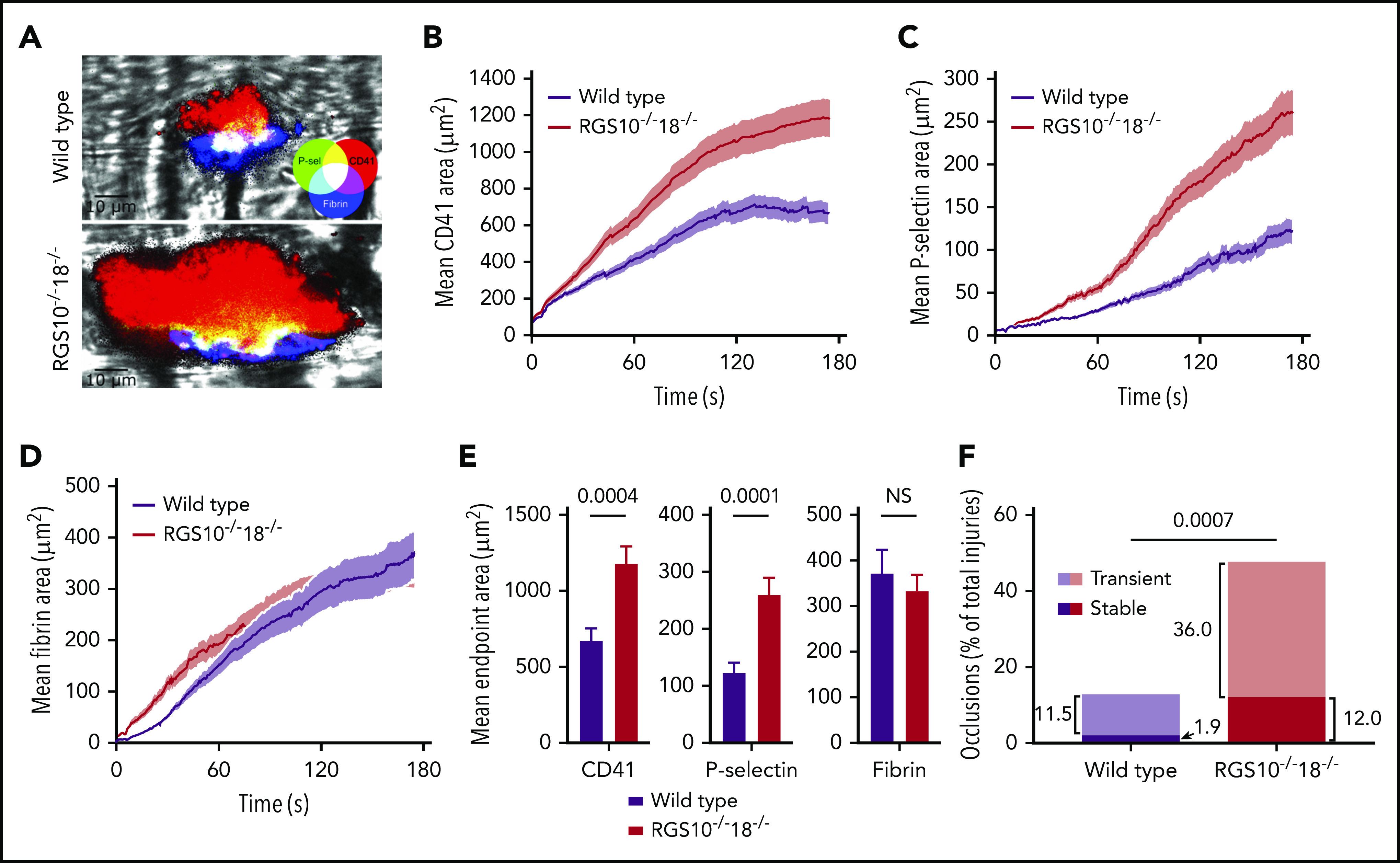
Hemostatic response to injury. Real-time confocal intravital microscopy following penetrating laser injuries in cremaster muscle arterioles in WT and RGS10−/−18−/− mice. (A) Representative end-point images of hemostatic plugs. Mean area of accumulation over time was measured for CD41 (αIIb integrin) (B), P-selectin (C), and fibrin (D). (E) Mean end-point area calculations. (F) The fraction of injuries that produced stable or transient occlusions. n = 52 injuries in 3 WT mice; n = 50 injuries in 4 RGS10−/−18−/− mice; mean ± SEM.
A second striking feature of the RGS10−/−18−/− mice in this model was in the consequences of the increased platelet accumulation. In contrast to the controls, many more of the RGS10−/−18−/− hemostatic thrombi grew to the point where they caused either stable or transient occlusion of the arteriole (Figure 3F; supplemental Video 2). This was not a phenomenon that we have observed with either RGS10−/− mice or the RGS-insensitive Gi2α(G184S) mice.20,21
Platelet production and platelet survival
Femurs obtained from RGS10−/−, RGS18−/−, RGS10−/−18−/−, and control mice were stained with anti-CD41 and examined by light microscopy. No measurable differences in the number of megakaryocytes were observed (Figure 4A-B). To assess platelet production, we used a GPIbα antibody to acutely deplete platelets and then tracked the platelet count as it recovered over the next 96 hours. During the first 48 hours, there were no significant differences among genotypes. After 72 hours, differences emerged, with the RGS10−/−18−/− and RGS18−/− mice taking longer to recover (Figure 4C). An in vitro analysis of bone marrow cells derived from each mouse line showed no differences in megakaryocyte progenitors or ploidy (supplemental Figure 4A-C). There was a small decrease in the proportion of RGS10−/−18−/− and RGS18−/− megakaryocytes with proplatelet extensions when the cells were maintained in culture. It did not, however, reach statistical significance (supplemental Figure 4D).
Figure 4.

Megakaryocytes and platelet production. (A) Cross-sectioned femurs harvested from WT, RGS18−/−, RGS10−/−, and RGS10−/−18−/− mice were stained for CD41 (αIIb integrin) and counterstained with hematoxylin. Large, multinucleate CD41+ cells were counted as megakaryocytes. White arrows point to representative examples. (B) Megakaryocyte counts from 5 randomly selected fields per mouse. n = 3; mean ± SEM. (C) Platelet depletion with an anti-GPIbα antibody followed by recovery over the course of 96 hours represented as a percentage of the baseline for each genotype. **P ≤ .05 for WT vs RGS18−/− and RGS10−/−18−/−. n = 6; mean ± SEM.
To see if there is a platelet survival difference, mice of each genotype were injected with a nonsaturating concentration of a GPIbβ antibody that has no effect on platelet activation.29 The percentage of anti-GPIbβ+ platelets remaining was determined every 24 hours for up to 96 hours (Figure 5A). The results show that RGS10−/−18−/− platelets have a significantly reduced survival (t1/2 = 46 hours), clearing faster than either the single RGS protein knockouts or controls (t1/2 = ∼62 hours).
Figure 5.
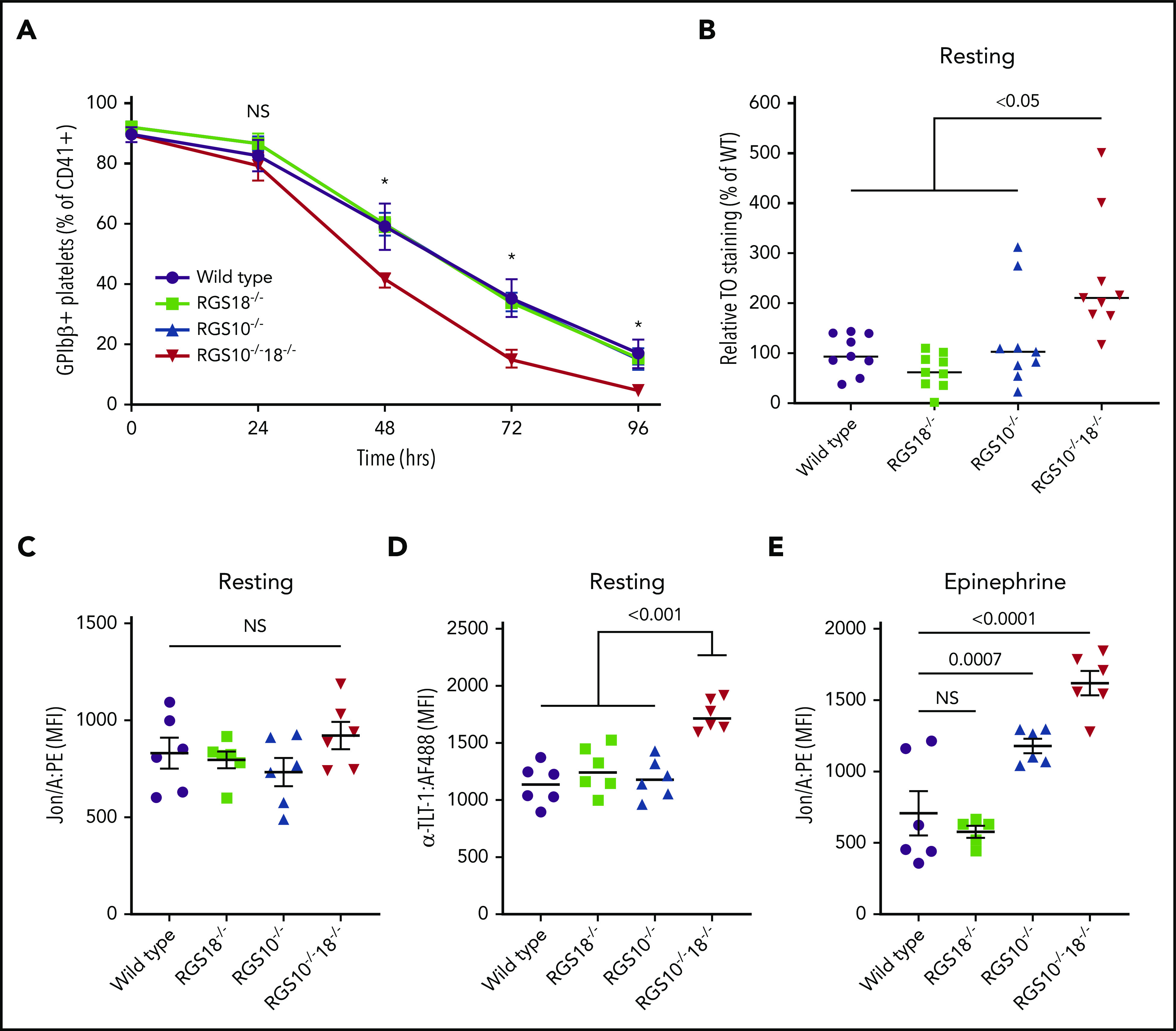
Platelet clearance and resting platelet activation markers. Flow cytometry analysis for WT, RGS18−/−, RGS10−/−, and RGS10−/−18−/− mice to measure clearance of anti-GPIbβ:DyLight488 in vivo–labeled platelets over the course of 96 hours (A). At baseline and every 24 hours thereafter, platelets were identified using anti-CD41 (αIIb integrin) and then analyzed for DyLight488+ by flow cytometry. *P ≤ .05 for WT vs RGS10−/−18−/−. n = 5; mean ± SEM. (B) Relative fraction of platelets that were positive for both anti-CD41 and TO. n = 9; mean ± SEM. Binding of Jon/A (C,E) and anti-TLT-1 (D) to resting (C-D) or 10 μM epinephrine–stimulated (E) platelets. n = 6; mean ± SEM.
Finally, we stained resting platelets from each genotype with TO, which binds to RNA. Since platelet RNA content declines as the cells age in the circulation, TO positivity reflects the proportion of younger platelets.30 The results show that a significantly higher proportion of RGS10−/−18−/− platelets were TO+ when compared with the other genotypes (Figure 5B).
Premature platelet activation
We next asked whether the hyperreactivity we observed in vitro in the absence of RGS10 and RGS18 leads to spontaneous platelet activation in the circulation. To do this, flow cytometry was used to simultaneously detect the binding of Jon/A, P-selectin, and TLT-1 antibodies to platelets in freshly isolated blood. As noted earlier, Jon/A detects the activated conformation of αIIbβ3. P-selectin expression is a marker for α-granule secretion. TLT-1 is highly expressed in platelets, at least partially stored in α-granules, and is reported to be an even more sensitive marker of platelet activation than P-selectin.31,32 As we observed in Figure 2, Jon/A and anti-P-selectin binding to resting platelets from all of the knockouts was indistinguishable from controls (Figure 5C; supplemental Figure 5A). However, there was an increase in TLT-1 expression on the RGS10−/−18−/− platelets that was not observed on either the RGS10−/− or the RGS18−/− platelets (Figure 5D), suggesting that RGS10−/−18−/− platelets are becoming activated as they circulate.
As an additional test for whether the platelets are preactivated, we stimulated WT and knockout platelets with epinephrine and measured Jon/A binding and anti-P-selectin expression. Epinephrine activates platelet α2A-adrenergic receptors coupled to the Gi family member, Gz.33 It has been shown that epinephrine alone does not normally cause platelet activation, but it can potentiate platelet activation when added with other agonists, particularly those whose receptors couple to Gq.33,34 The results in Figure 5E show that epinephrine causes an increase in αIIbβ3 activation in RGS10−/− platelets and an even greater increase on RGS10−/−18−/− platelets, supporting the conclusion that these platelets are already partially activated. There was also a trend toward an increase in P-selectin expression, but it did not reach statistical significance (supplemental Figure 5B).
We explored several potential mechanisms for increased clearance of RGS10−/−18−/− platelets, the first being premature desialylation leading to enhanced clearance by Ashwell-Morell receptors in the liver.35 RCA-I lectin was used as the probe, since it binds to galactose residues that become exposed by sialic acid removal.36 Control assays showed that RCA-I binding to resting WT platelets was increased by treating the platelets with a sialidase and reduced by including galactose in the medium (supplemental Figure 5C). However, we found no measurable differences in RCA-I binding to RGS10−/−18−/− and WT platelets (supplemental Figure 5D). We also found no evidence for increased phosphatidylserine exposure; Annexin V binding was the same for the RGS10−/−18−/− platelets and controls (supplemental Figure 5E).37,38
To determine whether the reduction in platelet counts in the RGS10−/−18−/− mice is due solely to changes in hematopoietic cells, we also prepared chimeras in which lethally irradiated WT mice were rescued with bone marrow from WT or RGS10−/−18−/− mice. After recovery, the reduction in platelet count in the RGS10−/−18−/− chimeras was the same as in RGS10−/−18−/− mice that had not been irradiated or reconstituted (supplemental Figure 5F).
The impact of antiplatelet agents
As a final step toward understanding the basis for thrombocytopenia in RGS10−/−18−/− mice and RGS18−/− mice, we gave aspirin and the P2Y12 antagonist, prasugrel, to all of the transgenic mouse lines and controls, using a dosing regimen sufficient to blunt platelet responses to both PAR4P and ADP (supplemental Figure 6). Platelet counts were measured before, during, and after treatment. While on treatment, RGS10−/−18−/− mice showed an increase in their platelet count, which fell back to pretreatment levels when the drugs were withdrawn (Figure 6A). Aspirin and prasugrel had no effect on the platelet counts of RGS10−/− mice, which were normal throughout, or RGS18−/− mice, which remained reduced. The increase in the platelet count in RGS10−/−18−/− mice raised it to approximately the same level as in RGS18−/− mice. It also normalized basal TLT-1 expression and the proportion of platelets that were TO+ (Figure 6B-C).
Figure 6.

In vitro and in vivo indicators of premature platelet activation. (A) Platelet counts in WT, RGS18−/−, RGS10−/−, and RGS10−/−18−/− mice treated daily for 10 days with 50 mg/kg aspirin (ASA; cyclooxygenase inhibitor) and 1.875 mg/kg prasugrel (pras; P2Y12 inhibitor) by oral gavage, followed by 5 days without treatment. n = 4; mean ± SEM. *P ≤ .05 for WT and RGS10−/− vs RGS18−/− vs RGS10−/−RGS18−/−. #P ≤ .05 for WT and RGS10−/− vs RGS18−/− and RGS10−/−RGS18−/−. Data showing the impact of these drugs on platelet activation is included in supplemental Figure 4. (B-C) Flow cytometric analysis of anti-TLT-1 binding (B) and TO staining (C) prior to and 5 days after drug treatment.
To see if activation of platelets in RGS10−/−18−/− mice reached the point of producing visible microvascular thrombi, we injected RGS10−/−18−/− mice and WT controls with DyLight488-labeled anti-GPIbβ 24 hours before harvesting their lungs for sectioning and fluorescence microscopy. As a positive control, we also injected control mice with collagen and epinephrine, a combination known to produce disseminated thrombosis. There were visible microaggregates in the lungs of the mice receiving collagen and epinephrine, but not in the untreated RGS10−/−18−/− mice (supplemental Figure 7).
Discussion
Platelets possess multiple receptors and signaling pathways through which they can respond to trauma and control bleeding, nearly all of which involve members of the GPCR superfamily. While these activating pathways have been mapped in detail, less is known about the intrinsic regulatory mechanisms that modulate the platelet signaling network to prevent unnecessary or premature platelet activation. Here, we sought to understand the collective impact of RGS proteins on platelet function, focusing for the first time on the consequences of deleting RGS10 and RGS18 at the same time.
The results show that preventing expression of both proteins produces a constellation of effects that is not fully recapitulated by removing either alone. The results also show that platelet RGS proteins affect signaling by different platelet agonists to different extents and highlight their role in preventing “spontaneous” platelet activation as well as sustaining a normal platelet count. The results also confirm an earlier observation that RGS18 promotes platelet production while RGS10 does not. These results lead us to conclude that RGS10 and RGS18 have distinct as well as overlapping roles in platelets such that the consequences of removing both is greater than the sum of removing either individually. In the remainder of the Discussion, we will briefly consider each of their contributions to platelet biology.
Modulating the platelet signaling network
Past studies on RGS10 and RGS18 suggest that both proteins serve as brakes on platelet activation. The direct comparisons performed here show that removing RGS10 has a greater impact than removing RGS18. Removing RGS10 increases platelet sensitivity to PAR4 activation and increases the magnitude of the response to ADP and TxA2. Removing RGS18 alone causes a smaller shift in the PAR4 dose/response but does not alter the response to ADP and TxA2. The molecular basis for the differences between agonists was not addressed in the present studies, but plausible explanations include differences between RGS10 and RGS18 expression levels (mouse platelets express approximately twice as many copies of RGS10 as RGS18),7 differences in their distribution within each platelet, and differences among agonist receptors in terms of their interactions with the RGS-binding protein spinophilin.39 Notably, when measured in vitro, removing both RGS10 and RGS18 had no greater effect than removing RGS10 alone, but this was not the case in vivo.
Preventing spontaneous activation, prolonging platelet survival, and maintaining a normal platelet count
We found substantial evidence that platelets lacking both RGS10 and RGS18 become activated as they circulate; surface expression of TLT-1 was increased, as was the fraction of platelets that were TO+30 and the response of the platelets to epinephrine in vitro in the absence of a second agonist. Survival studies showed a shortened platelet lifespan in RGS10−/−18−/− mice. The double knockouts were also thrombocytopenic, with a 40% reduction in their circulating platelet count that improved but did not fully normalize when the mice were given aspirin and prasugrel. Although the failure of the antiplatelet agents to fully restore the platelet count in the RGS10−/−18−/− mice could mean that the dosing was inadequate, we note that the platelet count in the RGS10−/−18−/− mice rose to approximately the same level as in the RGS18−/− mice. As reported by others,18 loss of RGS18 alone causes a 15% reduction in platelet count. We show here that this reduction is due at least in part to reduced platelet production (seen during the recovery from acute thrombocytopenia) and is unaffected by aspirin and prasugrel. We propose that the thrombocytopenia in the RGS10−/−18−/− mice is due to (1) the combined effects of increased turnover of overly reactive platelets (caused by the absence of RGS10 and, to a lesser, extent, RGS18) and (2) reduced platelet production capacity (caused by the absence of RGS18). In contrast to a previous report, we did not observe significant differences among the knockouts in the number of bone marrow megakaryocytes, the ploidy of the megakaryocytes, or the ability of the megakaryocytes to mature in culture. There was a small decrease in megakaryocytes with proplatelets in the RGS18−/− and RGS10−/−18−/− mice, but it did not reach significance. However, we did see slower recovery of the platelet count from acute thrombocytopenia in both RGS18−/− mice and RGS10−/−18−/− mice. Lastly, we note that while circulating platelets in RGS10−/−18−/− mice were TLT-1+ ex vivo, they were not measurably positive for markers of α-granule expression or integrin activation. Given the degree of thrombocytopenia that we observed, we propose that platelets that had gone too far down the path to full activation are removed from the circulation. If so, their removal may be a widely distributed process. We did not find evidence of increased desialylation, increased apoptosis, or formation of platelet aggregates in vivo large enough to cause visible microvascular thrombosis in the pulmonary circulation. Finally, while not previously recognized for RGS proteins, we note that the phenomenon of shortened platelet survival when a signaling regulator is removed is not unique to RGS10/18 dual deletion. Other examples include deletion of the catalytic subunit for protein kinase A, ablating its ability to phosphorylate substrates,40 and, most dramatically, loss of RASA3 function, which leads to increased integrin activation due to an increase in the amount of activated RAP1B.41
Restraining the hemostatic response in vivo
The published evidence that RGS proteins restrain platelet reactivity can reasonably be viewed at this point as convincing for both RGS10 and, to a lesser extent, RGS18. One of our goals was to understand what happens to the hemostatic response to injury when both proteins are removed. The most striking finding was the exaggerated growth of hemostatic thrombi produced by laser injury in the cremaster muscle microcirculation. Although we did not study the lineage-matched RGS10 and RGS18 knockouts as part of the present study, we have previously studied the effects of deleting RGS10.20 Comparing those observations with the present study suggests that the effects of the double knockout are greater than removing RGS10 alone, leading not only to greater platelet accumulation but also a greater incidence of vascular occlusion caused by unnecessary growth of the hemostatic thrombus. Put differently, the hemostatic thrombi in RGS10−/−18−/− mice continue to grow well beyond the mass needed to achieve hemostasis. At first this might seem a bit puzzling, since the in vitro comparisons performed here indicate no large differences in agonist responses between RGS10−/−18−/− and RGS10−/− platelets. We propose that the greater effect of the double knockout in vivo reflects not only increased responsiveness to thrombin, ADP, and TxA2, all of which are present at the same time in vivo, but also the presence in the circulation of platelets that are preactivated, some of which may be lost during the isolation procedure. RGS10−/− platelets have the enhanced responsiveness, but not the same preactivation, and therefore produce hemostatic thrombi that are bigger than in controls, but not as big as in RGS10−/−18−/− mice. Notably, we also found that removing both RGS proteins causes an expansion of both the P-selectin+ core of the hemostatic mass and the P-selectin− shell. This contrasts with what we observed in the Gi2α(G184S) mice.21 That substitution affects Gi2-dependent signaling that would be expected downstream of platelet receptors such as P2Y12. It does not affect Gq, which is the predominant mediator of events downstream of platelet thrombin receptors. Hemostatic thrombus expansion in the Gi2α(G184S) mice was largely due to an increase in the shell region, where ADP is one of the major drivers, and provides evidence that the effects of RGS proteins in platelets are exerted on Gqα as well as Gi2α.
In conclusion, the results presented here, along with those published previously, provide new insights into the regulation of the platelet signaling network. The data show that RGS10 and RGS18 have complementary rather than identical roles, jointly modulating the hemostatic response to injury, improving platelet production, prolonging platelet survival, and preventing unwarranted platelet activation in the absence of injury.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the Transgenic and Chimeric Mouse Facility at the University of Pennsylvania School of Medicine for assistance with mouse generation, the Flow Cytometry and Cell Sorting Facility at the University of Pennsylvania School of Medicine for providing core flow cytometry facilities, and the Comparative Pathology Core at the University of Pennsylvania School of Veterinary Medicine for processing of histological samples.
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute (grants T32 HL007439 and P01 HL40387 [D.D. and L.F.B.] and grants R01 HL089224 and K12 HL141954 [K.M.H. and M.M.L.-S.]). P.M. was supported by the American Heart Association (grant 14SDG20380473) and the National Institutes of Health, National Heart, Lung, and Blood Institute (grant R01 HL144574).
D.D. is a PhD candidate at the University of Pennsylvania, and this work is submitted in partial fulfillment of the thesis requirements.
Footnotes
For original data, please e-mail the corresponding author. The data sets, protocols, and mice in this study will be made available to other investigators per the policies of the National Institutes of Health, the University of Pennsylvania, and this journal.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.D. designed and performed research, collected and analyzed data, performed statistical analyses, and wrote the manuscript; P.M. contributed vital new mouse models and designed research; K.M.H., M.M.L.-S., and S.G. designed research; M.P. designed research and interpreted data; C.T. performed research and collected and analyzed data; B.E. designed and performed research and analyzed data; M.C., K.L., and J.W. performed research; and L.F.B. designed research, interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lawrence F. Brass, Department of Medicine, University of Pennsylvania, 815 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: brass@pennmedicine.upenn.edu.
REFERENCES
- 1.Druey KM, Blumer KJ, Kang VH, Kehrl JH. Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature. 1996;379(6567):742-746. [DOI] [PubMed] [Google Scholar]
- 2.Koelle MR, Horvitz HR. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell. 1996;84(1):115-125. [DOI] [PubMed] [Google Scholar]
- 3.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4–activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89(2):251-261. [DOI] [PubMed] [Google Scholar]
- 4.Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for heterotrimeric G-protein α-subunits. Nature. 1996;383(6596):172-175. [DOI] [PubMed] [Google Scholar]
- 5.Burkhart JM, Vaudel M, Gambaryan S, et al. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73-e82. [DOI] [PubMed] [Google Scholar]
- 6.Rowley JW, Oler AJ, Tolley ND, et al. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes [published correction appears in Blood. 2011;118(14):e101-e111]. Blood. 2011;118(14):3760-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13(12):3435-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gagnon AW, Murray DL, Leadley RJ. Cloning and characterization of a novel regulator of G protein signalling in human platelets. Cell Signal. 2002;14(7):595-606. [DOI] [PubMed] [Google Scholar]
- 9.Nagata Y, Oda M, Nakata H, Shozaki Y, Kozasa T, Todokoro K. A novel regulator of G-protein signaling bearing GAP activity for Galphai and Galphaq in megakaryocytes. Blood. 2001;97(10):3051-3060. [DOI] [PubMed] [Google Scholar]
- 10.Park IK, Klug CA, Li K, et al. Molecular cloning and characterization of a novel regulator of G-protein signaling from mouse hematopoietic stem cells. J Biol Chem. 2001;276(2):915-923. [DOI] [PubMed] [Google Scholar]
- 11.Yowe D, Weich N, Prabhudas M, et al. RGS18 is a myeloerythroid lineage-specific regulator of G-protein-signalling molecule highly expressed in megakaryocytes. Biochem J. 2001;359(Pt 1):109-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alqinyah M, Maganti N, Ali MW, et al. Regulator of G-protein signaling 10 (RGS10) expression is transcriptionally silenced in activated microglia by histone deacetylase activity. Mol Pharmacol. 2017;91(3):197-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.García-Bernal D, Dios-Esponera A, Sotillo-Mallo E, García-Verdugo R, Arellano-Sánchez N, Teixidó J. RGS10 restricts upregulation by chemokines of T cell adhesion mediated by α4β1 and αLβ2 integrins. J Immunol. 2011;187(3):1264-1272. [DOI] [PubMed] [Google Scholar]
- 14.Hooks SB, Callihan P, Altman MK, Hurst JH, Ali MW, Murph MM. Regulators of G-Protein signaling RGS10 and RGS17 regulate chemoresistance in ovarian cancer cells. Mol Cancer. 2010;9(1):289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao R, Lu Y, Xing X, et al. Regulator of G-protein signaling 10 negatively regulates cardiac remodeling by blocking mitogen-activated protein kinase-extracellular signal-regulated protein kinase 1/2 signaling. Hypertension. 2016;67(1):86-98. [DOI] [PubMed] [Google Scholar]
- 16.Yang S, Li Y-PP. RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2+]i oscillation regulation. Genes Dev. 2007;21(14):1803-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alshbool FZ, Karim ZA, Vemana HP, Conlon C, Lin OA, Khasawneh FT. The regulator of G-protein signaling 18 regulates platelet aggregation, hemostasis and thrombosis. Biochem Biophys Res Commun. 2015;462(4):378-382. [DOI] [PubMed] [Google Scholar]
- 18.Delesque-Touchard N, Pendaries C, Volle-Challier C, et al. Regulator of G-protein signaling 18 controls both platelet generation and function. PLoS One. 2014;9(11):e113215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hensch NR, Karim ZA, Druey KM, Tansey MGG, Khasawneh FT. RGS10 Negatively Regulates Platelet Activation and Thrombogenesis. PLoS One. 2016;11(11):e0165984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma P, Gupta S, Sampietro S, et al. RGS10 shapes the hemostatic response to injury through its differential effects on intracellular signaling by platelet agonists. Blood Adv. 2018;2(16):2145-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Signarvic RS, Cierniewska A, Stalker TJ, et al. RGS/Gi2alpha interactions modulate platelet accumulation and thrombus formation at sites of vascular injury. Blood. 2010;116(26):6092-6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Wu J, Jiang H, et al. Signaling through Gi family members in platelets. Redundancy and specificity in the regulation of adenylyl cyclase and other effectors. J Biol Chem. 2002;277(48):46035-46042. [DOI] [PubMed] [Google Scholar]
- 23.Henao-Mejia J, Williams A, Rongvaux A, Stein J, Hughes C, Flavell RA. Generation of genetically modified mice using the CRISPR-Cas9 genome-editing system. Cold Spring Harb Protoc. 2016;2016(2):t090704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stalker TJ, Traxler EA, Wu J, et al. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121(10):1875-1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berman CL, Yeo EL, Wencel-Drake JD, Furie BC, Ginsberg MH, Furie B. A platelet alpha granule membrane protein that is associated with the plasma membrane after activation. Characterization and subcellular localization of platelet activation-dependent granule-external membrane protein. J Clin Invest. 1986;78(1):130-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu-Lin S, Berman CL, Furie BC, August D, Furie B. A platelet membrane protein expressed during platelet activation and secretion. Studies using a monoclonal antibody specific for thrombin-activated platelets. J Biol Chem. 1984;259(14):9121-9126. [PubMed] [Google Scholar]
- 27.Bertoni A, Tadokoro S, Eto K, et al. Relationships between Rap1b, affinity modulation of integrin α IIbbeta 3, and the actin cytoskeleton. J Biol Chem. 2002;277(28):25715-25721. [DOI] [PubMed] [Google Scholar]
- 28.Shen J, Sampietro S, Wu J, et al. Coordination of platelet agonist signaling during the hemostatic response in vivo. Blood Adv. 2017;1(27):2767-2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Da Q, Derry PJ, Lam FW, Rumbaut RE. Fluorescent labeling of endogenous platelets for intravital microscopy: Effects on platelet function. Microcirculation. 2018;25(6):e12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee LG, Chen CH, Chiu LA. Thiazole orange: a new dye for reticulocyte analysis. Cytometry. 1986;7(6):508-517. [DOI] [PubMed] [Google Scholar]
- 31.Smith CW, Raslan Z, Parfitt L, et al. TREM-like transcript 1: a more sensitive marker of platelet activation than P-selectin in humans and mice. Blood Adv. 2018;2(16):2072-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Washington AV, Schubert RL, Quigley L, et al. A TREM family member, TLT-1, is found exclusively in the α-granules of megakaryocytes and platelets. Blood. 2004;104(4):1042-1047. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Wu J, Kowalska MA, et al. Loss of signaling through the G protein, Gz, results in abnormal platelet activation and altered responses to psychoactive drugs. Proc Natl Acad Sci USA. 2000;97(18):9984-9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lanza F, Beretz A, Stierlé A, Hanau D, Kubina M, Cazenave JP. Epinephrine potentiates human platelet activation but is not an aggregating agent. Am J Physiol. 1988;255(6 Pt 2):H1276-H1288. [DOI] [PubMed] [Google Scholar]
- 35.Grozovsky R, Begonja AJ, Liu K, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med. 2015;21(1):47-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green ED, Brodbeck RM, Baenziger JU. Lectin affinity high-performance liquid chromatography. Interactions of N-glycanase-released oligosaccharides with Ricinus communis agglutinin I and Ricinus communis agglutinin II. J Biol Chem. 1987;262(25):12030-12039. [PubMed] [Google Scholar]
- 37.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84(5):1415-1420. [PubMed] [Google Scholar]
- 38.Zhang H, Nimmer PM, Tahir SK, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14(5):943-951. [DOI] [PubMed] [Google Scholar]
- 39.Ma P, Cierniewska A, Signarvic R, et al. A newly identified complex of spinophilin and the tyrosine phosphatase, SHP-1, modulates platelet activation by regulating G protein-dependent signaling. Blood. 2012;119(8):1935-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao L, Liu J, He C, et al. Protein kinase A determines platelet life span and survival by regulating apoptosis. J Clin Invest. 2017;127(12):4338-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stefanini L, Paul DS, Robledo RF, et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J Clin Invest. 2015;125(4):1419-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.