

Abstract
The cell regulates complicated signaling and metabolism through strict transcriptional, translational, and post-translational regulation. Considerable advances have been made for monitoring transcription and translation over the past few decades. Until recently, there have not been generalizable methods for assessing the effects of post-translational regulation on enzymatic activity. Activity-based sensing (ABS) has emerged as a powerful approach for monitoring small-molecule and enzymatic activities within living systems. Initial examples of ABS were applied for measuring general enzymatic activity; however, a recent focus has been on increasing selectivity to monitor a single enzyme or isoform. The highest degree of selectivity is required for differentiating between isoforms, where the targets display significant structural similarities as a result of a gene duplication or alternative splicing. This review highlights key examples of small-molecule, isoform-selective probes with a focus on the relevance of isoform differentiation, design strategies to achieve selectivity, and applications in basic biology or in the clinic.
Keywords: Activity-based sensing, probe development, fluorescence, molecular imaging, rational design
Graphical Abstract
The development of isoform-selective activity-based probes enables the identification of enzymatic activity within living systems. Within this Minireview, we highlight key examples and discuss the relevance of isoform differentiation, design strategies, and applications.
1. Introduction
Enzymes catalyze a diverse range of chemical reactions that are essential for cellular metabolism, signaling, and defense. They achieve high catalytic proficiency, (kcat/Km)/kuncat, by selectively stabilizing the transition state, thereby reducing the activation barrier for the corresponding chemical transformation.[1,2] To exert control over specific biological processes, the cell can modulate the transcription and translation of key proteins. Alternatively, it can directly regulate protein activity via post-translational modifications, intracellular availability of cofactors and substrates, or feedback mechanisms (e.g., activators or inhibitors).[3–5] When enzyme networks become dysregulated, a variety of disease states can emerge. Since it is ultimately the activity and not the concentration of an enzyme that dictates function, it is crucial to be able to directly monitor this within its native cellular environment.[6]
Although reliable methods exist for quantifying transcription (e.g., Northern blotting, quantitative polymerase chain reaction) and translation (e.g., Western blotting, enzyme-linked immunosorbent assay), direct detection of enzymatic activity remains a significant challenge. Activity-based sensing (ABS) is a powerful approach to facilitate post-translational monitoring because it provides a direct read-out of enzymatic activity with a biocompatible detection method.[7] Therefore, ABS strategies can monitor regulation beyond concentration and can shed light on previously hidden regulatory mechanisms.[8]
The origins of enzyme ABS date back to early biochemical analyses, when substrates were prepared for measuring general enzymatic activities (e.g., esterases, phosphatases, galactosidases, lipases) in vitro.[9–12] More recently, the focus has shifted to providing probes with cellular and live animal compatibilities.[13] This has the potential to yield new, innovative methods for discovering biology[6] and to serve as diagnostic tools.[14] Regardless of the application, a general ABS probe utilizes enzyme recognition and catalysis to unmask a detectable handle. The most common handles are fluorescent dyes, however other detection strategies are possible (e.g., chemiluminescent, magnetic resonance, and photoacoustic readouts).[15] Strategic design is essential to impart both specificity and the signal enhancement induced by enzyme-catalyzed turn-over. As the focus moved from the test tube into complex biological systems, specificity has become the critical parameter. Probes with good selectivity facilitate direct evaluation of activity in vivo. This overcomes difficulties with protein purification and identification of optimal conditions.
ABS development becomes more complicated when several functionally related, but non-identical, enzymes exist. These proteins (isoforms) typically display high sequence and structural similarities and operate on near-identical substrates. Isoforms typically emerge via gene duplication or alternative splicing and enable differential regulation of the protein for increased phenotypic robustness.[16] The ability to distinguish between isoforms is critical for differentiating their function because of their distinct expression, activation, and/or substrate preferences. This manifests itself in physiological and pathological contributions that are simultaneously operative. Without isoform-selective ABS probes it would be difficult to decipher their relative contributions to a specific condition. Isoform-selective approaches are designed to harness isoform-specific substrate affinity, stability, interactions, and localization to achieve selective detection (Figure 1). Herein, we will focus on reviewing small-molecule ABS approaches for imaging the enzymatic activity of a single isoform. The discussion focuses on the rationale for isoform selectivity, design principles for isoform-selective ABS, and applications. While not comprehensive, this review highlights key examples that illustrate current approaches and suggests potential future directions.
Figure 1.

General schematic depicting enzyme-catalyzed activation of an isoform-selective ABS probe to afford a fluorescent product.
2. Probe Design Criteria and Strategies
Careful selection of the substrate component is critical to ensure a robust, isoform-selective ABS design. First, a comprehensive literature search should be conducted to identify any known substrates that exhibit high reactivity (kcat) and binding affinity (Kd or Km) for the desired isoform. We recommend prioritizing substrates that bear an aromatic substituent and display 2-fold selectivity over the other isoforms. This is because aromatic groups are a feature of most optical sensors and 2-fold selectivity provides a promising initial scaffold for building selectivity. If aromatic moieties are not tolerated by the enzyme, this can be circumvented by employing self-immolative linker strategies. In the absence of good selectivity data, small substrate screens can be performed. Molecular docking and in silico screening efforts can be used to exploit differences in active site topology and identify an ideal substrate scaffold for the ABS probe (see sections 3.1 — 3.4 and 3.6). Along the same lines, steric interactions can be employed to exploit differences in the active site architecture (see sections 3.1 — 3.7). If this approach is not applicable, it is possible to consider installing functionalities that form or remove important hydrogen bonding or ionic contacts to afford selectivity (see section 3.1 and 3.3). Finally, differences in enzyme mechanism can provide unique opportunities for differentiating between enzyme activity, both in terms of regioselectivity and enantioselectivity (see section 3.5 and 3.7).
After designing the substrate component, it is necessary to select a reporter (e.g., fluorophore) to obtain a prominent response following activation. Based on our experience, we recommend an in vitro signal enhancement of at least 10-fold because the corresponding response will be lower in cells. ABS probes that do not meet this suggested threshold can still be useful for sensing applications; however, rigorous validation including siRNA knockdown and isoform-selective inhibition may be required. Additionally, the fluorophore should not adversely impact substrate binding and probe turn-over. When appropriate, the reporter can be selected to provide additional binding interactions and/or preclude undesirable binding to isoforms featuring a smaller active site.
Like inhibitor design, the development of isoform-selective ABS probes often requires several iterations of optimization to identify an ideal probe. This is because the detectable handle and substrate are not orthogonal. The sterics, electronics, and connectivity alter substrate binding, redox properties, and photophysical characteristics. Alternative strategies and possible future directions for the field are included in the outlook (see section 4).
3. Isoform-Selective ABS Probes
3.1. Aldehyde Dehydrogenase
Aldehyde dehydrogenases (ALDH, EC 1.2.1.4) are a family of 19 isoforms in humans. These enzymes catalyze the oxidation of reactive aldehyde species to their corresponding carboxylic acid products via a conserved mechanism. First, the aldehyde is activated as a thiohemiacetal and then oxidized in a NAD(P)H-dependent manner.[17] Cellular maintenance of aldehyde levels is critical due to their potent crosslinking activity and propensity to form adducts/polymers. Interestingly, ALDH1A1 is upregulated in normal stem cells and cancer stem cells.[18] This increase in activity is presumably required to detoxify excess reactive aldehydes and regulate differentiation via retinaldehyde metabolism. For this reason, ALDH1A1 isoform-selective ABS probes would be powerful chemical tools for identifying, isolating, and studying stem cells. Due to overlapping substrate scopes and high isoform homology, only a few marginally selective accumulation-based probes (no turn-on) have been reported, which require the tandem application of ion pump inhibitors.[19,20]
Our group overcame this challenge by developing AlDeSense, the first ABS probe for ALDH1A1 (Figure 2a). Initially, we surveyed the literature to identify benzaldehydes as preferential substrates for class 1 isoforms (i.e., 1A1, 1A2, and 1A3).[21] Structural data demonstrates that small deviations are found between isoform substrate pockets[22] and ALDH1A1 contains a wider active site channel relative to the other ALDHs.[23] Thus, we hypothesized that reactivity with the other 16 isoforms could be prevented by installing a bulky xanthene-based dye para to the reactive aldehyde group. This aldehyde would also serve to quench the fluorescence through a d-PeT mechanism and yield an off-on response following turn-over. We predicted that reducing the phenolic pKa with fluoro substituents could eliminate the requirement of ion pump inhibitors by trapping the dianionic product within the cell. Additionally, this modification could afford selectivity for ALDH1A1 over the other class 1 isoforms by enhancing an additional ionic interaction that is unique to ALDH1A1. We observed a 20-fold fluorescent enhancement when AlDeSense was treated with recombinant ALDH1A1. Moreover, the probe was selective over a panel of common ALDH isoforms (Figure 2b). AlDeSense was then applied to visualize ALDH1A1 activity in cancer cells (Figure 2c), tumorspheres, pulmonary metastatic lesions (ex vivo), and solid tumors (in vivo). In each model, AlDeSense identified the subpopulation of cells with the highest enzymatic activity. These are anticipated to be the cancer stem cells. Results were validated using siRNA knockdown and with a small-molecule inhibitor.[24] Using the same design strategy, we recently developed a red-shifted analog (red-AlDeSense, Figure 2d). Replacement of the endocyclic oxygen with the dimethyl-silicon afforded the desired circa 100 nm red-shift, but additional hydrophobic contact sites on the benzaldehyde substrate were required to achieve isoform selectivity. Multicolor imaging with red-AlDeSense demonstrated that the canonical CD44 marker stains independently of ALDH1A1 in non-small cell lung cancer stem cells.[25]
Figure 2.

a) Chemical structure of AlDeSense. b) Normalized fluorescence turn-on of AlDeSense after incubation with ALDH isoforms. c) Confocal images of K562 cells stained with AlDeSense. Further permissions related to the material excerpted should be directed to the American Chemical Society. https://pubs.acs.org/doi/10.1021/acscentsci.8b00313. d) Chemical structure of red-AlDeSense.
3.2. Carboxylesterase
Carboxylesterase (CE, EC 3.1.1.1) catalyzes the hydrolysis or transesterification of endogenous (e.g., lipids) and exogenous (e.g., gemcitabine prodrug) esters.[26] CE is a member of the serine hydrolase family, which employ a catalytic triad to facilitate hydrolysis through an enzyme-linked intermediate.[27] While CE’s metabolism of xenobiotic compounds is well studied, much less is known about the endogenous substrates. To date, the two major isoforms, CE1 and CE2, have been identified with distinct expression profiles and substrates. CE1 is expressed within the liver and brain and preferentially hydrolyzes bulky acyl compounds conjugated to a small alcohol. On the other hand, CE2 is expressed at higher levels in the intestine and lower levels in cancer cells. CE2 generally prefers compounds with a small acyl component and bulky alcohol.[26,28] CE ABS probes promise to provide a means for identifying compatible prodrugs, patient screening for drug selection, and evaluating the basic biological contributions of each isoform.
After surveying the literature for known CE2 substrates, Yang and coworkers identified bulky benzoyl esters as possible selective substrates.[28] This led to several generations of fluorescent probes. The first example utilized the 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran dye platform, to form the dye TCFB (Figure 3a), for the first live cell imaging of endogenous CE2 activity in A549 cells. The activation mechanism was confirmed with molecular dynamics and docking, where the most favorable binding interactions were observed with the benzoyl ester near the active serine.[29] Next, a ratiometric analog was prepared from the 4-ethylbenzyl functionalized 3-hydroxylflavone backbone (Figure 3b) and applied for an inhibitor screen.[30] To increase the accessible imaging depths, Kim and coworkers prepared a two-photon compatible probe, Probe 2, containing a succinate ester to mimic the CE2-selective inhibitor 18β-glycyrrhetinic acid. This alkyl ester proved stable and optically confirmed the decreased enzymatic activity in cancer as compared to healthy tissue with a BTDAN-based probe.[31] Finally, Yang and coworkers leveraged a NIR dye, DDAO, that was capped with a benzoyl ester (DDAB, Figure 3c) to confirm that the elevated expression of CE2 in the liver and intestine correspond with increased in vivo activity.[32]
Figure 3.

The chemical structures of a) TCFB; b) 4-ethylbenzyl substrate on a 3-hydroxylflavone backbone; c) DDAB; d) 4-MOMMP; and e) MMB. f) MMB docked into the active site of CE1. Reprinted (adapted) with permission from ref. 36. Copright 2019 American Chemical Society.
CE1 selective probes have also been prepared to compliment the CE2 probes. For example, the methyl ester of D-luciferin was identified as a convenient bioluminescent probe for CE1 activity.[33] On the other hand, benzoylation of the HMBT dye platform yielded a ratiometric fluorescent probe for CE1 rather than CE2. This highlights the importance of proper characterization because unexpected changes can result as the substrate and optical handles are not orthogonal.[34] Since the HMBT probe displayed solvolysis, Morisseau and coworkers prepared a medium sized panel of aryl amide probes which were inspired by CE1’s activity towards lidocaine and prilocaine. The lead compound, 4-MOMMP (Figure 3d), is a fatty acid mimic employing a 2-methyl-6-methoxy-3-pyridinyl amide fluorophore with a long, non-linear octanoic tail. This substrate-derived specificity is unique compared to the previous designs; however, the cellular compatibility was not assessed.[35] Next, Ge, Cui, and coworkers developed a panel of ester-BODIPY and halogen decorated derivates. These probes undergo a considerable electronic change upon hydrolysis with a corresponding increase in emission. Molecular dynamics and docking were used to identify promising structures before advancing to the synthesis (Figure 3f). The authors identified the lead compound, MMB, which contains a methyl ester and dichlorofunctionalization of the BODIPY (Figure 3e). MMB was applied for live-cell imaging, tissue ex vivo analysis of CE1 activity (tissue slices and whole organ), and live animal imaging in zebrafish.[36] Altogether, these works highlight the utility of searching the literature for existing substrates/inhibitors and employing molecular docking to design isoform-selective ABS probes.
3.3. Cyclooxygenase
Two isoforms of cyclooxygenase (COX, EC 1.14.99.1) have been identified. Both COX-1 and COX-2 catalyze the dioxygenation and cyclization of arachidonic acid to the organic peroxide intermediate, PGG2.[37] The reaction is initiated by an active site tyrosyl radical and the desired stereochemistry is templated by the cyclooxygenase active site to afford one major product.[38] Subsequently, the intermediate translocates to the heme-containing peroxidase active site for reduction to PGH2. The resulting prostaglandin can then be elaborated by a range of prostaglandin synthases or thromboxane synthase to afford the prostanoids (arachidonic acid derived lipid signaling molecules). These lipids function in a wide range of physiological processes, such as the immune response, blood clotting, and sodium metabolism.[39] When dysregulated, COX and the prostanoids contribute to many pathologies. For example COX expression is correlated with poor prognoses in cancer.[40,41] While the two isoforms share a large degree of sequence identity (~60%), there are small structural differences.[42] These manifest with minor differences in the cyclooxygenase active site and ligand specificity. Of the available biocompatible technologies (e.g., fluorescence, PET) for COX detection, all have depended on utilizing inhibitor scaffolds that display imperfect selectivity[43] and only report on concentration.[44–46] Therefore, we sought to develop the first ABS approach for COX-2 imaging in live cells.
CoxFluor was designed through the strategic linkage between arachidonic acid and a fluorophore precursor (Figure 4a). We proposed that CoxFluor could function as a substrate for the cyclooxygenase activity to yield a PGG2-like intermediate. This intermediate could then serve as a selective reductant for the peroxidase activity. We hypothesized that the probe would be selective for COX-2 due to the arachidonic acid substrate, steric constraints, and lack of a critical salt bridge in COX-1’s active site. Experimentally, CoxFluor displayed a 40-fold turn on after incubation with recombinant COX-2. Under similar conditions, we observed no cross-reactivity against a panel of related enzyme (Figure 4c) nor with a range of bioanalytes. The mechanism was then supported using molecular dynamics and ensemble docking (Figure 4b). CoxFluor was validated as a model for inhibitor validation and for the detection of COX-2 activity within live cells using confocal microscopy and flow cytometry. Finally, CoxFluor was employed for the identification of oxygen-dependent activity changes that where not a result of protein expression (Figure 4d).[47] This example highlights the importance of ABS for studying protein function and regulation beyond expression and translation.
Figure 4.

a) The chemical structure of CoxFluor. b) CoxFluor docked into the active site of COX-2. c) Normalized fluorescent turn-on of CoxFluor with various enzymes. d) Normalized fluorescent turn-on of CoxFluor under 0.1, 1, or 21% oxygen concentrations. Reprinted (adapted) from ref. 47 with permission from John Wiley and Sons, Inc. Copyright 2020.
3.4. Cytochrome P450
Cytochrome P450s (CYPs) catalyze the heme-dependent oxygenation of a variety of metabolites and xenobiotics.[48–50] To date, 57 human isoforms have been identified that differ in substrate scope, reaction, and expression profile.[51–54] The metabolic activity of CYPs increases the water solubility of the substrate for phase 1 xenobiotic metabolism.[50] An undesirable consequence of this activity is the inadvertent production of carcinogenic intermediates.[55] This associates particular CYP isoforms with cancer; however, no major correlations between expression and predisposition can be made at this time.[56] Isoform-selective ABS probes have potential applications in cancer imaging and for identifying native CYP isoform signaling.
Due to the characteristically broad CYP substrate scope, probe design relied largely on structural data and in silico docking. Yang and coworkers hypothesized large O-alkyl substrates would yield CYP1A1 (EC 1.14.14.1)-selective substrate by biasing against narrower binding pockets. Initial work by the group screened several 1,8-naphthalimide probes containing a range of O-alkyl substrates, where enzyme-catalyzed O-dealkylation would uncap the dye, red-shift the emission, and enable ratiometric detection of CYP1A1 activity. Prior to synthesis, 13 probes were docked into CYP1A1 and CYP1A2. Only the alkyl substrates could orient properly for catalysis (Figure 5d). These hits were prepared and screened for activity against a range of CYPs. The chloroethoxy probe, NBCeN, was identified as ideal in terms of selectivity and sensitivity (Figure 5a). Treatment of NBCeN with CYP1A1 yielded a 32-fold ratiometric response with minimal activation by other CYP isoforms. NBCeN was further validated using α-naphthoflavone, a CYP1A1 inhibitor, before application to live-cell imaging and two photon microscopy of rat livers.[57] The Qi group leveraged an isopropyl substrate on a similar dye platform (the N-butyl was replaced with a solubilizing PEG) to prepare NEiPN. This probe afforded a 133-fold ratiometric response in vitro and was applied for confocal microscopy in live cells and zebrafish (Figure 5b).[58] Presumably, the increased sensitivity was due to increased water solubility. It would be important to confirm NEiPN’s selectivity against CYP2B6 and CYP3A4.[57]
Figure 5.

Chemical structures of a) NBCeN; b) NeiPN; and c) BnXPI. Docking of d) NBCeN in the CYP1A1 active site. Reprinted (adapted) from the Royal Society of Chemistry from ref. 57.; e) BnXPI bound in the CYP2J2 active site. Reprinted (adapted) with permission from ref. 59. Copyright 2019 American Chemical Society.
In contrast to the previous examples, CYP2J2 (EC 1.14.14.1) has a narrow, cylindrical active site. This hinders the development of isoform-selective probes because minimizing steric interactions will enable catalysis but is not expected to increase selectivity. Ma and coworkers utilized molecular docking strategies to select the dye, linker, and substrate components of the probe (Figure 5e). From this initial screen, the HD dye (HXPI) was identified as the optimal fluorophore due to its flexibility, structure, and photophysical properties. Next a small panel of O-alkyl substrates were prepared with or without a self-immolative p-hydroxybenzyl linker. It was hypothesized that this may enable efficient binding within the long active site. Of all the screened probes (in silico and in vitro), Ma and coworkers identified the linker containing O-methylated HXPI probe as the most selective and reactive probe, BnXPI (Figure 5c). Upon demethylation BnXPI decomposes to release HXPI, formaldehyde, and the linker byproduct with a 48-fold fluorescent response (minimal to no change with other isoforms). This probe was validated with an inhibitor and siRNA experiments prior to application in live-cell imaging, detection of CYP2J2 activity in patient blood samples, and in murine tumor models.[59] These works emphasize the utility of leveraging active site topology with molecular docking for ABS design.
3.5. Methionine Sulfoxide Reductase
Methionine sulfoxide reductase (Msr, EC 1.8.4.11) catalyzes the reduction of protein methionine sulfoxides to methionine.[60,61] This is essential for mitigating the effects of protein damage that results from cellular oxidative stress.[62,63] Reduction is believed to proceed through a cysteine-linked intermediate, followed by disulfide exchange with adjacent cysteines, and ultimately reduction (from the thioredoxin–thioredoxin reductase system) to afford the product.[64] Through this activity, MsrA and MsrB are intrinsically tied to recovery from oxidative stress and aging. MsrA and MrsB are responsible for the reduction of the S- and R-epimers of methionine sulfoxide, respectively.[61]
Misek and coworkers sought to delineate the effects of each isoform by leveraging a mechanism-based approach. First, a viable MsrA-selective substrate, methyl p-tolyl sulfoxide, was identified from the literature[60] and appended onto the BODIPY dye platform (Figure 6c). This compound, (S)-Sulfox-1, undergoes enzyme-catalyzed reduction to a methylphenyl sulfide with a corresponding red-shift. In vitro, a robust 13-fold ratiometric response was observed upon incubation with recombinant MsrA, whereas no change was observed from the R-epimer. The steady-state characterization concluded similar activity as is observed for methionine sulfoxide. While no selectivity is included in the paper, the selectivity of MsrA for the Sepimer suggests that (S)-Sulfox-1 and (R)-Sulfox-1 are most likely isoform-selective probes for MsrA and MsrB, respectively. (S)-Sulfox- 1 was then validated with inhibitor studies in E. coli (Figure 6d).[65] This example shows the potential of utilizing stereospecificity for probe development.
Figure 6.

a) The chemical structure of (S)-Sulfox-1. b) Images of E. coli cells at OD600 treated with (S)-Sulfox-1 (left) or treated with (S)-Sulfox-1 and inhibitor (right). Reprinted (adapted) with permission from ref. 65 with permission from John Wiley and Sons, Inc. Copyright 2016.
3.6. Monoamine Oxidase
Monoamine oxidases (MAO, EC 1.4.3.4) consist of two isoforms (MAO-A and MAO-B) that catalyze the conversion of monoamines to their corresponding aldehyde product, ammonia, and hydrogen peroxide in a flavin-dependent manner.[66] Since monoaminergic neurotransmitters (e.g., dopamine) are MAO substrates, the protein’s overexpression or overactivation can lead to aberrant hydrogen peroxide production and neurological disorders.[67] In support of this, MAO-B inhibitors have successfully protect neurons from damage in Parkinson’s disease models.[68] While non-selective MAO probes have existed for over a decade,[69] isoform-selective ABS probes present a promising approach to decipher the individual isoform contributions in neuronal health and cancer.
Yao and coworkers developed the first isoform-selective ABS probes for MAO-B to facilitate direct enzymatic detection within live cells. First, the authors identified primary, secondary, and tertiary propylamines as tentative substrates for MAO-B. These substrates were converted into a small panel of probes by linking the substrate to the two-photon acetyl-naphthalene or conjugated pyrimidine dye platforms through a self-immolative carbamic acid linker. Activation requires enzymatic oxidation of the alkylamine to the corresponding aldehyde followed by a β-elimination and release of acrolein along and the unstable dye-carbamate intermediate. Spontaneous decarboxylation uncaps the dye with a concomitant red-shift and fluorescent turn-on response. The 9 probes were assayed against both MAO-A and MAO-B, and only MAO-B activity was observed. The best probe, U1 (Figure 7a), was selected due to the largest fluorescent response. The isoform selectivity was rationalized using docking experiments which indicated that the small nature of the probe was optimal for binding within the long, narrow substrate cavity of MAO-B. U1 was applied in live-cell imaging, two-photon imaging of parkin-null Drosophila brain, and Parkinson’s patient-derived lysates. This work established cell type MAO-B activity differences between Parkinson’s and healthy patients. Moreover, U1 suggested peripheral blood cell MAO-B activity as a possible biomarker for Parkinson’s disease.[70] This was later expanded by the Yao group, where U1 was equipped with an alkyne handle for protein profiling. The resulting dual-purpose ABS probe (M2) was applied to live cells where MAO-B antibodies colocalized with the probe.[71]
Figure 7.

Chemical structures of a) U1 and b) Probe 1. c) Confocal images of HeLa and NIH-3T3 cells after incubation with Probe 1 for 1 hour. False coloring for signal from the initial probe (blue) and activated probe (red). d) Relative ratio of turned-over probe to initial fluorescence. Reprinted (adapted) with permission from ref. 72. Copyright 2018 American Chemical Society.
From the previous work, it was clear that a different dye scaffold would be required for MAO-A detection. The Ma group chose a 1,8-napthalimide based probe for two photon detection of MAO activity due to its small size, stability, and cell-permeability. The probes differed by displaying either methylamine (Probe 1, Figure 7b) or a primary amine (Probe 2, not shown) as the substrate. In silico modeling was applied by Ma and coworkers to computationally compare binding affinity. Both probes utilized the same activation strategy as the MAO-B probes and yielded good selectivity. The ideal probe, Probe 1, was selected for its higher dynamic range (greater than 40-fold ratiometric response) and was then applied for imaging MAO-A activity across a panel of cell lines (Figure 7c–d) and further validated with clorgyline, MAO-A-selective inhibitor.[72] These works further exemplify the importance of structural data and docking experiments for designing isoform-selective ABS probes.
3.7. Nucleotide pyrophosphatase/phosphodiesterase
Lysophospholipase C (NPP6, EC 3.1.1.5) is a member of the nucleotide pyrophosphatase/phosphodiesterase (NPP) enzyme family. This family includes a range of membrane-associated or secreted enzymes that regulate the concentrations of nucleotides (e.g., ATP) and phospholipids via hydrolysis promoted by a catalytic threonine or serine and proceeding through an enzyme-linked intermediate.[73,74] NPP isoforms are characterized by substrate class. NPP6 is a type 1 membrane protein which displays selectivity for choline substrates (e.g., lsyophosphatidylcholine, glycerophosphodiesters) and has been implicated in choline uptake.[73]
Given the well-defined substrate scope differences between the NPP family, Nagano and coworkers developed TG-mPC, the first ABS probe for imaging NPP6 activity (Figure 6a). TG-mPC consists of a TokyoGreen dye with a phosphoryl choline capping group. It was hypothesized that the dye could mimic the native substrate’s non-polar tail which flanks the zwitterionic capping group. Importantly, the authors bestow selectivity to NPP6 over the closely related isoform, NPP2, through a strategic mechanistic design.[75] NPP2 is a particularly efficient phosphodiesterase that promotes cell migration at low (pM) concentrations.[76] To achieve selectivity, Nagano and coworkers require hydrolysis proximal to the dye for fluorescent activation. NPP2, on the other hand, would catalyze the release of choline and a non-fluorescent, capped TokyoGreen dye. TG-mPC, was selected from a panel of choline-mimicking analogs because it displayed the highest selectivity. The authors confirm no cross-reactivity across the entire nucleotide pyrophosphatase/phosphodiesterase family (Figure 6b). With the probe in hand, the authors screened 80,000 compounds to identify a scaffold for NPP6 selective inhibitors. Structure activity relationship efforts yielded T11, an isoform-selective inhibitor.[75] This work emphasizes the utility of employing the enzyme mechanism for selectivity. Additionally, the ABS approach was employed to develop isoform-selective inhibitors that can be used with or without the probe for studying NPP6 activity.
3.8. Protein Serine/Threonine Phosphatase
Protein serine/threonine phosphatase (PSP, EC 3.1.3.16) is a family of about 30 enzymes that mediate dephosphorylation in a metal-dependent manner.[77] These enzymes regulate a wide range of peptides and proteins by modulating phosphorylation state.[78] In contrast to protein kinases, PSP have been understudied due to a poor understanding of substrate recognition. More recent data suggests that PSP selectivity and regulation requires complex assembly with regulatory domains.[77,79] These have been difficult to reconstitute in recombinant assays.
To overcome this limitation, the Stain’s lab sought to develop an ABS approach for PSP activity. The sulfonamido-oxine (Sox) fluorophore was selected as the phosphorylation-responsive moiety. The CSox detection strategy utilizes chelation-enhanced fluorescence to report on phosphorylation state, where a Sox-modified cysteine is fluorescent only when complexed with magnesium and the phosphate of interest. This technology has been employed for studying a wide range of protein kinases and probe selectivity is derived from the peptide substrate.[80] PSPtide was designed on a truncated peptide substrate to monitor PP2B activity (Figure 8a). Interestingly, while the probe was efficiently dephosphorylated in cell extracts, it was not modulated by calcium or calmodulin, two well-known activators of PP2B. To understand the source of the activity, the Stains lab underwent a series of inhibition studies (Figure 8b) and siRNA knockdown (Figure 8c) experiments to identify PP2AC as the source of most of the activity (greater than 80% based on pull-down experiments). This was then applied to detect insulin-dependent PP2AC activation in HepG2 liver cell lysates. This illuminates the difficulty associated with designing isoform-selective probes. While it is reasonable to assume that substrate-derived probe design will work, even small perturbations in structure can alter the selectivity or specificity. The Stains lab demonstrated the importance of rigorous probe evaluation, as the unexpected changes yielded a PP2AC selective probe from a PP2B substrate.[81] It will be interesting to see whether PSPtide or a second generation can be used to detect activity within live cells.
Figure 8.
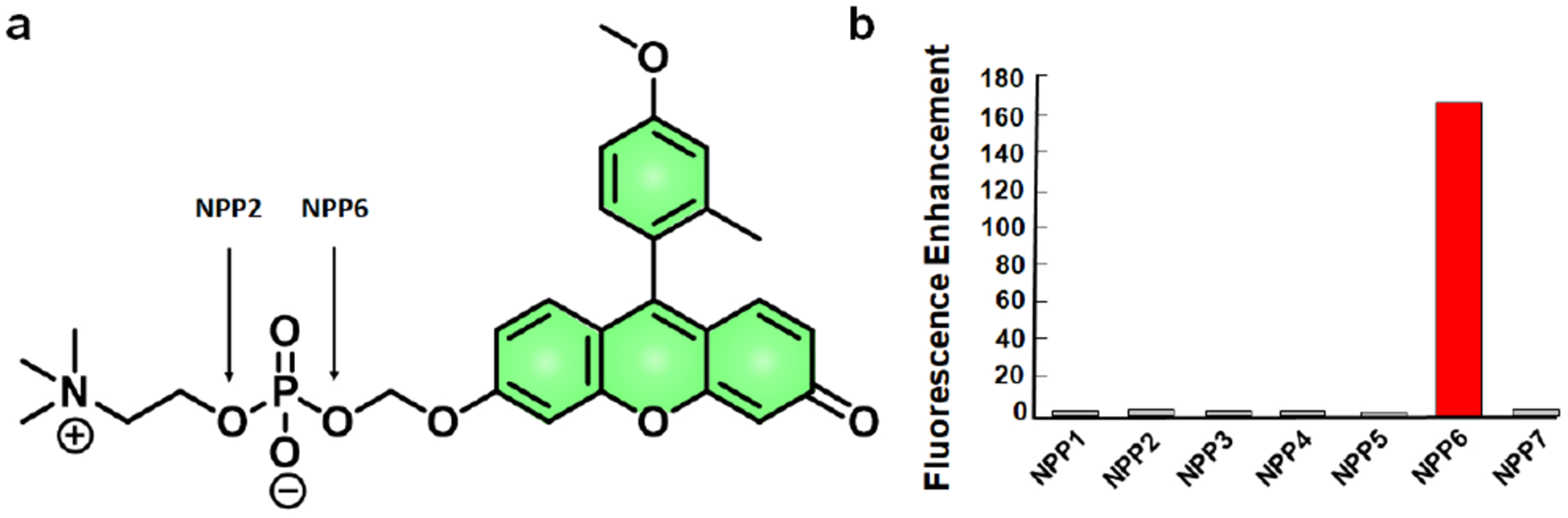
a) Chemical structure of TG-mPC. b) TG-mPC fluorescence enhancement after incubation with various NNPs. Reprinted (adapted) with permission from ref. 75. Copyright 2011 American Chemical Society.
4. Outlook
ABS represents a powerful detection strategy that relies on selective chemical reactivity rather than traditional supramolecular chemistry and molecular recognition. The development of ABS probes has profoundly impacted how biological analytes (e.g., metal ions, gasotransmitters) are monitored in living systems.[15] In this minireview, we focused our discussion on small-molecule ABS probes for isoform-selective imaging of enzymatic activity. Prior to the advent of ABS probes, studies relied almost exclusively on transcriptomic and proteomic methods. This required the assumption that expression correlated with activity. In recent years, both activity-based protein profiling and ABS technologies have demonstrated this is not always true.[47,82] We have summarized key design criteria and strategies that guide the development of probes for selectively targeting new isoforms.
We would like to note the parallels between ABS probe design and small-molecule drug development, both of which aim to maximize efficacy while limiting off-target effects. Moving forward, we believe that it is vital to consider various medicinal chemistry approaches, such as combinatorial synthesis,[83] fragment-based discovery,[84] and computational screening[85] to streamline isoform-selective ABS probe development. For example, fluorescent probe library development has been successful for identifying analyte-responsive probes.[86–88] Additionally, the development of genetically encoded ABS probes would enhance the detection of tightly regulated enzymatic pathways by utilizing cellular trafficking for probe localization.[89] In the context of small-molecule approaches, this could be envisioned with SNAP or HaloTag technologies.[90,91] It is also important to note, most ABS probes have relied on fluorescence imaging with rare examples employing luminescent approaches and potentially photoacoustic[92] or magnetic resonance imaging.[93] Future work should expand the scope and imaging capabilities of the available probes. This will allow the transition from cellular systems to live animals. Finally, the application of isoform-selective probes for identifying biological regulation, drug discovery, and/or applications in the clinic are essential for advancing the field, where we envision a move towards quantitative analysis of activity (rather than relying on relative differences).
Figure 9.

a) Schematic of PSPtide sensing mechanism. b) The change in fluorescence as a result of PP2A, in lysates, can be resolved using calyculin A. c) The change in probe fluorescence decreases when the PP2ACα subunit is knocked down with siRNA. Reprinted (adapted) with permission from ref. 81. Copyright 2016 American Chemical Society.
Acknowledgements
C.J.R. thanks the Chemistry-Biology Interface Training Grant (T32 GM070421) and the Seemon Pines Graduate Fellowship for support. J.C. acknowledges the National Institutes of Health (R35 GM133581) for funding. Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.[94]
Biography
Sarah Gardner graduated from the University of Iowa in 2017 with a BSc in Biochemistry. With financial support from the Fight for Sight organization, Sarah performed research in the laboratory of Prof. Sheila Baker. She is currently pursuing a PhD in Biochemistry with Prof. Jeff Chan at UIUC. Her research interests include the development and evaluation of chemical tools to study the roles of reactive aldehydes in the context of cancer and aging.

Christopher Reinhardt graduated from SUNY Buffalo in 2015 with a BSc in Chemistry and Biology. During this time, he conducted undergraduate research with Prof. John Richard. Chris is currently pursuing his PhD with Prof. Jeff Chan in the Department of Chemistry at UIUC as a CBI-TP trainee and Pines Graduate Fellow. His research interests include the development of ABS probes for nitric oxide and cyclooxygenase-2 activity.

Prof. Jeff Chan completed his PhD at Simon Fraser University with Prof. Andrew Bennet and received his postdoctoral training at UC Berkeley with Prof. Chris Chang. He began his independent career in 2014 at UIUC where he is currently an Assistant Professor of Chemistry. His research interests include developing acoustic-based imaging agents, chemical tools to study the role of reactive aldehydes in aging, and ABS probes for various sensing applications.

Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
References
- [1].Amyes TL, Richard JP, Biochemistry 2013, 52, 2021–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Radzicka A, Wolfenden R, Science 1995, 267, 90–93. [DOI] [PubMed] [Google Scholar]
- [3].Pawson T, Scott JD, Trends Biochem. Sci 2005, 30, 283–286. [DOI] [PubMed] [Google Scholar]
- [4].Beltrao P, Bork P, Krogan NJ, Van Noort V, Mol. Syst. Biol 2013, 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zotter A, Bäuerle F, Dey D, Kiss V, Schreiber G, J. Biol. Chem 2017, 292, 15838–15848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chan J, Dodani SC, Chang CJ, Nat. Chem 2012, 4, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Aron AT, Reeves AG, Chang CJ, Curr. Opin. Chem. Biol 2018, 43, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Baruch A, Jeffery DA, Bogyo M, Trends Cell Biol 2004, 14, 29–35. [DOI] [PubMed] [Google Scholar]
- [9].Rotman B, Zderic JA, Edelstein M, Proc. Natl. Acad. Sci. USA 1963, 50, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rotman B, Proc. Natl. Acad. Sci. USA 1961, 47, 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Morillas M, Goble ML, Virden R, Biochem. J 1999, 338, 235–239. [PMC free article] [PubMed] [Google Scholar]
- [12].Bier M, in Methods Enzym I, 1955, pp. 627–642. [Google Scholar]
- [13].Chyan W, Raines RT, ACS Chem. Biol 2018, 13, 1810–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang J, Chai X, He XP, Kim HJ, Yoon J, Tian H, Chem. Soc. Rev 2019, 48, 683–722. [DOI] [PubMed] [Google Scholar]
- [15].Bruemmer KJ, Crossley SWM, Chang CJ, Angew. Chemie Int. Ed 2020, 10.1002/anie.201909690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tomaiuolo M, Bertram R, Houle D, Evolution (N. Y) 2008, 62, 2884–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hempel J, Perozich J, Chapman T, Rose J, Boesch JS, Liu Z-J, Lindahl R, Wang B-C, in Enzymol. Mol. Biol. Carbonyl Metab 7, 1999, pp. 53–59. [DOI] [PubMed] [Google Scholar]
- [18].Tomita H, Tanaka K, Tanaka T, Hara A, Oncotarget 2016, 7, 11018–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, Smith C, Proc. Natl. Acad. Sci. U. S. A 1999, 96, 9118–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yagishita A, Ueno T, Esumi H, Saya H, Kaneko K, Tsuchihara K, Urano Y, Bioconjug. Chem 2017, 28, 302–306. [DOI] [PubMed] [Google Scholar]
- [21].Wang MF, Han CL, Yin SJ, Chem. Biol. Interact 2009, 178, 36–39. [DOI] [PubMed] [Google Scholar]
- [22].Cynthia A. Morgan, Thomas D.Hurley, Chem. Biol. Interact 2015, 234, 29–37.25450233 [Google Scholar]
- [23].Morgan CA, Hurley TD, J. Med. Chem 2015, 58, 1964–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Anorma C, Hedhli J, Bearrood TE, Pino NW, Gardner SH, Inaba H, Zhang P, Li Y, Feng D, Dibrell SE, et al. , ACS Cent. Sci 2018, 4, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bearrood TE, Aguirre-Figueroa G, Chan J, Bioconjug. Chem 2019, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hosokawa M, Molecules 2008, 13, 412–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang D, Zou L, Jin Q, Hou J, Ge G, Yang L, Acta Pharm. Sin. B 2018, 8, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pindel EV, Kedishvili NY, Abraham TL, Brzezinski MR, Zhang J, Dean RA, Bosron WF, J. Biol. Chem 1997, 272, 14769–14775. [DOI] [PubMed] [Google Scholar]
- [29].Feng L, Liu ZM, Xu L, Lv X, Ning J, Hou J, Ge GB, Cui JN, Yang L, Chem. Commun 2014, 50, 14519–14522. [DOI] [PubMed] [Google Scholar]
- [30].Feng L, Liu ZM, Hou J, Lv X, Ning J, Ge GB, Cui JN, Yang L, Biosens. Bioelectron 2015, 65, 9–15. [DOI] [PubMed] [Google Scholar]
- [31].Park SJ, Kim YJ, Kang JS, Kim IY, Choi KS, Kim HM, Anal. Chem 2018, 90, 9465–9471. [DOI] [PubMed] [Google Scholar]
- [32].Jin Q, Feng L, Wang DD, Wu JJ, Hou J, Dai ZR, Sun SG, Wang JY, Ge GB, Cui JN, et al. , Biosens. Bioelectron 2016, 83, 193–199. [DOI] [PubMed] [Google Scholar]
- [33].Wang DD, Jin Q, Zou LW, Hou J, Lv X, Lei W, Cheng HL, Ge GB, Yang L, Chem. Commun 2016, 52, 3183–3186. [DOI] [PubMed] [Google Scholar]
- [34].Liu ZM, Feng L, Ge GB, Lv X, Hou J, Cao YF, Cui JN, Yang L, Biosens. Bioelectron 2014, 57, 30–35. [DOI] [PubMed] [Google Scholar]
- [35].Kodani SD, Barthélemy M, Kamita SG, Hammock B, Morisseau C, Anal. Biochem 2017, 539, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tian Z, Ding L, Li K, Song Y, Dou T, Hou J, Tian X, Feng L, Ge G, Cui J, Anal. Chem 2019, 91, 5638–5645. [DOI] [PubMed] [Google Scholar]
- [37].Rouzer CA, Marnett LJ, J. Lipid Res 2009, 50, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kulmacz RJ, van der Donk WA, Tsai AL, Prog. Lipid Res 2003, 42, 377–404. [DOI] [PubMed] [Google Scholar]
- [39].Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA, J. Lipid Res 2009, 50, 423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Howe LR, Chang SH, Tolle KC, Dillon R, Young LJT, Cardiff RD, Newman RA, Yang P, Thaler HT, Muller WJ, et al. , Cancer Res 2005, 65, 10113–10119. [DOI] [PubMed] [Google Scholar]
- [41].Xu L, Stevens J, Hilton MB, Seaman S, Conrads TP, Veenstra TD, Logsdon D, Morris H, Swing DA, Patel NL, et al. , Sci. Transl. Med 2014, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Appleby SB, Ristimaki A, Neilson K, Narko K, Hla T, Biochem. J 1994, 302, 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McAdam BF, Mardini IA, Habib A, Burke A, Lawson JA, Kapoor S, FitzGerald GA, J. Clin. Invest 2000, 105, 1473–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].McCarthy TJ, Sheriff AU, Graneto MJ, Talley JJ, Welch MJ, J. Nucl. Med 2002, 43, 117–124. [PubMed] [Google Scholar]
- [45].Uddin MJ, Crews BC, Blobaum AL, Kingsley PJ, Gorden DL, McIntyre JO, Matrisian LM, Subbaramaiah K, Dannenberg AJ, Piston DW, et al. , Cancer Res 2010, 70, 3618–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang H, Fan J, Wang J, Zhang S, Dou B, Peng X, J. Am. Chem. Soc 2013, 135, 11663–11669. [DOI] [PubMed] [Google Scholar]
- [47].Yadav AK, Reinhardt CJ, Arango AS, Huff HC, Dong L, Malkowski MG, Das A, Tajkhorshid E, Chan J, Angew. Chemie Int. Ed 2020, 59, 3307–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Capdevila JH, Falck JR, Harris RC, J. Lipid Res 2000, 41, 163–181. [PubMed] [Google Scholar]
- [49].Meunier B, de Visser SP, Shaik S, Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- [50].Nebert DW, Dalton TP, Nat. Rev. Cancer 2006, 6, 947–960. [DOI] [PubMed] [Google Scholar]
- [51].Guengerich FP, Waterman MR, Egli M, Trends Pharmacol. Sci 2016, 37, 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bièche I, Narjoz C, Asselah T, Vacher S, Marcellin P, Lidereau R, Beaune P, De Waziers I, Pharmacogenet. Genomics 2007, 17, 731–742. [DOI] [PubMed] [Google Scholar]
- [53].Rendic S, Guengerich FP, Chem. Res. Toxicol 2012, 25, 1316–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shimada T, Yun CH, Yamazaki H, Gautier JC, Beaune PH, Guengerich FP, Mol. Pharmacol 1992, 41, 856–864. [PubMed] [Google Scholar]
- [55].Shimada T, Peter Guengerich F, Cancer Res 1991, 51, 5284–5291. [PubMed] [Google Scholar]
- [56].Rodriguez-Antona C, Ingelman-Sundberg M, Oncogene 2006, 25, 1679–1691. [DOI] [PubMed] [Google Scholar]
- [57].Dai ZR, Feng L, Jin Q, Cheng H, Li Y, Ning J, Yu Y, Ge GB, Cui JN, Yang L, Chem. Sci 2017, 8, 2795–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ji H, Zhang X, Dai Y, Xue T, Misal S, Qi Z, Analyst 2019, 144, 7390–7397. [DOI] [PubMed] [Google Scholar]
- [59].Ning J, Liu T, Dong P, Wang W, Ge G, Wang B, Yu Z, Shi L, Tian X, Huo X, et al. , J. Am. Chem. Soc 2019, 141, 1126–1134. [DOI] [PubMed] [Google Scholar]
- [60].Moskovitz J, Weissbach H, Brot N, Proc. Natl. Acad. Sci. U. S. A 1996, 93, 2095–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Moskovitz J, Biochim. Biophys. Acta - Proteins Proteomics 2005, 1703, 213–219. [DOI] [PubMed] [Google Scholar]
- [62].Jiang B, Moskovitz J, Antioxidants 2018, 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Brot N, Weissbach L, Werth J, Weissbach H, Proc. Natl. Acad. Sci. U. S. A 1981, 78, 2155–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lowther WT, Brot N, Weissbach H, Matthews BW, Biochemistry 2000, 39, 13307–13312. [DOI] [PubMed] [Google Scholar]
- [65].Makukhin N, Tretyachenko V, Moskovitz J, Míšek J, Angew. Chemie Int. Ed 2016, 128, 12919–12922. [DOI] [PubMed] [Google Scholar]
- [66].Gaweska H, Fitzpatrick PF, Biomol. Concepts 2011, 2, 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gorman AM, McGowan A, O’Neill C, Cotter T, J. Neurol. Sci 1996, 45–50. [DOI] [PubMed] [Google Scholar]
- [68].Heikkila RE, Manzino L, Cabbat FS, Duvoisin RC, Nature 1984, 311, 467–469. [DOI] [PubMed] [Google Scholar]
- [69].Albers AE, Rawls KA, Chang CJ, Chem. Commun 2007, 1, 4647–4649. [DOI] [PubMed] [Google Scholar]
- [70].Li L, Zhang CW, Chen GYJ, Zhu B, Chai C, Xu QH, Tan EK, Zhu Q, Lim KL, Yao SQ, Nat. Commun 2014, 5, 3276. [DOI] [PubMed] [Google Scholar]
- [71].Li L, Zhang C-W, Ge J, Qian L, Chai B-H, Zhu Q, Lee J-S, Lim K-L, Yao SQ, Angew. Chemie Int. Ed 2015, 54, 10821–10825. [DOI] [PubMed] [Google Scholar]
- [72].Wu X, Li L, Shi W, Gong Q, Li X, Ma H, Anal. Chem 2016, 88, 1440–1446. [DOI] [PubMed] [Google Scholar]
- [73].Sakagami H, Aoki J, Natori Y, Nishikawa K, Kakehi Y, Natori Y, Arai H, J. Biol. Chem 2005, 280, 23084–23093. [DOI] [PubMed] [Google Scholar]
- [74].Zalatan JG, Fenn TD, Brunger AT, Herschlag D, Biochemistry 2006, 45, 9788–9803. [DOI] [PubMed] [Google Scholar]
- [75].Kawaguchi M, Okabe T, Okudaira S, Hanaoka K, Fujikawa Y, Terai T, Komatsu T, Kojima H, Aoki J, Nagano T, J. Am. Chem. Soc 2011, 133, 12021–12030. [DOI] [PubMed] [Google Scholar]
- [76].Bollen M, Gijsbers R, Ceulemans H, Stalmans W, Stefan C, Crit. Rev. Biochem. Mol. Biol 2000, 35, 393–432. [DOI] [PubMed] [Google Scholar]
- [77].Shi Y, Cell 2009, 139, 468–484. [DOI] [PubMed] [Google Scholar]
- [78].Millward TA, Zolnierowicz S, Hemmings BA, Trends Biochem. Sci 1999, 24, 186–191. [DOI] [PubMed] [Google Scholar]
- [79].Virshup DM, Shenolikar S, Mol. Cell 2009, 33, 537–545. [DOI] [PubMed] [Google Scholar]
- [80].Stains CI, Luković E, Imperiali B, ACS Chem. Biol 2011, 6, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Beck JR, Truong T, Stains CI, ACS Chem. Biol 2016, 11, 3284–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Cravatt BF, Wright AT, Kozarich JW, Annu. Rev. Biochem 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- [83].Thompson LA, Ellman JA, Chem. Rev 1996, 96, 555–600. [DOI] [PubMed] [Google Scholar]
- [84].Congreve M, Chessari G, Tisi D, Woodhead AJ, J. Med. Chem 2008, 51, 3661–3680. [DOI] [PubMed] [Google Scholar]
- [85].Jorgensen WL, Science 2004, 303, 1813–1818. [DOI] [PubMed] [Google Scholar]
- [86].Rhee HW, Lee SW, Lee JS, Chang YT, Hong JI, ACS Comb. Sci 2013, 15, 483–490. [DOI] [PubMed] [Google Scholar]
- [87].Lee JS, Kang NY, Yun KK, Samanta A, Feng S, Hyeong KK, Vendrell M, Jung HP, Chang YT, J. Am. Chem. Soc 2009, 131, 10077–10082. [DOI] [PubMed] [Google Scholar]
- [88].Lee JS, Kim YK, Vendrell M, Chang YT, Mol. Biosyst 2009, 5, 411–421. [DOI] [PubMed] [Google Scholar]
- [89].Song L, Bachert C, Schjoldager KT, Clausen H, Linstedt AD, J. Biol. Chem 2014, 289, 30556–30566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K, Nat. Biotechnol 2003, 21, 86–89. [DOI] [PubMed] [Google Scholar]
- [91].Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. , ACS Chem. Biol 2008, 3, 373–382. [DOI] [PubMed] [Google Scholar]
- [92].Levi J, Kothapalli SR, Ma TJ, Hartman K, Khuri-Yakub BT, Gambhir SS, J. Am. Chem. Soc 2010, 132, 11264–11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yue X, Wang Z, Zhu L, Wang Y, Qian C, Ma Y, Kiesewetter DO, Niu G, Chen X, Mol. Pharm 2014, 11, 4208–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]