

Abstract
Phosphodiesterase type 4 (PDE4) inhibitors prevent hydrolysis of cyclic adenosine monophosphate and increase protein kinase A (PKA)-mediated phosphorylation. PDE4 inhibitors also regulate responses to ethanol and GABAergic drugs. We investigated mechanisms by which the PDE4 inhibitor, apremilast, regulates acute effects of ethanol and GABAergic drugs in male and female mice. Apremilast prolonged the sedative-hypnotic effects of gaboxadol, zolpidem, and propofol but did not alter etomidate effects, and unexpectedly shortened the sedative-hypnotic effects of diazepam. Apremilast prolonged rotarod ataxia induced by zolpidem, propofol, and loreclezole, shortened recovery from diazepam, but had no effect on ataxia induced by gaboxadol or etomidate. The PKA inhibitor H89 blocked apremilast’s ability to prolong the sedative-hypnotic effects of ethanol, gaboxadol, and propofol and to prolong ethanol- and propofol-induced ataxia. H89 also blocked apremilast’s ability to shorten the sedative-hypnotic and ataxic effects of diazepam. The β1-specific antagonist, salicylidene salicylhydrazide (SCS), produced faster recovery from ethanol- and diazepam-induced ataxia, but did not alter propofol- or etomidate-induced ataxia. SCS shortened the sedative-hypnotic effects of ethanol and diazepam but not of propofol. In Xenopus oocytes, a phosphomimetic (aspartate) mutation at the PKA phosphorylation site in β1 subunits decreased the maximal GABA current in receptors containing α1 or α3, but not α2 subunits. In contrast, phosphomimetic mutations at PKA sites in β3 subunits increased the maximal GABA current in receptors containing α1 or α2, but not α3 subunits. The GABA potency and allosteric modulation by ethanol, propofol, etomidate, zolpidem, flunitrazepam, or diazepam were not altered by these mutations. We propose a model whereby apremilast increases PKA-mediated phosphorylation of β1- and β3-containing GABAA receptors and selectively alters acute tolerance to ethanol and GABAergic drugs.
Keywords: PDE4 inhibitor apremilast, β1 and β3 GABAA receptor subunits, protein kinase A, acute tolerance to alcohol and GABAergic drugs, loss of righting reflex, rotarod ataxia
1. INTRODUCTION
Phosphodiesterases (PDEs) catalyze the hydrolysis of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate and play a key role in regulating intracellular levels of these cyclic nucleotides. Of the 11 different families of PDEs, PDE4 is the most important for controlling cAMP levels, is expressed in the brain, and is involved in alcohol and drug dependence as well as in the regulation of inflammatory and neuroimmune responses (Wen et al., 2018). Chronic alcohol intake (liquid diet model) increases neuroimmune signaling, including activation of astrocytes and microglia, and these effects are attenuated by a PDE4 inhibitor (rolipram) or genetic deletion of Pde4b (Avila et al., 2017). In view of the role of neuroimmune activation in regulating alcohol consumption and other behavioral effects (Erickson et al., 2019), the anti-inflammatory actions of PDE4 inhibitors may be an important part of their mechanism of action in these responses.
PDE4 inhibitors also decrease ethanol seeking and consumption in rodents (Blednov et al., 2014; Franklin et al., 2015; Hu et al., 2011; Liu et al., 2017; Wen et al., 2012). In a large genetic association study in humans, PDE4B was identified as a locus associated with all tobacco and alcohol use phenotypes examined (Liu et al., 2019). We recently reported that apremilast, a selective FDA-approved PDE4 inhibitor, produced stable decreases in ethanol intake in male and female mice in different drinking tests (Blednov et al., 2018b) and altered other behaviors that are correlated with ethanol consumption (Blednov et al., 2018a). For example, apremilast prolonged the acute sedative-hypnotic and ataxic effects of ethanol and decreased acute functional tolerance to ethanol. Acute functional tolerance is behavioral tolerance that occurs within an individual test session, and is distinguished from rapid tolerance which develops over 8–72 h after ethanol or drug exposure (Pietrzykowski and Treistman, 2008).
Understanding how PDE4 inhibitors decrease ethanol consumption could be beneficial for drug development to treat alcohol use disorder. We are particularly interested in apremilast because of its low side effect profile and clinical success. Apremilast, like other PDE4 inhibitors, reduces hydrolysis of cAMP leading to increased activation of protein kinase A (PKA). Current evidence indicates that PKA regulates γ-aminobutyric acid type A (GABAA) receptor function. For example, intracerebroventricular administration of the PKA activator Sp-cAMP increases the sedative-hypnotic effects of ethanol and the GABAA receptor agonist muscimol (Kumar et al., 2012). PKA is able to phosphorylate the large intracellular loops of β1 and β3 subunits (McDonald et al., 1998). Phosphorylation of β3-containing receptors at S408 and S409 enhances GABA-stimulated responses, but phosphorylation of S409 alone inhibits responses, similar to effects found with β1 subunits, which are phosphorylated solely on S409. Similar to neuronal receptors, GABAA receptors expressed in HEK293 cells and phosphorylated by PKA on β1 and β3 subunits show opposing effects on GABA-stimulated currents (i.e., phosphorylation increases α1β3γ2 responses and decreases α1β1γ2 responses) (McDonald et al., 1998).
In this study, we examined mechanisms by which apremilast regulates behavioral responses to ethanol and different GABAergic drugs in mice. We found that apremilast altered recovery from the ataxic and sedative-hypnotic effects of ethanol and GABAergic drugs in a PKA-dependent manner. We also used two-electrode voltage clamp of αβγ GABAA receptors expressed in Xenopus laevis oocytes to show that the phosphorylation states of β1 and β3 differentially alter receptor function depending on the type of co-expressed α subunit. Our findings suggest that apremilast-induced increases in PKA-dependent phosphorylation of β1-and β3-containing GABAA receptors in the brain alter acute tolerance to ethanol and GABAergic drugs.
2. MATERIALS AND METHODS
2.1. Mice
Male and female C57BL/6J mice were from a colony maintained in the Animal Resources Center at The University of Texas at Austin. Original breeders were purchased and replenished every 6 months from The Jackson Laboratory (Bar Harbor, ME). Mice were group-housed by sex (4 or 5 per cage) in temperature- and humidity-controlled rooms with free access to food and water using a 12-h light/dark cycle (lights on at 7:00 a.m.). Experiments began when the mice were 2–3 months old. Mice were allowed to adapt to the testing rooms for about one week before behavioral testing. Experiments were approved by the Institutional Animal Care and Use Committee at The University of Texas at Austin and comply with the ARRIVE guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2. Drug Administration
Ethanol (100% stock, Aaper Alcohol and Chemical, Shelbyville, KY) solutions were prepared in 0.9% saline (20%, v/v) and injected i.p. Apremilast (Toronto Research Chemicals Inc., North York, ON, Canada) was freshly prepared as a suspension in saline with 3–4 drops of Tween-80, and 20 mg/kg p.o. was administered once daily in a volume 0.05 ml/10 g of body weight 1 h before experiments. This timeframe was chosen based on our previous working showing peak levels of apremilast in plasma, liver, and brain 1 h after administration (Blednov et al., 2018b). Gaboxadol (10 and 55 mg/kg), diazepam (6 and 50 mg/kg), and propofol (30 and 120 mg/kg) were purchased from Sigma-Aldrich (St. Louis, MO) and administered by i.p. injection (0.1 ml/10 g body weight). Gaboxadol was dissolved in saline, and diazepam and propofol were suspended in saline with 3–4 drops of Tween-80. The propofol suspension was also sonicated for 10 min. Zolpidem (5 and 60 mg/kg), loreclezole (60 mg/kg), salicylidene salicylhydrazide (SCS) (40 mg/kg), and H89 (10 mg/kg) were purchased from Tocris Bioscience (Minneapolis, MN), and etomidate (10, 15, 25, and 50 mg/kg) was purchased from Toronto Research Chemicals Inc. These drugs were freshly prepared in 0.9% saline with 3–4 drops of Tween-80 and injected at 0.1 ml/10 g of body weight for i.p. administration or at 0.05 ml/10 g of body weight for s.c. administration of H89. SCS and H89 were injected 15 min before drug treatment based on previous findings (Kumar et al., 2012) and our preliminary experiments.
2.3. Loss of the Righting Reflex
Responses to sedative-hypnotic doses of ethanol and other drugs were measured as the duration of the loss of righting reflex (LORR). When mice became ataxic, they were placed in the supine position in V-shaped plastic troughs until they were able to right themselves three times within 30 s. The duration of the LORR was defined as the time elapsed between being placed in the supine position until recovering the righting reflex. Saline or apremilast (20 mg/kg, p.o.) was injected once 1 h before i.p. injection of gaboxadol (55 mg/kg), zolpidem (60 mg/kg), propofol (120 mg/kg), etomidate (25 and 50 mg/kg), or diazepam (50 mg/kg). To study the role of PKA on apremilast responses, mice were treated with apremilast (20 mg/kg, p.o.) 1 h before testing and then treated with H89 (10 mg/kg, s.c.) 15 min before i.p. injection of ethanol (3.6 g/kg), gaboxadol (55 mg/kg), propofol (120 mg/kg), or diazepam (50 mg/kg). To study the role of β1 subunits, mice were pretreated with SCS (40 mg/kg, i.p.) 15 min before i.p. injection of ethanol (3.6 g/kg), propofol (120 mg/kg), or diazepam (50 mg/kg).
2.4. Rotarod Ataxia
Mice were trained on a fixed speed rotarod (Economex; Columbus Instruments, Columbus, OH) at 10 rpm, and training was considered complete when mice were able to remain on the rotarod for 60 s. Every 15 min after drug injection, each mouse was placed on the rotarod and latency to fall was measured until the mouse was able to remain on the rotarod for 60 s. Saline or apremilast (20 mg/kg, p.o.) was injected once 1 h before i.p. injection of gaboxadol (10 mg/kg), diazepam (6 mg/kg), zolpidem (5 mg/kg), propofol (30 mg/kg), loreclezole (60 mg/kg), or etomidate (10 mg/kg). To study effects of PKA inhibition, mice were treated with apremilast (20 mg/kg, p.o.) or saline (p.o.) 1 h before testing and then treated with saline or the PKA inhibitor H89 (10 mg/kg, s.c.) 15 min before injection of ethanol (2 g/kg), diazepam (6 mg/kg), or propofol (30 mg/kg). To investigate the role of β1-containing GABAA receptors, saline or the β1-specific antagonist SCS (40 mg/kg, i.p.) was injected 15 min before i.p. injection of ethanol (2 g/kg), diazepam (6 mg/kg), propofol (30 mg/kg), or etomidate (15 mg/kg).
2.5. Electrophysiology
Xenopus laevis frogs were obtained from Nasco (Fort Atkinson, WI). Experiments were approved by the Institutional Animal Care and Use Committee at The University of Texas at Austin and comply with the ARRIVE guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The complementary DNAs encoding the rat GABAA subunits α1, β1, β3, and γ2 were provided by Dr. M. H. Akabas (Albert Einstein College of Medicine); human α2 (provided by Dr. Neil Harrison, Columbia University) was subcloned into pGEMHE, and rat α3 was optimized and synthesized by GenScript (Piscataway, NJ). Mutations in the β cDNAs were made through site-directed mutagenesis using QuikChange (Agilent Technologies, Santa Clara, CA). The in vitro transcription of GABAA subunits was performed using mMessage mMachine (Life Technologies, Grand Island, NY). Manually isolated Xenopus laevis oocytes were injected with 50 nl capped complementary RNAs encoding wild-type or mutant subunits in different ratios, depending on the subunits: αβ3γ2, 0.5:0.3:0.5 ng/oocyte and αβ1γ2, 0.5:0.5:0.5 ng/oocyte. The injected oocytes were incubated at 15°C in sterilized Modified Barth’s solution for 1–4 days before recording.
The responses of GABAA receptors expressed in oocytes were studied using two-electrode voltage clamp. Oocytes were discarded if the maximal current was over 30 μA or if the baseline was unstable or drifted to positive values. Final drug dilutions were freshly prepared each day. GABA concentration-response curves were determined using increasing concentrations of GABA (0.1–3000 μM) applied for 20–30 s followed by a 5–15 min washout. The oocyte’s response to each concentration was expressed as the percentage of the maximal current produced by that oocyte. To verify the presence of the γ2 subunit in the expressed receptors, responses to GABA were evaluated in the presence of Zn++ (10 μM). Flunitrazepam, zolpidem, etomidate, and propofol stocks were prepared in DMSO. The maximal GABA concentration was applied for 20 s, and after a 15-min washout, the GABA concentration that produced 5% of the maximal response was applied. If the resulting current was not between 3 and 7% of the maximal response, the GABA concentration was adjusted accordingly until the response was within those parameters. This was defined as the nominal EC5 GABA. After two consecutive applications of EC5 GABA, the modulators were coapplied with EC5 GABA in between EC5 GABA alone applications. Ethanol or zinc were preapplied alone for 60 s immediately before their co-application with EC5 GABA.
2.6. Statistical Analysis
Statistical analyses were performed using Prism 8 (GraphPad Software, Inc., La Jolla, CA) software. Data are reported as mean ± S.E.M values (number of mice and oocytes used are reported in the figure legends). For behavioral tests, sex as a factor was not significant so we combined the data from male and female mice (with the exception of data in Figures 3D and 6 which were collected only in male mice). Data were analyzed by one- or two-way ANOVA and Tukey’s post hoc tests. For electrophysiology, GABA concentration-response curves were determined using non-linear fitting of a Hill equation with variable slope. Current values elicited by a maximal GABA concentration were analyzed over three consecutive days using two-way ANOVA (multiple comparisons with Sidak’s correction). Drug responses in the presence of EC5 GABA were quantified as the percent change in current from the average of the EC5 GABA alone responses obtained immediately before and after the drug. One-way ANOVA testing was used to detect significant differences in drug modulation between mutant receptors.
3. RESULTS
3.1. Apremilast prolongs duration of the LORR induced by gaboxadol, zolpidem, and propofol
We previously showed that apremilast (20 mg/kg, p.o.) prolonged the sedative-hypnotic effects of ethanol in male and female C57BL/6J mice (Blednov et al., 2018a). Here we measured the duration of the LORR following injection of different GABAergic sedative-hypnotic drugs in male and female C57BL/6J mice after pretreatment with saline or apremilast (20 mg/kg, p.o.). Apremilast significantly prolonged the duration of LORR induced by 55 mg/kg of gaboxadol [t(13) = 29.80, p < 0.0001)], 60 mg/kg of zolpidem [t(15) = 6.39, p < 0.0001], and 120 mg/kg of propofol [t(13) = 8.48, p < 0.0001)] (Figure 1A–C) but did not alter LORR induced by etomidate (Figure 1D). However, apremilast significantly shortened the duration of LORR induced by 50 mg/kg of diazepam [t(16) = 9.36, p < 0.0001)] (Figure 1E).
Figure 1.
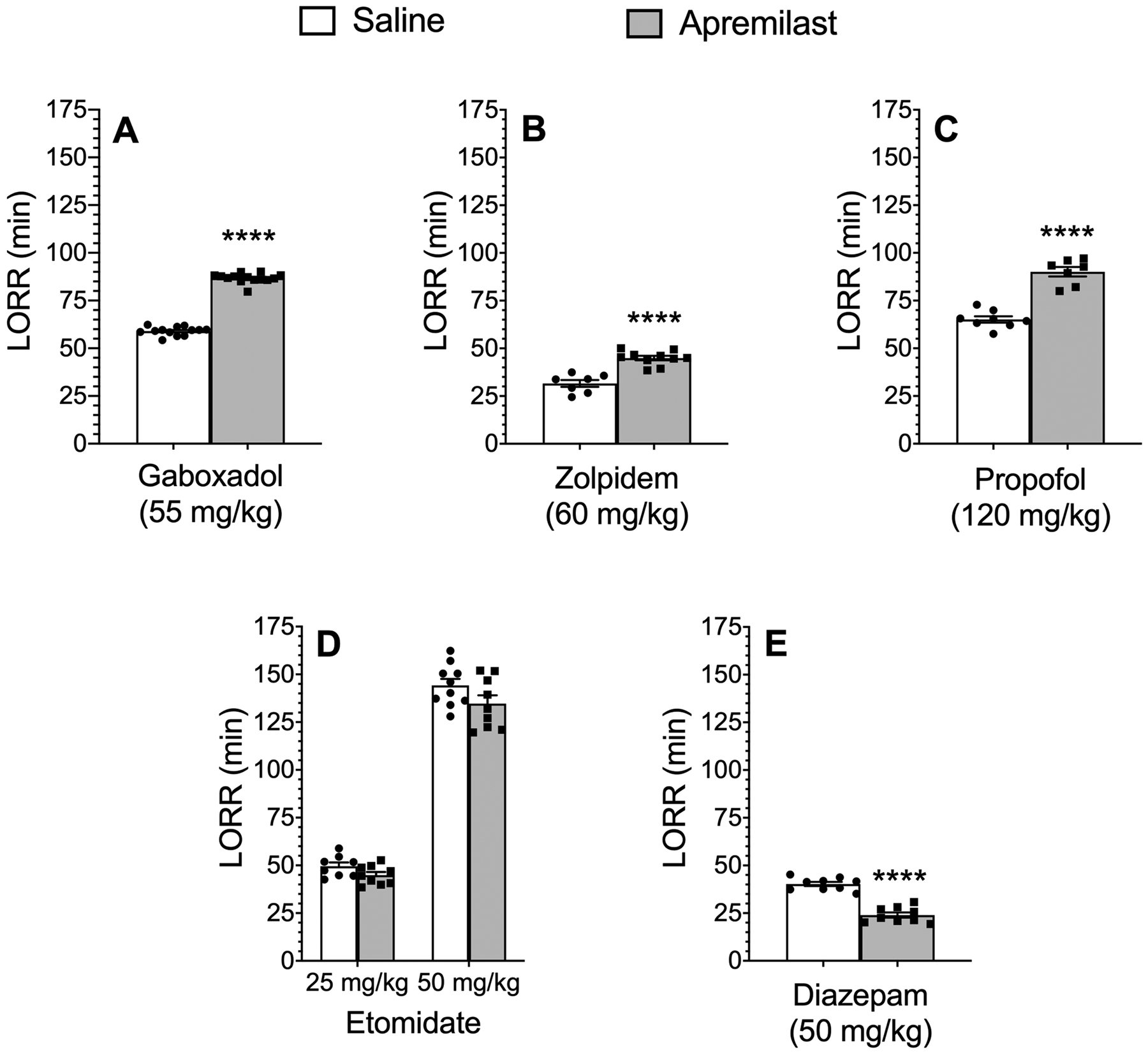
Effect of apremilast on the loss of righting reflex (LORR) induced by different sedativehypnotics. Duration of LORR in saline- vs. apremilast (20 mg/kg)-pretreated male and female C57BL/6J mice after i.p. injection of (A) gaboxadol (n = 13–14), (B) zolpidem (n = 7–10), (C) propofol (n = 7–8), (D) etomidate (n = 8–10), or (E) diazepam (n = 9). Data from male and female mice were combined. ****p < 0.0001 compared with the saline-treated group, two-tailed t-test.
3.2. Apremilast prolongs ataxia induced by zolpidem, loreclezole, and propofol
We previously showed that apremilast (20 mg/kg, p.o.) prolongs recovery from the acute ataxic effect of ethanol (Blednov et al., 2018a). Here, we investigated whether this effect occurs with other GABAergic drugs using lower doses that are selective for certain GABAA receptor subtypes. Apremilast (20 mg/kg, p.o.) did not alter recovery from rotarod ataxia induced by gaboxadol (10 mg/kg, i.p.), which is a selective agonist for receptors containing α4 and δ subunits (Figure 2A). However, it significantly prolonged recovery from the motor impairing effects of zolpidem (5 mg/kg) (F1,30 = 13.7, p < 0.001, effect of pretreatment; F7,210 = 144, p < 0.0001, effect of time; F7,210 = 9.5, p < 0.0001, pretreatment x time interaction), which is a positive allosteric modulator of receptors containing α1 and γ2 subunits (Figure 2B). Apremilast also prolonged recovery from loreclezole (60 mg/kg) (F1,20 = 101, p < 0.0001, effect of pretreatment; F9,180 = 68.6, p < 0.0001, effect of time; F9,180 = 23.8, p < 0.0001, pretreatment x time interaction) (Figure 2D), which is a positive allosteric modulator of receptors that contain β2 or β3 subunits. Additionally, apremilast prolonged recovery from propofol (30 mg/kg) (F1,29 = 11.9, p < 0.01, effect of pretreatment; F6,174 = 91.5, p < 0.0001, effect of time; F6,174 = 3.9, p < 0.01, pretreatment x time interaction) (Figure 2E), which is not selective for β subunits (Rudolph and Antkowiak, 2004). In contrast, pretreatment with apremilast produced faster recovery from the motor impairing effects of diazepam (6 mg/kg, Figure 2C) (F1,28 = 19.6, p < 0.001, effect of pretreatment; F8,224 = 136, p < 0.0001, effect of time; F8,224 = 7.8, p < 0.0001, pretreatment x time interaction), which is a positive allosteric modulator of receptors containing γ2 with α1, α2, α3, or α5 subunits. Apremilast did not alter recovery from ataxia induced by etomidate (10 mg/kg) (Figure 2F). At low doses, etomidate is a positive allosteric modulator of receptors that contain β2 or β3 subunits (Sieghart and Savic, 2018).
Figure 2.
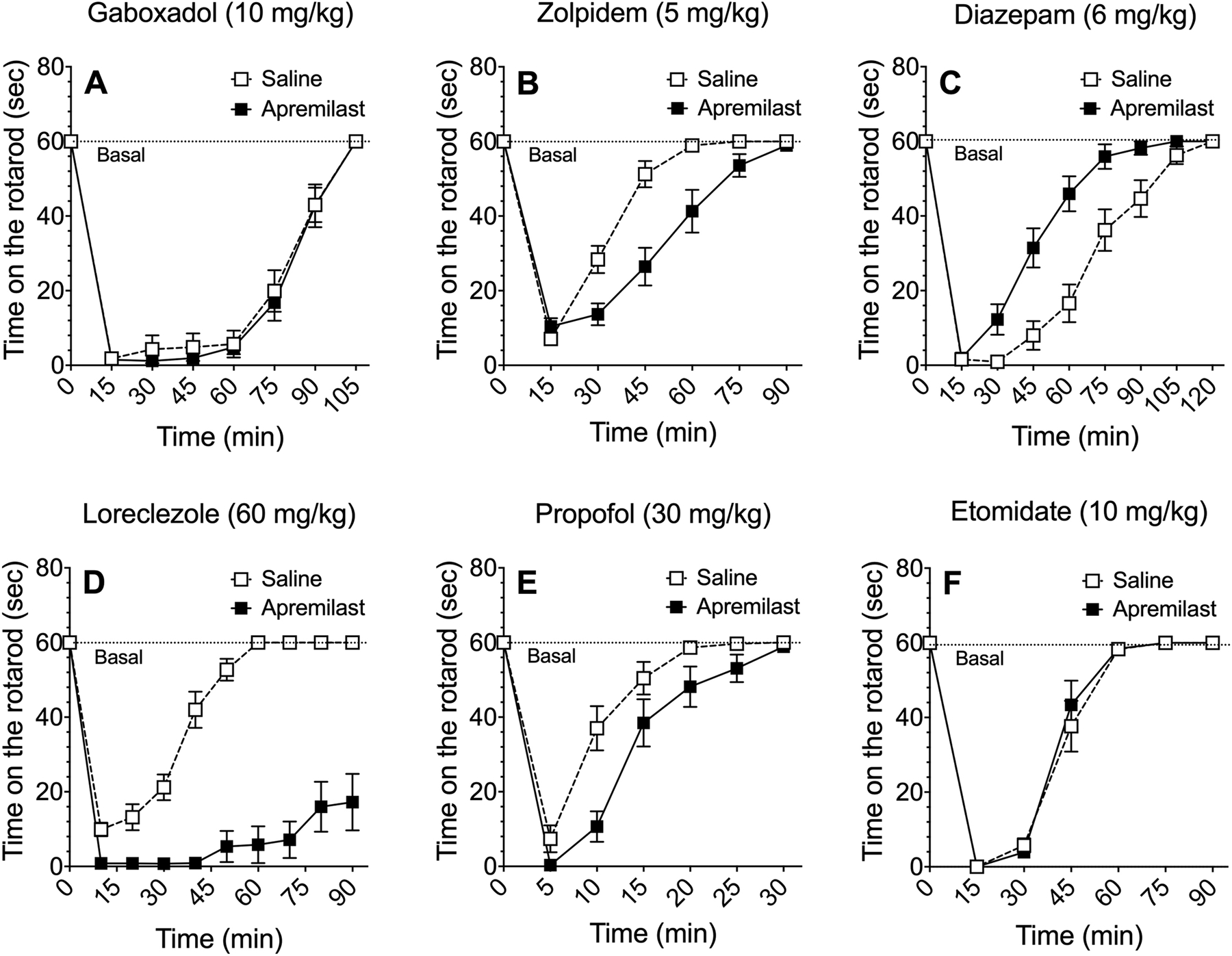
Effect of apremilast on recovery from ataxia induced by GABAergic drugs. Time on the rotarod in saline- vs. apremilast (20 mg/kg)-pretreated male and female C57BL/6J mice after i.p. injection of (A) gaboxadol (n = 16), (B) zolpidem (n = 16), (C) diazepam (n = 15), (D) loreclezole (n = 10–12), (E) propofol (n = 15–16), or (F) etomidate (n = 11–12). Data from male and female mice were combined and analyzed by two-way repeated measures ANOVA.
3.3. A PKA inhibitor prevents apremilast modulation of LORR induced by ethanol, gaboxadol, or propofol
Since apremilast is a PDE4 inhibitor and increases activation of PKA, we next used the kinase inhibitor H89 (Hidaka et al., 1984) to evaluate the role of PKA on apremilast-induced increases in LORR duration in male and female C57BL/6J mice. Pretreatment with H89 (10 mg/kg, s.c.) did not alter the duration of the LORR induced by ethanol (3.6 g/kg, i.p.), gaboxadol (55 mg/kg, i.p.), or propofol (120 mg/kg, i.p.), but it completely blocked the ability of apremilast (20 mg/kg, p.o.) to prolong the sedative-hypnotic effect of these drugs [effect of pretreatment on LORR induced by ethanol (F1,34 = 118.0, p < 0.0001), gaboxadol (F1,34 = 479.5, p < 0.0001), and propofol (F1,34 = 140.1, p < 0.0001)] (Figure 3A–C). Pretreatment with H89 also did not alter duration of the LORR induced by diazepam (50 mg/kg), but it blocked the ability of apremilast to shorten diazepam-induced LORR (F1,20 = 161.7, p < 0.0001) (Figure 3D).
Figure 3.
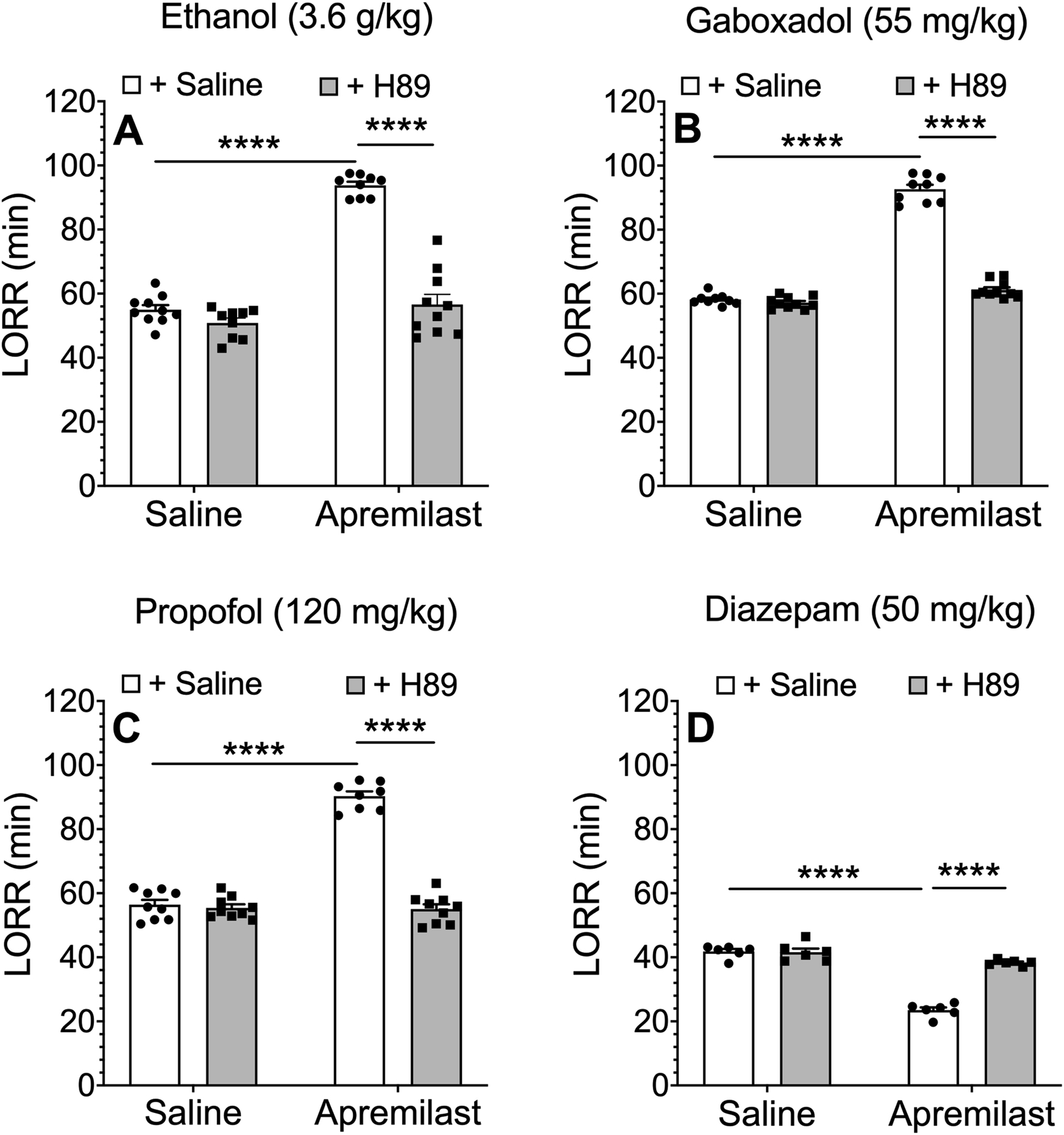
The PKA inhibitor H89 prevents apremilast-induced changes in duration of the loss of righting reflex (LORR) by ethanol, gaboxadol, propofol, and diazepam. Duration of LORR induced by i.p. injection of (A) ethanol (n = 9–10), (B) gaboxadol (n = 9–10), (C) propofol (n = 8–9), or (D) diazepam (n = 6) in C57BL/6J mice pretreated with saline (p.o.) or apremilast (20 mg/kg, p.o.) 1 h before LORR assay then saline (s.c.) or H89 (10 mg/kg, s.c.) was given 15 min before sedative-hypnotic drug. Data from male mice (D) or males and females combined (A-C) were analyzed by two-way ANOVA and Tukey’s post hoc tests, ****p < 0.0001.
3.4. Blockade of PKA prevents apremilast modulation of rotarod ataxia induced by ethanol, diazepam, or propofol
We next examined potential PKA-dependent effects of apremilast on ataxia induced by ethanol and GABAergic drugs. In male and female C57BL/6J mice, H89 (10 mg/kg, s.c.) reversed the ability of apremilast (20 mg/kg, p.o.) to prolong recovery from the ataxic effects of 2 g/kg of ethanol (F1,22 = 32.1, p < 0.0001, effect of pretreatment; F9,198 = 181, p < 0.0001, effect of time; F9,198 = 10.7, p < 0.0001, pretreatment x time interaction) (Figure 4A) and 30 mg/kg of propofol (F1,21 = 38.1, p < 0.0001, effect of pretreatment; F6,126 = 152, p < 0.0001, effect of time; F6,126 = 15.7, p < 0.0001, pretreatment x time interaction) (Figure 4B). H89 also reversed the ability of apremilast to speed recovery from ataxia induced by 6 mg/kg of diazepam (F1,21 = 97.9, p < 0.0001, effect of pretreatment; F8,168 = 447, p < 0.0001, effect of time; F8,168 = 35.9, p < 0.0001, pretreatment x time interaction) (Figure 4C).
Figure 4.
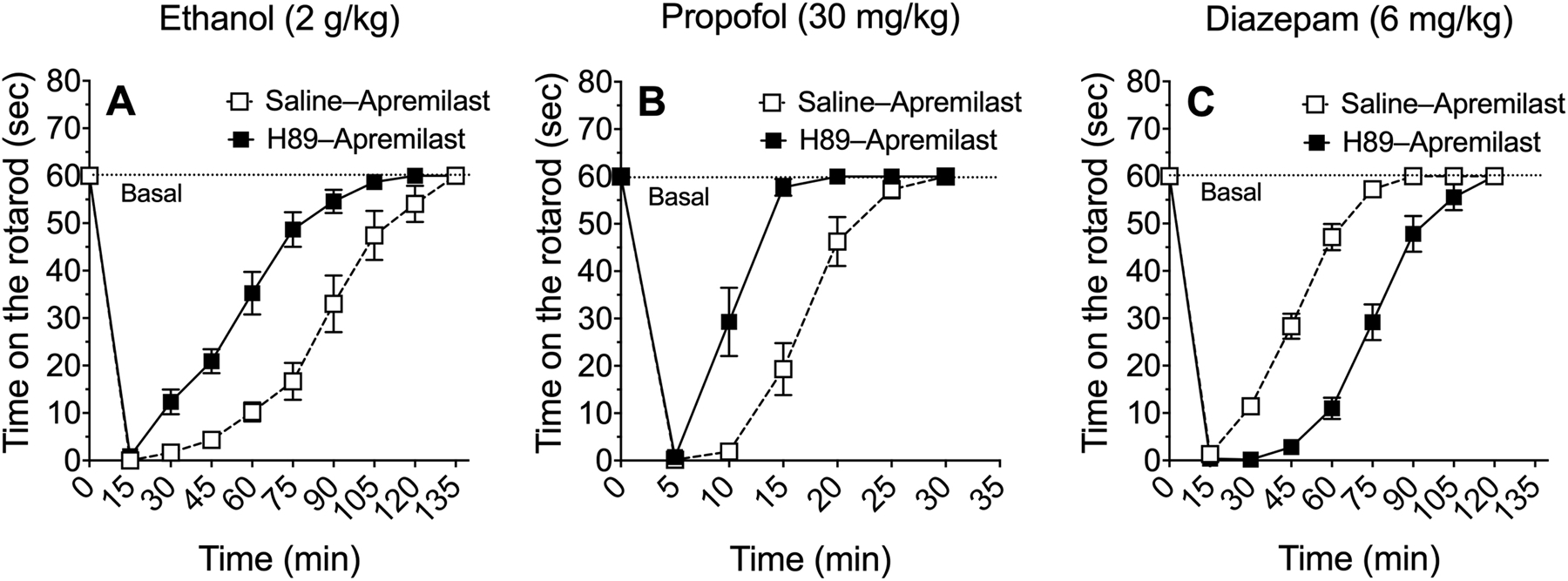
The PKA inhibitor H89 prevents apremilast modulation of rotarod ataxia induced by ethanol, diazepam, or propofol. Time on the rotarod after i.p. injection of (A) ethanol (n = 12), (B) propofol (n = 11–12), and (C) diazepam (n = 11–12) in male and female C57BL/6J mice pretreated with saline (s.c.) + apremilast (20 mg/kg, p.o.) or H89 (10 mg/kg, s.c.) + apremilast (20 mg/kg, p.o.). Data from male and female mice were combined and analyzed by two-way repeated measures ANOVA.
3.5. A GABAA receptor β1 subunit antagonist accelerates recovery from ataxia induced by ethanol or diazepam
Because PKA can regulate GABAA receptor function through phosphorylation of β1 and β3 subunits (McDonald et al., 1998), we first investigated the importance of β1-containing receptors using SCS, a β1-specific antagonist (Thompson et al., 2004). Pretreatment of C57BL/6J male mice with SCS (40 mg/kg, i.p.) induced faster recovery from the motor impairing effects of ethanol (F1,10 = 25.6, p < 0.001, effect of treatment; F7,70 = 191, p < 0.0001, effect of time; F7,70 = 8.5, p < 0.0001, treatment x time interaction) (Figure 5A) and diazepam (F1,10 = 22.6, p < 0.001, effect of treatment; F8,80 = 194, p < 0.0001, effect of time; F8,80 = 10.2, p < 0.0001, treatment x time interaction) (Figure 5B). As predicted, SCS did not change recovery from ataxia induced by propofol (F5,45 = 337, p < 0.0001, effect of time) or etomidate (F7,70 = 41, p < 0.0001, effect of time) (Figure 5C and D).
Figure 5.
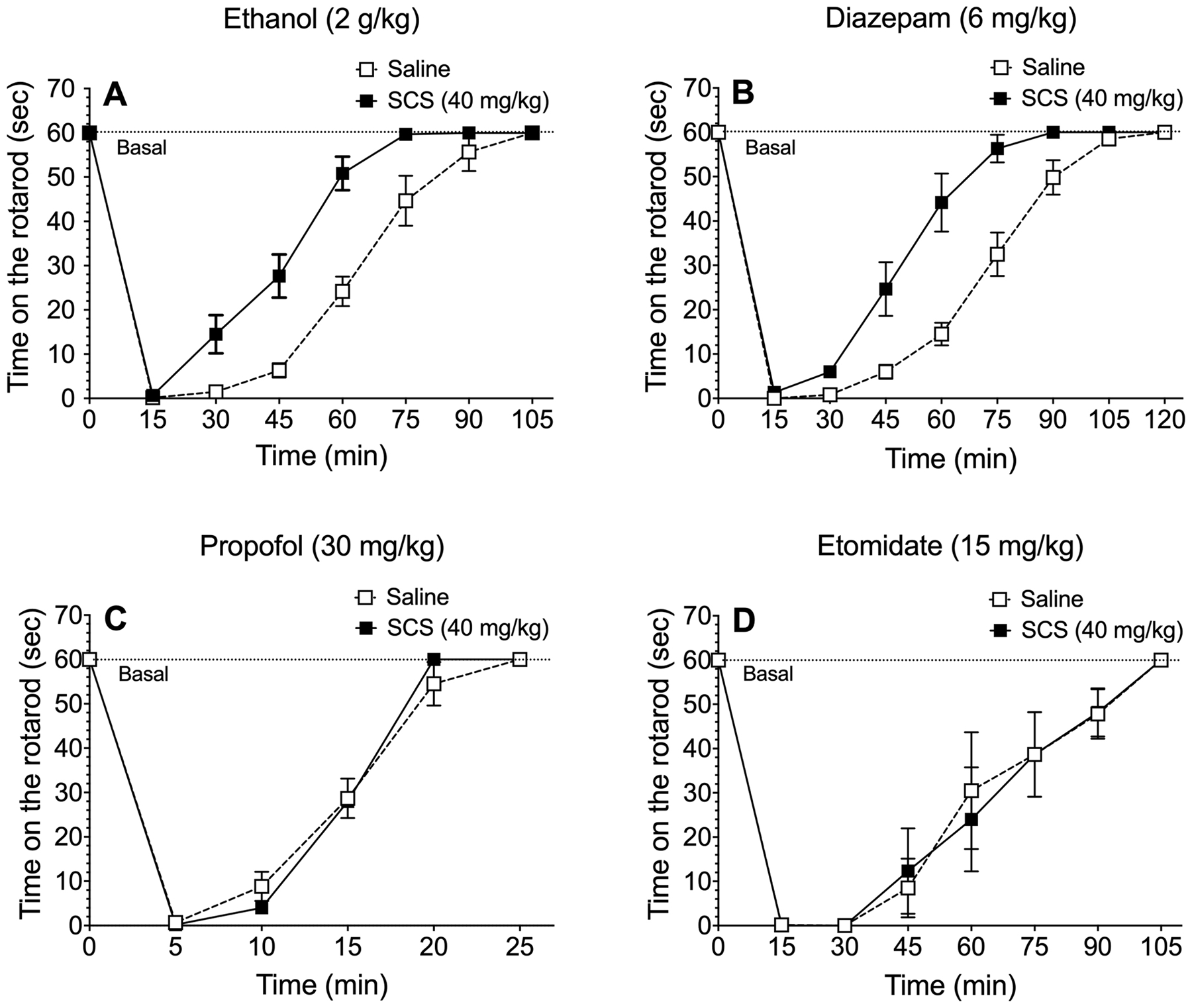
A GABAA receptor β1 subunit antagonist accelerates recovery from ataxia induced by ethanol or diazepam. Time on the rotarod after i.p. injection of (A) ethanol (n = 6), (B) diazepam (n = 6), (C) propofol (n= 5–6), and (D) etomidate (n = 6) in male C57BL/6J mice pretreated with saline (i.p.) or salicylidene salicylhydrazide (SCS, 40 mg/kg, i.p.). Data were analyzed by two-way repeated measures ANOVA.
3.6. A GABAA receptor β1 subunit antagonist shortens the duration of LORR induced by ethanol or diazepam, but not by propofol
We next examined the effect of SCS (40 mg/kg, i.p.) pretreatment on the sedative-hypnotic effects of ethanol, diazepam, and propofol in male C57BL/6J mice. SCS significantly shortened the duration of LORR induced by 3.6 g/kg of ethanol [t(10) = 12.9, p < 0.0001)] or 50 mg/kg of diazepam [t(10) = 12.3, p < 0.0001], but did not alter the sedative-hypnotic effect of 120 mg/kg of propofol (Figure 6A–C). A summary of results from our behavioral tests are shown in Table 1.
Figure 6.
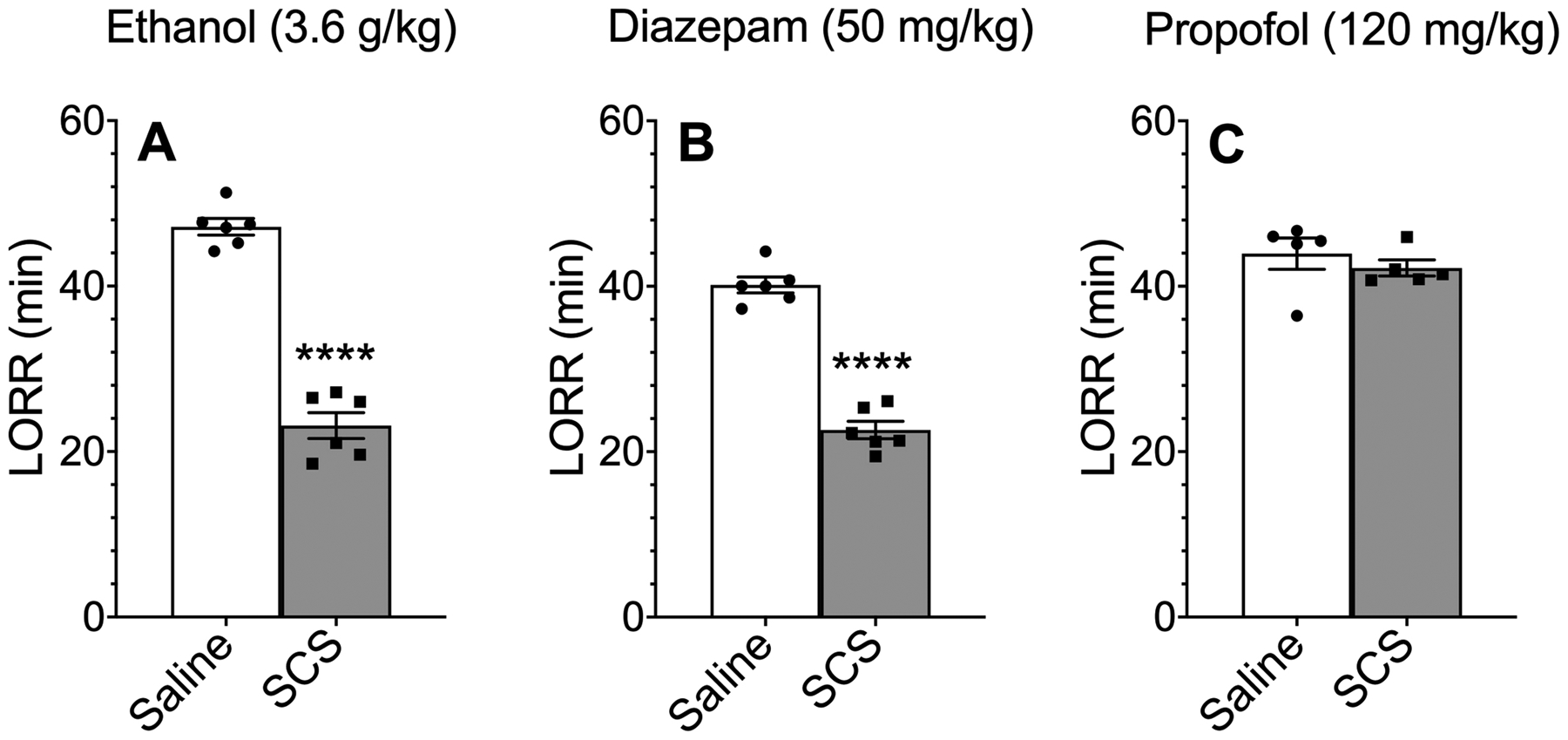
Effect of SCS on the loss of righting reflex (LORR) induced by ethanol, diazepam, or propofol. Duration of LORR after i.p. injection of (A) ethanol (n = 6), (B) diazepam (n= 6), or (C) propofol (n = 5) in male C57BL/6J mice pretreated with saline (i.p.) or salicylidene salicylhydrazide (SCS, 40 mg/kg, i.p.). ****p < 0.0001 compared with the saline-treated group, two-tailed t-test.
Table 1.
Summary of behavioral effects in male and female C57BL/6J mice.
| Behavior | Modulator | Dose | + Apremilast | + Apremilast + H-89 | + SCS a |
|---|---|---|---|---|---|
| Ethanol intake | Ethanol | ↓ b | |||
| LORR duration | Ethanol | 3.6 g/kg | ↑ b | ↓ | ↓ |
|
Gaboxadol Non-specific |
55 mg/kg | ↑ | ↓ | ||
|
Etomidate β2-β3 specific |
25 and 50 mg/kg | = | |||
|
Propofol Non β-specific |
120 mg/kg | ↑ | ↓ | = | |
| Diazepam α3/2/1/5 γ2 | 50 mg/kg | ↓ | ↑ | ↓ | |
|
Zolpidem Non-specific |
60 mg/kg | ↑ | |||
| Rotarod recovery | Ethanol | 2 g/kg | → b | ← | ← |
|
Gaboxadol α4 δ specific |
10 mg/kg | = | |||
|
Etomidate β2-β3 specific |
10 mg/kg | = | |||
|
Etomidate β2-β3 specific |
15 mg/kg | = | |||
|
Propofol Non β-specific |
30 mg/kg | → | ← | = | |
|
Diazepam α3/2/1/5 γ2 |
6 mg/kg | ← | → | ← | |
|
Zolpidem α1γ2-specific |
5 mg/kg | → | |||
|
Loreclezole β2-β3 specific |
60 mg/kg | → |
Effects of apremilast (20 mg/kg), apremilast (20 mg/kg) + PKA inhibitor H-89 (10 mg/kg), or a β1-specific antagonist salicylidene salicylhydrazide (SCS, 40 mg/kg) on ethanol- and GABAergic-mediated behaviors are summarized as follows: = no change from saline control; ↑ (increased) or ↓ (decreased) response from saline control; → (longer) or ← (shorter) recovery from rotarod ataxia compared with saline control. GABAergic drugs have subunit-specific or non-specific actions depending on the dose. Etomidate mediates ataxia mainly through β2-containing receptors.
Results are from male mice only.
Results are from (Blednov et al., 2018b). LORR, loss of the righting reflex.
3.7. Mutation of phosphorylation sites on β1 or β3 subunits alters the maximal response to GABA in heterologously expressed GABAA receptors
The direct application of apremilast (1–10 μM) to α1β2γ2 or α1β3γ2 GABAA receptors heterologously expressed in Xenopus oocytes did not alter receptor function (Figure S1).
In order to study a homogeneous population of receptors possessing a known phosphorylation state, we expressed mutated β1 and β3 subunits in combination with α1, α2, or α3, along with γ2 subunits. In these β subunits, the relevant serines were replaced by phosphomimetic or non-phosphorylatable residues. The GABA sensitivity of each subunit combination and the GABA-induced maximal current were determined over three consecutive days to control for changes in levels of receptor expression. The analysis of GABA-mediated maximal current yielded the same significant differences, whether the data were analyzed dayby-day by two-way ANOVA, or pooled and analyzed by one-way ANOVA.
The relevant serine residue in the intracellular loop of the β1 subunits (S409) was replaced by either an aspartate (phosphomimetic) or alanine (non-phosphorylatable) residue. When assessing the maximal GABA concentration-induced currents mediated by α1β1γ2 combinations, the receptors containing the non-phosphorylatable subunit [β1(A)] showed larger currents than the phosphomimetic subunit [β1(D)] (F2,102 = 21.93, p < 0.0001, effect of days after injection; F2,102 = 17.54, p < 0.0001, effect of phosphorylation state; F4,102 = 3.35, p < 0.05, days after injection × phosphorylation state interaction) (Figure 7A). The same result was observed for α3β1γ2 combinations, except that the difference was not yet significant on day 2 after injection (F2,107 = 40.24, p < 0.0001, effect of days after injection; F2,107 = 28.38, p < 0.0001, effect of phosphorylation state; F4,107 = 3.53, p < 0.001, days after injection × phosphorylation state interaction) (Figure 7E). Currents mediated by receptors containing the wild-type subunit [β1(S)] were not consistent compared with the phosphomimetic and non-phosphorylatable subunits, likely reflecting a variable endogenous phosphorylation state. When β1 was expressed with α2 and γ2 subunits, there were no differences in the maximal currents in β1-containing receptors with differing phosphorylation states (Figure 7C). The sensitivity to GABA was not affected by the phosphorylation state of the β1 409 residue in any subunit combination (Figure 7B, D and F, and Table 2).
Figure 7.
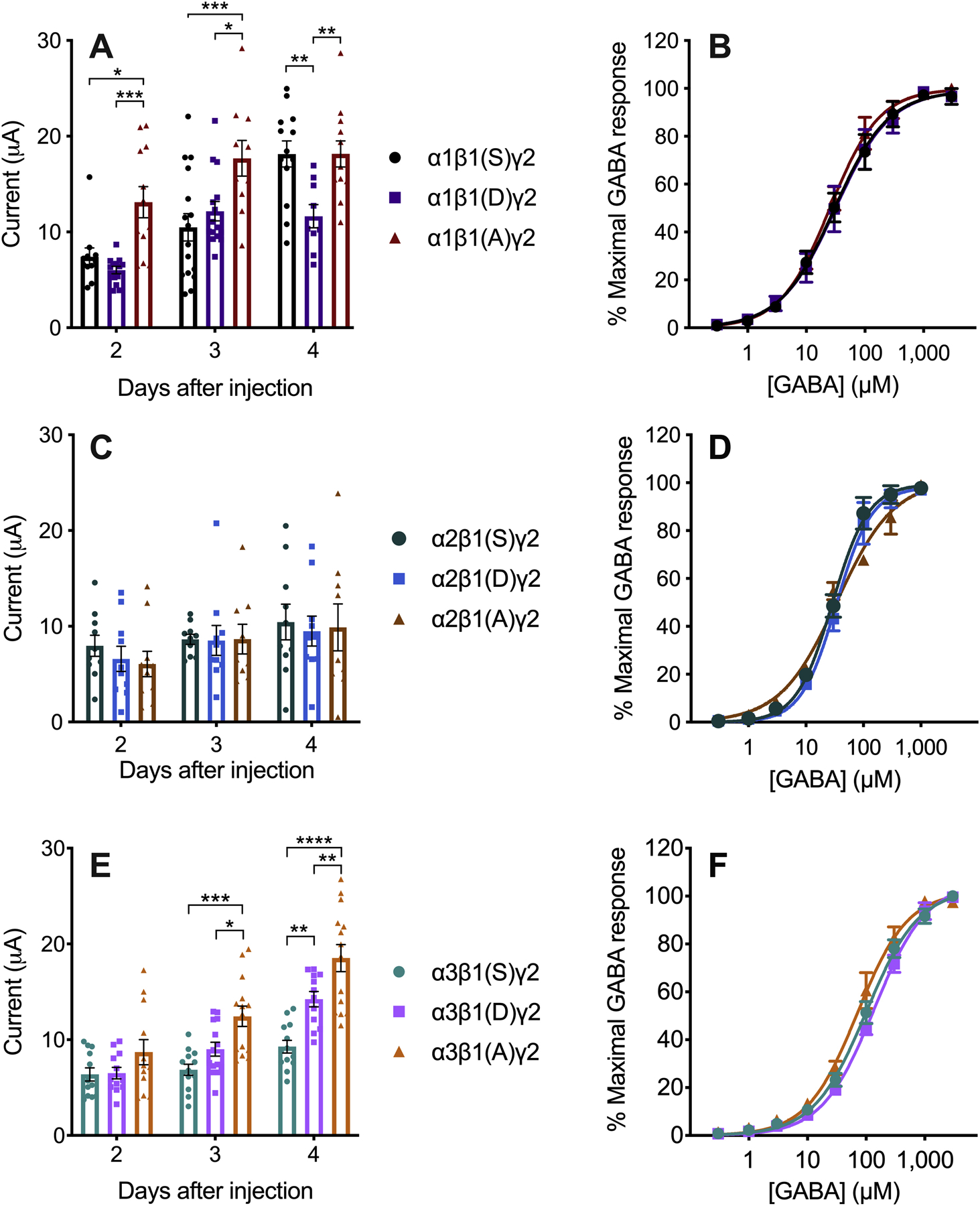
β1-containing GABAA receptors expressed in Xenopus laevis oocytes. The letters in parentheses in the legend indicate the residues in position 409 of β1. A) Currents induced by maximal GABA concentration (3 mM GABA, n = 9–16) and B) GABA concentration-response curves (n = 4–5) in α1β1γ2 GABAA receptors. C) Currents induced by maximal GABA concentration (300 μM GABA, n = 9–10) and D) GABA concentration-response curves (n = 4–5) in α2β1γ2 GABAA receptors. E) Currents induced by maximal GABA concentration (3 mM GABA, n = 12–14) and F) GABA concentration-response curves (n = 4–6) in α3β1γ2 GABAA receptors. Data were analyzed by two-way ANOVA followed by Sidak’s multiple comparisons test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Table 2.
Parameters determined by nonlinear regression of GABA concentration-response curves (Figures 7 and 8). GABA EC50, GABA effective concentration 50 (μM); nH, Hill slope; n, number of oocytes.
| Receptor | GABA EC50 (95% confidence intervals) | nH ± SEM | n |
|---|---|---|---|
| α1β1(S)γ2 | 31.1 (26.2 to 37.0) | 0.92 ± 0.05 | 5 |
| α1β1(D)γ2 | 25.3 (23.4 to 27.4) | 1.03 ± 0.03 | 5 |
| α1β1(A)γ2 | 29.8 (26.1 to 34.1) | 0.94 ± 0.04 | 4 |
| α2β1(S)γ2 | 28.8 (24.8 to 33.7) | 1.39 ± 0.10 | 4 |
| α2β1(D)γ2 | 33.7 (29.8 to 38.1) | 1.42 ± 0.08 | 5 |
| α2β1(A)γ2 | 34.9 (18.4 to 65.9) | 0.88 ± 0.14 | 4 |
| α3β1(S)γ2 | 100 (82 to 127) | 0.96 ± 0.07 | 6 |
| α3β1(D)γ2 | 137 (117 to 165) | 0.97 ± 0.06 | 4 |
| α3β1(A)γ2 | 70.4 (55.8 to 91.5) | 1.01 ± 0.10 | 5 |
| α1β3(SS)γ2 | 24.7 (21.4 to 28.6) | 1.50 ± 0.10 | 3 |
| α1β3(DD)γ2 | 28.2 (26.6 to 30.0) | 1.50 ± 0.05 | 6 |
| α1β3(AA)γ2 | 21.5 (18.7 to 24.8) | 1.77 ± 0.15 | 5 |
| α2β3(SS)γ2 | 42.3 (30.4 to 59.1) | 1.11 ± 0.12 | 5 |
| α2β3(DD)γ2 | 14.8 (12.5 to 17.6) | 1.53 ± 0.13 | 5 |
| α2β3(AA)γ2 | 23.6 (21.8 to 25.5) | 1.47 ± 0.06 | 7 |
| α3β3(SS)γ2 | 19.8 (17.4 to 22.5) | 1.57 ± 0.10 | 5 |
| α3β3(DD)γ2 | 15.3 (13.7 to 17.1) | 1.26 ± 0.06 | 5 |
| α3β3(AA)γ2 | 21.0 (18.0 to 24.5) | 1.47 ± 0.11 | 6 |
For α1β3γ2 combinations, both relevant serines in the intracellular loop of β3 (408 and 409) were replaced by either aspartates or alanines. The phosphomimetic [β3(DD)] subunit showed a larger maximal GABA-induced current than the non-phosphorylatable [β3(AA)] and wild-type [β3(SS)] subunits, except on day 1, when the differences were not yet significant (F2,153 = 71.0, p < 0.0001, effect of days after injection; F2,153 = 13.9, p < 0.0001, effect of phosphorylation state; F4,153 = 1.34, p > 0.05, days after injection × phosphorylation state interaction) (Figure 8A). The α2β3γ2 receptors showed a similar trend on days 1 and 2, but the only significant difference was on day 2 between the phosphomimetic [β3(DD)] and non-phosphorylatable [β3(AA)] subunits (F2,138 = 30.35, p < 0.0001, effect of days after injection; F2,138 = 6.07, p < 0.01, effect of phosphorylation state; F4,138 = 0.42, p > 0.05, days after injection × phosphorylation state interaction) (Figure 8C). We observed no differences in the maximal currents in α3β3γ2 combinations on any day (Figure 8E). In β3-containing receptors, the sensitivity to GABA was not affected by the phosphorylation state of the β3 408 and 409 residues in α1 and α3 combinations (Figure 8B and F, and Table 2). Small differences in the GABA EC50 values were observed in α2β3γ2 receptors (Figure 8D and Table 2).
Figure 8.
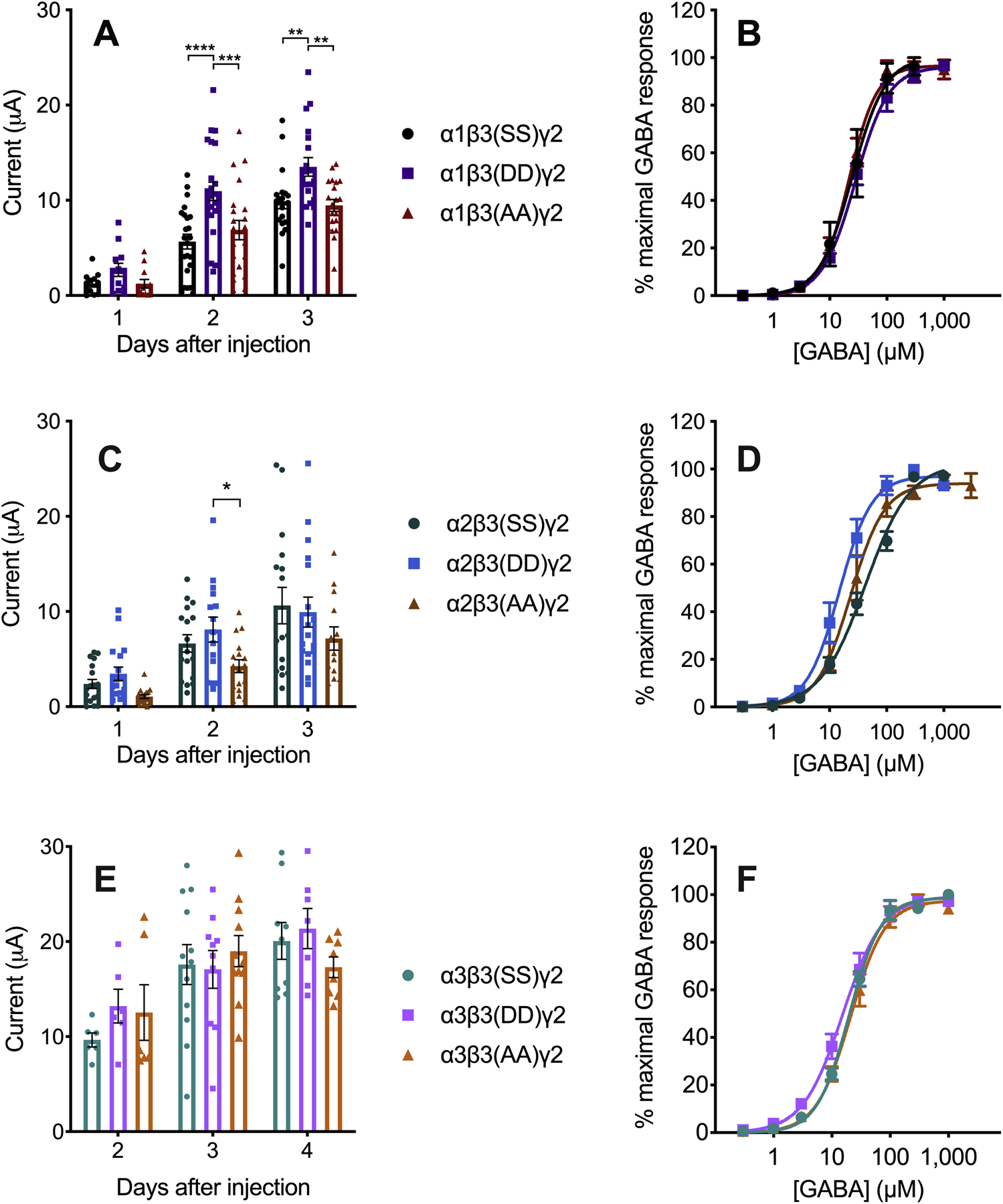
β3-containing GABAA receptors expressed in Xenopus laevis oocytes. The letters in parentheses in the legend indicate the residues in positions 408 and 409 of β3. A) Currents induced by maximal GABA concentration (1 mM GABA, n = 12–24) and B) GABA concentration-response curves (n = 3–5) in α1β3γ2 GABAA receptors. C) Currents induced by maximal GABA concentration (1 mM GABA, n = 13–18) and D) GABA concentration-response curves (n = 5–7) in α2β3γ2 GABAA receptors. E) Currents induced by maximal GABA concentration (300 μM GABA, n = 6–12) and F) GABA concentration-response curves in α3β3γ2 GABAA receptors (n = 5–6). Data were analyzed by two-way ANOVA followed by Sidak’s multiple comparisons test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
In order to determine if the phosphorylation state of β subunits affects GABAA receptor function by different allosteric modulators, we co-applied a submaximal GABA concentration (EC5) with ethanol (200 mM), propofol (2 μM), etomidate (1 μM) (Figure 9), zolpidem (0.1 μM), flunitrazepam (0.1 μM), or diazepam (0.1 and 3 μM) to oocytes expressing α1β3γ2 receptors (Figure 10). We also tested diazepam modulation of α1β1γ2 receptors (Figure 10). We corroborated expression of γ2 along with α and β subunits by measuring the effect of an endogenous modulator, zinc (10 μM), which inhibits αβ and αβγ receptors with different potencies (Figure 9). None of the allosteric modulators showed a differential effect that depended on the phosphorylation state of β subunits.
Figure 9.
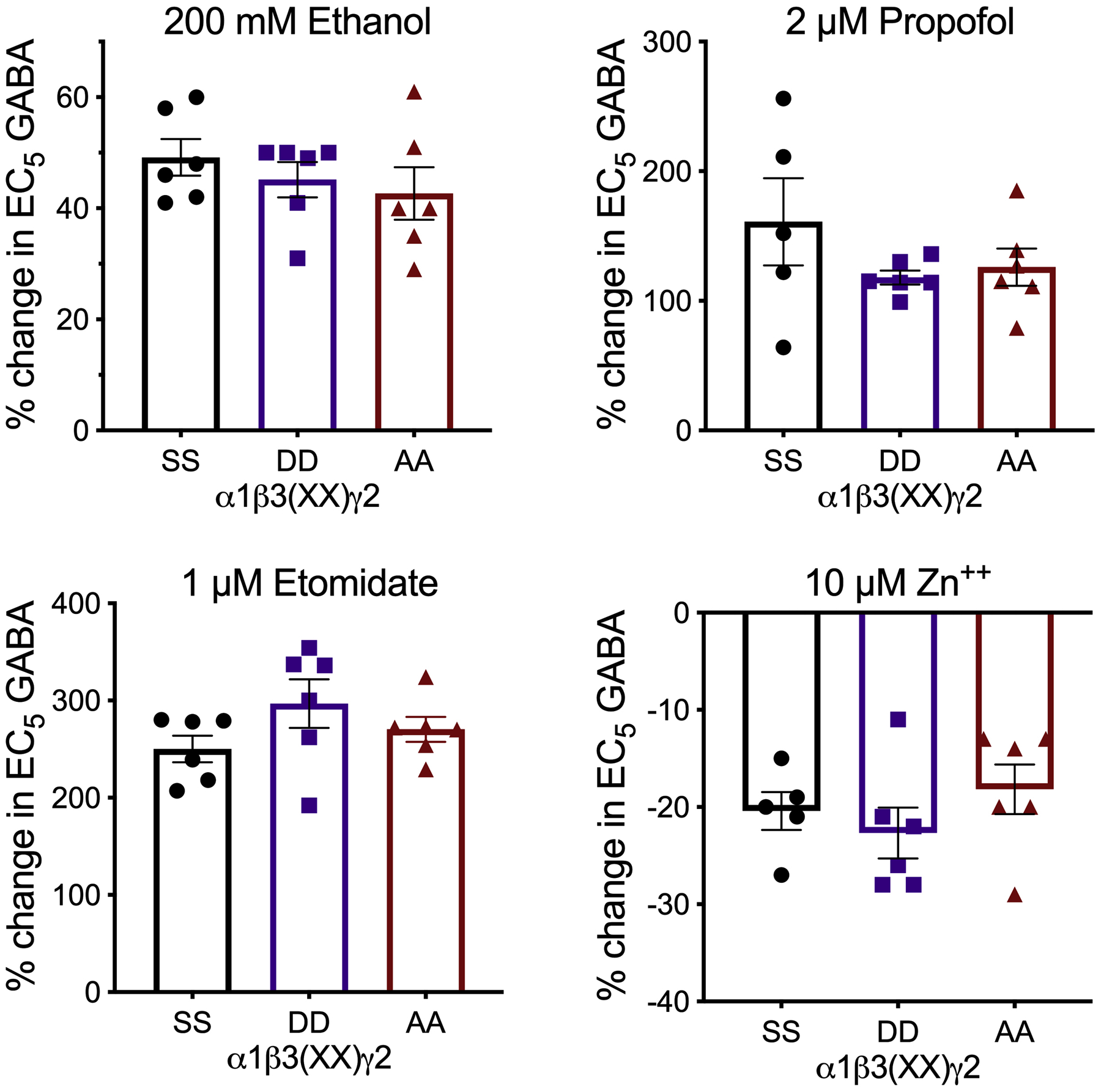
Allosteric modulators of submaximal GABA currents in α1β3γ2 GABAA receptors with different phosphorylation states of β3 subunits. Data were analyzed by one-way ANOVA (n = 5–6).
Figure 10.
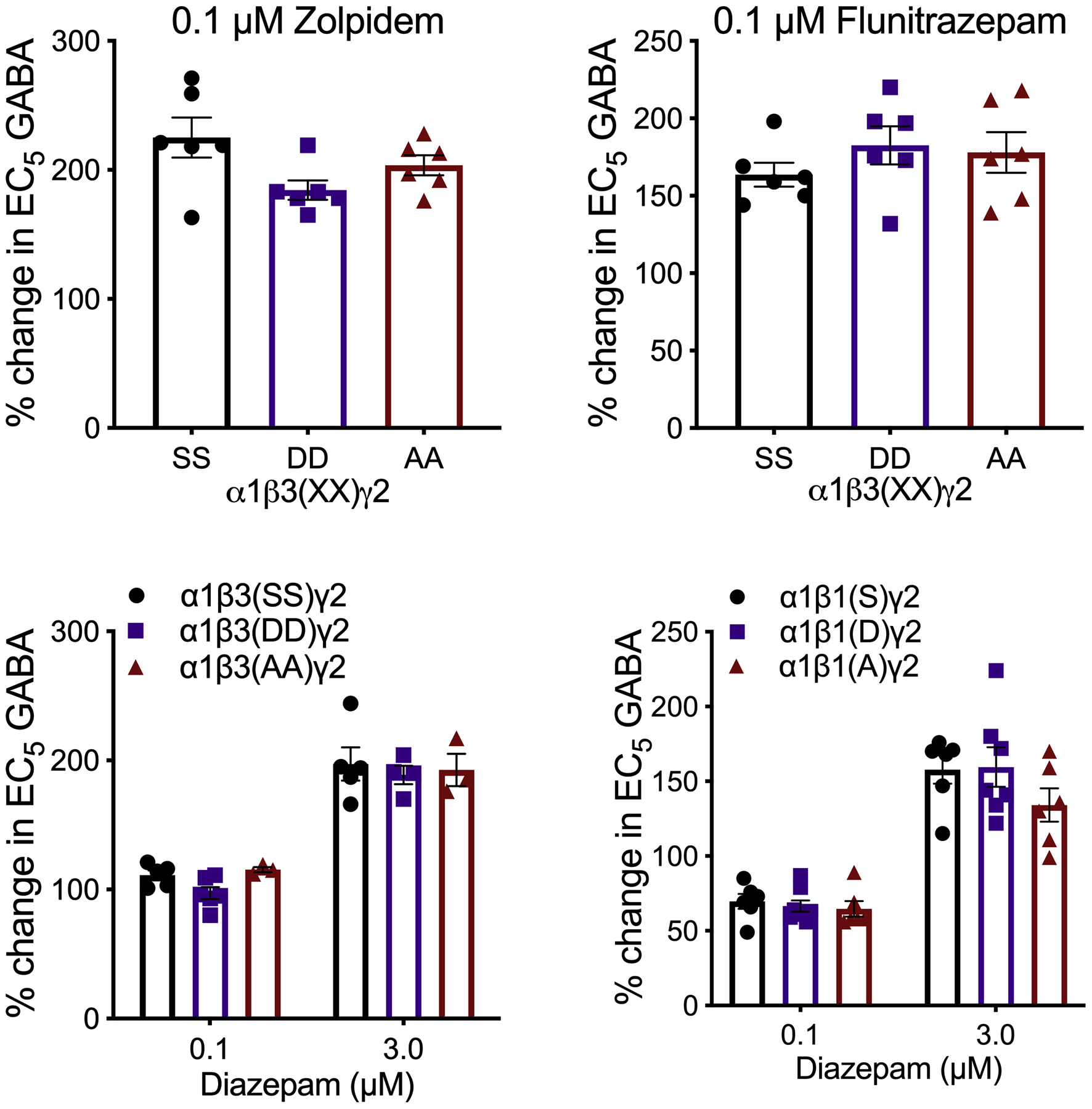
Modulation by ligands of the benzodiazepine binding site of submaximal GABA currents in α1βγ2 GABAA receptors with differing phosphorylation states of β subunits. Data were analyzed by one-way (flunitrazepam, zolpidem; n = 6) and two-way (diazepam) ANOVA (β3, n = 3–6; β1, n = 6–8).
4. DISCUSSION
Apremilast profoundly altered the behavioral effects of ethanol and GABAA receptor-specific drugs in male and female mice. Apremilast prolonged the duration of the LORR induced by ethanol, gaboxadol, zolpidem, and propofol and also prolonged ataxia induced by ethanol, zolpidem, propofol, and loreclezole. Surprisingly however, apremilast shortened the duration of the LORR and of ataxia induced by diazepam. The PKA inhibitor H89 blocked apremilast modulation of behavior by ethanol, propofol, gaboxadol, and diazepam, suggesting that apremilast alters acute tolerance to ethanol and other GABAergic drugs via PKA-mediated phosphorylation of GABAA receptors. Our results with apremilast are consistent with work showing that increasing PKA activity intracerebroventricularly increases the sedative-hypnotic effects of ethanol or the GABAA receptor agonist muscimol (Kumar et al., 2012). Conversely, other work has shown that inhibiting PKA decreases ethanol’s sedative-hypnotic effects (Thiele et al., 2000).
Our behavioral studies suggest that α, β1, and β3 subunits are important for apremilast modulation of GABAergic drugs. Therefore, we studied how the phosphorylation state of β1 or β3 expressed with different α subunits altered GABAA receptor function and modulation. In Xenopus oocytes, apremilast did not produce any direct changes in GABAA receptor function, suggesting a low level of PDE4 activity as previously reported (Stahl et al., 2015). To study β phosphorylation, we used mutated receptors expressed in oocytes. Decreased maximal GABA-induced currents were observed in phosphomimetic β1-containing subunits expressed with α1 or α3, but not α2 subunits. In contrast, increased maximal GABA-induced currents were observed in phosphomimetic β3-containing subunits expressed with α1 and α2. Our findings in oocytes agree with those in HEK293 cells (McDonald et al., 1998), showing that β1 phosphorylation decreases and β3 phosphorylation increases GABAA responses. These same β phosphorylation effects were observed in α1-containing receptors in oocytes, and thus apremilast prolongation of zolpidem (an α1-selective modulator) responses in vivo suggests that it acts by increasing phosphorylation of α1β3-containing receptors. Furthermore, the differential GABAA responses of α2 and α3 subunits in combination with phosphomimetic β1- or β3-containing receptors suggest that these α subunits can modulate ethanol- and diazepam-induced ataxia. Additional support for this comes from our previous study showing that deletion of α2 shortened recovery from ethanol and flurazepam, whereas deletion of α3 prolonged ataxia by both drugs (Blednov et al., 2013).
We also studied modulation of α1βγ2 GABAA receptors in different phosphorylation states by the GABAergic drugs used in the behavioral tests. We found that the allosteric effects of ethanol and other GABAergic drugs did not depend on β subunit phosphorylation. Thus, we propose that the behavioral effects of apremilast result from PKA-mediated alterations in GABAA receptor responses to GABA rather than to changes in allosteric modulation by these drugs.
We were particularly interested in the effects of apremilast on ataxia because the lower drug doses used for this behavior would be expected to have more GABAA receptor specificity than the higher doses required to induce LORR. Apremilast did not alter ataxia induced by gaboxadol, which in low doses selectively targets GABAA receptors containing α4 and δ subunits (Chandra et al., 2006). However, apremilast prolonged recovery from the ataxic effects of zolpidem, which has a relative preference for receptors containing α1 and γ2 subunits at low doses, but can also potentiate α2/3βγ2 receptors at higher concentrations (Sieghart and Savic, 2018). In contrast, we found that apremilast shortened recovery from LORR and from ataxia induced by diazepam, which acts at receptors containing α1, α2, α3, or α5 subunits in combination with γ2 subunits (efficacy α3 > α2 > α1 ~ α5) (Sieghart and Savic, 2018). These differential effects suggest that α subunits may influence apremilast modulation of certain GABAergic drugs.
It is not known which α subunits mediate the ataxic effect of diazepam since diazepam-induced rotarod ataxia is not altered in α1, α2, or α3 knock-in mice carrying a mutation that prevents benzodiazepine binding to the respective α subunit (Low et al., 2000; Rudolph et al., 1999). Instead it appears that benzodiazepine binding to any two of these subunits is sufficient for diazepam to produce ataxia. None of the knock-in mice have undergone LORR testing in response to GABAergic drugs.
The findings in α subunit knock-in mice raised the possibility that differential modulation of receptors containing α1 and α3 subunits may account for contrasting effects of apremilast on behavioral responses to zolpidem and diazepam. Based on our electrophysiological recordings, the α-selectivity shown by diazepam and zolpidem in heterologous systems (Sieghart and Savic, 2018), and the most common combinations of GABAA subunits apparently present in brain (Benke et al., 1994), we propose the following: PKA-mediated phosphorylation reduced the function of α3β1-containing GABAA receptors resulting in reduced diazepam-induced ataxia, while PKA-mediated phosphorylation of α1β3-containing receptors increased their function and drove the increase in zolpidem-induced ataxia. Other factors in play could be the selective expression of α subunits in different neuronal circuits (Pirker et al., 2000), or a differential selectivity for the pharmacologically active metabolites derived from diazepam (Nikas et al., 2015). Alternatively, in the case of diazepam, PKA modulation by apremilast may involve another target such as a protein that interacts with GABAA receptors rather than a specific GABAA receptor subunit.
Apremilast also prolonged ataxia induced by propofol (which shows no selectivity towards β subunits in trimeric receptors) (Rudolph and Antkowiak, 2004) and loreclezole, (which preferentially modulates GABAA receptors containing β2 or β3 subunits) (Sieghart and Savic, 2018). PKA activation increases phosphorylation of β1 and β3, but not β2 subunits (McDonald et al., 1998), suggesting that β3 phosphorylation is responsible for apremilast modulation of propofol and loreclezole responses in vivo. As demonstrated in HEK293 cells (McDonald et al., 1998) and here in oocytes, phosphorylation of β3 increases inhibitory GABAA receptor-induced currents, which would be expected to increase the intoxicating motor effects and decrease acute tolerance to these drugs. Our finding that apremilast prolongs ataxia induced by ethanol and GABAergic drugs that can potentiate β3-containing receptors (propofol and loreclezole) is consistent with phosphorylation of β3 subunits being important for development of acute tolerance to ethanol.
Apremilast did not alter ataxia by etomidate, which also acts on β2/β3 subunits (Sieghart and Savic, 2018), but only β2 subunits are critical for the ataxic effects of etomidate (Reynolds et al., 2003). Because β2 subunits are not a target for PKA-mediated phosphorylation, the etomidate ataxic effect was not modified after apremilast administration. This stands in contrast with the increase in the propofol ataxic effect. Although etomidate and propofol have many similarities, including sharing GABAA receptors as main pharmacological targets, they also have clear differences in their molecular pharmacology and behavioral effects that could be responsible for this divergence (Drexler et al., 2009; Rudolph and Antkowiak, 2004).
Our findings that apremilast accelerated recovery from diazepam-induced ataxia and shortened duration of diazepam-induced LORR are consistent with a role for β1 subunits. The function of β1 subunits in GABAA receptor responses has not been well characterized, and there is also limited information about their role in behavioral responses to ethanol. To determine if apremilast modulation could be mediated by β1 subunits, we used the β1-specific antagonist SCS (Thompson et al., 2004). SCS produced faster recovery from diazepam-induced ataxia and LORR, mimicking the effect of apremilast. These findings suggest that PKA-induced phosphorylation of β1-containing receptors, which would be expected to decrease neuronal GABAA responses (McDonald et al., 1998), is involved in the ability of apremilast to accelerate recovery from the behavioral effects of diazepam. Our results agree with work showing that allosteric GABAA modulators with limited activity at β1-containing GABAA receptors have reduced ability to cause ataxia (Gee et al., 2010).
When β1-containing receptors were blocked with SCS, the recovery from etomidate-induced ataxia was not affected given that this drug acts through β2-containing receptors to impair motor responses on the rotarod (Reynolds et al., 2003). Propofol-mediated ataxia was also unaffected, providing the first evidence that this propofol effect is not mediated by β1-containing receptors. Propofol-induced LORR was also not modified by blocking β1-containing receptors. The role of β3-containing receptors in both etomidate and propofol-induced LORR has already been shown (Jurd et al., 2003), and while the increase in GABA-mediated currents through β3-containing receptors after apremilast administration explains the increase in propofol-induced LORR, the absence of change in etomidate-induced LORR seems to indicate a more complex mechanism of action. Perhaps apremilast modifies one or more of the other pharmacological targets of etomidate (Rudolph and Antkowiak, 2004).
Limiting β1-mediated responses using SCS also reduced the ataxic and sedative-hypnotic effects of ethanol, but unlike diazepam responses, SCS did not mimic the effect of apremilast. While these findings indicate a contributory role for β1 subunits in ethanol-induced ataxia, under conditions of enhanced PKA activation by apremilast, the increased ataxic effects of ethanol appear to be mediated primarily by β3-containing receptors.
Other phosphorylation-dependent mechanisms that regulate GABAA receptors in neurons were not captured by our heterologous expression system. For example, PKC also phosphorylates β3 S408/S409 in cultured cortical neurons (Brandon et al., 2000), which modifies interaction with proteins like AP2 in the intracellular loop, ultimately interfering with the receptor’s clathrin-mediated endocytosis (Nakamura et al., 2015). Thus, the phosphorylation state of β subunits in neurons may be determined by additional factors that regulate GABAA receptor function and trafficking that are absent in the heterologous system. Furthermore, cAMP elevation and subsequent PKA activation have been shown to mediate the increase of GABA release from presynaptic terminals (Diao et al., 2017; Kelm et al., 2008; Lachamp et al., 2009), adding another possible apremilast mechanism for influencing GABAergic transmission. Despite the limitations of our oocyte studies, our behavioral results indicate that PKA-mediated phosphorylation of β1 or β3 subunits is important for the effects of apremilast on responses to GABAergic drugs.
In our hypothetical model shown in Figure 11, apremilast activates PKA-mediated phosphorylation of β1- and β3-containing GABAA receptors, producing differential regulation of ethanol and other GABAergic positive allosteric modulators. We propose that apremilast reduces the ataxic and sedative-hypnotic effects of diazepam via phosphorylation of β1 receptors, while its opposing effects on β3-containing receptors decrease acute tolerance to ethanol and other GABAergic drugs.
Figure 11.
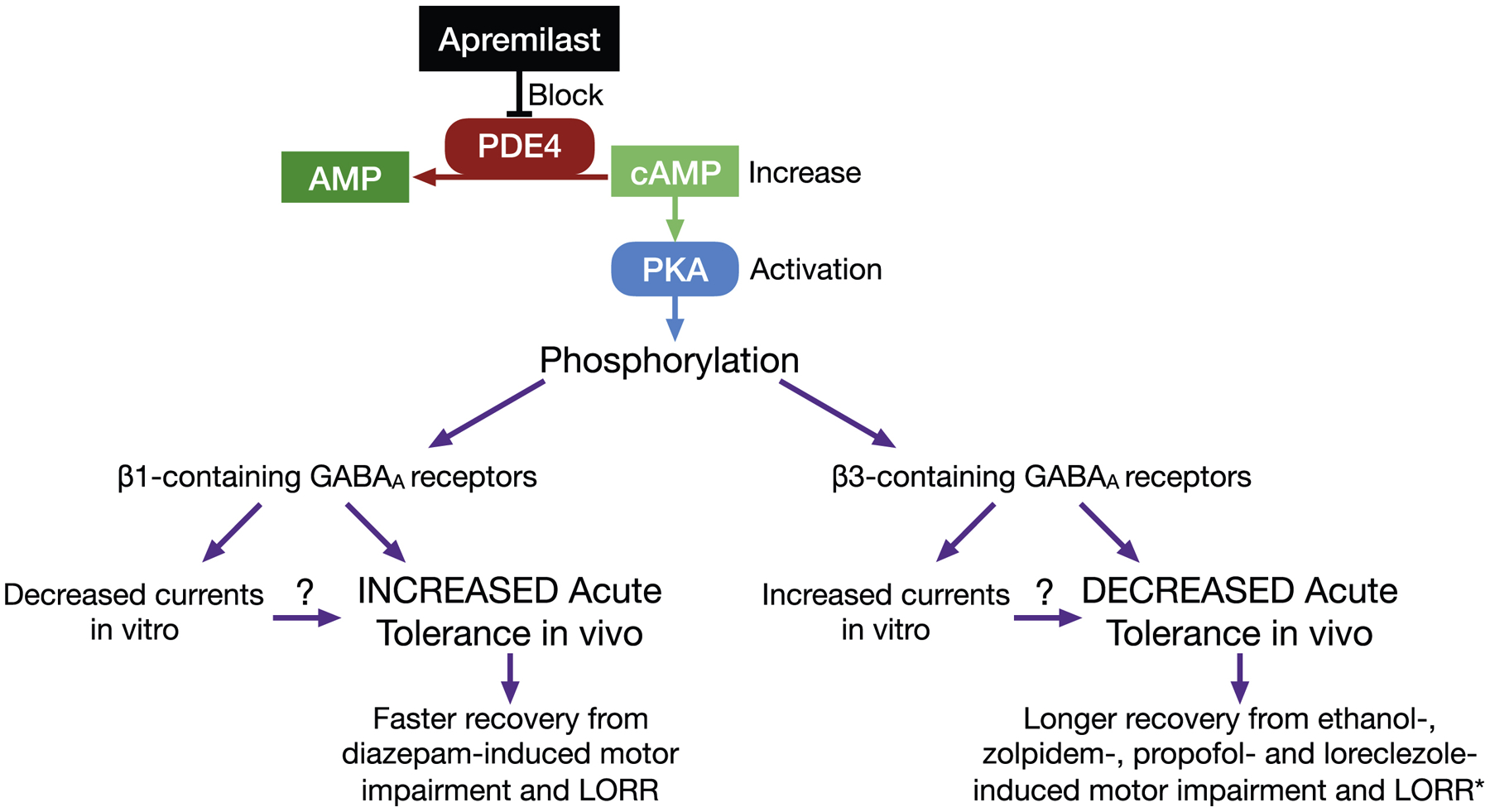
Hypothetical mechanism of action for apremilast. The phosphodiesterase type 4 (PDE4) inhibitor, apremilast, blocks hydrolysis of cAMP (cyclic adenosine monophosphate) to AMP, thus increasing levels of cAMP and activation of protein kinase A (PKA). PKA-induced phosphorylation differentially regulates β1- and β3-containing GABAA receptors (as shown in both HEK293 cells and Xenopus oocytes), producing specific effects on acute tolerance to the ataxic (rotarod) and sedative-hypnotic (LORR) effects of ethanol and other GABAergic drugs in mice. *Loreclezole-induced LORR could not be determined.
PDE4 is present throughout the brain and because apremilast acts as a nonselective inhibitor of all PDE4 subclasses, it can produce widespread effects that would depend on GABAA receptor composition and distribution. Phosphorylation of β3 subunits is consistent with the ability of apremilast to prolong ethanol-induced ataxia and decrease acute functional tolerance, as we observed in male and female mice (Blednov et al., 2018a), and is also consistent with the increased response to β3-acting drugs (propofol and loreclezole) by apremilast observed here. Given that β3 subunits are widely expressed in brain compared to the more discrete localization of β1 subunits (Hortnagl et al., 2013), and that expression levels of β1 are decreased in C57BL/6J mice compared with other strains (Mulligan et al., 2019), the net in vivo effects of apremilast on ethanol responses may be explained by actions on β3-containing GABAA receptors. Effects of the α1-selective modulator zolpidem on α1β3-containing receptors would also be consistent with its modulation by apremilast in vivo.
In summary, we propose that apremilast-induced phosphorylation of β3-containing GABAA subunits increases synaptic inhibition and decreases acute tolerance to ethanol and GABAergic drugs, which could contribute to the reduced alcohol drinking and related behaviors observed in mice (Blednov et al., 2018a; 2018b). Development of tolerance is one of the criteria for diagnosing alcohol dependence in humans, and our findings show that apremilast may be a promising candidate to reduce acute tolerance (and alcohol drinking) through PKA modulation of GABAergic signaling.
Supplementary Material
HIGHLIGHTS.
Apremilast regulates alcohol and GABAergic drug responses in vivo
Apremilast regulation occurs in a protein kinase A (PKA)-dependent manner
Phosphorylation of β1 and β3 subunits differentially alters GABAA receptor function
Apremilast acts via PKA to alter acute tolerance to alcohol and GABAergic drugs
Acknowledgements:
This work was supported by the National Institutes of Health grants U01 AA013520 to YAB and ROM and U24 AA025479 to RAH. The authors thank Jody Mayfield for contributing to the writing and editing of the manuscript and figure preparation.
ABBREVIATIONS
- cAMP
cyclic adenosine monophosphate
- EC5
effective concentration 5
- GABAA receptor
ɣ-aminobutyric acid type A receptor
- i.p.
intraperitoneal
- LORR
loss of righting reflex
- PDE4
phosphodiesterase type 4
- PKA
protein kinase A
- p.o.
per os
- SCS
salicylidene salicylhydrazide
- s.c.
subcutaneous
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no competing financial interests.
References
- Avila DV, Myers SA, Zhang J, Kharebava G, McClain CJ, Kim HY, Whittemore SR, Gobejishvili L, Barve S, 2017. Phosphodiesterase 4b expression plays a major role in alcohol-induced neuro-inflammation. Neuropharmacology 125, 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke D, Fritschy JM, Trzeciak A, Bannwarth W, Mohler H, 1994. Distribution, prevalence, and drug binding profile of gamma-aminobutyric acid type A receptor subtypes differing in the beta-subunit variant. J Biol Chem 269, 27100–27107. [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Black M, Chandra D, Homanics GE, Rudolph U, Harris RA, 2013. Linking GABA(A) receptor subunits to alcohol-induced conditioned taste aversion and recovery from acute alcohol intoxication. Neuropharmacology 67, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Black M, Harris RA, 2014. Inhibition of phosphodiesterase 4 reduces ethanol intake and preference in C57BL/6J mice. Front Neurosci 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Da Costa AJ, Harris RA, Messing RO, 2018a. Apremilast Alters Behavioral Responses to Ethanol in Mice: II. Increased Sedation, Intoxication, and Reduced Acute Functional Tolerance. Alcohol Clin Exp Res 42, 939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Da Costa AJ, Tarbox T, Ponomareva O, Messing RO, Harris RA, 2018b. Apremilast Alters Behavioral Responses to Ethanol in Mice: I. Reduced Consumption and Preference. Alcohol Clin Exp Res 42, 926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Kittler JT, McDonald BJ, Sieghart W, Brown DA, Smart TG, Moss SJ, 2000. GABAA receptor phosphorylation and functional modulation in cortical neurons by a protein kinase C-dependent pathway. J Biol Chem 275, 38856–38862. [DOI] [PubMed] [Google Scholar]
- Chandra D, Jia F, Liang J, Peng Z, Suryanarayanan A, Werner DF, Spigelman I, Houser CR, Olsen RW, Harrison NL, Homanics GE, 2006. GABAA receptor alpha 4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc Natl Acad Sci U S A 103, 15230–15235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao HL, Xue Y, Han XH, Wang SY, Liu C, Chen WF, Chen L, 2017. Adenosine A2A Receptor Modulates the Activity of Globus Pallidus Neurons in Rats. Front Physiol 8, 897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler B, Jurd R, Rudolph U, Antkowiak B, 2009. Distinct actions of etomidate and propofol at beta3-containing gamma-aminobutyric acid type A receptors. Neuropharmacology 57, 446–455. [DOI] [PubMed] [Google Scholar]
- Erickson EK, Grantham EK, Warden AS, Harris RA, 2019. Neuroimmune signaling in alcohol use disorder. Pharmacol Biochem Behav 177, 34–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KM, Hauser SR, Lasek AW, McClintick J, Ding ZM, McBride WJ, Bell RL, 2015. Reduction of alcohol drinking of alcohol-preferring (P) and high-alcohol drinking (HAD1) rats by targeting phosphodiesterase-4 (PDE4). Psychopharmacology (Berl) 232, 2251–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee KW, Tran MB, Hogenkamp DJ, Johnstone TB, Bagnera RE, Yoshimura RF, Huang JC, Belluzzi JD, Whittemore ER, 2010. Limiting activity at beta1-subunit-containing GABAA receptor subtypes reduces ataxia. J Pharmacol Exp Ther 332, 1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y, 1984. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry 23, 5036–5041. [DOI] [PubMed] [Google Scholar]
- Hortnagl H, Tasan RO, Wieselthaler A, Kirchmair E, Sieghart W, Sperk G, 2013. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience 236, 345–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Lu T, Chen A, Huang Y, Hansen R, Chandler LJ, Zhang HT, 2011. Inhibition of phosphodiesterase-4 decreases ethanol intake in mice. Psychopharmacology (Berl) 218, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U, 2003. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J 17, 250–252. [DOI] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR, 2008. The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. J Neurophysiol 100, 3417–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Ren Q, Beckley JH, O’Buckley TK, Gigante ED, Santerre JL, Werner DF, Morrow AL, 2012. Ethanol Activation of Protein Kinase A Regulates GABA(A) Receptor Subunit Expression in the Cerebral Cortex and Contributes to Ethanol-Induced Hypnosis. Front Neurosci 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachamp PM, Liu Y, Liu SJ, 2009. Glutamatergic modulation of cerebellar interneuron activity is mediated by an enhancement of GABA release and requires protein kinase A/RIM1alpha signaling. J Neurosci 29, 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, Datta G, Davila-Velderrain J, McGuire D, Tian C, Zhan X, andMe Research T, Psychiatry HA-I, Choquet H, Docherty AR, Faul JD, Foerster JR, Fritsche LG, Gabrielsen ME, Gordon SD, Haessler J, Hottenga JJ, Huang H, Jang SK, Jansen PR, Ling Y, Magi R, Matoba N, McMahon G, Mulas A, Orru V, Palviainen T, Pandit A, Reginsson GW, Skogholt AH, Smith JA, Taylor AE, Turman C, Willemsen G, Young H, Young KA, Zajac GJM, Zhao W, Zhou W, Bjornsdottir G, Boardman JD, Boehnke M, Boomsma DI, Chen C, Cucca F, Davies GE, Eaton CB, Ehringer MA, Esko T, Fiorillo E, Gillespie NA, Gudbjartsson DF, Haller T, Harris KM, Heath AC, Hewitt JK, Hickie IB, Hokanson JE, Hopfer CJ, Hunter DJ, Iacono WG, Johnson EO, Kamatani Y, Kardia SLR, Keller MC, Kellis M, Kooperberg C, Kraft P, Krauter KS, Laakso M, Lind PA, Loukola A, Lutz SM, Madden PAF, Martin NG, McGue M, McQueen MB, Medland SE, Metspalu A, Mohlke KL, Nielsen JB, Okada Y, Peters U, Polderman TJC, Posthuma D, Reiner AP, Rice JP, Rimm E, Rose RJ, Runarsdottir V, Stallings MC, Stancakova A, Stefansson H, Thai KK, Tindle HA, Tyrfingsson T, Wall TL, Weir DR, Weisner C, Whitfield JB, Winsvold BS, Yin J, Zuccolo L, Bierut LJ, Hveem K, Lee JJ, Munafo MR, Saccone NL, Willer CJ, Cornelis MC, David SP, Hinds DA, Jorgenson E, Kaprio J, Stitzel JA, Stefansson K, Thorgeirsson TE, Abecasis G, Liu DJ, Vrieze S, 2019. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 51, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hao PD, Yang MF, Sun JY, Mao LL, Fan CD, Zhang ZY, Li DW, Yang XY, Sun BL, Zhang HT, 2017. The phosphodiesterase-4 inhibitor roflumilast decreases ethanol consumption in C57BL/6J mice. Psychopharmacology (Berl) 234, 2409–2419. [DOI] [PubMed] [Google Scholar]
- Low K, Crestani F, Keist R, Benke D, Brunig I, Benson JA, Fritschy JM, Rulicke T, Bluethmann H, Mohler H, Rudolph U, 2000. Molecular and neuronal substrate for the selective attenuation of anxiety. Science 290, 131–134. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Amato A, Connolly CN, Benke D, Moss SJ, Smart TG, 1998. Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat Neurosci 1, 23–28. [DOI] [PubMed] [Google Scholar]
- Mulligan MK, Abreo T, Neuner SM, Parks C, Watkins CE, Houseal MT, Shapaker TM, Hook M, Tan H, Wang X, Ingels J, Peng J, Lu L, Kaczorowski CC, Bryant CD, Homanics GE, Williams RW, 2019. Identification of a Functional Non-coding Variant in the GABA A Receptor alpha2 Subunit of the C57BL/6J Mouse Reference Genome: Major Implications for Neuroscience Research. Front Genet 10, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Darnieder LM, Deeb TZ, Moss SJ, 2015. Regulation of GABAARs by phosphorylation. Adv Pharmacol 72, 97–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikas P, Gatta E, Cupello A, Di Braccio M, Grossi G, Pellistri F, Robello M, 2015. Study of the Interaction of 1,4- and 1,5-Benzodiazepines with GABAA Receptors of Rat Cerebellum Granule Cells in Culture. J Mol Neurosci 56, 768–772. [DOI] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Treistman SN, 2008. The molecular basis of tolerance. Alcohol Res Health 31, 298–309. [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G, 2000. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101, 815–850. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, Rosahl TW, Cirone J, O’Meara GF, Haythornthwaite A, Newman RJ, Myers J, Sur C, Howell O, Rutter AR, Atack J, Macaulay AJ, Hadingham KL, Hutson PH, Belelli D, Lambert JJ, Dawson GR, McKernan R, Whiting PJ, Wafford KA, 2003. Sedation and anesthesia mediated by distinct GABA(A) receptor isoforms. J Neurosci 23, 8608–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Antkowiak B, 2004. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci 5, 709–720. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Mohler H, 1999. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature 401, 796–800. [DOI] [PubMed] [Google Scholar]
- Sieghart W, Savic MM, 2018. International Union of Basic and Clinical Pharmacology. CVI: GABAA Receptor Subtype- and Function-selective Ligands: Key Issues in Translation to Humans. Pharmacol Rev 70, 836–878. [DOI] [PubMed] [Google Scholar]
- Stahl K, Stahl M, de Jonge HR, Forrest JN Jr., 2015. ANP and CNP activate CFTR expressed in Xenopus laevis oocytes by direct activation of PKA. J Recept Signal Transduct Res 35, 493–504. [DOI] [PubMed] [Google Scholar]
- Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS, 2000. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci 20, RC75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, Wheat L, Brown NA, Wingrove PB, Pillai GV, Whiting PJ, Adkins C, Woodward CH, Smith AJ, Simpson PB, Collins I, Wafford KA, 2004. Salicylidene salicylhydrazide, a selective inhibitor of beta 1-containing GABAA receptors. Br J Pharmacol 142, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen RT, Zhang FF, Zhang HT, 2018. Cyclic nucleotide phosphodiesterases: potential therapeutic targets for alcohol use disorder. psychopharmacol 235, 1793–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen RT, Zhang M, Qin WJ, Liu Q, Wang WP, Lawrence AJ, Zhang HT, Liang JH, 2012. The phosphodiesterase-4 (PDE4) inhibitor rolipram decreases ethanol seeking and consumption in alcohol-preferring Fawn-Hooded rats. Alcohol Clin Exp Res 36, 2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.