

Abstract
Mitogen activated protein kinases (MAPKs) control essential eukaryotic signaling pathways. While much has been learned about MAPK activation, much less is known about substrate recruitment and specificity. MAPK substrates may be other kinases that are crucial to promote a further diversification of the signaling outcomes. Here, we used a variety of molecular and cellular tools to investigate the recruitment of two substrate kinases, RSK1 and MK2, to three MAPKs (ERK2, p38α, ERK5). Unexpectedly, we identified that kinase heterodimers form structurally and functionally distinct complexes depending on the activation state of the MAPK. These may be incompatible with downstream signaling, but naturally they may also form structures that are compatible with the phosphorylation of the downstream kinase at the activation loop, or alternatively at other allosteric sites. Furthermore, we show that small molecule inhibitors may affect the quaternary arrangement of kinase heterodimers and thus influence downstream signaling in a specific manner.
Keywords: cellular signaling, MAP kinase, protein kinase complex, SAXS, X-ray crystallography, NMR
eTOC Blurb
Cellular signaling pathways depend on protein kinase cascades. Sok et al studied how MAP kinases bind and activate downstream substrate kinases. They found that the activation state of the upstream kinase affects the quaternary structure of the kinase heterodimer and small molecules can block the formation of specific heterodimers.
Graphical Abstract
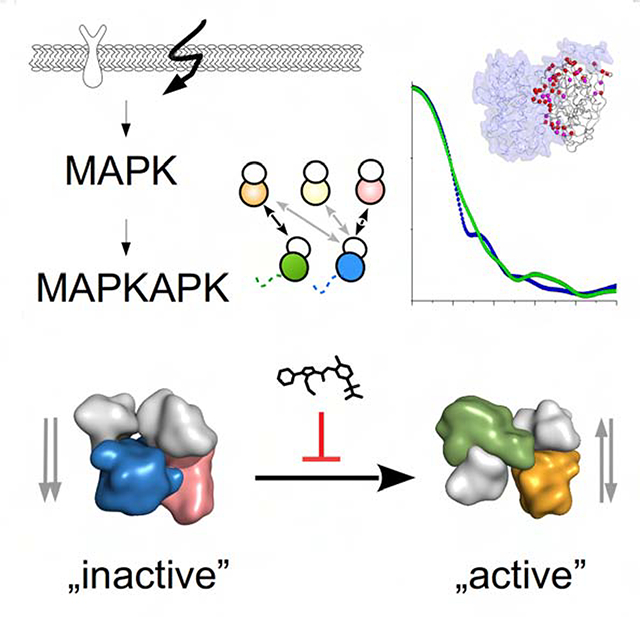
Introduction
Mitogen-activated protein kinases (MAPKs) are the essential components of hierarchically organized protein kinase cascades (Pearson et al., 2001; Cargnello and Roux, 2011). These cascades are critical for a large number of signaling events that drive all biological functions in higher eukaryotes. MAPKs are specifically activated by dual-specificity MAPK kinases (MKKs) that phosphorylate a threonine and a tyrosine residue on their MAPK activation loop (AL), which is commonly used as a measure of MAPK activation (Canagarajah et al., 1997). Interestingly, both activating MKKs and downstream substrates use linear binding motifs to anchor to the MAPKs. These sites have been referred to as the D(ocking)-motif (or Kinase interaction motif/KIM-motif) and the F-motif, both of which bind on their respective D/F-motif docking grooves on the MAPKs (Gavin and Nebreda, 1999; Smith et al., 2000; Lee et al., 2004; Zeke et al., 2015). While most substrate phosphorylation sites are part of intrinsically disordered regions (IDRs), some substrates are folded proteins. Indeed, one set of such substrates are MAPK activated protein kinases (Figure 1A). Specifically, it has been shown that the MAPKs ERK2 and p38α phosphorylate the kinases RSK1 or MK2, which have critical roles in the regulation of cell growth and apoptosis, respectively, and thus regulate central biological processes (Cargnello and Roux, 2011). How the activation of RSK1 and MK2 is achieved is currently not understood.
Figure 1. MAPKs and their substrate kinases, MAPKAPKs, form distinct heterodimers.

(A) Independently from phosphorylation state, all MAPK-MAPKAPK heterodimers depend on the D-motif mediated interaction between the disordered C-terminal MK2/RSK1 tail and the MAPK docking groove. Schematic of three MAPKs (ERK2, p38α and JNK1) and two MAPKAPKs (RSK1 and MK2) and their binding. Naturally, only double-phosphorylated (pp) MAPK binding will result in MAPKAPK activation. The binding of 4 nonphosphorylated (inactive) and 4 phosphorylated (active) MAPK-MAPKAPK heterodimers were studied and these were also structurally analyzed (see arrows). “BC” or “D” indicate that these complexes are shown in panels B/C or D with more structural details. How phosphorylated MAPKs form signaling competent MAPK-MAPKAPK heterodimers is not understood.
(B,C) Crystal structure of the nonphosphorylated p38α-MK2 (PDB ID: 2OZA; MK2 is shown in blue, p38α in salmon) and ERK2-RSK1 (PDB ID: 4NIF; RSK1 is shown in green, ERK2 in orange). The N-terminal kinase lobes are colored in white on panel C showing these two structures in a rough surface representation. Note that the N-to-C terminal direction of the interacting kinases is different: antiparallel vs parallel. Panel B: The MAPKAPK activation loop is at the other side of the MAPK in the parallel complex and it is inaccessible for the MAPK catalytic site. The C-terminal MAPK binding docking region (D-motif) and the activation loop (AL) of the kinases are shown in thicker ribbon and the latter is colored red. The catalytic sites (Asp150, Asp149 in p38α or ERK2, respectively) and the MAPK phosphorylation target sites on the AL of MK2 (Thr222) or on RSK1 (Thr573) are colored in magenta and shown in stick representation. N and C denote N-terminal and C-terminal lobes of the kinases, respectively. The boxed regions highlight the distinct kinase domain-domain contacts in the parallel (MK2 inhibitory helix, αINH, and p38 β3-αC loop) or in the antiparallel (RSK1 APE motif and ERK2 Gly-rich loop) crystallographic heterodimers, which are shown in a view 180°rotated on the right. In addition to the specific kinase domain-domain contacts, the intervening region connecting the D-motif and the MK2/RSK1 kinase domain (Linker) plays an important role in orienting the two D-motif tethered kinase domains.
(D) Model of the catalytic pp-ERK2-RSK1 complex (AL-T573). The flexible AL of RSK1 (colored in red containing the MAPK target site, Thr-573, shown with a green sphere) can reach to the pp-ERK2 active site where its phosphorylated activation loop, pp-AL, has a different conformation compared to the nonphosphorylated enzyme. The model was generated based on the antiparallel nonphosphorylated ERK2-RSK1 (PDB ID: 4NIF) and the pp-ERK2 (PDB ID: 2ERK) crystal structures using multidomain flexible docking combined with directed modeling of the RSK1 activation loop. The structure of the signaling competent pp-p38-MK2 heterodimer (MM-AL-T222) is enigmatic as the parallel arrangement is incompatible with AL phosphorylation.
(E) Calculated SAXS plots of p38α-MK2 (in blue) and ERK2-RSK1 (in green) complexes based on their crystal structures. Arrows show the N-to-C terminal direction of the kinases in the parallel vs antiparallel heterodimers. The active site on the MAPK is shown with an ellipsoid and * indicates the MAPK phosphorylation target site on the MAPKAPK in magenta. Because of their different shapes, the D-motif tethered antiparallel or parallel heterodimers have distinct scattering (I: intensity).
The structures of the nonphosphorylated p38α-MK2 and the ERK2-RSK1 complex have been previously determined. These crystal structures showed how a docking interaction between the D- motif in the C-terminal tail of MK2/RSK1 and the MAPK docking groove tethers the two kinase domains together through a short intervening region (Linker) that connects the D-motif to the kinase domain in MK2/RSK1 (Figure 1B) (White et al., 2007; ter Haar et al., 2007; Alexa et al., 2015). The ERK2-RSK1 complex adopted an antiparallel (head-to-toe) heterodimeric quaternary structure, where the activation loop of RSK1 may directly engage the ERK2 active site, explaining how phosphor-transfer can readily occur (Alexa et al., 2015; Turjanski et al., 2009; Ghose, 2019) (Figure 1C,D). Unexpectedly, the p38α-MK2 complex adopted a parallel (head-to-head) heterodimeric quaternary structure, which made it sterically impossible for the MK2 activation loop to reach the p38 active site (Figure 1C,D). This led to the idea that the adoption of specific quaternary structure might be important to understanding MAPK signaling specificity.
For this reason, we systematically investigated binding and activation of three MAPKs (ERK2, p38α, ERK5) with two MAPKAPKs (RSK1, MK2). In order to determine their solution heterodimer structure we used Small angle X-ray scattering (SAXS), which enables a structural understanding of dynamic ensembles of complexes, particularly when there are differences in the quaternary structure (Figure 1E) (Bernadó et al., 2007; Mertens and Svergun, 2010; Tsutakawa et al., 2007). This analysis was complemented by nuclear magnetic resonance (NMR) and X-ray crystallographic analysis on the p38-MK2 heterodimer in different MAPK phosphorylation states. We show that all MAPK-MAPKAPK pairs can form an antiparellel complex in which the phosphorylated MAPK is poised to phosphorylate the downstream kinase at its activation loop. Most critically our data shows that the p38-MK2 heterodimer is unique: it can form both antiparallel and parallel complexes. This unique behavior provides the structural basis for a recently discovered MK2 substrate specific p38 inhibitor (Cumming et al., 2015). Taken together, our data highlights how the quaternary structure of MAPK-substrate kinase complexes contributes to cognate signaling in ubiquitous MAP kinase signaling pathways.
Systematic characterization of MAPK-MAPKAPK complexes: ERK2 binds to both RSK1 and MK2, while p38α exclusively binds to MK2
MAPK-MAPKAPK binding engages the C-terminal disordered tail of the substrate kinase and the docking groove of the MAPK to form complexes with sub-micromolar binding affinity. The binding affinity of nonphosphorylated (np) or double-phosphorylated (pp) p38 to MK2 is similar (~5 or ~20 nM, respectively) (Figure S1). For the ERK2-RSK1 complex the binding affinity was earlier found to be ~100 nM (Alexa et al., 2015). RSK1 is a two kinase domain containing signaling protein and its C-terminal kinase (CTK) domain is phosphorylated by ERK2. This phosphorylation event at the activation loop of the CTK (Thr-573) fully activates the kinase (Dalby et al., 1998), which in turn will activate the N-terminal kinase (NTK) domain responsible for RSK substrate phosphorylation (Gógl et al., 2019). RSK1 refers to the CTK throughout the text.
In an active heterodimer the activation loop (AL) of the downstream kinase needs to reach the active site of the upstream kinase. Due to the lack of phosphorylated MAPK-MAPKAPK structures, how kinase domain-domain contacts form and in turn lead to signaling is not understood. The two nonphosphorylated crystal structures that are currently available (ERK2-RSK1, PDB ID: 4NIF; p38α-MK2, PDB ID: 2OZA) give only limited insights into MAPK mediated MAPKAPK activation, as these complexes do not contain active, phosphorylated MAPKs. Moreover, the two nonphosphorylated heterodimers are dramatically different: they show a parallel or an antiparallel quaternary structure (Figure 1B). Formation of the AL phosphorylation competent catalytic heterodimer is only possible in the antiparallel arrangement of the ERK2-RSK1 complex. In order to understand how the nonphosphorylated parallel p38-MK2 heterodimer may adopt a phosphorylated antiparallel complex, we wanted to structurally characterize ERK2-RSK1 and p38-MK2 complexes containing double-phosphorylated MAPKs. First, we set out to characterize protein-protein binding and signaling between three MAPKs (ERK2, p38α, ERK5) and two MAPKAPKs (MK2, RSK1) in order to be able to identify more MAPK-MAPKAPK heterodimers that are amenable for structural investigation (see Figure 1A).
We used a luciferase complementation assay to systematically explore the MAPK (p38α, ERK2, ERK5) binding specificity for RSK1 and MK2 in HEK293T cells (Figure 2A) (Dixon et al., 2016). Our data showed that p38α exclusively bound to MK2, while ERK2 and ERK5 bound to both RSK1 and MK2. RSK1 and MK2 have a well-known D-motif in their C-terminal tail (~20 amino acid long). As expected, recruitment of RSK1 and MK2 with MAPKs required the D-motif (Garai et al., 2012).
Figure 2. Complex formation between ERK2, p38α, ERK5 and MK2 or RSK1.

(A) Binding of MAPKs and MAPKAPKs was monitored by using the NanoBit luciferase complementation assay in HEK293T cells. MAPKAPK_dC constructs lack the C-terminal MAPK binding D-motif.
(B) Results of in vitro phosphorylation assays using recombinant expressed and purified kinases. MK2 and RSK1 phosphorylation by MAPKs were monitored by phosphorimaging of SDS-PAGE gels. Activated p38α, ERK2 and ERK5 (*) were made by adding their cognate constitutively active MAP2Ks (MKK6, MKK1 and MKK5, respectively) into the kinase reaction mix (5 μM MAPKAPK, 1 μM MAPK and 0.5 μM MAP2K). Note that despite that ERK5 and RSK1 showed some interaction on Panel A, ERK5 did not phosphorylate RSK1 but only MK2. Moreover, ERK5 could not be gel-filtrated together with RSK1 (see panel C), so this potential complex was not pursued further.
(C) Complex formation in vitro was monitored by size exclusion chromatography (SEC) using purified proteins. Eluted fractions were run on SDS-PAGE (MAPK alone on top, MAPKAPK alone in the middle and 1:1 mix of the two at the bottom). Gels show 0.5 ml fractions from 14 to 20 ml. See also Figure S1–3.
To test if RSK1 and MK2 recruitment also meant activation we used both in vitro and cellular assays. Based on in vitro kinase assays using purified proteins we demonstrated that ERK2 phosphorylates both RSK1 and MK2, while p38 and ERK5 are specific to MK2 (Figure 2B). Next, we tested the phosphorylation specificity of p38 and ERK2 in cells. We used “designer” HEK293T cells that allow for endogenous MAPKs to be selectively turned on (Figure S2) (Bach et al., 2007). All assays showed that ERK2 activated RSK1, while p38 activated MK2 (Figure 2B).
As we expected that heterodimer formation is important for MK2 and RSK1 activation, we tested complex formation between nonphosphorylated and phosphorylated (not shown) p38α, ERK2, ERK5 with RSK1 and MK2 by size-exclusion chromatography (SEC) (Figure 2C). SEC chromatograms showed that p38α-MK2, ERK2-MK2, ERK2-RSK1 and ERK5-MK2 form stable complexes in vitro (independent if nonphosphorylated or phosphorylated), which we used for further structural studies.
Structural analysis of MAPK-MAPKAPK heterodimers in solution: the quaternary structures of np- and pp-p38-MK2 complexes differ
Although we focused on p38-MK2, we also analyzed other MAPK-MAPKAPK heterodimers so that to be able to compare them and establish a more complete model of MAPK-MAPKAPK heterodimerization based on all structurally tractable complexes. We could create 8 complexes by either using nonphosphorylated or phosphorylated ERK2, p38α and ERK5 with RSK1 and MK2. The phosphorylated/activated forms of ERK2, p38α and ERK5 were prepared by phosphorylation with their respective MAP2Ks (MKK1, MKK6 and MKK5) (Figure S3). The MAPKs were mixed with RSK1 and MK2 and subjected to size exclusion chromatography in order to ensure homogeneous samples for the subsequent SAXS data collection and analysis.
To determine the correlation between our SAXS data and the two known crystal structures, we compared our experimental SAXS data with SAXS plots calculated using the ERK2-RSK1 or p38α-MK2 crystal structures (PDB ID: 4NIF and 2OZA, respectively; Figure 3A) (7–9), which showed good agreements. Next we compared the SAXS data for the nonphosphorylated or phosphorylated ERK2-RSK1 complex, which overlapped well (Figure 3B, left panel), showing that both complexes adopt similar structures in solution. This was in contrast with the nonphosphorylated or phosphorylated p38α-MK2 complex, which showed significant differences, indicating that their solution structures differ (Figure 3B, right panel).
Figure 3. Structures of nonphosphorylated and phosphorylated heterodimers.

(A) Fit between the experimental scattering plots (ERK2-RSK1 in green, p38-MK2 in blue) and the simulated curves calculated with nonphosphorylated ERK2-RSK1 and p38α-MK2 crystal structures (in brown).
(B) Experimental SAXS plots for nonphosphorylated (green or blue) and active (pp) complexes (orange or salmon).
(C) Summary of SAXS analysis on four nonphosphorylated and four phosphorylated (pp) MAPK-MAPKAPK heterodimers. Rg: radius of gyration; Dmax: maximum dimension; χ2 : discrepancy value for the fit to the simulated solution scattering curve calculated with the crystal structure (4NIF or 2OZA) or to models chosen from the MD ensemble (MD 4NIF or MD 2OZA). Quality of the fit is color-coded: red – bad (χ2 > 3); orange – moderate (χ2 = 2–3); green – good (χ2 < 2). MD models used for ensemble modeling are shown below. The starting model for the MD runs is shown on the left and the MD generated ensemble (150 structures) is on the right. * highlights that the nonphosphorylated p38-MK2 complex has a different quaternary structure compared to all other nonphosphorylated or phosphorylated MAPK-MAPKAPK heterodimers. The last 4 rows in the table are boxed and shown in gray and contain results for ERK2-MK2 and ERK5-MK2, which were not analyzed further in this study but the data were used for comparison to ERK-RSK1 and p38-MK2 (shown in the upper 4 rows). See also Figure S4,5.
Thus, we expanded our SAXS measurements to see if we can see these structural differences for other complexes (Figure 3C; Figure S4A). In order to allow for a more quantitative comparison, we calculated the discrepancy value (χ2) against the nonphosphorylated ERK2-RSK1 or p38α-MK2 crystal structures using CRYSOL (Svergun et al., 1995). A low χ2 (≤2) indicates that the crystallographic structures agree well with the solution structure, higher values indicate significant differences. Using this analysis it became evident that the SAXS profiles of ERK2-MK2 and ERK5-MK2 (independent if nonphosphorylated or phosphorylated) matched well to the antiparallel nonphosphorylated ERK2-RSK1 crystal structure.
To gain further insights into the structural differences between these complexes we generated free MD simulation runs (150 ns) using the crystal structures as starting models. Here we collected a single structure at each ns to generate 150 stereochemically feasible MAPK-MAPKAPK heterodimers. These models were used as starting models for the Ensemble Optimization Method (EOM) (Bernadó et al., 2007). Lastly, we used the Genetic Algorithm Judging Optimization of Ensembles (GAJOE) to select those MD models that best matched the experimental SAXS data (Figure S4B) (Franke et al., 2017). GAJOE selected 2–3 different MD models that likely represent the major conformers of these complexes in solution, with which the experimental SAXS plots fit with a criterion of χ2 ~ 1. Taken together, these data suggest that the MAPK-MAPKAPK complexes exist as heterodimeric ensembles in which complexes differ in their quaternary arrangements and the crystal structures represent one conformer of this ensemble.
NMR analysis on p38-MK2 complexes confirms the differences between np- and pp-p38-MK2
Our SAXS analysis showed that p38-MK2 complex can adopt a variety of confirmation depending of the phosphorylation state of the p38 activation loop. Thus, to gain additional insights into np- and pp-p38-MK2 complexes we used solution state biomolecular NMR spectroscopy. Due to the large molecular size of these complexes (~80 kDa) we used 2D [1H,15N] TROSY spectra and (2H,15N)-labeled np-p38 or (2H,15N)-labeled pp-p38 in complex with (2H)-labeled MK2. Since we have previously determined the sequence-specific backbone assignment of np/pp-p38, we were able to map the chemical shift perturbations (CSPs) to determine the solution complex interface on p38 (Figure 4A) (Kumar et al., 2018). The detected p38 CSPs included p38 residues belonging to the D-motif docking groove, showing that the D-motif of MK2 engages the p38 D-motif docking groove in a highly similar manner for np-p38 and pp-p38. Overall, most p38 CSPs agreed well with residues that are part of the protein-protein interface as described in the np-p38-MK2 crystal structure (Figure 4B). However, in the pp-p38-MK2 complex we were able to identify additional CSPs, particularly in p38 helix αc-sheet β4, helix αe and helix αf. These CSPs are likely due to indirect conformational changes, or as we have recently described, are indicative of allosteric changes characteristic to the assembly of the catalytic MAPK-substrate complex (Kumar et al., 2018).
Figure 4. NMR analysis on MK2 binding of nonphosphorylated (np) or phosphorylated (pp) p38α.

(A) Histograms showing the 1H/15N chemical shift perturbations (CSPs) upon MK2 binding vs residue number for np-p38 (left) and pp-p38 (right). Peaks that are broadened beyond detection are shown in brown. Dashed lines indicate CSPs corresponding to 1ϭ, 2ϭ or 3ϭ changes. Biggest changes map to the Gly-rich loop (a), the MAPK docking groove: hydrophobic (b) or the charged CD groove (c), and to the activation loop (d) for both complexes. Regions displaying the greatest CSP differences between the two complexes are boxed in green (e.g. β0a-β0b, αC-β4, αe, or αf).
(B) Backbone nitrogen atoms displaying line broadening (red: np-p38; blue: pp-p38) or CSPs above 2ϭ (magenta) are shown in the parallel np-p38-MK2 crystal structure (PDB ID: 2OZA; top) or in the antiparallel pp-p38-MK2 model (bottom) with spheres. MK2 is colored blue and shown in transparent surface representation. * indicates the position of the p38 catalytic site. ^ this antiparallel model was built by multi-domain flexible docking. D-motif: MK2 docking motif binding in the MAPK docking groove. AL: MK2 activation loop. Note the different AL conformation in the parallel or in the antiparallel complex. Panels on the right show the same structure as on the left but the view is 90° rotated so that to see p38 from the top.
Unique vs universal kinase regions govern parallel/antiparallel complex formation
To better understand the large conformational changes in the p38-MK2 complex in the different signaling stages, we carefully analyzed the amino acid interface between the two proteins. Indeed, the p38-MK2 crystal structure revealed negatively charged residues (Asp351, Glu354 and Glu355) in the so-called MK2 inhibitory helix, a structural element of MK2 that is N-terminal to the D-motif sequence, that are critical for binding (Figure 1B; Figure S5A). These residues form a strong salt-bridge with p38 residues Arg57. Next we tested if mutating these residues (MK2mut: D351A/E354R/E355R; 2RA) impact heterodimer formation (Figure S4C). Indeed, the p38α-mutMK2 complex only adopt an antiparallel complex as tested by SAXS (Figure S5B; table, row 1), in contrast to p38-MK2 which is parallel (see Figure 3C; table, row1). Conversely, when p38 Arg57 was changed to glutamate, the resulting p38mut(R57E)-MK2 complex can clearly adopt a variety of either parallel or antiparallel complexes (Figure S4D). This highlights that these residues, located in a unique MK2 region (inhibitory helix) and in a p38 specific loop (Figure S5A), have the ability to lock the parallel confirmation of the nonphosphorylated p38-MK2 complex.
Next, we performed a similar analysis for ERK2-RSK1. The ERK2-RSK1 crystal structure showed that domain-domain contacts form in this complex via strictly conserved kinase regions: RSK1 residues at the C-terminal end of helix F (αF) contain the conserved APE motif (Ala582, Pro583 and Glu584) that interacts with the Gly-rich loop residues of ERK2 (34GAYG37) (Figure 1B; Figure S5A). Changing the RSK1 APE motif to bulky arginine or glutamate residues (mutRSK1: A582R/P583E/E584R; RER) had been previously shown to impair ERK2 mediated RSK1 activation (Alexa et al., 2015), but globally SAXS data for pp-ERK2-mutRSK1 or for the wild-type complex did not differ (Figure S5B, Figure 3C), albeit ensemble modeling chose different models from the MD generated antiparallel set (not shown). This suggests that APE motif mutations cause only small level changes, nonetheless a properly aligned antiparallel pp-ERK2-RSK1 heterodimer is key to form an AL phosphorylation competent catalytic complex.
An MK2 substrate-specific p38 inhibitor converts the phosphorylated antiparallel heterodimer into parallel
A high-throughput screening (HTS) effort that was designed to specifically identify compounds that inhibited p38 mediated MK2 phosphorylation discovered a new class of inhibitors (Cumming et al., 2015). While general p38 inhibitors (e.g. SB202190) interfere with the activity of the kinase directly (inhibitors of catalysis, IoC) and thus affect all p38 substrates, the new compounds were selective only towards MK2 and did not block the phosphorylation of another p38 substrate kinase, MSK1. These compounds, termed as prevention of (MK2) activation (PoA) drugs, were suggested to have fewer side effects compared to broad-spectrum p38 inhibiting drugs that function as generic inhibitors of catalysis (IoC).
In order to further examine the mode of action of MK2 PoA inhibitors, we tested compound 12, hereinafter referred to as “PoA”, from (Cumming et al., 2015) in vitro and in cell-based kinase assays where phosphorylation of MK2 and other p38 substrates were simultaneously monitored (Figure 5A). This characterization showed that PoA, in contrast to an IoC inhibitor (SB202190), indeed specifically blocked MK2 activation. For example, when p38 activation is turned on in HT-M6 cells (engineered for p38 specific activation after adding doxycycline), ATF2, a downstream p38-regulated transcription factor (Raingeaud et al., 1995), and MK2 phosphorylation were both increased, but PoA inhibitor treatment left ATF2 phosphorylation intact, while MK2 phosphorylation was blocked. This was in sharp contrast to IoC/SB202190 treatment, which blocked ATF2 and MK2 phosphorylation. Furthermore, selective blocking of MK2 activation was also confirmed using in vitro kinase assays with purified proteins (Figure 5B).
Figure 5. An MK2 substrate-specific p38 inhibitor blocks antiparalell pp-p38-MK2 heterodimer formation.

(A) Inhibition of MK2 and ATF2 by PoA or a generic (IoC/SB202190) p38 inhibitor was tested using MK2 and ATF2 phospho-specific antibodies in Western-blots. p38 activation in HT-M6 cells was initiated by doxycycline (DOX) addition. MKK6EE-FLAG expression was monitored by using anti-FLAG antibody and concomitant p38 activation was confirmed by using a pp-p38 specific antibody. (Inhibitors were added in 1 μM concentration an hour ago before sample collection.)
(B) The impact of inhibitors (1μM) on p38 mediated substrate phosphorylation was tested in in vitro kinase assays. Phosphorylation of a reporter construct (2 μM) - containing a p38 binding docking motif (MEF2A) and a MAPK phosphorylation target site (Garai et al., 2012) - was compared to MK2 (2 μM) phosphorylation. Concentration of pp-p38 was 10 nM. Kinase reactions were initiated by the addition of ATP(γ)32P, reactions were stopped by the addition of loading buffer at different time points, samples then were loaded onto SDS-PAGE gels, and analyzed by phosphorimaging. Error bars show SD of the phosphorylation rates calculated based on three independent experiments.
(C) SAXS analysis of p38-MK2 complexes bound to the PoA inhibitor. SAXS data were collected on p38-MK2 and pp-p38-MK2 bound to PoA, and these were compared to binary complexes without the compound. The schematic below shows that PoA binding only affects the quaternary structure of the pp-p38-MK2 heterodimer. Inhibitor binding promotes the formation of the “inactive-like“ parallel complex from an antiparallel phosphorylated heterodimer, while the parallel nonphosphorylated p38-MK2 complex is unaffected. See also Figure S6.
The PoA inhibitor binds in the ATP binding pocket of p38 and its binding affinity to p38 alone is several hundred times weaker than binding to the nonphosphorylated p38-MK2 complex. However, the crystal structure of the np-p38-MK2-PoA ternary complex showed no structural differences when compared to the np-p38-MK2 complex. Although PoA inhibitors also bind to activated p38 (pp-p38), their impact on the active pp-p38-MK2 complex has not been addressed. Based on our earlier results that showed that the dominating structures of nonphosphorylated and phosphorylated p38-MK2 binary complexes differ in solution, we examined the effect of PoA on the assembly of the signaling competent pp-p38-MK2 heterodimer. SAXS analysis of pp-p38-MK2 and pp-p38-MK2-PoA samples showed that the solution structure of the inhibitor bound ternary complex markedly differs from the phosphorylated binary complex. While the latter fit to antiparallel, the inhibitor bound complex fit to parallel heterodimers, suggesting that inhibitor binding interferes with the formation of the AL phosphorylation competent antiparallel complex, providing the molecular basis for this class of specific p38 inhibitors (Figure 5C; Figure S6).
In order to elucidate the structure of the pp-p38-MK2-PoA ternary complex further, we crystallized this complex and determined its structure by X-ray crystallography to 3.7 Å resolution (Table 1; Figure S7A). The asymmetric unit contains four different pp-p38-MK2 heterodimers each binding a PoA inhibitor molecule and the four crystallographic models capture slightly different parallel arrangements of the kinase domains tethered by D-motif mediated docking. When the four heterodimers are superimposed on p38, the MK2 molecules are offset by 2–6 Å compared to the nonphosphorylated p38-MK2 crystal structure (Figure 6A). The four heterodimers differ similarly from each other and these differences match well to the scale of quaternary structural differences found between the structures of the MD ensemble (see Figure 3C). Since the crystallographic model of the nonphosphorylated p38-MK2-PoA ternary complex (PDB ID: 4TYH) contains only one PoA bound parallel heterodimer in the asymmetric unit (Cumming et al., 2015), flexibility in global structure seems to be a hallmark of the pp-p38-MK2 containing complex. The crystal structure of the new PoA bound complex is in excellent agreement with the results of SAXS experiments: solution scattering of the pp-p38-MK2-PoA showed an excellent fit to parallel MD ensemble structures (Figure S7B). The activation loop of p38 is markedly different in the pp-p38-MK2-PoA ternary complex compared to the nonphosphorylated p38-MK2-PoA ternary complex (PDB ID: 4TYH). The phosphoamino acids in the p38 AL are coordinated by arginine residues and make this region well-defined and greatly different from the nonphosphorylated AL (Figure 6B). AL conformation and contacts between phosphoamino acid and arginine residues are the same as in the homologous pp-ERK2 crystal structure, but surprisingly somewhat different compared to the apo pp-p38 crystal structure (Figure S7C) (Canagarajah et al., 1997; Zhang et al., 2011).
Table 1.
Crystallographic data collection and refinement statistics
| Data collection | |
| Space group | C 1 2 1 |
| a, b, c (Å) | 279.35 69.27 221.11 |
| α, β, γ (°) | 90.00 123.85 90.00 |
| Resolution range (Å) | 70.0 – 3.7 (3.86–3.70) |
| CC1/2 | 0.997 (0.841) |
| Rmerge† | 0.123 (0.725) |
| <I/ϭ(I)> | 6.4 (1.5) |
| Completeness (%) | 99.4 (99.6) |
| Redundancy | 3.8 (3.9) |
| No. reflections | 144336 (18144) |
| Refinement | |
| Rwork/ Rfree | 0.2514 /0.2972 |
| Number of atoms | 21798 |
| Protein | 21678 |
| Ligand/ion | 120 |
| B-factors (Å2) | 149.6 |
| Protein | 149.7 |
| Ligand | 123.6 |
| Ramachandran | |
| Favored (%) | 95.08 % |
| Allowed (%) | 4.69 % |
| Outliers (%) | 0.23 % |
| Rotamer outliers (%) | 0.09 % |
| R.m.s deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.625 |
Rmerge = ΣhklΣi |Ii(hkl)-<I(hkl)>|/ΣhklΣi Ii(hkl); Data for the highest resolution bin is shown in brackets.
Figure 6. Crystal structure of the pp-p38-MK2-PoA ternary complex.

(A) Comparison of the nonphosphorylated p38-MK2-PoA (PDB ID: 4TYH) and the pp-p38-MK2-PoA crystal structures. The four heterodimers from the pp-p38 complex were superimposed through the MAPK (colored in salmon) and MK2 molecules are colored in blue (panel on the left corner). One of the heterodimers from this complex was similarly superimposed to the nonphosphorylated heterodimer (main panel). The PoA compound and the p38 catalytic site (Asp150) is colored in yellow, and the flexible loop region containing the Ser272 regulatory site in MK2 is colored red. Dotted lines indicate disordered regions. The inset on the right shows a zoomed-in view of the Docking motif-Linker-Inhibitory helix region of MK2, but the view is 180º vertically rotated compared to the other panels. Key contact residues, responsible for high affinity p38-MK2 binding are colored yellow (e.g. Ile730, Ile732, Ile375, Leu382 and Arg386). In addition, a pivotal residue in the Linker connecting the docking motif and the inhibitory helix is also highlighted (Gln269). This region serves as a pivot through which the kinase domains may bind differently but still held together similarly by the D-motif mediated interaction. Notice that the p38 Hinge (connecting the N- and C-lobes of the MAPK) and the MK2 Linker both contribute to PoA inhibitor binding.
(B) Structural comparison of the nonphosphorylated vs phosphorylated p38 activation loop. Panels on the top or below show a zoomed-in view around the p38 catalytic site (Asp150) from the nonphosphorylated p38-MK2-PoA ternary complex (PDB ID: 4TYH) or from the activated pp-p38-MK2-PoA complex, respectively. Notice that the nonphosphorylated p38 activation loop is wedged in-between the MK2 inhibitory helix and the structured part of Loop-S272. The activation loop is fixed in this inhibitory position by several H-bonds (shown with dashed lines). The activation loop of p38 between the DFG loop and the APE motif are colored in magenta. Disordered segments are indicated with a dotted line. Phosphates of the double-phosphorylated p38 AL (p-Thr180 and p-Tyr182) are coordinated by arginines (Arg149, Arg173, Arg186 and Arg189), which pulls the AL down and opens up the catalytic site. See also Figure S7.
Overall, SAXS and crystallographic structural analysis suggest that PoA binding affects the quaternary structure and/or dynamics of the pp-p38-MK2 heterodimer differently compared to the nonphosphorylated complex, allowing for the high specificity of the PoA inhibitors.
Parallel vs antiparallel models of pp-p38-MK2: phosphorylation of auxiliary sites depends on a parallel heterodimer
Our SAXS analysis showed that the dominating conformer of the pp-p38-MK2 complex is antiparallel, which is compatible with AL phosphorylation. In contrast to RSK1 that is known to be phosphorylated only on its AL (Thr573) and then it becomes fully activated, MK2 apart from AL phosphorylation (Thr-222) also depends on the phosphorylation of either of two additional sites to get fully activated: Ser-272 or Thr-334 (Figure 7A) (Ben-Levy et al., 1995; Engel et al., 1995). Phosphorylation of these auxiliary sites cannot happen in the antiparallel heterodimer, since these sites cannot reach to the p38 active site (HRD motif). Interestingly, the pp-p38-MK2-PoA ternary complex crystal structure captured an “active” parallel heterodimer in which phosphorylation of Ser-272 is feasible: the pp-p38 active site is open and the flexible MK2 loop containing Ser-272 can be docked into the pp-p38 substrate binding pocket. This latter model provides the structural basis on how alternative regulatory sites on MK2, apart from the AL, can get phosphorylated (Figure 7B). This model postulates that Ser-272 phosphorylation is impaired if parallel pp-p38-MK2 heterodimer formation were blocked. The earlier described mutMK2 protein has its parallel heterodimer-specific protein-protein contacts disrupted and thus forms an antiparallel complex (see Figure S5). In excellent agreement to our model, Ser-272 (or Thr-334, Figure S8A,B) phosphorylation in mutMK2 is greatly diminished compared to wild-type MK2 (Figure 7C; Figure S8C).
Fig. 7. Phosphorylation of MK2 at different sites requires the formation of different pp-p38-MK2 heterodimers.

(A) Schematic of MK2 regulatory sites and their 3D location (lower panel) relative to the p38 catalytic site in different p38-MK2 heterodimers. The structural panel below shows the position of the three phosphorylation sites (Thr222, Ser272 and Thr334) in the parallel nonphosphorylated p38-MK2 heterodimer. The catalytic HRD region is colored red and the inhibitory helix is magenta. NES: nuclear export signal.
(B) Structural models of the antiparallel and parallel catalytic pp-p38-MK2 heterodimers (AL-T222 or S272, respectively; * indicates complexes with pp-p38). The models were generated using multidomain flexible docking by starting out from the nonphosphorylated parallel p38-MK2 or from the new parallel crystal structure of the pp-p38-MK2-PoA complex. The MK2 activation loop (AL) or Loop-272 (colored in red) were docked into the pp-p38 active site by directed docking. Note that the nonphosphorylated (see Panel A) and the phosphorylated parallel p38-MK2 heterodimers have the same global quaternary structure, but these parallel complexes differ locally at the p38 active site: the nonphosphorylated p38 AL blocks while phosphorylated p38 AL allows access for Ser-272 to the active site (compare the upper and lower panels on Figure 6B).
(C) Phosphorylation of Ser272 in MK2 vs mutMK2 by pp-p38. The panel shows the results of in vitro kinase assays in which the relative phosphorylation of Ser272 vs Thr222 was monitored by using phosphosite-specific antibodies (Figure S8C). See also Figure S8.
Next, we examined if there is hierarchy in MK2 phosphorylation at different sites. MK2 was phosphorylated by pp-p38 in vitro and the kinetics of phosphorylation at these sites were monitored by using MK2 phosphosite-specific antibodies. This experiment showed that Thr-222 phosphorylation happens ahead of Ser-272/Thr-334 phosphorylation (Figure S8D). This finding supports a model where AL phosphorylation is the gate-keeper for MK2 auxiliary site phosphorylation: pp-p38 forms a universal antiparallel heterodimer so that to be able to activate MK2 at its activation loop, and then the complex, possibly through a distributive process, forms an active parallel heterodimer in which Ser-272/Thr-334 may get phosphorylated.
Discussion
Considerable molecular and cellular data have been reported that enables us to understand the activation and regulation of kinases, which are critical for virtually all cellular signal transduction. However, much less is known how kinases recruit their substrates. While over 10,000 kinase substrate have been reported, it has become apparent that kinase active site recognition sequences cannot alone account towards the specificity of kinases (Miller and Turk, 2018).
Thus, here we set out to understand how MAPKs recruit and activate their specific substrates, the MAPK activated protein kinases RSK1 and MK2. While crystal structures of these complexes have been reported – they did not provide an understanding on how RSK1 and MK2 were activated and how they function. To understand this substrate interaction is important for multiple reasons: 1) as kinase:kinase interaction/activation are critical for many signaling events and thus a detailed molecular understanding can lead to further biological insights; 2) MK2 and RSK1 have critical roles in a number of key biological processes and 3) a highly specific inhibitor has been reported for the p38:MK2 interaction – but without understanding its mode of action the inhibitor cannot be optimized nor can this approach be transferred to other kinase:kinase interaction modules.
We show that the interaction of p38/ERK2/ERK5 are mediated via multiple interactions, including D-motif mediated binding, but more importantly interactions that regulate the relative orientation of the heterodimer – either parallel or antiparallel. Indeed, our molecular and cellular data show that AL phosphorylation for example requires antiparallel heterodimers. Furthermore, we also demonstrated that MAPK-MAPKAPK heterodimers exist as dynamic ensembles in solution, while former crystal structures of nonphosphorylated p38-MK2 and ERK2-RSK1 heterodimers captured only two distinct states. Despite the inherently low resolution of SAXS based structural analysis, it is complementary to X-ray crystallography that can give only static structures. We postulate that both nonphosphorylated and phosphorylated heterodimers display similar D-motif mediated binding but kinase domain-domain contacts differ.
MKK3/6 phosphorylate p38 at its activation loop. Interestingly, MKK3/6 and MK2 binding to p38 are mutually exclusive: they both depend on the MAPK docking groove (Garai et al, 2012). Because of this, it is unlikely that MK2 phosphorylation by pp-p38 proceeds within an MKK3/6-p38-MK2 ternary complex (Gaestel, 2016). It is more likely that p38 forms highly dynamic binary complexes with its substrate (MK2) or with its activator (MKK3/6), and depending on the phosphorylation state of p38 some of these complexes are catalytically productive or unproductive. Despite the strong steady state binding affinity of the p38-MK2 complex (KD ~5 nM), fast dissociation (high koff) allows efficient turnover of the enzymatic reactions (see Figure S1).
It is easy to understand why the pp-p38-MK2 complex needs to form the antiparallel heterodimer based on biochemical logic, namely for MK2 AL phosphorylation. However, it is enigmatic why p38 and MK2 form a parallel heterodimer that is clearly incompatible with AL phosphorylation. We postulate that the parallel complex may have two distinct functions, one is related to nonphosphorylated and the other to phosphorylated p38: 1) On MK2 the nuclear exit signal (NES) is located on the solvent exposed side of the inhibitory helix and binding to nonphosphorylated p38 masks this and keeps the complex inside the nucleus. In agreement to this, it had been reported that protein levels of p38 and MK2 are dependent on each other and nucleocytoplamic shuttling of MK2 also depends on its complex formation with p38 (Kotlyarov et al., 2002; Ronkina et al., 2007; Meng et al., 2002; Engel et al., 1998; Ben-Levy et al., 1998). The nonphosphorylated parallel p38-MK2 heterodimer is a stable decoy complex and is important for the sequestration of nonphosphorylated p38. 2) When MK2 binds phosphorylated p38, Ser-272 and/or Thr-334 on MK2 also become phosphorylated, which further increases its enzymatic activity (Ben-Levy et al., 1995). Phosphorylation at these auxiliary sites cannot happen in the AL phosphorylation competent antiparallel arrangement and it requires a parallel one. It is interesting to note that the nonphosphorylated p38-MK2 decoy complex and the alternative phosphorylated heterodimer share a related global quaternary structure (parallel). This suggests that the capacity to form a nonphosphorylated parallel decoy complex and the capacity to regulate additional regulatory sites on the substrate kinase domain may be structurally linked. It is interesting to note that regions responsible for parallel p38-MK2 heterodimerization are strictly conserved in metazoans among p38/MK2 orthologs but clearly distinct from other MAPK/MAPKAPK paralogs (see Figure S5A). Functionally, the parallel p38-MK2 heterodimer interface may have co-evolved in these two proteins to acquire new complex-specific signaling functions (Caffrey et al., 1999). This sets it apart from the ERK2-MK2 complex for example that can only make an antiparallel heterodimer (Stokoe et al., 1992; Coxon et al., 2003). In contrast to this, regions responsible for antiparallel heterodimerization and required for universal AL phosphorylation are universally conserved basically in all protein kinases (Gly-rich loop and the APE motif) (Huse and Kuriyan, 2002).
Here we confirmed that an MK2 substrate-specific p38 inhibitor (PoA) indeed had the unique capacity to inhibit p38 to signal towards MK2, but not to other differentiation or development related p38 substrates such as ATF2 or MEF2A transcription factors (Yang et al., 1999). We demonstrated that PoA interferes with antiparallel heterodimer formation and the mechanistic basis of this is the following. The active pp-p38-MK2 heterodimer is dynamic and form antiparallel and parallel structures – since full MK2 activation requires auxiliary site phosphorylation in addition to AL phosphorylation. In contrast to nonphosphorylated p38, pp-p38 can make an antiparallel heterodimer that seems to be the dominating quaternary conformer in solution and where pp-p38 is poised to phosphorylate MK2 on its AL. Crystal structures of PoA-bound p38-MK2 heterodimers revealed that the phenyl group of this compound fits into a pocket formed by the Hinge of p38, connecting the N- and C-lobe, and the Linker of MK2, connecting the C-terminal D-motif and the MK2 kinase domain. We postulate that PoA binding favors the parallel heterodimer-specific arrangement between the p38 Hinge and the MK2 Linker (see Figure 6A). Although binding of the C-terminal MK2 D-motif to p38 is the same in both heterodimers, the conformation of the Linker must be different in the two complexes. PoA binding favors the formation of parallel heterodimers, and thus indirectly prevents MK2 AL phosphorylation occurring in the antiparallel complex.
MAPKs are activated by their respective MAPK kinases (MAP2K) at one level higher in the signaling cascade. As all MAP2Ks have a D-motif required to bind MAPKs, MAP2K-MAPK heterodimerization likely proceeds similar to what described in this study for MAPK-MAPKAPK heterodimers (Garai et al., 2012; Zeke et al., 2015). Moreover, because of the structural similarity of protein kinase domain cores and the universal dependence of individual kinase domain activities on AL phosphorylation, other kinase cascade heterodimers apart from MAPKs may also be similar.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Attila Reményi (remenyi.attila@ttk.hu).
Materials Availability
Plasmids, cells and materials generated in this study are available upon request.
Data and Code Availability
The Protein Data Bank accession number is 6TCA for the atomic coordinates and structure factors of the pp-p38-MK2-PoA complex. SAXS data is available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains
Insect cell expression strains
MKK5DD was expressed in SF9 cells.
Human HEK293T Cells
Cell line based measurements were carried out in female HEK293T cells cultured in DMEM with 10% FBS.
METHOD DETAILS
Protein constructs, expression and purification
All proteins were from human. For MK2, which contains only one CAMK-type kinase domain and a C-terminal MAPK binding linear motif, an N-terminally truncated construct (41–400) was expressed. For RSK1, an N-terminally truncated construct lacking the AGC kinase domain was used (411–735) (Alexa et al., 2015). Full-length ERK2, p38α and a MAPK domain containing ERK5 construct was expressed in E. coli using standard procedures as earlier described (Garai et al., 2012; Glatz et al., 2013). MK2 and RSK1 were expressed in bacteria as GST fusion proteins with an N-terminal GST and C-terminal hexa-histidine tag and double-affinity purified (on Ni-NTA and glutathione resin). After cleaving the GST-tag by the tobacco etch virus (TEV) protease, RSK1 was further purified on a HiTrap Blue-Sepharose column (GE Healthcare). MK2 and RSK1 mutants were purified the same way. Phosphorylated ERK2 and p38 were produced by co-expressing them with constitutively active GST-tagged MAP2Ks. Phosphorylated ERK5 (1–431) was prepared by incubating the bacterially expressed protein with active MKK5 - produced in SF9 cells – and with ATP in vitro. The MKK5 construct containing the kinase domain was expressed as an MBP fusion protein in SF9 cells with the Bac-to-Bac baculoviral expression system (Invitrogen). The SF9 cell lysate was double-affinity purified with Ni-NTA and maltose resin. Phosphorylated MK2 was produced by activating it with pp-p38 in vitro. Phosho-MK2 was further purified on a Resource S column. Mass spectometry analysis of these samples confirmed phosphorylation of these sites in our hands, too (Figure S3C). Biotinylated p38, ERK2 and pp-p38 for the SPR experiments were expressed with N-terminal AviTag and co-expressed with the BirA ligase in E. coli (Fairhead and Howarth, 2015), and purified similarly to other MAPK samples. For the analytical size-exclusion chromatography experiments, purified MAPK and MAPKAPK samples were mixed in a 1:1 ratio and gel-filtrated on a Superdex 200 10/300 (GE Healthcare) column; 0.5 ml fractions were collected and run on SDS-PAGE.
Protein sample preparation for SAXS, data analysis, and modeling
Proteins were mixed in a 1:1 stoichiometric ratio and gel-filtrated on a Superdex 200 Hiload 16/600 (GE Healthcare) column, concentrated and dialyzed in 20mM Tris-HCL pH 8.0, 150mM NaCl, 1mM DTT, 1mM TCEP, 10% glycerol using small volume dialysis buttons. In order to preclude buffer scattering subtraction problems, equilibrated dialysis buffer was used as the control buffer in the SAXS measurement as well as in the dilution series. Control buffers were always handled parallel to protein samples (e.g. freezing and thawing the same time), as this was important for best buffer correction for good SAXS measurements. The PoA inhibitor (100 μM dissolved in DMSO), DMSO (2%) or AMPPNP (1 mM) were dialyzed into concentrated protein samples using dialysis buttons, where compounds were added into the dialysis buffer (~10 ml). The equilibrated buffers were used for making the corresponding dilution series and for buffer correction. Protein complexes for SAXS were analyzed by Western blotting using pp-ERK2 antibody (Phospho-p44/42 MAPK T202/Y204; Cell Signaling #9101), pp-p38 antibody (Phospho-p38 MAPK T180/Y182; Cell Signaling #9215), pp-ERK5 antibody (Phospho-Erk5 Thr218/Tyr220; Cell Signaling #3371), phospho-MK2 antibody (Phospho-MAPKAPK-2 T222 Rabbit; Cell Signaling #3316S), and phospho-RSK1 antibody (phospho-p90RSK Thr573; Cell Signaling #9346). The phosphorylation state of each samples were analyzed by mass spectrometry. The mass measurement of intact proteins was performed by HPLC-MS using a Q-TOF Premier mass spectrometer in positive electrospray ionization mode coupled to an Acquity UPLC System (Waters Corporation, Milford, MA, USA). To determine the site of phosphorylation, trypsin digested proteins were analyzed by nanoUHPLC/MS/MS using a Bruker Maxis II ETD Q-TOF (Bremen, Germany) mass spectrometer with CaptiveSpray nanoBooster ionization source coupled to a Dionex Ultimate 3000 NanoLC System (Sunnyvale, CA, USA). SAXS measurements were performed at the BM29 beamline at ESRF or at the P12 beamline at EMBL-Hamburg (PETRA). Data were analyzed using the ATSAS program package (Franke et al., 2017). Primary data analysis was performed in PRIMUS. To minimize the inter-particle effect on the scattering curve, a dilution series on concentrated stoichiometric complexes from ~10–15 to ~0.2 mg/ml were measured. Only minor concentration effect was observed during the measurement that was excluded by manual merging (Figure S4A). Fit of the crystal structures were determined by CRYSOL. Ensemble modeling was done with GAJOE using the externally generated MD structural pools, which were generated as described previously (Alexa et al., 2015).
Protein crystallization and X-ray structure determination
The gel-filtrated pp-p38-MK2 complex was concentrated to 11 mg/ml. Commercial and in-house custom crystallization trials gave no results with this complex. Next, samples were mixed with 250 μM PoA inhibitor. Crystallization was carried out in hanging drop diffusion setups having 1.0 M (NH4)2SO4 in the reservoir. Crystals finally grew in 0.1M HEPES pH 7.3, 3.5 % PEG 8000, 1% MPD and 1% DMSO. Crystals were flash cooled in liquid nitrogen after supplementing the drop with 25% glycerol as cryoprotectant. Data were collected at PETRA III beam lines, Hamburg. The crystal structure of the pp-p38-MK2-PoA complex was solved by molecular replacement using PHASER (McCoy et al., 2007). The p38 search model contained the full polypeptide chain of the MAPK from the p38-pepMKK6 protein-peptide complex (PDB ID: 2YO8), while the MK2 search model was from the nonphosphorylated p38-MK2 complex (PDB ID: 2OZA) but lacked the C-terminal docking motif region (365–400) and the activation loop (219–236). The MR solution contains four different p38-MK2 binary complexes (8 polypeptide chains) bound via the C-terminal MK2 docking motif region. The MK2 activation loop, the phosphorylated p38 activation loop, the C-terminal MK2 docking motif region and PoA inhibitors were manually built in Coot (Emsley et al., 2010). Structure refinement was done using PHENIX with NCS restraints for corresponding polypeptides (Adams et al., 2011). All four binary pp-p38-MK2 complexes contain a PoA inhibitor located in the p38 nucleotide binding pocket and the p38 activation loop is well-defined and shows clear electron density for the phospho-tyrosine (Tyr182) and phospho-threonine (Thr180) residues (Figure S7A). Details of the structure solution and refinement are found in Table 1.
NMR experiments
Expression and purification of human p38 was performed as described earlier (Kumar et al., 2018). For NMR measurements, expression of uniformly [2H,15N]-labeled p38 and [2H]-labeled MK2 was achieved by growing cells in D2O based M9 minimal media containing 1 g/L 15NH4Cl and 14NH4Cl, respectively, and D-glucose as the sole nitrogen and carbon sources. Multiple rounds (0%, 30%, 50%, 70% and 100%) of D2O adaptation was necessary for high-yield expression. The interaction between nonphosphorylated/phosphorylated p38 and MK2 was studied by direct comparison of 2D [1H,15N] TROSY spectra of free and [2H]-MK2-bound [2H,15N]-labeled np-/ppp38. The final concentration used was 0.05–0.1 mM for the np-/pp-p38:MK2 complex in 10 mM HEPES pH 7.4, 0.15 M NaCl, 5 mM DTT and 90% H2O/10% D2O. The spectra was processed using Topspin 4.0.6 and analyzed using NMRFAM-SPARKY. The NMR spectra were acquired on a Bruker Avance NEO 600 MHz 1H Larmor frequency NMR spectrometer equipped with a TCI-active HCN cooled z-gradient cryoprobe at 308K.
Modeling by CORAL and HADDOCK
Rigid body modeling was done with CORAL (Franke et al., 2017). Here the MAPK domain bound to the C-terminal docking motif of the MAPKAPK and the kinase domain of the MAPKAPK were treated as two rigid bodies. The Linker between the D-motif motif and the kinase domain, the terminal elements and the relative orientation of the kinase domains were allowed to change freely.
The antiparallel pp-p38-MK2 model (AL-T222) was generated by applying multidomain flexible docking with HADDOCK2.2 on one of the parallel heterodimers from the pp-p38-MK2-PoA crystal structure (Karaca and Bonvin, 2011). The extended MK2 activation loop from this structural template was remodeled based on the MAPKAPK activation loop from the ERK2-RSK1 (PDB ID: 4NIF) crystal structure, the MK2 Linker was defined as fully flexible and the MK2 APE motif region was restrained to be close to the MAPK Gly-rich loop as in the ERK2-RSK1 crystal structure: the distance between Cα of Pro232(MK2) and Tyr35(p38) was set to ~5Å. The HADDOCK models were re-ranked according to the SAXS data; the final model had a χ2 value of 1.1. The model of the parallel pp-p38-MK2 complex (S72) was generated by completing the unstructured part of Loop-272 in the pp-p38-MK2-PoA crystal structure. The model for the antiparallel pp-ERK2-RSK1 complex (AL-T573) was made by superimposing pp-ERK2 (PDB: 2ERK) on the MAPK of the nonphosphorylated ERK-RSK1 crystal structure (PDB ID:4NIF) and the short unstructured part of RSK1 AL was completed. After energy minimization, models were submitted to directed docking to model MAPKAPK activation loop confirmation if it were to bind at the MAPK catalytic site (van Zundert et al., 2016; Piserchio et al., 2017). The quaternary arrangement was allowed to change only minimally, but the RSK1 or MK2 AL or Loop-272 from MK2 were flexibly docked by applying restraints on the distance of the MAPK phosphorylation target site (Thr573 in RSK1 and Thr222 or Ser272 in MK2) and the catalytic residue (ERK2: Asp149 or p38: Asp150) based on the crystal structure of the DYRK1A-substrate peptide complex (PDB ID: 2WO6) (Alexa et al., 2015; Soundararajan et al., 2013).
Protein kinase assays
Radioactivity based kinase reactions were carried out in 50 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 0.05% IGEPAL, 5% glycerol, 2 mM DTT using recombinant expressed and purified proteins in the presence of 400 μM ATP and ~5 μCi of [γ−32P]ATP. Reactions were stopped with protein loading sample buffer complemented with 20 mM EDTA, boiled, and then subjected to SDS-PAGE. Gels were dried before phosphorimaging by a Typhoon Trio+ scanner (GE Healthcare). Western-blot based reactions were carried out in the same buffer but using 1 mM ATP. Western-blot results were analyzed using Odyssey CLx imaging system (Li-Cor) and fluorescently labeled secondary antibodies (IRDye 680 RD goat anti-Rabbit 925–32211, IRDye 800 CW goat anti-Rabbit 925–32211, or IRDye 680 RD goat anti-Mouse 925–68070; Li-Cor). FLAG tagged protein, total p38, MK2 or ATF2 phosphorylation levels were monitored by using the following antibodies: anti-FLAG (Sigma #F1804), anti-p38 (Cell Signaling #9228), phosphoMK2 (Phospho-MAPKAPK-2 T222 Rabbit, Cell signaling #3316S; Phospho-MAPKAPK-2 (Thr334), Cell Signaling #3007; or Phospho-MAPKAPK2(Ser272), Invitrogen PAS-39792), and phosphoATF2 (T69/T71, Cell Signaling #9225). Western-blots based on ECL detection were analyzed on a Fluorchem FC2 camera (Cell Bioscience) using anti-rabbit IgG HRP-linked antibody (Cell Signaling #7074S) and anti-mouse IgG (Millipore #401215). SB202190 generic p38 inhibitor was purchased from Sigma (# S7067), and the PoA inhibitor ((N-[5-(Dimethylsulfamoyl)-2-methylphenyl]-1-phenyl-5-propyl-1H-pyrazole-4-carboxamide); compound 12 from (Cumming et al., 2015) was synthesized as described.
Protein-protein interaction assays
For surface plasmon resonance (SPR) measurements, biotinylated ERK2, ERK5, p38 and pp-p38 were captured on a Biacore CAP sensor chip using a Biacore S200 instrument (GE-Healthcare). All measurements were done at room temperature using single-cycle setup with the standard Biacore method for CAP chip including double referencing. MK2 binding was measured with a 3X dilution series with two replicates. Sensorgrams and steady state affinity binding fits were processed with the BiaEvaluation software.
For luciferase complementation NanoBiT assays in live cells, HEK293T cells were transfected with Lbit and Sbit containing plasmids using Lipofectamin 2000 in DMEM. cDNAs were sub-cloned into Lbit and Sbit expression vectors: MAP kinase constructs were expressed (ERK2, ERK5 and p38) as N- or C-terminal Lbit fusions and MAPKAP kinases (MK2 and the C terminal kinase domain of RSK1) as C-terminal or N-terminal Sbit fusion proteins. Cells were serum-starved for 20 hrs and luciferase activity was measured (for 10–15 minutes) in 96 well plates (Greiner 657160) in a luminescence plate reader (Cytation 3, BioTek) after the addition of 10 μM Coelenterazine h.
Cell Culture
For making the HT-M cell lines, HEK293T cells were transfected with pEBTetD vectors containing constitutively activated versions of MAP2Ks with phosphor-mimicking activation loop residues: MKK1EE, MKK6EE, MKK7EE, MKK5DD, which turn on ERK1/2, p38, JNK and ERK5, respectively (Bach et al., 2007). HEK293T Tet-on (HT) stable cell lines (HT-M1, HT-M6, HT-M7 and HT-M5) were established by keeping the cells under puromycine for at least one week, then expression of FLAG-tagged MAP2Ks and concomitant specific MAPK activation were monitored by Western-blots after doxycycline (DOX) treatment (2 μg/mL) in DMEM containing 10 % FBS. Cells were transfected with Lipofectamine 2000 reagent and were kept under puromycine (5 μg/mL) selection.
QUANTIFICATION AND STATTISTICAL ANALYSIS
Refinement statistics of atomic model 6TCA is summarized in Table 1. Representative densities are shown in Figure S7. Cell-based assays and in vitro kinase assays are shown with SD from three independent measurements. Western-blots were analyzed and quantified with Image Studio Lite (LI-COR). Relevant details of in vitro kinase and cell-based assays can be found in the respective figure legend.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| pp-ERK2 antibody (T202/Y204) | Cell Signaling | Cat#9101 |
| pp-p38 antibody (T180/Y182) | Cell Signaling | Cat #9215 |
| pp-ERK5 antibody (Thr218/Tyr220) | Cell Signaling | Cat #3371 |
| Phospho-MAPKAPK-2 (T222) | Cell Signaling | Cat#3316S |
| Phospho-MAPKAPK-2 (S272) | Invitrogen | Cat#PAS-39792 |
| Phospho-MAPKAPK-2 (T334) | Cell Signaling | Cat #3007S |
| phospho-RSK1 antibody (T573) | Cell Signaling | Cat #9346 |
| phospho-ATF2 (T69/T71) | Cell Signaling | Cat #9225 |
| FLAG | Sigma Aldrich | Cat #F1804 |
| GAPDH | Sigma Aldrich | Cat#G9545 |
| IRDye 680 RD goat anti-Rabbit | Li-Cor | Cat#925–32211 |
| IRDye 680 RD goat anti-Mouse | Li-Cor | Cat#925–68070 |
| IRDye 800 CW goat anti-Rabbit | Li-Cor | Cat#925–32211 |
| IgG HRP-linked antibody anti-rabbit | Cell Signaling | Cat#7074S |
| IgG HRP-linked antibody anti-mouse | Millipore | Cat #401215 |
| MK2 (total) | Cell Signaling | Cat#3042 |
| p38 (total) | Cell Signaling | Cat#9228 |
| Bacterial and Virus Strains | ||
| E. coli Rosetta(DE3) | Merck (Novagen) | Cat#70954–3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TEV protease | Produced in house | Reményi Lab |
| Puromycin | Sigma Aldrich | Cat#P8833 |
| Doxycycline | Sigma Aldrich | Cat#D9891 |
| SB202190 | Sigma Aldrich | Cat#S7067 |
| PoA inhibitor | Synthesized in house | Reményi Lab |
| PEG 8000 | Sigma Aldrich | Cat#P2139 |
| HEPES | Sigma Aldrich | Cat#H3375 |
| MPD | Sigma Aldrich | Cat#112100 |
| ATP | Sigma-Aldrich | Cat#A26209 |
| AMPPNP | Sigma-Aldrich | Cat#10102547001 |
| D2O | Sigma-Aldrich | Cat#151882 |
| Coelenterazine h | NanoLight Technology | Cat#50909–86-9 |
| Lipofectamin 2000 | ThermoFisher Scientific | Cat#11668019 |
| Deposited Data | ||
| pp-p38-MK2 structure with PoA inhibitor | This paper | PDB: 6TCA |
| p38-MK2 complex | White at al. | PDB: 2OZA |
| p38-MK2 complex with PoA inhibitor | Cumming et al. | PDB: 4TYH |
| ERK2-RSK1 complex | Alexa et al. | PDB: 4NIF |
| Experimental Models: Cell Lines | ||
| SF9 cells | Invitrogen | Cat#LSB82501 |
| Experimental Models: Organisms/Strains | ||
| HEK293T | ATCC | Cat#CRL-3216 |
| Recombinant DNA | ||
| pBit1.1N (p38) | Promega | Cat#N2014 |
| pBit1.1C (ERK2) | Promega | Cat#N2014 |
| pBit1.1C (ERK5) | Promega | Cat#N2014 |
| pBit2.1C (MK2) | Promega | Cat#N2014 |
| pBit2.1N (RSK) | Promega | Cat#N2014 |
| pEBTetD (MKK1EE) | Bach et al, 2007 | Reményi Lab |
| pEBTetD (MKK6EE) | Bach et al, 2007 | Reményi Lab |
| pEBTetD (MKK7EE) | Bach et al, 2007 | Reményi Lab |
| pEBTetD (MKK5DD) | Bach et al, 2007 | Reményi Lab |
| pETARA-MK2 (modified pET15b vector) | Novagen | Reményi Lab |
| pETARA-RSK1 (modified pET15b vector) | Novagen | Reményi Lab |
| PETARA-ERK5 (modified pET15b vector) | Novagen | Reményi Lab |
| MG950-p38/λ-phosphatase (modified bicistronic pET15b vector) | Novagen | Reményi Lab |
| MG950-ERK2/λ-phosphatase (modified bicistronic pET15b vector) | Novagen | Reményi Lab |
| pFastBac-MKK5DD | ThermoFisher Scientific | Cat#10360014 |
| MG950-p38/MKK6EE (modified bicistronic pET15b vector) | Novagen | Reményi Lab |
| MG950-ERK2/MKK1EE (modified bicistronic pET15b vector) | Novagen | Reményi Lab |
| pET28a-BirA | Addgene | Cat#20857 |
| Software and Algorithms | ||
| Origin | OriginLab | www.originlab.com |
| Image Studio Lite | LI-COR | www.licor.com/bio/image-studio-lite/ |
| Coot | Emsley et al., 2010 | www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Phenix | Liebschner et al., 2019 | www.phenix-online.org |
| XDS | Kabsch, 2010 | xds.mpimf-heidelberg.mpg.de/ |
| Phaser | McCoy et al., 2007 | www.phaser.cimr.cam.ac.uk |
| Aimless | Evans and Murshudov, 2013 | www.ccp4.ac.uk/html/aimless.html |
| Modeller | B. Webb, A. Sali, 2016 | salilab.org/modeller/ |
| Haddock | van Zundert et al., 2016 | haddock.science.uu.nl/services/HADDOCK2.2/ |
| Pymol | PyMOL Molecular Graphics System | pymol.org |
| Atsas package | Franke et al., 2017 | www.embl-hamburg.de/biosaxs/software.html |
| Topspin | Bruker | www.bruker.com |
| NMRFAM-SPARKY | Lee et al., 2009 | nmrfam.wisc.edu/nmrfam-sparky-distribution/ |
| BiaEvaluation software | GE Healthcare | https://www.biacore.com |
Highlights.
MAPK-substrate kinases form antiparallel or parallel heterodimers
RSK1 and MK2 are phosphorylated at the activation loop in an antiparallel heterodimer
p38-MK2 forms different heterodimers depending on the activation state of p38
A small molecule stabilizes the parallel p38-MK2 heterodimer and blocks signaling
Acknowledgments
We are grateful for Csaba Hetényi and Krisztina Paál for their help in MD simulations and mass spectometry analysis, respectively, and for SAXS and X-ray beam line scientists at ESRF and PETRA, particularly for Isabel Bento for her help in X-ray diffraction data collection. We are grateful for Gergely Katona for his advice on SAXS data analysis, and for György Kardos for the synthesis of the PoA inhibitor. We thank Steven J. Nick for help with the purification of MK2 for NMR, and for Rebecca Page for critical reading of the manuscript.
Funding: This work was supported by the National Research Development and Innovation Office (NKFIH) grants (NN 114309, KKP 126963 awarded to AR), the National Institute of Medicine grant 1R01GM100910 to W.P, VEKOP-2.3.3-15-2016-00011 and NVKP_16-1-2016-0037 grants to A.R.
Footnotes
Declaration of interests: Authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, Hung L-W, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, Gógl G, Glatz G, Garai Á, Zeke A, Varga J, Dudás E, Jeszenői N, Bodor A, Hetényi C, et al. (2015). Structural assembly of the signaling competent ERK2–RSK1 heterodimeric protein kinase complex. Proc. Natl. Acad. Sci. 112, 201417571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach M, Grigat S, Pawlik B, Fork C, Utermöhlen O, Pal S, Banczyk D, Lazar A, Schömig E, and Gründemann D (2007). Fast set-up of doxycycline-inducible protein expression in human cell lines with a single plasmid based on Epstein-Barr virus replication and the simple tetracycline repressor. FEBS J. 274, 783–790. [DOI] [PubMed] [Google Scholar]
- Ben-Levy R, Leighton IA, Doza YN, Attwood P, Morrice N, Marshall CJ, and Cohen P (1995). Identification of novel phosphorylation sites required for activation of MAPKAP kinase-2. EMBO J. 14, 5920–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Levy R, Hooper S, Wilson R, Paterson HF, and Marshall CJ (1998). Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol. 8, 1049–1057. [DOI] [PubMed] [Google Scholar]
- Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, and Svergun DI (2007). Structural Characterization of Flexible Proteins Using Small-Angle X-ray Scattering. J. Am. Chem. Soc. 129, 5656–5664. [DOI] [PubMed] [Google Scholar]
- Caffrey DR, O’Neill LA, and Shields DC (1999). The evolution of the MAP kinase pathways: coduplication of interacting proteins leads to new signaling cascades. J. Mol. Evol. 49, 567–582. [DOI] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev a, Cobb, M.H., and Goldsmith EJ (1997). Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 90, 859–869. [DOI] [PubMed] [Google Scholar]
- Cargnello M, and Roux PP (2011). Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coxon PY, Rane MJ, Uriarte S, Powell DW, Singh S, Butt W, Chen Q, and McLeish KR (2003). MAPK-activated protein kinase-2 participates in p38 MAPK-dependent and ERK-dependent functions in human neutrophils. Cell. Signal. 15, 993–1001. [DOI] [PubMed] [Google Scholar]
- Cumming JG, Debreczeni JÉ, Edfeldt F, Evertsson E, Harrison M, Holdgate GA, James MJ, Lamont SG, Oldham K, Sullivan JE, et al. (2015). Discovery and Characterization of MAPK-activated Protein Kinase-2 Prevention of Activation Inhibitors. J. Med. Chem. 58, 278–293. [DOI] [PubMed] [Google Scholar]
- Dalby KN, Morrice N, Caudwell FB, Avruch J, and Cohen P (1998). Identification of Regulatory Phosphorylation Sites in Mitogen-activated Protein Kinase (MAPK)-activated Protein Kinase-1a/p90 rsk That Are Inducible by MAPK. J. Biol. Chem. 273, 1496–1505. [DOI] [PubMed] [Google Scholar]
- Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, Butler BL, Binkowski BF, Machleidt T, Kirkland TA, et al. (2016). NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 11, 400–408. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K, Schultz H, Martin F, Kotlyarov A, Plath K, Hahn M, Heinemann U, and Gaestel M (1995). Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J. Biol. Chem. 270, 27213–27221. [DOI] [PubMed] [Google Scholar]
- Engel K, Kotlyarov A, and Gaestel M (1998). Leptomycin B-sensitive nuclear export of MAPKAP kinase 2is regulated by phosphorylation. EMBO J. 17, 3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhead M, and Howarth M (2015). Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 1266, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, et al. (2017). ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garai Á, Zeke A, Gógl G, Törő I, Ferenc F, Blankenburg H, Bárkai T, Varga J, Alexa A, Emig D, et al. (2012). Specificity of linear motifs that bind to a common mitogen-activated protein kinase docking groove. Sci Signal 5, ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, and Nebreda AR (1999). A MAP kinase docking site is required for phosphorylation and activation of p90(rsk)/MAPKAP kinase-1. Curr. Biol. 9, 281–284. [DOI] [PubMed] [Google Scholar]
- Ghose R (2019). Nature of the Pre-Chemistry Ensemble in Mitogen-Activated Protein Kinases. J. Mol. Biol. 431, 145–157. [DOI] [PubMed] [Google Scholar]
- Gaestel M (2016). MAPK-Activated Protein Kinases (MKs): Novel Insights and Challenges. Front. Cell Dev. Biol. 3: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatz G, Gogl G, Alexa A, and Remenyi A (2013). Structural Mechanism for the Specific Assembly and Activation of the Extracellular Signal Regulated Kinase 5 (ERK5) Module. J. Biol. Chem. 288, 8596–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gógl G, Kornev AP, Reményi A, and Taylor SS (2019). Disordered Protein Kinase Regions in Regulation of Kinase Domain Cores. Trends Biochem. Sci. 44, 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Haar E, Prabhakar P, Prabakhar P, Liu X, and Lepre C (2007). Crystal structure of the p38 alpha-MAPKAP kinase 2 heterodimer. J. Biol. Chem. 282, 9733–9739. [DOI] [PubMed] [Google Scholar]
- Huse M, and Kuriyan J (2002). The conformational plasticity of protein kinases. Cell 109, 275–282. [DOI] [PubMed] [Google Scholar]
- Karaca E, and Bonvin AMJJ (2011). A Multidomain Flexible Docking Approach to Deal with Large Conformational Changes in the Modeling of Biomolecular Complexes. Structure 19, 555–565. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Yannoni Y, Fritz S, Laass K, Telliez J-B, Pitman D, Lin L-L, and Gaestel M (2002). Distinct cellular functions of MK2. Mol. Cell. Biol. 22, 4827–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar GS, Clarkson MW, Kunze MBA, Granata D, Wand AJ, Lindorff-Larsen K, Page R, and Peti W (2018). Dynamic activation and regulation of the mitogen-activated protein kinase p38. Proc. Natl. Acad. Sci. 115, 4655–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, and Ahn NG (2004). Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell 14, 43–55. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng W, Swenson LL, Fitzgibbon MJ, Hayakawa K, Ter Haar E, Behrens AE, Fulghum JR, and Lippke J. a (2002). Structure of mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 suggests a bifunctional switch that couples kinase activation with nuclear export. J. Biol. Chem. 277, 37401–37405. [DOI] [PubMed] [Google Scholar]
- Mertens HDT, and Svergun DI (2010). Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 172, 128–141. [DOI] [PubMed] [Google Scholar]
- Miller CJ, and Turk BE (2018). Homing in: Mechanisms of Substrate Targeting by Protein Kinases. Trends Biochem. Sci. 43, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, and Cobb MH (2001). Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22, 153–183. [DOI] [PubMed] [Google Scholar]
- Piserchio A, Warthaka M, Kaoud TS, Callaway K, Dalby KN, and Ghose R (2017). Local destabilization, rigid body, and fuzzy docking facilitate the phosphorylation of the transcription factor Ets-1 by the mitogen-activated protein kinase ERK2. Proc. Natl. Acad. Sci. 114, E6287–E6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, and Davis RJ (1995). Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 270, 7420–7426. [DOI] [PubMed] [Google Scholar]
- Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin L-L, Gaestel M, et al. (2007). The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol. Cell. Biol. 27, 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Poteet-Smith CE, Lannigan DA, Freed TA, Zoltoski AJ, and Sturgill TW (2000). Creation of a stress-activated p90 ribosomal S6 kinase. The carboxyl-terminal tail of the MAPK-activated protein kinases dictates the signal transduction pathway in which they function. J. Biol. Chem. 275, 31588–31593. [DOI] [PubMed] [Google Scholar]
- Soundararajan M, Roos AK, Savitsky P, Filippakopoulos P, Kettenbach AN, Olsen JV, Gerber S. a, Eswaran J, Knapp S, and Elkins JM (2013). Structures of Down syndrome kinases, DYRKs, reveal mechanisms of kinase activation and substrate recognition. Structure 21, 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D, Campbell DG, Nakielny S, Hidaka H, Leevers SJ, Marshall C, and Cohen P (1992). MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. EMBO J. 11, 3985–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D, Barberato C, and Koch M (1995). CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Crystallogr. 28, 768–771. [Google Scholar]
- Tsutakawa SE, Hura GL, Frankel KA, Cooper PK, and Tainer JA (2007). Structural analysis of flexible proteins in solution by small angle X-ray scattering combined with crystallography. J. Struct. Biol. 158, 214–223. [DOI] [PubMed] [Google Scholar]
- Turjanski AG, Hummer G, and Gutkind JS (2009). How Mitogen-Activated Protein Kinases Recognize and Phosphorylate Their Targets: A QM/MM Study. J. Am. Chem. Soc. 131, 6141–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A, Pargellis C. a, Studts JM, Werneburg BG, and Farmer BT (2007). Molecular basis of MAPK-activated protein kinase 2:p38 assembly. Proc. Natl. Acad. Sci. U. S. A. 104, 6353–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Galanis A, and Sharrocks AD (1999). Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol. Cell. Biol. 19, 4028–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeke A, Bastys T, Alexa A, Garai Á, Mészáros B, Kirsch K, Dosztányi Z, Kalinina OV, and Reményi A (2015). Systematic discovery of linear binding motifs targeting an ancient protein interaction surface on MAP kinases. Mol. Syst. Biol. 11, 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-Y, Wu J-W, and Wang Z-X (2011). Mitogen-activated protein kinase (MAPK) phosphatase 3-mediated cross-talk between MAPKs ERK2 and p38alpha. J. Biol. Chem. 286, 16150–16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, and Bonvin AMJJ (2016). The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 428, 720–725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Protein Data Bank accession number is 6TCA for the atomic coordinates and structure factors of the pp-p38-MK2-PoA complex. SAXS data is available upon request.