

Abstract
The ABCA4 protein (then called a “rim protein”) was first identified in 1978 in the rims and incisures of rod photoreceptors. The corresponding gene, ABCA4, was cloned in 1997, and variants were identified as the cause of autosomal recessive Stargardt disease (STGD1). Over the next two decades, variation in ABCA4 has been attributed to phenotypes other than the classically defined STGD1 or fundus flavimaculatus, ranging from early onset and fast progressing cone-rod dystrophy and retinitis pigmentosa-like phenotypes to very late onset cases of mostly mild disease sometimes resembling, and confused with, age-related macular degeneration. Similarly, analysis of the ABCA4 locus uncovered a trove of genetic information, including >1200 disease-causing mutations of varying severity, and of all types – missense, nonsense, small deletions/insertions, and splicing affecting variants, of which many are located deep-intronic. Altogether, this has greatly expanded our understanding of complexity not only of the diseases caused by ABCA4 mutations, but of all Mendelian diseases in general. This review provides an in depth assessment of the cumulative knowledge of ABCA4-associated retinopathy – clinical manifestations, genetic complexity, pathophysiology as well as current and proposed therapeutic approaches.
Keywords: Stargardt disease, ABCA4-associated retinopathy, Allelic heterogeneity, Autofluorescence, Phenocopies, Hypomorphic variant, Penetrance, Splice defects, Pseudoexon, Structural variant, Therapy
1. Historical perspective
Hereditary dystrophies of the macula reminiscent of autosomal recessive Stargardt disease (STGD1) have been documented from as early as the end of the 19th century (Lang, 1885). However, Karl Bruno Stargardt, of the University of Strasbourg, is recognized as having first published the most comprehensive clinical description, including fundus drawings, of seven patients from two families in 1909 (Stargardt, 1909). In this seminal article, Stargardt concluded that the patients had a genetic, neuroepithelial disease that initially affected cones, followed by the retinal pigment epithelium (RPE) and subsequently, the underlying choroid.
Decades later, in 1962, the Swiss ophthalmologist Adolph Franceschetti coined the term “fundus flavimaculatus” in a cohort of patients he described as having a “peculiar fundus affection”, to colleagues at a meeting of the German Ophthalmological Society in Hamburg. In collaboration with Jules Francois from Ghent University, Franceschetti further described 36 cases in two articles published in 1965 (Franceschetti, 1965; Franceschetti and Francois, 1965). In the latter article, Franceschetti suspected that these patients had the same condition as earlier described by Stargardt opining, “if the foci (flecks) are localized at the posterior pole of the eye and accompanied by macular affection, the distinction from Stargardt disease may be difficult or even impossible”. Further evidence arrived two years later when Alex E. Krill and Bertha A. Klien documented the presence of delayed dark adaptation in a similar cohort of patients whom they described as having “flecked retina syndrome” (Klien and Krill, 1967). They presented the first histopathological analysis of an eye from a patient in the third decade of life concluding that the primary abnormality of this condition lies within the RPE. In 1971, August F. Deutman in great detail described 25 STGD1 and six fundus flavimaculatus families (Deutman, 1971). In 1975, Francois confirmed the connection between all of these disorders citing characteristic heritable, clinical and electrophysiological features (François et al., 1975). Although regarded as a distinct disease entity, Gerald Fishman, of the Illinois Eye and Ear Infirmary in Chicago, recognized differences in clinical expression and accordingly established a four-tier classification system (Fishman, 1976) that to this day remains influential to ophthalmologists around the world. From his investigation of fundoscopic and electrophysiological findings of 38 patients, Fishman classified the severity of STGD1 across the following stages:
Stage I: Confined central macular lesions ranging from irregular pigmentary mottling to well-defined lesions of RPE atrophy with a characteristic “beaten-bronze” or “snail-slime” appearance underlying central or paracentral scotomas.
Stage 2: Presence of yellow fundus flecks, some of which may be resorbed, beyond 1 disc diameter from the fovea extending beyond the vascular arcades and regions nasal to the optic disc.
Stage 3: Diffusely resorbed flecks and choriocapillaris atrophy within the macula.
Stage 4: Extensive choriocapillaris atrophy throughout the posterior pole resulting in moderate to severe restriction of peripheral fields.
Numerous clinical studies have since been published adding to the growing body of knowledge. However, the “modern era” of our understanding of STGD1 was precipitated by parallel breakthroughs in the basic sciences beginning with the initial characterization of the ABCA4 protein in 1978, which was initially referred to as “rim” protein for its localization in rod photoreceptor outer segments and incisures (Papermaster et al., 1976). The genetic locus was mapped to 1p13 in mid-1990s (Anderson et al., 1995; Gerber et al., 1995; Kaplan et al., 1993) and, finally, the gene was cloned in 1997 (Allikmets et al., 1997). Taken together, cumulative advances over the last three decades have provided a defining foundation for understanding what we now know to be the most common inherited Mendelian eye disorder in the world.
2. Clinical hallmarks of ABCA4-associated retinopathy
2.1. Ophthalmic examination
In the “classic” presentation of STGD1, central vision loss typically becomes apparent between adolescence and young adulthood. However, the age of onset varies extensively, where a proportion of individuals start experiencing delayed vision loss between the 4th and 7th decades of life (Gerber et al., 1995; Lambertus et al., 2016; Runhart et al., 2018; Runhart et al., 2019; Westeneng-van Haaften et al., 2012; Yatsenko et al., 2001; Zernant et al., 2017; Zernant et al., 2018). The exact age of disease onset is often difficult to determine, as many patients—particularly children—may be unaware of their visual impairment or have preserved central vision due to functional sparing of the fovea (Bax et al., 2019b; Fujinami et al., 2013b; Nakao et al., 2012; Runhart et al., 2019; van Huet et al., 2014). In general, ABCA4-associated retinopathy subtypes that manifest early in life tend to progress more rapidly, while a later age of onset is associated with a milder prognosis (Fujinami et al., 2015; Tanaka et al., 2018; Zernant et al., 2017). The initial symptoms of ABCA4-associated retinopathy typically begin with central or pericentral vision loss and may include difficulty with dark adaptation as the disease progresses in severity (Fishman et al., 1999; Kang Derwent et al., 2004; Klien and Krill, 1967; Salvatore et al., 2014; Scholl et al., 2002). The latter is seldomly reported by patients but generally recognized upon careful inquiry in the clinic. Other symptoms may include impaired color discrimination and photophobia (Klevering et al., 2002; Rotenstreich et al., 2003). Examination of the anterior segment and vitreous is generally unremarkable. As most patients present to the clinic in relatively early disease stages, fundoscopic findings may be subtle and rarely include typical features of advanced retinal degeneration such as a pale, disc pallor or extensive attenuation of the retinal vessels.
2.2. Family history and inheritance patterns
Although STGD1 is an autosomal recessive disease caused by bi-allelic variants in ABCA4 (Allikmets et al., 1997), several factors need to be considered when taking a thorough family history and constructing a pedigree. Due to its clinical heterogeneity, many phenocopies—retinal disease caused by other genes resembling ABCA4-associated retinopathy—exist, including dominantly inherited conditions. Furthermore, some of the more prevalent dominant masqueraders exhibit incomplete penetrance across generations which may further simulate autosomal recessive inheritance in a pedigree (Chapi et al., 2019; Michaelides et al., 2005; Shankar et al., 2016; Sohocki et al., 1998). One should be aware of including relatives with age-related macular degeneration (AMD) in the assessment of family history due to its overlapping symptoms with late-onset STGD1 including central vision loss, especially as it has been reported that the prevalence of AMD is higher in families with STGD1 (Allikmets, 2000; Souied et al., 1999). Lastly, pseudodominant inheritance has been extensively reported in families segregating three (Beit-Ya’acov et al., 2007; Huckfeldt et al., 2016; Shroyer et al., 2000), or even four ABCA4 alleles within a single family (Klevering et al., 2002; Lee et al., 2016; Runhart et al., 2018). A further heightened awareness is warranted when working with patients of consanguineous families, mostly from culturally and geographically isolated populations due to the founder alleles resulting in frequent enrichment of homozygosity (Ducroq et al., 2006; Falfoul et al., 2018; Lee et al., 2017).
2.3. The diagnostic triad
Despite the breadth of clinical heterogeneity associated with ABCA4-associated retinopathy, all patients share a common genetic etiology caused by mutations in a single gene, and a set of near ubiquitous clinical features. The following are three diagnostic clinical findings that, when occurring together in a patient, are highly indicative of ABCA4-associated retinopathy:
2.3.1. Macular affection
Progressive deterioration of cellular layers originating in the central macula is a canonical feature of ABCA4-associated retinopathy and the principal cause of visual deterioration over time. The cellular origin of ABCA4 dysfunction and temporal sequence of cellular degeneration has been and remains a contentious issue (Duncker et al., 2014; Greenstein et al., 2017; Lenis et al., 2018; Song et al., 2015a; Sparrow et al., 2012). Clinically, atrophic changes typically begin with loss of the outer retinal layers (retinal pigment epithelium - RPE and photoreceptor-attributable ellipsoid zone) and, in cases that progress to more advanced stages, invariably involve the choriocapillaris. Further deterioration of the underlying choroidal layers (Sattler and Haller) may occur in response to the rapid demise of RPE (Bertelsen et al., 2014; Muller et al., 2017; Tanaka et al., 2018) or progressively to a stage at which the underlying sclera may be visible on fundoscopy depending on the duration of disease (Fig. 1) (Lee et al., 2018).
Fig. 1.

Stages of atrophy progression in ABCA4-associated retinopathy. Fundus photographs with corresponding short wavelength autofluorescence (SW-AF) images and foveal spectral domain-optical coherence tomography (SD-OCT) scans depicting the progressive stages of macular atrophy in ABCA4-associated retinopathy. (A) Early lesions exhibit a mottled appearance on funduscopy and (B) diffusely decreased autofluorescence on SW-AF imaging. (C) An apparent loss of the photoreceptor-attributable ellipsoid zone (EZ) band and appearance of hyper-reflective debris can be observed by SD-OCT within the lesion at this stage. (D) Lesions in the chorioretinal atrophy stage exhibit the canonical beaten-bronze appearance, are well-delineated and enable visibility of underlying choroidal vessels. (E) This stage is also uniquely characterized by a homogeneous and complete loss of autofluorescence; (F) A marked thinning of the retinal pigment epithelium (RPE) layer resulting in an increased transmission of the SD-OCT signal (F, inset) is typically present at this stage. (G, H) Continued progression of atrophy extends across the macula and posteriorly, sequentially involving the choriocapillaris, Sattler and Haller layers of the choroid (I, inset). (J, K) The end-stage of widespread degeneration results in a complete loss of outer retinal and choroidal layers (L) resulting in a visibility of the underlying sclera. The discernible edge of the atrophic lesion and its corresponding position on SD-OCT are denoted by yellow arrows (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article).
2.3.2. Fundus flecks
The augmented accumulation of RPE lipofuscin occurs throughout the retina; however, this process can also manifest locally giving rise to one of the most recognizable features of ABCA4-associated retinopathy—flecks. This feature is most conspicuous on short-wavelength autofluorescence (SW-AF) as intensely fluorescent foci distributed across the macula or extending far across the posterior pole at more advanced disease stages (Chen et al., 2019; Cideciyan et al., 2015). Histopathological observations have attributed flecks to engorged RPE cells, stacked aggregations of RPE cells or remnants of dying RPE (Eagle et al., 1980; Lopez et al., 1990). However more recently, Sparrow et al. proposed that flecks may be extracellular accumulations of unphagocytized outer segments from longitudinal observations of their structural (SD-OCT) and autofluorescence (SW-AF and near infrared-AF) characteristics over time (Sparrow et al., 2015). The ubiquitous presence of flecks in ABCA4-associated retinopathy is an invaluable diagnostic asset in the clinic; however, the inherent variation in their individual size and morphology, irregular patterns across the fundus and spatial evolution over time, may conceal crucial information about disease etiology and prognosis. The most frequent fleck patterns of ABCA4-associated retinopathy are illustrated in Fig. 2. Unfortunately, very few studies have comprehensively explored the significance of fleck characteristics to date. This is likely due to the community’s preoccupation with lesion-centric characteristics and atrophy progression. Functionally, patients exhibit decreased visual sensitivity over flecked areas but their contribution to the centrally progressing atrophy is uncertain (Querques et al., 2006; Verdina et al., 2012). Flecks are generally regarded as a biomarker of disease severity based on their emergence along the central and peripheral axis of the ABCA4-associated retinopathy retina (Cukras et al., 2012). Teussink et al. sought to study the effect of light on the progression of ABCA4-associated retinopathy by comparing fleck accumulation in an eye of patients compared to the fellow eye that was continuously patched over one year, but the results were variable (Teussink et al., 2015). The causal role of flecks in the pathophysiology of ABCA4-associated retinopathy is thus far uncertain and much remains to be elucidated. Particular insight may lie in their longitudinal patterns—centrifugal (Cukras et al., 2012) versus zonal (Paavo et al., 2019; Sparrow et al., 2015). Furthermore, flecks are highly dynamic and exhibit rapid changes, which may better define the “leading disease front” of ABCA4-associated retinopathy as compared to the central lesion of atrophy.
Fig. 2.

Morphological spectrum of fundus flecks in ABCA4-associated retinopathy. Lipofuscin-laden flecks deposited across the fundus exhibit a yellow appearance on fundoscopy and an intense autofluorescent signal on short wavelength autofluorescence (SW-AF) imaging. The collective spatio-temporal pattern of flecks and their individual morphology (A–G) vary across disease stage and genotypic trajectories. Areas of resorbing flecks become hypoautofluorescent and coalescence into a heterogeneous pattern across the posterior pole.
2.3.3. Peripapillary sparing
Perhaps the most unusual feature amongst the triad is the observation that the proximal tissue surrounding the optic nerve is spared of disease changes in ABCA4-associated retinopathy (Fig. 3) (Cideciyan et al., 2005). Other diseases, most notably PRPH2- and ROM1-associated pattern dystrophy and RDH12-associated Leber congenital amaurosis (LCA), have been reported to exhibit a similar manifestation although not as consistent as in ABCA4-associated retinopathy (Duncker et al., 2015c; Garg et al., 2017; Ma et al., 2019). Sparing of this region often persists in ABCA4-associated retinopathy, both structurally and functionally but progressively lost at later disease stages; however, its etiological basis is unknown. Several theories have been proposed including the disc membrane–load hypothesis, light-load hypothesis, lipofuscin-clearance hypothesis and neurotrophic factors hypothesis, although all are largely inferential (Cideciyan et al., 2005).
Fig. 3.

Disease-sparing of the peripapillary region in ABCA4-associated retinopathy. Sparing of the peripapillary region around the optic nerve (magenta arrowheads) from disease changes is a characteristic feature of ABCA4-associated retinopathy and becomes apparent as flecks extend centripetally across the posterior pole of the retina (A, B). Resistance of this region persists into the late atrophic stages and gradually becomes affected (C). The presence of residual circumpapillary tissue may be discernible despite the widespread atrophy (D).
Each feature of the ABCA4-associated retinopathy triad exhibits stage-dependent changes, which have been documented independently. Effective modeling of ABCA4-associated retinopathy should encompass the variability of all three features and their relationship to one another in order to acquire a deeper understanding of the condition.
2.4. Bull’s eye maculopathy
A diagnostic exception to the triad is the Bull’s eye maculopathy (BEM) stage which accounts for up to ~20% of ABCA4-associated retinopathy cases presenting to the clinic and is over-represented by the c.5882G>A, p.(G1961E) variant (Celia et al., 2009). BEM is defined as a circularly confined region of atrophy beginning in the central macula. The BEM stage precedes the development of all triad features making this stage the most challenging to diagnose. Furthermore, overlapping variations of the BEM phenotype are found in many inherited macular as well as non-genetic conditions including hydroxychloroquine toxicity, infectious diseases and acute injuries, although BEM observed in ABCA4-associated retinopathy can be often distinguished by quantifying macular levels of autofluorescence (RPE lipofuscin) (Duncker et al., 2015b). A detailed discussion of differential diagnoses is presented in section 3, Phenocopies of ABCA4-associated retinopathy. Representative manifestations of BEM in ABCA4-associated retinopathy are provided in Fig. 4.
Fig. 4.

Autofluorescence subtypes of the bull’s eye maculopathy (BEM) stage of ABCA4-associated retinopathy. Confined BEM lesions are generally the earliest manifestation of macular affection in ABCA4-associated retinopathy and are highly associated with the c.5882G>A, p.(Gly1961Glu) mutation. (A–C) Small, focal lesions are typically associated with a loss of the ellipsoid zone (EZ) band and subsequent cavitation of this space in the fovea (“optical gap”). (D–F) Uniformly round BEM lesions exhibit continuous autofluorescence borders and punctate debris within the atrophy region. (G–I) Elliptical BEM lesions also exhibit smooth, continuous autofluorescent borders; however, the region inside the lesion contains less debris and are marked by a central patch of autofluorescence (“bull’s eye”) indicating prior sparing of the fovea. Much less common are centrally mottled BEM lesions (J–L) which are distinct in that they lack a hyperautofluorescent perimeter and are almost exclusive to adolescent patients.
2.5. Early perturbations in young patients
Studies to date indicate that visual function loss may precede readily detectable fundus features in young patients on clinical exam, although asymptomatic cases due to foveal sparing can be incidentally encountered by routine examination (Khan et al., 2018; Lee et al., 2014). In a cohort of 50 young ABCA4-associated retinopathy patients (age ≤ 10 years), Lambertus and colleagues identified 10 individuals with visual function loss in the absence of discernible fundus abnormalities at the time of examination and reported the onset of visual acuity decline from as early as 3 years of age (Lambertus et al., 2015). Defects in color vision have also been reported in patients with early to no detectable fundus changes (Bax et al., 2019a; Vandenbroucke et al., 2015). The youngest documented case of ABCA4-associated retinopathy was an asymptomatic 5-year-old girl in a pseudodominant family harboring the c.5018 + 2T>C, p.(?) and c.5882G>A, p.(G1961E) alleles. At the time of examination, the girl had mildly decreased visual acuities and no fundus changes except a prominent thickening of the external limiting membrane (ELM) on OCT (Burke et al., 2013). A subsequent study (Lee et al., 2014) and several others thereafter (Bax et al., 2019a; Melillo et al., 2016; Pang et al., 2015; Park et al., 2015) corroborated the observation of ELM thickening to be a prominent feature of early stage ABCA4-associated retinopathy while more recently, this thickening has been attributed to the adjacent outer nuclear layer (ONL) (Khan et al., 2018). It is possible that structural changes occur prior to functional loss in patients. Considering the pathophysiology of STGD1, marginal increases in autofluorescence or microscopic perturbations in the cone or rod mosaic may be plausible outcomes to pursue in the future with advances in quantitative autofluorescence (qAF) imaging and adaptive optics-scanning laser ophthalmoscopy (AO-SLO), respectively. Doing so will shed light on the anatomic origin of STGD1 and ultimately shape therapeutic approaches.
2.6. Characteristics of advanced stages
Progression to the advanced stages of ABCA4-associated retinopathy varies in accordance with the age of disease onset. An earlier onset of disease is usually associated with a poorer prognosis in patients. In the most severe cases, generalized rod and cone function is unrecordable by ffERG, vision deteriorates to hand motion or light perception and chorioretinal atrophy extends as far as the equatorial regions of the eye and as deep as the underlying sclera. The emergence of pigmentary changes is also highly associated with advanced disease and often accompanies other indicators of retina-wide degeneration such as waxy optic disc pallor and severe attenuation of the retinal vasculature. The appearance of the pigment deposits ranges from nummular aggregations that co-localize with atrophic lesions to extensive bone spiculeshaped depositions. The appearance of the latter is identical to the pathognomonic bone-spicule pigment seen in the fundus of patients with retinitis pigmentosa (RP).
3. Phenocopies of ABCA4-associated retinopathy
The clinical expression of STGD1, an autosomal recessive disease, is extremely variable (see above). There have been three more loci, called STGD2-4, which describe genetically and phenotypically different diseases. Historically, the term “Stargardt-like macular dystrophy” was introduced in 1994 for a dominantly inherited maculopathy, which mapped to a locus on 6q (Stone et al., 1994). The ELOVL4 gene was later cloned from this locus, which is also called STGD3. Phenotypes caused by ELOVL4 mutations are clinically, genetically and pathophysiologically very different from the “real” Stargardt disease due to mutations in ABCA4 (STGD1). While the macular dystrophy phenotype of the dominant forms of the disease may somewhat resemble STGD1, the recessive forms express spinocerebellar ataxia 34 (Giroux and Barbeau, 1972; Turcotte Gauthier, 2010), ichthyosis, spastic quadriplegia and, mental retardation (Aldahmesh et al., 2011), among others. All forms of the disease are due to defects in fatty acid metabolism (Agbaga et al., 2008). Another suggested independent locus for STGD2 disease was eventually discarded and included in STGD3. The locus for another “Stargardt-like” phenotype, STGD4 (Kniazeva et al., 1999), contains the PROM1 gene. While the phenotype of patients caused by dominant PROM1 mutations sometimes resembles ABCA4-associated retinopathy (Wolock et al., 2019), the recessive form resembles RP (Maw et al., 2000; Zhang et al., 2007). These terms have, unfortunately, remained in the literature and even evolved into “dominant Stargardt disease”, which is misleading and incorrect.
The common denominator for STGD1/ABCA4-associated retinopathy is maculopathy; i.e., the disease invariably begins in the central macula; however, as described below, the age of onset and progression are highly variable depending on the combination of specific disease-causing alleles and modifiers. Monogenic maculopathies collectively comprise a larger group; currently variants in ~38 genes are known to cause macular disease (Table 1). Some of these, e.g., CRX, MT-TL1, PPRH2, RDH12 and RPGR-associated diseases, can be challenging to distinguish from various stages of ABCA4-associated retinopathy (Fig. 5). Moreover, some maculopathies are also caused by environmental factors, such as drug-toxicity (e.g. hydroxychloroquine) (Noupuu et al., 2016; Shroyer et al., 2001a), light damage, etc. Therefore, it is often practically impossible to determine the cause of a maculopathy without comprehensive genetic screening. Even at the most advanced retinal centers, about 10–15% of cases, who are clinically diagnosed with ABCA4-associated retinopathy, do not harbor disease-causing ABCA4 variants. Most of these cases, called phenocopies, are solved by more thorough clinical assessment, including a careful examination of family history, knowledge of environmental exposure and, eventually, by genetic screening, usually by whole exome sequencing (WES) (Wolock et al., 2019).
Table 1.
Clinical summary of ABCA4-associated retinopathy phenocopying genes and their respective phenotypes.
| Phenocopy | Gene(s) | Disease | Inheritance |
ABCA4-associated
retinopathy triad |
Auxiliary ABCA4-associated retinopathy feature | Pathognomonic feature (Non-ABCA4-associated retinopathy) | ||
|---|---|---|---|---|---|---|---|---|
| Macular affection | Flecks | Peripapillary sparing | ||||||
| Tier 1 | PRPH2 | Pattern macular dystrophy | AD | x | x | x* | ||
| ROM1 | Pattern macular dystrophy | AR | x | x | x | |||
| Tier 2 | ABCC6 | Pseudoxanthoma elastieum (PXE) | AR | x | x | Angioid streaks; systemic features | ||
| ALDH3A2 | Sjögren-Larsson syndrome | AR | x | x | Systemic features | |||
| BEST1 | Vitelliform macular dystrophy | AR, AD | x | x* | Vitelliform | Multifocal vitelliform lesions in AR disease | ||
| CDHR1 | Cone-rod dystrophy | AR | x | x* | BEM | |||
| CHM | Choroideremia | XL | x | x* | Chorioretinal atrophy; foveal sparing* | |||
| COL4A3, COL4A4, COL4A5 | Alport syndrome | XL* | x | x | BEM | Systemic features | ||
| CTNNA1 | Butterfly-shaped pigment dystrophy | AD | x | x | ||||
| ELOVL4 | Stargardt disease 3 (STGD3) | AD | x | x* | BEM | |||
| MT-TL1, MT-TK, or MT-TE | Maternally-inherited diabetes and deafness (MIDD) | Maternal | X | X | Foveal sparing | |||
| PROM1 | Stargardt disease 4 (STGD4) | AD | x | x* | BEM | |||
| RDH12 | Leber congenital amaurosis (LCA) | AR | x | x | Phenocopies advanced stage STGD1 | Variegated watercolour-like pattern of atrophy | ||
| TIMP3 | Sorsby fundus dystrophy | AD | x | x* | Chorioretinal atrophy | Nummular atrophy | ||
| ZFYVE26 | Kjellin syndrome | AR | x | x | Systemic features | |||
| Tier 3 | C1QTNF5 | Late-onset retinal degeneration (L-ORD) | AD | x | Chorioretinal atrophy | |||
| CERKL | Retinitis pigmentosa with macular involvement | AR | x | Increased AF | ||||
| CNGA3 | Achromatopsia | AR | x | BEM, optical gap | Severely impaired color distinction; nystagmus | |||
| CNGB3 | Achromatopsia | AR | x | BEM, optical gap | Severely impaired color distinction; nystagmus | |||
| CRB1 | Macular dystrophy | AR | x | foveal sparing* | ||||
| CRX | Cone-rod dystrophy | AD | x | BEM; foveal sparing | ||||
| DRAM2 | Cone-rod dystrophy | AR | x | BEM | ||||
| EFEMP1 | Doyne honeycomb retinal dystrophy; malattia leventinese | AD | x | x* | Drusen (autofluorescent) | |||
| GUCA1A | Cone/Cone-rod dystrophy | AD | x | BEM | ||||
| GUCY2D | Cone-rod dystrophy | AD, AR | x | BEM | ||||
| IMPG1 | Vitelliform macular dystrophy | AD | x | Vitelliform | ||||
| KCNV2 | Cone dystrophy with supernormal rod response (CDSRR) | AR | x | BEM | Supernormal rod response | |||
| MFSD8 | Macular dystrophy | AR | x | BEM; foveal sparing* | ||||
| OPN1LW | Blue cone monochromacy | XL | x | Optical gap | Severely impaired color distinction | |||
| OPN1MW | Blue cone monochromacy | XL | x | Optical gap | Severely impaired color distinction | |||
| PDE6C | Cone dystrophy | AR | x | Optical gap; BEM | ||||
| PLA2G5 | Benign fleck | AR | x | Macular sparing | ||||
| POC1B | Cone-rod dystrophy | AR | x | Optical gap | ||||
| RDH5 | Fundus albipunetatis | AR | x | Macular sparing | ||||
| RIMS1 | Cone-rod dystrophy | ? | x | BEM | ||||
| RLBP1 | Retinitis punctata albescens | AR | x | Macular sparing | ||||
| RP1L1 | Occult macular dystrophy | AD | x | Optical gap | ||||
| RPE65 | Retinitis pigmentosa/Leber congenital amaurosis | AR | x | Chorioretinal atrophy | ||||
| RPGR | Cone-rod dystrophy | XL | x | BEM; foveal sparing* | Lyonization in female relatives | |||
| TTLL5 | Cone-rod dystrophy | AR | X | BEM; foveal sparing* | ||||
Phenocopying genes are grouped into three tiers according to their shared phenotypic features with ABCA4-associated retinopathy. Auxiliary ABCA4-associated retinopathy features include stage-dependent characteristics or phenotypes belonging to clinical or genetic subgroups of ABCA4-associated retinopathy. Genes that exhibit the full ABCA4-associated retinopathy diagnostic triad represent Tier 1. Genes in Tier 2 exhibit two of the three triad features and Tier 3 consists of genes exhibiting one triad and one auxiliary ABCA4-associated retinopathy. Genes within each tier are listed alphabetically.
Asterisks (*) indicate variability in the degree to which the indicated feature overlaps with corresponding feature in ABCA4-associated retinopathy. Abbreviations: AD, autosomal dominant; AR, autosomal recessive; XL, X-linked recessive; BEM, bull’s eye maculopathy; AF, autofluorescence.
Fig. 5.

Common ABCA4-associated retinopathy phenocopying genes and masquerading phenotypes. (A) A 35-year-old woman harboring the c.638G>C, p.(Cys213Ser) variant in PRPH2 with autofluorescent flecks across the posterior pole and peripapillary sparing phenocopying a 29-year-old ABCA4-associated retinopathy patient harboring with the C.302 + 1G> A, p.(?) variant of ABCA4. (B) A 62-year-old woman harboring a canonical splice site variant, c.582-1G>A, p.(?), in PRPH2 with a confluent distribution of autofluorescent flecks across the posterior pole and “peninsular” sparing of the fovea phenocopying a 41-year-old woman harboring the c.4457C>T, p.(Pro1486Leu) and c.4793C>A, p.(Ala1486Asp) variants of ABCA4. (C) A 60-year-old man with pattern dystrophy harboring the c.584G>A, p.(Arg195Gln) missense variant in PRPH2 with foveal sparing phenocopying a 40-year-old ABCA4 disease patient harboring the hypomorphic variant, c.5603A>T, p.(Asn1868Leu), and c.4670A>G, p.(Tyr1557Cys) variants of ABCA4. (D) A 54-year-old man with a large, circular lesion of chorioretinal atrophy and autofluorescent flecks harboring the c.571G>T, p.(Glu191*) nonsense variant in PRPH2 phenocopying a 44-year-old ABCA4 disease patient harboring the hypomorphic variant, c.5603A>T, p.(Asn1868Ile), and c.4670A>G, p.(Tyr1557Cys) variants of ABCA4. (E) A 37-year-old man with maternally inherited diabetes and deafness (MIDD) with granular autofluorescent fleck-like depositions and “bridged” sparing of the fovea phenocopying a 42-year-old ABCA4-associated retinopathy patient harboring a missense, c.2971G>C, p.(Gly991Arg), and a deep-intronic, C.570 + 1798A>G, p.(Phe191Leufs*6), variant in ABCA4. (F) A 51-year-old woman with an elliptical BEM lesion caused by the c.449C>G, p.(Ser150*) variant in CRX phenocopying a 17-year-old boy with ABCA4 disease harboring a mild missense variant, c.3113C>T, p.(Ala1038Val), and a known exon-skipping intronic variant, c.5461–10T>C, p.[Thr1821Aspfs*6,Thr1821Valfs*13] (Sangermano et al., 2016) variant in ABCA4. (G) A 57-year-old man with a uniform BEM lesion caused by the c.3423G>T, p.(Trp1141Cys) missense variant in RPGR phenocopying a 15-year-old boy with ABCA4-associated retinopathy harboring the c.5882G>A, p.(Gly1961Glu) and c.45G>A, p.(Trp15*) variant of ABCA4. (H) A 5-year-old girl with RDHl2-associated Leber congenital amaurosis (LCA) and peripapillary sparing homozygous for the missense variant, c.698T> A, p.(Val233Asp), phenocopying a 60-year-old man with end-stage ABCA4-associated retinopathy harboring the c.4139C>T, p.(Pro1380Leu) and c.4601del p.(Leu1534Trpfs*1) variants in ABCA4.
The gene that is most often carrying (dominant) variants, which are associated with phenotypes that are indistinguishable from ABCA4-associated retinopathy, is PRPH2. Variants in PRPH2 cause autosomal dominant pattern dystrophy and often exhibit the triad of ABCA4-associated retinopathy features. Since ABCA4-associated retinopathy is recessive, it should be quite straightforward to distinguish the two by family history. However, PRPH2 variants often exhibit variable penetrance across generations in a family simulating recessive inheritance patterns. In addition, pattern dystrophy is a late-onset disease, which may also lead to an incorrect inference of AMD. Furthermore, pseudodominant families are frequent in ABCA4-associated retinopathy due to high allelic load of ABCA4 variants in the general population (Beit-Ya’acov et al., 2007; Huckfeldt et al., 2016; Lee et al., 2016; Maugeri et al., 1999; Shroyer et al., 2000; Tracewska et al., 2019) and late onset disease expression due to reduced penetrance of variants in both PRPH2 and ABCA4 is well documented (Runhart et al., 2018; Yatsenko et al., 2001; Zernant et al., 2017, 2018). Therefore, also taking into account phenotype similarities, only comprehensive genetic screening can solve the causality in these cases.
In a recent study we investigated a cohort of cases where phenotypes were consistent with ABCA4-associated retinopathy but no disease-causing variants were found in ABCA4 even after complete locus sequencing (Wolock et al., 2019). While variants in PRPH2 were the most frequent cause of disease in this cohort (5–10% of all cases), and variants in another plausible gene, PROM1, came close second, this study also revealed variants in several other genes, usually not considered obvious causal candidates for phenocopies, including CDHR1, CERKL, CRX and RPE65 (Wolock et al., 2019).
These findings are not surprising, since genetic studies have recently significantly expanded phenotype heterogeneity in many retinal diseases in addition to those caused by ABCA4 variants. Variants in many genes, such as CRB1 and CRX, cause drastically different phenotypes depending on specific mutations and inheritance pattern, where some variants cause recessive disease while others cause dominant. The tiered list of genes (based on the diagnostic triad given above), mutations in which could lead to ABCA4 phenocopies, are given in Table 1. The important caveat, however, is the depth of clinical analysis, since some of these cases could have been distinguished from ABCA4-associated retinopathy already by comprehensive clinical assessment.
4. ABCA4 structure and function
The ABCA4 protein is an ATP-binding cassette (ABC) transporter in photoreceptor outer segments that functions in the visual cycle. More specifically, it is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer (Quazi et al., 2012; Quazi and Molday, 2013, 2014), the only known importer among mammalian ABC transporters. ABCA4 dysfunction results in accumulation of all-trans and 11-cis retinoids in photoreceptors (PRs), formation of A2E (and other bisretinoids) cumulatively called “lipofuscin”, and their accumulation mostly in the RPE (Sparrow and Boulton, 2005; Sparrow et al., 2010; Sparrow and Yamamoto, 2012). This accumulation of cytotoxic products is a hallmark, and often also the cause, of most phenotypes resulting from dysfunctional ABCA4 (Burke et al., 2014). More recently, expression of ABCA4 has also been reported in the RPE, suggesting an additional role of the protein in this cell type that, when disturbed, could somehow contribute to ABCA4-associated retinopathy (Lenis et al., 2018).
The structure of ABCA4 at a high resolution is not yet described. Mammalian ABC transporters are notoriously difficult substrates for determining crystal structure (Dahl et al., 2004). The best published resolution of the native ABCA4 protein and some mutants, is 18 Å (Tsybovsky et al., 2010, 2013; Tsybovsky and Palczewski, 2014). The lack of high-resolution ABCA4 structure makes functional studies challenging, limiting experimental systems to animal models (Makelainen et al., 2019; Molday et al., 2018; Molday and Molday RS, 2016; Zhang et al., 2015) and in vitro assays, including ATP binding, ATPase activity and vesicular transport studies (Ahn and Molday, 2000; Beharry et al., 2004; Sun et al., 1999). The current status of the structure and (biochemical) function of ABCA4 protein is outside of the scope of this review. These aspects have been described in depth in manuscripts from the laboratories of Robert Molday and Krzysztof Palczewski, and we direct the reader to these papers for excellent overviews of the status of structure and function correlations in ABCA4 (Molday, 2015; Tsybovsky et al., 2013; Tsybovsky and Palczewski, 2014). We will address the functional studies of ABCA4 variants affecting splicing in depth below.
5. Disease-causing variants in the ABCA4 locus
Disease-causing variation in the ABCA4 locus is extensive; there are currently >1200 disease-causing variants known and the number is rapidly growing as new cohorts, especially those of non-European descent, are screened (www.lovd.nl/ABCA4). When classified by a variant effect, the locus contains all classes of variants – missense, nonsense, indels, canonical and noncanonical splice site (NCSS) defects, deep-intronic variants and structural variants (SVs). These can be further grouped according to the proposed severity of the variant, including deleterious (i.e., complete null), severe, moderate, mild, and hypomorphic categories. The specific distinction between groups is not always unequivocal since, due to the lack of the high-resolution structure of the ABCA4 protein, the functional consequences derived from indirect assays (protein yield, ATPase and transport activity, etc.) do not always correlate exactly with resulting disease phenotypes and progression. For example, many missense alleles are deleterious (Molday et al., 2018; Zernant et al., 2017; Zhang et al., 2015), while other seemingly deleterious alleles (stop-gained and indel variants) sometimes do not result in a complete lack of function. Therefore, variant severity assignments are often not straightforward and these are constantly updated as new information becomes available from both genotype/phenotype analysis of extensive cohorts and functional studies, including those involving animal models. In section 6, we do classify variants based on pre-mRNA splicing defects.
5.1. Distribution of types of variants
As depicted in Fig. 6, most of the ABCA4 variants/alleles, both in number of unique variants/alleles (50%) (Fig. 6A) and total variant/allele numbers (61%) (Fig. 6B) are missense mutations. The relatively low contribution of protein truncating mutations (23% of total and 33% of unique alleles) probably can be explained through the genotype-phenotype correlation model in which all ABCA4-associated retinopathy cases, except those with early-onset disease, carry at least one non-truncating mutation. The latter may also explain the relatively high frequency of NCSS variants (~5%) as their effects range from mild to deleterious. As described in more detail below, 46 different structural variants (SVs) (in ~1% of all alleles) and 35 different causal deep-intronic (DI) variants (in 4% of all alleles), have been identified in ABCA4-associated retinopathy cases. As the majority of genotyped ABCA4-associated retinopathy cases has not yet been screened for the presence of SVs and DI variants, we estimate that ~2% of all ABCA4 alleles are SVs and ~10% are DI variants.
Fig. 6.

Distribution of different types of ABCA4-alleles. Unique (A) and all (B) ABCA4 variants or alleles based on data collected by Cornelis et al. (2017), supplemented with deep-intronic variant and structural variant data published since then (listed in Tables 3 and 4). The contribution of each type of variant or allele is represented. Protein truncating variants comprise nonsense, frameshift and canonical splice site variants. The complex alleles represented in these pie-charts only consist of combinations of missense variants, the most frequent of which were c.[1622T>C;3113C>T] and c.[4469G>A;5603A>T]. They do not include the complex alleles that contain noncanonical splice site variants, deep-intronic variants or protein truncating variants, when present in cis with other variants. If these had been included, ~10% of the alleles would consist of complex alleles. Most of the structural variants, deep-intronic variants and noncanonical splice site variants also result in protein truncation.
A significant fraction of ABCA4 alleles (~10%) consists of more than one variant (Shroyer et al., 2001b; Zhang et al., 2015). Two of these ‘complex alleles’ are conspicuously prevalent. The c.[2588G>C; 5603A>T], p.[Gly863A1a,Gly863del;Asnl868Ile] allele is present in ~50% of all complex alleles and its relevance only came to light after the significance of p.(Asn1868Ile) was appreciated as a single causal allele, when found in trans with a severe or moderately severe ABCA4 allele, see below (Runhart et al., 2018; Zernant et al., 2017). As shown previously, the c.2588G>C variant was always found in cis with c.5603A>T in ABCA4-associated retinopathy cases (Maugeri et al., 1999, 2002). Both variants are required to render the complex allele fully penetrant (Zernant et al., 2017). The c.[1622T>C;3113C>T], p.[Leu541Pro;Ala1038Val] complex allele constitutes 34% of all complex alleles. Interestingly, the single variants p.(Leu541Pro) and p.(Ala1038Val) were also found in 43 and 78 alleles, respectively, in ABCA4-associated retinopathy cases (Cornells et al., 2017). Based on the combinations of variants identified in ABCA4-associated retinopathy cases and their allele frequencies in healthy individuals, they are considered to be (moderately) severe and mild, respectively (Cornells et al., 2017). The complex allele p.[Leu541Pro;Ala1038Val] is deleterious, a complete loss-of-function allele (Zernant et al., 2017; Zhang et al., 2015).
5.2. Founder mutations in different populations
The ABCA4 locus is informative with regard to multiple founder alleles; i.e. variants, which initially occurred in one geographical locale, a well-known phenomenon for geographically or culturally isolated populations (Table 2). What makes the ABCA4 locus especially interesting is that, in addition to expected significant differences of disease-causing ABCA4 alleles in various racial and ethnic groups, almost every nation in Europe has its “own” ABCA4 mutation, which is much higher in frequency than in neighboring countries. Examples include the C.768G>T allele, which is very frequent in the Netherlands, but almost absent in neighboring Germany (Cremers et al., 2004), the complex allele p.[Leu541Pro;Alal038Val] in Germany (Rivera et al., 2000), which has, however, spread especially throughout Eastern Europe due to geopolitical events (Sciezynska et al., 2016; Tracewska et al., 2019; Zolnikova et al., 2017), and the p.(Arg1129Leu) variant in Spain (Valverde et al., 2006). Most of these variants have occurred once; an interesting deviation from this is the severe p.(Asn965Ser) variant, which was first described as a founder mutation in Denmark (Rosenberg et al., 2007) but subsequently also was found at a very high frequency in China (Jiang et al., 2016). Haplotype analysis confirmed that the variant occurred independently in the two regions (Jiang et al., 2016). The p.[Gly863Ala,Gly863del] variant is frequent in Western/Northern Europe (Maugeri et al., 1999, 2002) and the most frequent disease-causing ABCA4 allele, p.(Gly1961Glu), originates from Eastern Africa, where it is found in ~10% of Somalis (Burke et al., 2012; Guymer et al., 2001). Subsequent population migration has spread p.(Gly1961Glu) throughout the world, but the allele frequency has dropped dramatically during evolution (Burke et al., 2012). The population frequency in Europe is ~0.4%, suggesting that the variant is causal in all, or at least most, populations. Other population-specific alleles are the deep-intronic c.4539 + 2001G>A variant, which is frequent in the Belgian population, comprising ~25% of all deep-intronic variants, but interestingly, is more rare in the Netherlands (Bauwens et al., 2015, 2019; Bax et al., 2015). The p.(Ala1773Val) allele is a founder variant in Mexico (Chacon-Camacho et al., 2013; Lopez-Rubio et al., 2018). The Ashkenazi Jewish population has several founder alleles, including c.4254-37_4254-15del (Beit-Ya’acov et al., 2007) and p.(Pro1380Leu) (Sharon et al., 2020). In summary, the genetic screening for pathogenic ABCA4 alleles can often identify the ethnicity, and even the nationality, of a patient.
Table 2.
ABCA4 founder variants in various populations.
| DNA variant | Protein variant | Population | Allele frequency in ABCA4-associated retinopathy (in the founder population) | Allele frequency in the founder population, if known | Reference |
|---|---|---|---|---|---|
| c.5882G> A | p.(Gly1961Glu) | Somali | N/A | 0.1 | Guymer et al. (2001); Burke et al. (2012) |
| c.[2588G>C;5603A>T] | p.[(Gly863Ala,Gly863del;Asn1868Ile)] | Western Europe | 0.15 | 0.015 | Maugeri et al. (1999) |
| c.768G>T | p.(Leu257Valfs*17) | Dutch | 0.08 | 0.00019 | Maugeri et al. (1999); Cremers et al. (2004) |
| c.[1622T>C;3113C>T] | p.[(Leu541Pro;Ala1038Val)] | German | 0.13 | 0.0003 | Rivera et al. (2000) |
| c.3386G>T | p.(Arg1129Leu) | Spanish | 0.24 | 0.002 | Valverde et al. (2006) |
| C.4539+2001G>A | p.[=,Arg1514Leufs*36] | Belgian | 0.025 | <0.0001 | Bauwens et al. (2015) |
| c.2894A>G | p.(Asn965Ser) | Danish | 0.16 | 0.0002 | Rosenberg et al. (2007) |
| c.2894A>G | p.(Asn965Ser) | Chinese | 0.03 | 0.0004 | Jiang et al. (2016) |
| c.101_106delCTTTAT | p.(Ser34_Leu35del) | Chinese | 0.03 | 0.0003 | Jiang et al. (2016) |
| c.2424C>G | p.(Tyr808*) | Chinese | 0.05 | <0.0001 | Hu et al. (2019) |
| c.6320G>A | p.(Arg2107His) | African American | 0.19 | 0.02 | Zernant et al. (2014a) |
| C.2966T>C | p.(Val989Ala) | African American | 0.07 | 0.0025 | Zernant et al. (2014a) |
| c.2971G> C | p.(Gly991Arg) | African American | 0.035 | 0.0064 | Zernant et al. (2014a) |
| c.4139C>T | p.(Pro1380Leu) | Ashkenazi Jewish | 0.035 | 0.002 | Sharon et al. (2020) |
| c.4254–37_4254-15del | p.(Ser1418_Pro1451delinsArg) | Arab-Muslim | 0.018 | <0.0001 | Beit-Ya’acov et al. (2007); Sharon et al. (2020) |
| c.5318C>T | p.(Ala1773Val) | Mexican | 0.17 | 0.00045 | Chacon-Camacho et al. (2013) |
Frequency of ABCA4 variants in ABCA4-associated retinopathy cohorts was determined in cited studies. Population frequency in respective populations was determined in the same studies or from gnomAD database. N/A - data not available.
Although the founder alleles are already frequent within the population of European descent, they are much more prominent in various racial groups. While there is some overlap with the Caucasian population, possibly due to admixture, the disease-causing ABCA4 mutation spectrum is significantly different, for example, in African American (Zernant et al., 2014a) and in East Asian (Hu et al., 2019; Jiang et al., 2016) populations. In both populations, the most prominent and frequent founder alleles, the c.101_106delCTTTAT, p.(Ser34_Leu35del), c.2424C>G, p.(Tyr808*), and c.6563T>C p.(Phe2188Ser), in China (Hu et al., 2019; Jiang et al., 2016) and the p.(Val989Ala), p.(Gly991Arg) and p.(Arg2107His) variants in African Americans, are almost absent from European populations (Zernant et al., 2014a). The only exception is the p.(Asn965Ser) variant, which is an independent founder allele in both Denmark and China. The population frequency of the most frequent disease-causing variant in African Americans, p.(Arg2107His), is ~2% in the general population of African American descent, suggesting that this allele can be considered hypomorphic, which is also supported by late-onset and mild disease in patients harboring this mutation (Zernant et al., 2014a). Another interesting observation is that the p.(Gly1961Glu) variant, which originates from East Africa and has a very high population frequency in Somalia, Kenya and Ethiopia (Burke et al., 2012), is almost absent in African Americans; i.e. in people of West African descent (Zernant et al., 2014a). Some populations, e.g., South Asian (Indian), have stronger admixture of European alleles (Lee et al., 2017). Most other racial and ethnic groups have not been screened in sufficient numbers of cases for valid statistical conclusions at this time.
5.3. Missing heritability
Recent advances in the genetic analysis of the ABCA4 locus, including complete locus sequencing, functional assays, and introduction of the concept of hypomorphic alleles, have significantly reduced the fraction of “missing alleles”. Another important aspect in this regard is the much better clinical characterization of patients due to major advances in imaging technologies and increased experience of retinal specialists. In our centers in Nijmegen and New York, the fraction of “unsolved” cases (i.e., those with certain ABCA4-associated retinopathy diagnosis and one definite pathogenic allele) is <5%. The fraction of phenocopies, i.e., cases with ABCA4-associated retinopathy-like phenotypes but with causal mutations in genes other than ABCA4, is still 10–15% of all cases, even in the most advanced centers, but these are most often solved with WES, as described above (Wolock et al., 2019). So where are the remaining pathogenic ABCA4 alleles? There are several possible scenarios:
Some variants can be in regulatory regions of ABCA4, promoter andenhancer sequences, which can affect ABCA4 expression (Bauwens et al., 2019; Cherry et al., 2020). Some of the possibly regulatory variants have been identified, e.g., c.768 + 3223C>T and c.2919–383C>T (Bauwens et al., 2019), but more comprehensive searches and functional characterization are necessary for this category of possible mutations.
Yet unidentified, and/or unconfirmed, deep-intronic variants. Sequencing of the entire ABCA4 genomic locus identifies in each patient, on average, 40 variants with an allele frequency <0.5% in population-matched control individuals, several of which could be considered causal even after thorough filtering for allele frequencies and comprehensive in silico analyses. Studies of monoallelic cases have identified and functionally proven 35 different deep-intronic variants to be causal, most of which are detected in more than one patient. However, since the ABCA4 locus is extremely heterogeneous and single cases of pathogenic variants are often identified in coding sequences, the comprehensive analysis of all possibly pathogenic deep-intronic variation remains a challenging task. As described below, some deep-intronic variants cause retina-specific splicing defects (Albert et al., 2018), and almost all putative splicing variants thus far were tested in human embryonic kidney cells (HEK293T) which do not recapitulate the retina-specific splicing factors.
Structural variants (SVs) are (very) rare in the ABCA4 locus (see below for deletions and duplications); however, it is likely that a small fraction of SVs is yet to be identified as short-read sequencing strategies will not identify inversions and insertions. Another class of very rare genetic events, such as uniparental isodisomy, has also been identified in three probands with ABCA4-associated retinopathy (Fingert et al., 2006; Khan et al., 2020; Riveiro-Alvarez et al., 2007).
It has also been suggested that some of the ABCA4-associated retinopathy could be dominant, or di- or polygenic. While theoretically possible, there is currently no evidence for either scenario. While clinically dominant phenotypes, such as those caused by the p.(Gly1961Glu) mutation, are documented in ABCA4-associated retinopathy, there is no reason to expect any genetically dominant cases since, based on our current knowledge of the ABCA4 function, a dominant-negative effect is not expected for any ABCA4 allele. The entire ABCA4-associated retinopathy continuum is based on a loss-of-function mechanism, whether complete, or partial; i.e., haploinsufficiency, as in carriers of ABCA4 variants. Whether the latter mechanism results in a late-onset macular disease is still open for debate (Duncker et al., 2015a; Kjellstrom, 2015; Lee et al., 2019; Maia-Lopes et al., 2008). However, we postulate that, based on pre-mRNA splice assay data (see below), all recessive ABCA4-associated retinopathy cases (together from both alleles) have no more total residual ABCA4 activity than 40% (Sangermano et al., 2018, 2019).
6. ABCA4 pre-mRNA splicing defects
6.1. In vitro splice assays in HEK293T cells
The analysis of putative splice defects ideally is performed using patient cells in which the gene of interest is endogenously expressed to perform reverse transcription-PCR analysis of the mRNA. In the absence of patient cells or when the gene of interest is not expressed in accessible human tissues, in vitro splice assays have traditionally been performed in human cell lines such as human embryonic kidney (HEK293T) cells. To this aim, small genomic fragments (<1 kb) were cloned in splicing vectors. As the ABCA4 gene is expressed at a very low level in non-ocular human tissues, we also employed minigenes to analyze NCSS variants previously identified in ABCA4-associated retinopathy cases. Due to the strong splice sites of vector exons that flank the cloned segments, splicing artefacts were observed (Sangermano et al., 2016). To systematically test the effect of NCSS ABCA4 variants, we cloned large wild-type genomic fragments (4.0–11.7 kb) of the ABCA4 gene into a Gateway splicing vector containing RHO exons 3 and 5 flanking the region of interest. The resulting splicing constructs were coined ‘midigenes’. Apart from fragments containing parts of two very large introns (introns 6 and 11), all ABCA4 cloned segments contained at least 3 exons, enabling us to perform RT-PCR using primers annealing to ABCA4 exons, minimizing the occurrence of artefacts (Sangermano et al., 2018). As the first and last exon do not contain a splice acceptor or donor site, respectively, exon 1, intron 1, intron 49 and exon 50 are not or only partially represented in midigenes.
6.2. Causal noncanonical splice site variants in ABCA4
Upon testing all published NCSS variants, 64 showed a wide spectrum of splicing defects, including single exon skipping, multiple exon skipping, exon elongation, intron retention, and partial exon skipping (Table 3) (Bauwens et al., 2019; Fadaie et al., 2019; Khan et al. (2020); Khan et al., 2019; Sangermano et al., 2019; Sangermano et al., 2018; Schulz et al., 2017). The majority of these variants (35/64) resulted in 100% aberrantly spliced RNA and are considered deleterious. Another 12 variants showed >0 and ≤ 30% of normal splice products and can be classified as severe splice variants. Twelve variants showed >30% and ≤70% wild-type RNA and were classified to have a moderate effect, three variants were classified as mild (>70% and ≤80%) and one (C.3608G>A) was classified as benign as 95% of the RNA was correctly spliced. Finally, c.2588G>C resulted in a 3-nt deletion, p.(Gly863del), and a normally splice product that can be translated in ABCA4 protein carrying a missense variant, i.e., p.(Gly863Ala) (Maugeri et al., 1999; Sangermano et al., 2018). In a subsequent study, this variant was found only to be causal (as a mild-moderate allele) when in cis with c.5603A>T, p.(Asn1868Ile) (Zernant et al., 2017).
Table 3.
Noncanonical splice site variants in ABCA4 and their RNA splice defect assessments in HEK293T cells.
| DNA variant | RNA variant | Protein variant | % correct RNA | RNA defect severity | Reference(s) |
|---|---|---|---|---|---|
| c.160+5G>C | r.[67_160del,=,161_302delinsl61+1_161+14] | p.[Ile23Alafs*24,=,His55Asnfs*63] | 34 | Moderate | Sangermano et al. (2018) |
| c.161G>A | r.[161_302del,=] | p.[Cys54Serfs*14,Cys54Tyr] | 44@ | Moderate | Fadaie et al. (2019) |
| c.161G>T | r.161_302del | p.(Cys54Serfs*14) | 0 | Deleterious | Sangermano et al. (2018) |
| C.302+4A>C | r.161_302del | p.(Cys54Serfs*14) | 0 | Deleterious | Sangermano et al. (2018) |
| c.303-3C>G | r.[161_302delins303-2_303–1,302_303ins302-2_302–1] | p.[ Cys54*,Leu102Alafs*14] | 0 | Deleterious | Sangermano et al. (2018) |
| c.768G>T | r.768_769ins769+1_769+30 | p.(Leu257Valfs*17) | 0 | Deleterious | Sangermano et al. (2018) |
| C.859-9T>C | r.[=,859_1356del] | p.[=,Phe287_Arg452del] | 76 | Mild | Sangermano et al. (2018) |
| c.1100-6T>A | r.1099_1100ins1099-4_1099-1 | p.(Thr367Serfs*6) | 0 | Deleterious | Sangermano et al. (2018) |
| c.1554+3A>T | r.[=,1357_1554del] | p.[=,Asp453_Glu518del] | 51 | Moderate | Khan et al. (2020) |
| c.1937+5G>A | r.1806_1937del | p.(Tyr603_Ser646del) | 0 | Deleterious | Fadaie et al. (2019) |
| c.2161-8G>A | r.2161_2382del | p.(His721_Val794del) | 0 | Deleterious | Fadaie et al. (2019) |
| C.2382+5G>C | r.[2161_2382del,=] | p.[His721_Val794del,=] | 48 | Moderate | Sangermano et al. (2018) |
| c.2588G>C | r.[2588G>C,2588_2590del] | p.[Gly863Ala,Gly863del] | 60 # | Mild-Moderate | Maugeri et al. (1999); Sangermano et al. (2018) |
| c.2654-8T>G# | r.[2653_2654ins2654-40_2654-1,=] | p.[Gly863Valfs*47,=] | 13 | Severe | Khan et al. (2020) |
| c.2919-10T> C | r.[=,2919_3050del] | p.[=,Leu97 3_His1017 delinsPhe] | 61 | Moderate | Sangermano et al. (2018) |
| c.2919-6C> A | r.[=,2919_3050del] | p.[=,Leu97 3_His1017 delinsPhe] | 80 | Mild | Sangermano et al. (2018) |
| c.3050+5G>A | r.2919_3050del | p.(Leu973_His1017delinsPhe) | 0 | Deleterious | Sangermano et al. (2018) |
| c.3191-11T>A | r.3190_3191ins3191-l_3191-9 | p.(Gly1064delinsValProProGly) | n.a. | Deleterious$ | Bauwens et al. (2019) |
| C.3522+5del | r.[=,3329_3522del] | p.[=,Arg1111 Aspfs*7] | 53 | Moderate | Sangermano et al. (2018) |
| c.3607G>A | r.3523_3607del | p.(Thr1176Metfs*2) | 11 | Severe | Sangermano et al. (2018) |
| c.3607+3A>T | r.3523_3607del | p.(Thr1176Metfs*2) | 0 | Deleterious | Sangermano et al. (2018) |
| c.3608G> A | r.[=,3608_3813del] | p.[Gly1203Glu,Gly1203Aspfs*10] | 95 | Benign | Khan et al. (2019) |
| c.3812A> G | r.3608_3813del | p.(Glyl203Aspfs*10) | 0 | Deleterious | Sangermano et al. (2018) |
| c.3813G> C | r.3608_3813del | p.(Glyl203Aspfs*10) | 0 | Deleterious | Sangermano et al. (2018) |
| c.3862G> A | r.[=3863g>a,3814_3862del] | p.[=,Gly1288Ser,Ile1272Valfs*l01] | 69 | Moderate | Khan et al. (2020) |
| C.3862+ 3A> G | r.[=,3814_3862del] | p.[=,Ilel272Valfs*101] | 53 | Moderate | Sangermano et al. (2018) |
| c.4128G>A | r.4128_4129ins4128 +1_4128 +12 | p.(Gln1376_Ile1377insValLeuLeuSer) | 0 | Deleterious$ | Sangermano et al. (2018) |
| c.4128G>C | r.4128_4129ins4128 +1_4128 +12 | p.(Gln1376_Ile1377insValLeuLeuSer) | 0 | Deleterious$ | Khan et al. (2019) |
| c.4129-3C>T | r.[=,3864_4128del,4129_4253del,3864_4253del] | p.[=,Ile1377Hisfs*3,Gly1288Aspfs*45] | 76 | Mild | Khan et al. (2020) |
| c.4253+4C>T | r.4129_4253del | p.(Ilel377Hisfs*3) | 8 | Severe | Sangermano et al. (2018) |
| c.4253+5G>A | r.4129_4253del | p.(Ilel377Hisfs*3) | 0 | Deleterious | Sangermano et al. (2018) |
| c.4253+5G>T | r.4129_4253del | p.(Ilel377Hisfs*3) | 5 | Severe | Sangermano et al. (2018) |
| c.4538A>C | r.[4539_4540ins4540+1_4530+30>4467_4539del,4538a>c] | p.[Pro1513_Arg1 514ins10,Cys1490 Glufs*12,Gln1513Pro] | 4 | Severe | Sangermano et al. (2018) |
| c.4538A>G | r.[4539_4540ins4540+1_4530+30,4467_4539del] | p.(Arg1513_Arg1514ins10,Cys1490Glufs*12) | 0 | Deleterious | Sangermano et al. (2018) |
| c.4539G>A | r.4467_4539del | p.(Cys1490Glufs*12) | 5 | Severe | Khan et al. (2019) |
| c.4540-8T>A | r.4539_4540ins4540-6_4540-l | p.(Gln1513insProGln) | 0 | Deleterious | Khan et al. (2020) |
| c.4667G> A | r.4635_4667del | p.(Ser1 545_Gln1555del) | 0 | Deleterious | Fadaie et al. (2019) |
| c.4667G>C | r.4635_4667del | p.(Ser1545_Gln1555del) | 0 | Deleterious | Sangermano et al. (2018) |
| c.4773G>C | r.[4668_5018del,4668_4773del] | p.(Tyr1557_Val1673del,Tyr1557Alafs*18) | 0 | Deleterious | Sangermano et al. (2018) |
| c.4773+3A>G | r.[4668_4773del,=] | p.[Tyr1557Alafs*18,=] | 25 | Severe | Schulz et al. (2017); Sangermano et al. (2018) |
| c.4773+5G>A | r.[4668_4773del,4668_5018del] | p.[Tyr1557Alafs*18,Tyr1557_Val1673del] | 29 | Severe | Sangermano et al. (2018) |
| C.4848+3A>G | r.[4774_4848del,=] | p.[Glyl592_Lysl616del,=] | 10 | Severe | Khan et al. (2020) |
| c.4849G>A | r.[4849_5018del,4849_5109del,=] | p.[Vai1617 Alafs*113,Val1617Met,=] | 60 | Moderate | Khan et al. (2019) |
| C.5018+5G>A | r.4849_5018del | p.(Vai1617Alafs*113) | 0 | Deleterious | Fadaie et al. (2019) |
| C.5196+3_5196+6del | r.4849_5196del | p.(Vall617_Ilel732del) | 0 | Deleterious | Sangermano et al. (2018) |
| C.5312+3A>T | r.5197_5312del | p.(Asnl734Glyfs*14) | 0 | Deleterious | Sangermano et al. (2018) |
| c.5313-3C> G | r.5312_5313ins5312-2_5312-1 | p.(Trpl772Aspfs*7) | 0 | Deleterious | Sangermano et al. (2018) |
| c.5460+5G>A | r.5313_5460del | p.(Trp177 2Argfs * 9) | 0 | Deleterious | Sangermano et al. (2018) |
| c.5461-10T>C | r.[5461_5714del,5461_5584del] | p.[Thr1821Aspfs*6,Thr1821Valfs*13] | 0 | Deleterious | Sangermano et al. (2016); Sangermano et al. (2018) |
| C.5461-6T>G | r.5461_5714del | p.(Thr1821Aspfs*6) | 0 | Deleterious | Khan et al. (2020) |
| c.5584G>C | r.5461_5714del | p.(Thr1821Aspfs*6) | 0 | Deleterious | Sangermano et al. (2018) |
| c.5584+5G>A | r.[5461_5714del,5461_5584del] | p.[Thr1821Aspfs*6,Thr1821Valfs*13] | 0 | Deleterious | Sangermano et al. (2018) |
| c.5584+6T>C | r.[5461_5714del,5461_5584del,5585.5714] | p.[Thr1821Aspfs*6,Thr1821Valfs*13,Glul863Leufs*33] | 0 | Deleterious | Sangermano et al. (2018) |
| c.5714 + 5G>A | r.[=,5585_5714del] | p.[=,Glul863Leufs*33] | 40 | Moderate | Sangermano et al. (2018) |
| c.5715-5T>G | r.5461_5714delins5715-4_5715-1 | p.(Thr1821Serfs*34) | 2^ | Severe | Fadaie et al. (2019) |
| c.5836-3C>A | r.5835_5836ins5836+1_5836+30 | p.(Lysl945Jlel946Pheinsl0) | 0 | Deleterious | Sangermano et al. (2018) |
| c.5898+5G>A | r.[5898_5899ins_5899 +1_5890-1,5898_5899ins5899+1_5899+170,=] | p.[Cysl967Valfs424,=] | 48 | Moderate | Khan et al. (2020) |
| c.5898+5del | r.[5898_5899ins_5899 +1_5890-1,5898_5899ins5899+1_5899+170] | p.(Cysl967Valfs424) | 5 | Severe | Sangermano et al. (2018) |
| c.6147G>A | r.6006_6147del | p.(Ser2002Argfs*ll) | 0 | Deleterious | Fadaie et al. (2019) |
| c.6385A>G | r.6340_6386del | p.(Val2114Hisfs*4) | 0 | Deleterious | Fadaie et al. (2019) |
| C.6386+3A>G | r.[6386_6387ins6386+1_6387-1,6340_6386del,=] | p.[Ser2129Serfs429,Val2114Hisfs*4,=] | 26 | Severe | Khan et al. (2020) |
| c.6478A>G | r.[6478a>g,6387_6479del] | p.[Lys2160Glu,Ser2129_Lys2160delinsArg] | 55 | Moderate | Sangermano et al. (2018) |
| C.6479+4A>G | r.6387_6479del | p.(Ser2129_Lys2160delinsArg) | 0 | Deleterious | Sangermano et al. (2018) |
| C.6729+5_6729+19del | r.6480_6729del | p.(Phe2161 Cysfs*3) | 0 | Deleterious | Sangermano et al. (2018) |
The severity assessment is based on RNA splice defects in HEK293T cells, as follows: 0% correct RNA, deleterious (complete null); >0% and ≤30% correct RNA, severe; >30% and ≤70% correct RNA, moderate; >70% and ≤80% correct RNA, mild; >80% correct RNA, benign. n.a., no quantification shown;
The wild-type midigene shows 14% natural 3 exon skipping;
For variant c.2588G>C, a rough quantification was based on Sanger sequence traces as the two splice products (3-nt difference) could not be separated;
Variants with in-frame small amino acid insertions that may not act deleterious in protein function.
The wild-type and a mutant (4-nt insertion) fragment co-migrate and together constitute 4% of the total RNA. For variants with multiple effects at the mRNA, the most prevalent product is listed first.
6.3. Causal near-exon variants in ABCA4
Seven variants located near exons were found to result in splicing defects that affect neighboring exons (Table 4). Variants c.1937 + 13T>G, c.1937 + 37C>G, c.3191-11T>A and c.4352+61G>A create or strengthen intronic splice sites. They thereby result in 12-nt, 36-nt, 9-nt and 57-nt exon elongations, respectively, leading to nonsense mutations in the corresponding RNA products (c.1937 + 13T>G, c.1937 + 37C>G, c.4352 + 61G>A) or a deletion of one amino acid and the insertion of four amino acids (c.3191-11T>A) (Fadaie et al., 2019; Sangermano et al., 2018). Variants c.161–23T>G and c.4253 + 43G>A result in partial exon 3 and exon 28 skipping, respectively, rendering them mild variants based on genotype/phenotype analyses (Zernant et al., 2018) and in vitro splice assays (Bauwens et al., 2019; Sangermano et al., 2019). The c.4253 + 43G>A variant is the most frequent intronic ABCA4 variant that is not residing in NCSS sequences. Variant c.6148-84A>T resulted in a wildtype and three mutant cDNAs, one of which carried a pseudoexon (PE), one carried a partially overlapping PE and was missing exon 44, and one did not contain exon 45 (Khan et al. (2020)).
Table 4.
Causal deep-intronic ABCA4 variants and their splice defects based on splice assays in HEK293T cells or analysis of patient-derived photoreceptor progenitor cells.
| DNA variant | RNA effect | Protein variant | % correct RNA | Severity based on RNA defect | Number of alleles | Reference(s) |
|---|---|---|---|---|---|---|
| C.67-2023T>G | r.[66_67ins67-2266_67-2024,=] | p.[IIe23IIefs*30,=] | 33 | Moderate | 4 | Zernant et al. (2014a,b); Khan et al. (2020) |
| c.161-23T>G | r.[=,161_302del] | p.[=,Cys54Serfs*14] | 50 | Moderate | 2 | Bauwens et al. (2019); Khan et al. (2020) |
| c.570+1798A>G | r.570_571ins570+1733_570+1797 | p.(Phel91Leufs*6) | 0 | Deleterious | 3 | Zernant et al. (2014a,b); Khan et al. (2020) |
| c.769–788A>T | r.768_769ins769–778_769-617 | p.[Leu257Aspfs*3,=] | 4 | Severe | 1 | Khan et al. (2020) |
| c.769–784C>T | r.[=,768_769ins769-617_769-778] | p.[=,Leu257Aspfs*3] | 70 | Moderate@ | 22 | Bauwens et al. (2019); Khan et al. (2019); Sangermano et al. (2019); Runhart et al. (2019); Khan et al. (2020) |
| c.859-640A>G | r.858_859ins859-685_859-640 | p.(Phe287Tyrfs*69) | 0 | Deleterious | 2 | Khan et al. (2020) |
| c.859-546G>A | r.[858_859ins859-545_859-685,=] | p.[Phe287Tyrfs*33,=] | 36 | Moderate | 1 | Khan et al. (2020) |
| c.859-540C>G | r.858_859ins859-545_859-685 | p.(Phe287Tyrfs*33) | 0 | Deleterious | 1 | Bauwens et al. (2019) |
| c.859-506G>C | r.[858_859ins859-503_859-447,=] | p.[Phe287Thrfs*32,=] | 24 | Severe | 6 | Sangermano et al. (2019); Khan et al. (2020) |
| c.1937+13T>G | r.[1937_1938ins_1938+1_1938+12,=] | p.[Phe647*,=] | 14 | Severe | 1 | Sangermano et al. (2018) |
| c.1937+37C>G | r.l937_1938insl938+1_1938+36 | p.(Phe647*) | 0 | Deleterious | 2 | Khan et al. (2020) |
| c.1937+435C>G | r.[=,1937_1938ins1937+396_1937+529] | p.[=,Ser646Serfs*25] | 91 | Benign# | 4 | Sangermano et al. (2019); Khan et al. (2020) |
| c.1938-621G>A | r.[=,1937_1938insl938-797_1938-624,1937+396_1937+529,1938-797_1938-624] | p.[=,Phe647Alafs*22,Phe647Serfs*22] | 93 | Benign^ | 1 | Khan et al., 2020 |
| c.1938-619A>G | r.1937_1938ins[1938-797_1938-624,1937+396_1937+529,1938-797_1938-624] | p.[Phe647Alafs*22,Phe647Serfs*22] | 12 | Severe | 2 | Zernant et al. (2014a,b); Fadaie et al. (2019); Khan et al. (2020) |
| c.1938-514A>G | r.[1937_1938insl938-623_1938-515,1937+396_1937+529,1938-623_1938-515,=] | p.[Phe647Serfs*155,Phe647Serfs*22,=] | 13 | Severe | 1 | Khan et al. (2020) |
| c.2588-706C>T | r.[2587_2588ins2588-839_2588-708,=] | p.[Gly863Alafs*3,=] | 5 | Severe | 1 | Khan et al. (2020) |
| c.2919-826T>A | r.[2918_2919ins2919-957_2919-825,=] | p.[Leu973Phefs*1,=] | 17 | Severe | 2 | Zernant et al. (2014a,b); Fadaie et al. (2019); Khan et al. (2020) |
| c.3050+370C>T | r.3050_3051ins3050+164_3050+368 | p.(Leu1018Glufs*4) | 0 | Deleterious | 2 | Zernant et al. (2014a,b); Fadaie et al. (2019); Khan et al. (2020) |
| c.3863-1064A>G | r.?% | p.(?)% | 70 | Moderate | 1 | Khan et al. (2020) |
| c.3191-11T> A | r.3190_3191ins3191-1_3191-9 | p.(Gly1064delinsValProProGly) | 0 | Deleterious | 1 | Bauwens et al. (2019) |
| c.4253+43G>A | r.[=,4129_4253del] | p.[=,Ile1377Hisfs*3] | 64 | Moderate | 100 | Zernant et al. (2018); Sangermano et al. (2019); Bauwens et al. (2019); Khan et al. (2019); Nassisi et al. (2019); Khan et al. (2020) |
| C.4352+61G>A | r.[4352_4353ins4352+1_4352+57,=] | p.[Glul452*,=] | 16 | Severe | 2 | Zernant et al. (2014a,b); Fadaie et al. (2019); Khan et al. (2020) |
| C.4539+1100A>G | r.[4539_4540ins4539+1033_4539+1100,4539_4540ins4539+989_4539+1100,=] | p.[Arg1514Valfs*31,Arg1514Glyfs*3,=] | 19 | Severe | 2 | Sangermano et al. (2019) |
| C.4539+1106C>T | r.[4539_4540ins4539+1033_4539+1100,4539_4540ins4539+989_4539+1100] | p.[Arg1514Glyfs*3,Arg1514Valfs*31] | 3 | Severe | 4 | Bauwens et al. (2019); Khan et al. (2019); Sangermano et al. (2019) |
| C.4539+2001G>A | r.[=,4539_4540ins4539+1891_4540-2162] | p.[=,Argl514Leufs*36] | 50 | Moderate$ | 64 | Braun et al. (2013); Zernant et al. (2014a,b), Bauwens et al. (2015); Bax et al. (2015); Albert et al. (2018); Sangermano et al. (2019); Bauwens et al. (2019); Khan et al. (2019); Khan et al. (2020) |
| C.4539+2028C>T | r.[=,4539_4540ins4539+1891_4540-2162] | p.[=,Argl514Leufs*36] | 70 | Moderate$ | 20 | Braun et al. (2013); Zernant et al. (2014a,b); Schulz et al. (2017); Albert et al. (2018); Khan et al. (2019); Khan et al. (2020) |
| C.4539+2064OT | r.[4539_4540ins4539 +1891_4540-2162,=] | p.[Arg1514Leufs*36,=] | 25 | Severe | 27 | Zernant et al. (2014a,b); Bauwens et al. (2019); Khan et al. (2019); Nassisi et al. (2019); Khan et al. (2020) |
| C.4539+2065C>G | r.[4539_4540ins4539+1891_4539+2060,=] | p.[Argl514Lysfs*35,=] | 50 | Moderate | 1 | Khan et al. (2019) |
| c.4634+741A>G | r.[4634_46354ins4634+614_4634+740,=] | p.[Seri 545Serfs*51,=] | 11 | Severe | 1 | Khan et al. (2020) |
| C.5196+1056A>G | r.5196_5197ins5196+880_5196+1056 | p.(Metl733Valfs*2) | 2 | Severe& | 22 | Braun et al. (2013); Zernant et al. (2014a,b); Schulz et al. (2017); Zernant et al. (2018); Khan et al. (2019); Khan et al. (2020); Khan et al. unpublished |
| C.5196+1137G>A | r.[=,5196_5197ins5196+1140_5196+1212] | p.[=,Metl733Glufs*78] | 55 | Moderate& | 47 | Braun et al. (2013); Zernant et al. (2014a,b); Bax et al. (2015); Sangermano et al. (2019); Khan et al. (2019); Nassisi et al. (2019); Khan et al. unpublished |
| C.5196+1216C>A | r.[=,5196_5197ins5196+1140_5196+1212] | p.[=,Met1733Glufs*78] | 33 | Moderate& | 1 | Bauwens et al. (2019); Khan et al. unpublished |
| c.5197-557G>T | r.5196_5197ins5197-563_5197-750 | p.(Metl733*) | 0 | Deleterious | 1 | Bauwens et al. (2019); Khan et al. unpublished |
| c.6148-84A>T | r.[6147_6148ins6148-262_6148-90,6006_6147delins6148-310_6148-90,6148_6149del,=] | p.[Val2050Valfs*68,Ile2003Hisfs*30, Val2050_Leu2094del,=] | 43 | Moderate | 1 | Khan et al. (2020) |
| c.6283-78G>T | r.[=,6283_6283ins6283-282_6283-80] | p.[=,Asp2095Aspfs*12] | 75 | Mild | 2 | Khan et al. (2020) |
| Total: | 355 | Khan et al. (2020) |
Definition of deep-intronic variants: all variants outside the splice site consensus sequences. The severity assessment is based on splice defects observed in transfected HEK293T cells or patient-derived photoreceptor progenitor cells: 0% correct RNA, deleterious (complete null); >0% and ≤30% correct RNA, severe; > 30% and ≤ 70% correct RNA, moderate; > 70% and ≤80% correct RNA, mild; > 80% correct RNA, benign.
Based on RT-PCR analysis of patient-derived photoreceptor progenitor cells.
Variant does not affect splice sites and is presumed to have a more severe effect in the retina.
Variant has small effect in HEK293T cells but may have a stronger effect in the retina.
Due to technical reasons exact boundaries of PE are not determined yet.
The RNA splicing defect of these intron 30 variants were analyzed in patient-derived photoreceptor progenitor cells. Based on genotype-phenotype correlations, they are presumed to have a severe (c.4539+2001G>A) and moderate effect (c.4539+2028C>T) on the function of ABCA4.
Based on midigene in vitro splice assays or on RT-PCR analysis of patient-derived photoreceptor progenitor cells (M. Khan et al. unpublished data). For variants with multiple effects at the mRNA, the most prevalent product is listed first.
6.4. Causal deep-intronic variants in ABCA4
The first five causal deep-intronic variants in ABCA4 were discovered based on the hypothesis that they may strengthen cryptic splice sites flanking PEs that are present in a small fraction of ABCA4 transcripts within a normal retina (Braun et al., 2013). In this way, small regions of the ABCA4 locus were sequenced in genetically unsolved ABCA4-associated retinopathy cases. Sequencing of the entire ABCA4 locus (Zernant et al., 2014b) in 114 monoallelic patients revealed another 16 possibly disease-associated variants, followed by variant-specific (Bauwens et al., 2015; Bax et al., 2015; Khan et al., 2019; Schulz et al., 2017; Zernant et al., 2017), or complete locus analysis in several other ABCA4-associated retinopathy cohorts (Bauwens et al., 2019; Khan et al. (2020); Sangermano et al., 2019). Finally, functional studies with midigene-based splice assays (Bauwens et al., 2019; Fadaie et al., 2019; Khan et al. (in press); Khan et al., 2019; Sangermano et al., 2019) were used to determine the effect of deep-intronic variants on splicing of 35 deep-intronic variants (Table 4).
In vitro splice assays were able to determine a partial or complete picture of the splicing defects for all deep-intronic variants except two. The effects of two neighboring variants in intron 30, i.e. c.4539+2001G>A and c.4539+2028C>T, were only shown using patient-derived photoreceptor progenitor cells (PPCs) (Albert et al., 2018). Both variants do not affect the strength of splice sites flanking a 345-nt PE, but strengthen and/or create exonic splice enhancer motifs inside the pseudo-exon; i.e., have a different disease-causing mechanism than most other deep-intronic pathogenic variants. Based on the in vitro splice assays and PPC analysis, 7/35 deep-intronic and near-exon variants had a deleterious effect (i.e. no correct RNA), 13 showed a severe effect, 12 showed a moderate effect, one showed a mild effect, and two (c.1937+435C>G, c.1938-621G>A) were classified as benign as 95% of the RNA was correctly spliced. However, we do consider the latter variants to be causal and attribute the low percentage of mutant transcripts to the cell type used in the splice assay (HEK293T cells). Fig. 7 displays the location of the deep-intronic and near-exon variants. There is a clustering of different deep-intronic variants in introns 7 (n = 4), 13 (n = 4), 30 (n = 6) and 36 (n = 4). The total number of alleles carrying deep-intronic and near-exon variants is 355 (Table 4) (Bauwens et al., 2015; Bauwens et al., 2019; Bax et al., 2015; Braun et al., 2013; Khan et al. (2020); Khan et al., 2019; Nassisi et al., 2019; Sangermano et al., 2019; Schulz et al., 2017; Zernant et al., 2018; Zernant et al., 2014b). Only seven variants were found in more than 10 alleles, i.e. c.4253+43G>A (n = 100), c.4539+2001G>A (n = 64), c.5196+1137G>A (n = 47), c.[769–784C>T;5603A>T] (n = 22), c.4539+2064C>T (n = 27), c.4539+2028C>T (n = 20) and c.5196+1056A>G (n = 22). It is difficult to estimate the frequency of these variants compared to all ABCA4 variants identified thus far, as not all ABCA4-associated retinopathy probands have been analyzed for variants in the entire genomic locus. We estimate that ~5% of all alleles (~10% of probands) carry causal deep-intronic or near-exon variants.
Fig. 7.
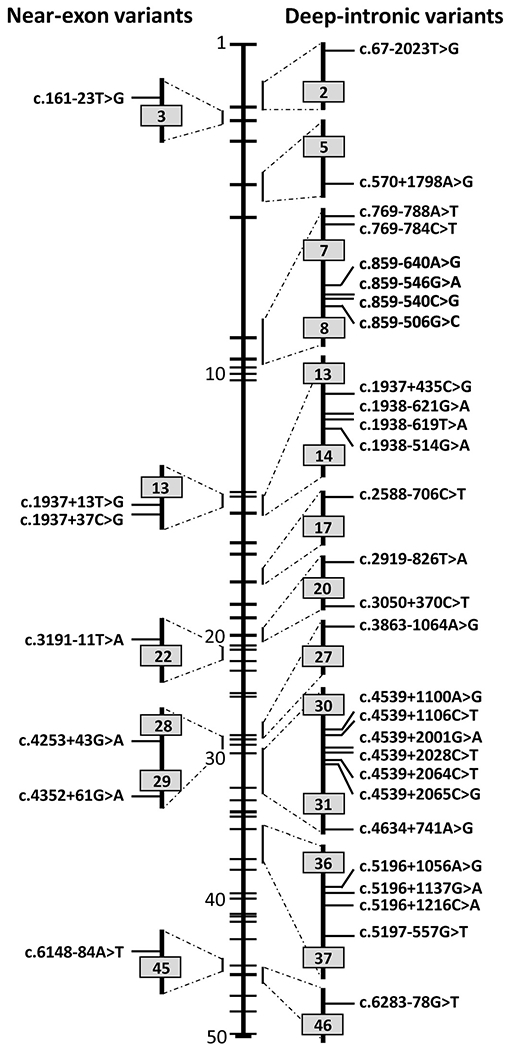
Location of deep-intronic variants in ABCA4. Left part shows variants located near exons that result in exon skipping or exon elongation. Right part shows deep-intronic variants that invariably result in the generation of pseudoexons (see Table 4).
7. Structural variants in the ABCA4 locus
Structural variants (SVs) in the ABCA4 gene/locus are relatively rare based on Southern blot analysis (Maugeri et al., 1999; Yatsenko et al., 2003), array-comparative genome hybridization (aCGH) assays (Zernant et al., 2014b) and multiplex ligation-dependent probe amplification (MLPA) analysis (Bauwens et al., 2019; Bax et al., 2015; Sangermano et al., 2019; Zernant et al., 2014b). Table 5 lists all 46 reported SVs larger than 20 bp, including 35 deletions, 6 duplications, 2 deletions-insertions, 2 deletions with internal inversions, and 1 insertion. Of the 23 SVs for which the size is known, seven are smaller than 100 bp. However, he predicted effect is severe for all SVs, except for a 7-kb intron 1 duplication, for which the predicted effect is unknown. Only seven SVs have been found in more than one case. Based on nested RT-PCR studies of lymphoblast RNA of a homozygous proband, the intron 28 deletion c.4254–37_4254-15del resulted in the skipping of exons 29 or 28 and 29. The variant was found in homozygosity in 14 cases and in heterozygosity in one case in six families of an Arab-Muslim village in Israel (Beit-Ya’acov et al., 2007). The second most frequent SV is an exon 20–22 deletion that was reported in eight probands originating from Belgium, Germany and the Netherlands (Table 5). Based on these published data, we estimate that 1–2% of ABCA4-associated retinopathy probands carry a causal SV.
Table 5.
Structural variants in ABCA4-associated retinopathy patients.
| Genomic position (hg 19) | DNA variant | Protein variant | Type of SV | Location | Exact size (if known) | Number of STGD1 cases carrying SV | Reference(s) |
|---|---|---|---|---|---|---|---|
| 94586601_94458796 | c.(?,−1),(*1,?)del | p.(?) | del | complete gene | n.a. | 1 | Valverde et al. (2006) |
| 94586536_94586601 | c.(?_−1)_(66+1,67–1)del | p.(?) | del | exon 1 | n.a. | 1 | Khan et al. (2020) |
| 94579011_94586016 | c.66+520_67-389dup | p.(?) | dup | intron 1 | 7006 bp | 1 | Bauwens et al. (2019) |
| 94553579_94579597 | c.67–975_769-4582dup{insA} | p.(Ile23_Val256dup) | dup | intron 1-6 | 26,019 bp | 1 | Bauwens et al. (2019) |
| 94568030_94573334 | c.442+799_570+541del | p.(Gly148Valfs*89) | del | intron 4-intron 5 | 5305 bp | 3 | Lambertus et al. (2015); Bax et al. (2015); Bauwens et al. (2019) |
| 94569917-94562911 | c.443–1219,768+ 1439del | p.(Gly148Alafs*23) | del | exons 5-6 | 7007 bp | 1 | Khan et al. (2020) |
| 94565348-94561288 | c.571–801_768+3062del | p.(Phe191_Val256del) | del | exon 6 | 4061 bp | 1 | Khan et al. (2020) |
| 94564419-94564009 | c.699_768+341del | p.(Gln234Phefs*5) | del | partial exon 6 | 411 bp | 6 | Khan et al. (2020) |
| 94564321_94564376 | c.742,768+29del | p.(Val248_Val256del) | del | exon 6 | 56 bp | 1 | Riveiro-Alvarez et al. (2013) |
| 94564350_94564547 | c.(570+1,571–1)_(768+1,769–1)del | p.(Phe191_Val256del) | del | exon 6 | n.a. | 2 | Khan et al. (2020) |
| 94548997_94548908 | c.(768+1,769–1)_(858+1,859–1)del | p.(Leu257_Glu286del) | del | exon 7 | n.a. | 1 | Fujinami et al. (2019) |
| 94546319_94546181 | c.859–45_952delinsTCTGACC | p.(?) | del/ins | intron 7-exon | n.a. | 1 | Fukui et al. (2002) |
| 94534447_94544587 | C.1239+291_1555-5574del | p.(Ala414_Glu518del) | del | intron 9-intron 11 | 10,141 bp | 1 | Bauwens et al. (2019) |
| 94543443_94520667 | c.(1356+1,1357–1)_(2587+1_2588–1)del | p.(Asp453Glufs*38) | del | exon 11-16 | n.a. | 1 | Rozet et al. (1999) |
| 94528873_94528133 | c.l555-983_1937+720del | p.(Cys519Phefs*119) | del | exon 12-13 | 2444 bp | 1 | Birtel et al. (2018) |
| 94529906-94527518 | c.1555–1033,1937+615delinsAGC | p.(Cys519Phefs*119) | del | exon 12-13 | 2389 bp | 1 | Khan et al. (2020) |
| 94532364-94526398 | c.1555–3491_1938-83delins1734,1761-107inv | p.(Cys519Phefs*22) | del/inv | exon 12-13 | 5967 bp | 1 | Khan et al. (2020) |
| 94531301-94522479 | c.1555–2428,2161-101delins2160+7,2160+230invATGAATGins | p.(?) | del/inv | exon 12-14 | 8588 bp | 1 | Khan et al. (2020) |
| 94528133_94528309 | c.(1760+1,1761–1)_(1937+1,1938–1)del | p.(Arg587_Asp645del) | del | exon 13 | n.a. | 3 | Muller et al. (2015); Birtel et al. (2018) |
| 94520667_94520871 | c.(2382+1,2383–1)_(2587+1_2588–1)del | p.(Ser795Glufs*38) | del | exon 16 | n.a. | 1 | Khan et al. (2020) |
| 94514513-? | c.(2653+1,2654–1)_(*1_?)del | p.(Gly885Valfs*71) | del | exon 18-50 | n.a. | 1 | Khan et al. (2020) |
| 94514389_94515418 | c.2654–905_2743+35del | p.(Gly885_His914del) | del | exon 18 | 1030 bp | 2 | Yatsenko et al. (2003) |
| 94510300_94508317 | c. 2918 + 775,3328+ 640del | p.(Ser974Glnfs*64) | del | exon 20-22 | 8 | Maugeri et al. (1999); Bax et al. (2015); Lambertus et al. (2015); Muller et al. (2015); Birtel et al. (2018); Bauwens et al. (2019) | |
| 94506923_94510186 | c.3033_3364del | p.(His1011 Glnfs*53) | del | exon 20-23 | n.a. | 1 | Khan et al. (2020) |
| 94505683_94458796 | c.(3522+1,3523–1)_(*l_?)del | p.(Gly1175*) | del | exon 24-50 | n.a. | 1 | Carss et al. (2017) |
| 94497418_94497441 | c.4021ins24 | p.(?) | ins | exon 27 | 24 bp | 1 | Passerini et al. (2010) |
| 94496676_94495001 | c.(4128+1,4129–1)_(4539+1_4540–1)del | p.(Ile1377_Gln1513del) | del | exon 28-30 | n.a. | 1 | Khan et al. (2020) |
| 94496096_94496118 | c. 4 254-37,4 254-15del | p.(Ser1418_Pro1451delinsArg) | del | intron 28 | 23 bp, skipping e29 and e28-29 | 14 hom, 1 het | Beit Ya’acov et al. (2007) |
| 94496279-94487503 | c.4254–197,4672delinsGCTTTTT | p.(?) | del | exon 29-33 | 8770 bp | 1 | Khan et al. (2020) |
| 94495005_94495030 | c.4510_4535del | p.(Glul504Profs*42) | del | exon 30 | n.a. | 1 | Nassisi et al. (2019) |
| 94495187_94486796 | c.(4352+1,4353–1)_(5018+1_5019–l)dup | p.(?) | dup | exon 30-35 | n.a. | 1 | Khan et al. (2020) |
| 94480099,94495187 | c.4353_5460del | p.(Glul452Argfs*9) | del | exon 30-38 | n.a. | 1 | Khan et al. (2020) |
| 94486911_94486934 | c.4880_4903dup | p.(Leu1627_Ala1634dup) | dup | exon 35 | 24 bp | 1 | Kellner et al. (2009) |
| 94476941,94461665 | c.(5460+ 1,5461 +1)_(6816+1_6817–1)del | p.(Thr1821_Gln2272del) | del | exon 39-49 | n.a. | 1 | Khan et al. (2020) |
| 94457537,94476649 | c.5585–166_*1254del | p.(Gly1862Valfs*71) | del | intron 39-3TJTR | 19,113 bp | 1 | Bauwens et al. (2019) |
| 94476485,94458796 | c.(5584+1,5585–1)_(*1_?)del | p.(Gly1862_Asp2273delins69) | del | exon 40-50 | n.a. | 1 | Khan et al. (2020) |
| 94471138_94467414 | c.(6005+1,6006–1)_(6282+1,6283–1)dup | p.(Asp2095Tyrfs*7) | dup | exon 44-45 | n.a. | 1 | Khan et al. (2020) |
| 94472532-94470240 | c.6005+658,6147+757delinsTTTAACAGTGTT | p.(Ser2002Argfs*12) | del | exon 44 | 2284 bp | 1 | Khan et al. (2020) |
| 94468246_94463476 | c.6148–698_6670del/insTGTGCACCTCCCrAG | p.(?) | del/ins | intron 44-exon 48 | n.a. | 1 | Lee et al. (2016) |
| 94467548_94466392 | c.(6147+1,6148–1)_(6479+1,6480–1)del | p.(Val2050Ilefs*21) | del | exon 45-47 | n.a. | 1 | Schorderet et al. (2013) |
| 94467351–94463600 | c.6282+63_6546del | p.(Asp2095_Leu2182del) | del | exon 46 to part of exon 48 | 3752 bp | 1 | Khan et al. (2020) |
| 94463566_94463601 | c.65456580del | p.(Leu2182_Phe2193del) | del | exon 48 | 36 bp | 1 | Lewis et al. (1999); Birtel et al. (2018) |
| 94461751-? | c.(6729+1_6730–1)_(*1_?)del | p.(Val2244*) | del | exon 49 to 50 | n.a. | 1 | Khan et al. (in press) |
| 94461722_94461765 | c.6730–14_6759del | p.(?) | del | intron 48-exon 49 | 44 bp | 1 | Stenirri et al. (2006) |
| 94461716_94461760 | c.6730–9_6765dup | p.(His2256_Asp2273delinsTyrLeu) | dup | exon 49 | 45 bp | 1 | Jiang et al. (2016) |
| 94458798_94458796 | c.(6816+16817–1)_(*1_?)del | p.(Asp2273*) | del | exon 50 | n.a. | 1 | Jespersgaard et al. (2019) |
Structural variants reported in PubMed, HGMD (Human Gene Mutation Database) and LOVD (Leiden Open Variation Database). HGVS (Human Genome Variation Society) nomenclature was used. An estimated allele number in STGD1 cases is indicated according to the number of cases described in the publications. SV, structural variant; PMID, PubMed unique identifier number; del, deletion; dup, duplication; ins, insertion; bp, base pair; n.a., not applicable; hom, homozygous; het, heterozygous.
8. Genotype-phenotype correlations
The extensive clinical heterogeneity of ABCA4-associated retinopathy is primarily caused by the similarly significant genetic variability in the ABCA4 locus. Shortly after the discovery of the role of ABCA4 in various maculopathies, we proposed a somewhat simplistic genotype/phenotype association model suggesting a correlation between the continuum of disease phenotypes and residual ABCA4 activity/function (Cremers et al., 1998; Lewis et al., 1999; van Driel et al., 1998). According to that model, different combinations of “mild”, “moderate”, and “severe” ABCA4 mutant alleles were suggested to result in distinct phenotypes. While still valid in some, especially extreme, cases, this model underestimates the phenotypic heterogeneity of ABCA4-associated retinopathy. The current model is derived from the analysis of large, comprehensively characterized cohorts of patients which have revealed an immensely complex landscape of independent disease trajectories that appear to be unrepresented in the ABCA4 genotype alone. The combinations of currently known >1200 disease-associated ABCA4 variants (Allikmets, 2007; Cornells et al., 2017; Maugeri et al., 1999) (www.lovd.nl/ABCA4) explain some, but definitely not all, disease phenotypes (Burke et al., 2012). The population frequency of potentially pathogenic ABCA4 alleles is 1:20 (Jaakson et al., 2003; Maugeri et al., 1999; Yatsenko et al., 2001), underscoring the substantial impact for the amount of retinal pathology attributable to ABCA4 variation. Some more recent developments in ABCA4 genetics, which have allowed us to make more precise genotype/phenotype correlations, are listed below.
8.1. Extremely hypomorphic and modifier variants
The emergence of ‘extremely’ hypomorphic and modifier alleles (Bauwens et al., 2019; Runhart et al., 2018; Zernant et al., 2017, 2018), most of which are still unknown, adds another layer of genetic and phenotypic heterogeneity. The most prominent of these variants is p.(Asn1868Ile), with population allele frequencies close to 7% in Europe. It had been shown in many studies to be more frequent in cases vs controls (Aguirre-Lamban et al., 2011; Maugeri et al., 2002; Webster et al., 2001) and speculated (Webster et al., 2001) or suggested (Schulz et al., 2017), that it may be associated with the disease. While most of the association is due to linkage disequilibrium (LD) with highly penetrant pathogenic variants (Schulz et al., 2017; Zernant et al., 2017), we proved the pathogenicity of p.(Asn1868Ile) under a specific condition – it is penetrant when in trans from a deleterious ABCA4 mutation (Zernant et al., 2017). Patients harboring the p.(Asn1868Ile) variant exhibit distinct clinical characteristics, including a very late disease onset with mean age at onset of 36.3 (Zernant et al., 2017) and 41.8 years (Runhart et al., 2018). Affected individuals often show foveal sparing, defined as the structural and function preservation of outer retinal layers in the fovea despite the progressing atrophy of the macula, in ~85% of cases. When the p.(Asn1868Ile) allele resides in cis with other mutations (e.g., c.5461-10T>C or p.(Cys1490Tyr)), the phenotypes are consistent with the overall genotype effect and presented with mostly early onset, severe phenotypes. Interestingly, when in cis with the p.[Gly863Ala,Gly863del] variant as a complex allele, p.(Asn1868Ile) acted as a fully penetrant allele with a mild to moderate effect and resulted in variable phenotypes, reflective of the variant on the opposite allele. Confirming the full penetrance of the complex p.[Gly863Ala,Gly863del;Asn1868Ile] allele, two homozygous patients presented with later onset and milder disease and foveal sparing in one of the two cases (Zernant et al., 2017). The discovery of the p.(Asn1868Ile) pathogenicity when in trans with a deleterious ABCA4 allele solved ~50% of cases who were considered monoallelic at the time and, therefore, explained the missing heritability in ~10% of the entire ABCA4-associated retinopathy population (Zernant et al., 2017). These studies also defined the frequent p.[Gly863Ala,Gly863del] variant not as a pathogenic allele on its own, but rather as the first major modifier allele in the ABCA4 locus.
8.2. The c.5882G>A, p.(Gly1961Glu) allele is associated with mild disease and other specific subphenotypes
Patients harboring the p.(Gly1961Glu) allele in homozygosity or in compound heterozygosity have a substantially different phenotype. Age at onset (mean 22.7 years) is somewhat later than in ABCA4-associated retinopathy cases not carrying the p.(Gly1916Glu) or p.(Asn1868Ile) alleles (mean 19.7 years) (Zernant et al., 2017). Patients with the p.(Gly1961Glu) allele exhibit milder disease expression (Burke et al., 2012; Celia et al., 2009), although not in the overall rate of disease progression but in a distinct phenotypic pattern that, interestingly, overlaps with patients harboring the p.(Asn1868Ile) allele. ABCA4-associated retinopathy invariably begins as a maculopathy with an enlarging lesion of outer retinal atrophy and accumulation of yellow foci, or flecks, at the level of the RPE. Patients with most other ABCA4 variants exhibit progressively severe fleck patterns from very few in the macula to a stage of “absolute confluence” across the posterior pole between the ages 30–40 years (Fig. 8).
Fig. 8.

Genotype-phenotype correlations for ABCA4-associated retinopathy. Summary of disease trajectories associated variants and genotypes of ABCA4. Overall disease severity is defined according to the spatial extent of the disease: Macular stage, disease changes are confined to the central macula; Extramacular stage, disease changes extend beyond the vascular arcades and regions nasal to the optic disc; Transitional stage, disease changes become confluent across the posterior pole initiating peripheral involvement and outer retinal atrophy; Advanced stage, multiple lesions occur and coalesce across the posterior pole. Disease trajectories are defined by the average age at which patients progress through each severity milestone. Three allele-specific trajectories are represented including patients with hypomorphic alleles, c.4253+ 43G> A, p.[ =,Ile1377Hisfs*3] and c.5603A>T, p.(Asn1868Ile) which occur only in trans with a deleterious allele, homozygous and compound heterozygous c.5882G> A, p.(Gly1961Glu) alleles, two deleterious alleles and Other two alleles which consist of all other combinations of ABCA4 alleles. The length of color-coded arrows represents the beginning and duration of defined disease severity stage. Representative autofluorescence images of patients within each trajectory group are arranged according to the age of the depicted phenotype along the time line (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article).
Patients harboring p.(Gly1961Glu) or p.(Asn1868Ile) consistently exhibit milder spatio-temporal fleck patterns even at advanced age, which never progresses to the absolute confluence stage as illustrated by fundus autofluorescence imaging which detects lipofuscin accumulation (Fig. 8). The resistance to fleck accumulation and lower rate of lipofuscin accumulation seen in bi-allelic cases carrying p.(Gly1961Glu) or p.(Asn1868Ile) (Burke et al., 2014), is further exemplified by the occurrence of a particular phenotype that is seen only in this patient group where a well-defined, unifocal lesion of dark atrophy with proximally-bordering, lesion-centric flecks appears at an early stage of the disease. Similar lesions of chorioretinal atrophy are typically numerous (multifocal) and observed in the background of more advanced fleck stages with most other ABCA4-associated retinopathy cases (Fig. 8). Additionally, the individual morphology of flecks in patients with p.(Gly1961Glu) or p.(Asn1868Ile) appears to be distinct in that they are predominantly larger in size and are well defined in shape and generally more sparsely distributed. Of specific interest is the fact that patients with the p.(Gly1961Glu) allele typically present with the ‘bull’s eye maculopathy’ phenotype regardless of the allele in trans, thereby acting as ‘clinically dominant’ alleles (Celia et al., 2009). This is especially apparent in multi-generation families, where the other variables (genetic background, age, etc.) are largely the same (Lee et al., 2016). The pathophysiological reasons of this phenomenon remain to be elucidated; however, it is very likely that p.(Gly1961Glu) mutation selectively affects ABCA4 function of foveal cones, often resulting in the ‘optical gap’ phenotype at early disease stages (Noupuu et al., 2014).
8.3. Two deleterious ABCA4 alleles result in severe cone-rod dystrophy
Two deleterious alleles in ABCA4 consistently result in early, childhood-onset of disease symptoms and a rapid progression to advanced disease stages characterized by profound visual impairment and deep, retinal wide degeneration extending towards the equatorial limits of the retina. Such patients have been historically classified as cone-rod dystrophy (MIM# 604116) (Cremers et al., 1998; Maugeri et al., 2000), due to considerable attenuations of cone amplitude on ffERG, or retinitis pigmentosa (RP19, MIM# 601718) (Cremers et al., 1998; Martinez-Mir et al., 1998; Verbakel et al., 2018). Cases of the latter diagnosis indeed exhibit features associated with RP including deposition of bone-spicule pigment, severe retinal vessel attenuation and extinguished ffERG responses, but also atypical features such as early macular involvement. Clinically distinguishing true RP (i.e. rod-cone dystrophy) from severe cone-rod dystrophy at such an advanced stage of disease can be challenging. Distinctions in the extent of residual rod or cone function would not be reliably detectable on ffERG due to widespread degeneration of both systems. Identifying indicators in the ocular history of night blindness and progressive constriction of the visual field may in such cases be more informative. An etiological connection between RP and ABCA4 thus remains to be established. Alternatively, the commonalities between pathophysiology of deleterious ABCA4 mutations and RP may result in the manifestation of overlapping features. Other characterizations of biallelic null ABCA4 phenotypes include the generalized choriocapillaris dystrophy (Bertelsen et al., 2014) and rapid-onset chorioretinopathy (ROC) (Tanaka et al., 2018). ROC is specifically characterized by disease onset within the first decade, a short interval of profoundly increased autofluorescence in the macula followed by the rapid development of degenerative lesions, which enlarge and coalesce across the posterior pole within the third decade of life (Fig. 8).
9. Variable expression and penetrance of ABCA4 alleles
As described above, ABCA4-associated retinopathy shows a broad spectrum of clinical expression, with onset ranging from as early as 5 years of age to as late as 70 years. Although genotype-phenotype correlations are apparent, differences in clinical expression between individuals with the same combination of ABCA4 variants have often been observed, suggesting the involvement of both cis- and trans-modifier alleles (Lee et al., 2019; Runhart et al., 2018). Recently, the discussion, also between the authors of this review, has focused on late onset cases carrying a deleterious variant on one ABCA4 allele and the p.(Asn1868Ile) on the other allele.
Researchers at the Nijmegen center calculated the penetrance of the p.(Asn1868Ile) allele, when in trans with a deleterious or severe allele, to be ~5% in the general population (Cremers et al., 2018; Runhart et al., 2018) and documented unaffected male patients who carried the same combination of variants as their affected siblings, in three families (Runhart et al., 2018). The time elapsed between the age at onset in the affected siblings and the current age of the unaffected siblings ranged between 14 and 37 years. However, it cannot be excluded that the unaffected bi-allelic individuals could still develop ABCA4-associated retinopathy later in life. In the same study, 22/34 (65%) affected persons were female and 12/34 (35%) were male, which was suggestive (p = 0.0615) for a sex imbalance; i.e. it seemed as if late-onset ABCA4-associated retinopathy affects more females than males (Cremers et al., 2018; Runhart et al., 2018). In a larger study of 125 international ABCA4-associated retinopathy cases carrying p.(Asn1868Ile) in trans with a severe or deleterious allele and fully sequenced for variants in the ABCA4 locus, 79 (63%) were female and 46 (37%) were male. In comparison with a perfect gender balance (50%/50%) observed in 284 bi-allelic ABCA4-associated retinopathy probands not carrying presumed hypomorphic alleles, this was statistically significant (p = 0.018) (E. Runhart, M. Khan, F.P.M. Cremers, C-M. Dhaenens, unpublished data).
Researchers at Columbia argued against these observations (Allikmets et al., 2018) by suggesting that the disease prevalence (see below in the “Future research” section), and the “strength” of mutations (deleterious vs. severe, etc.) is largely unknown, so one has to be careful with using these variables in statistical calculations. Furthermore, the female-to-male ratio among all 1060 ABCA4-associated retinopathy cases at the Columbia center is 54:46 in the entire disease cohort and 57:43 in the sub-cohort with the p.(Asn1868Ile) allele (100 cases) (J. Zernant, W. Lee, R. Allikmets, unpublished data). Excluding the hypomorphic alleles from the entire ABCA4-associated retinopathy cohort did not affect the overall 54:46 ratio, i.e. the difference was not statistically significant. Interestingly, the only two “non-penetrant” cases (i.e. older members in families with affected younger cases) with the p.(Asn1868Ile) allele at Columbia are female and carry the p.(Pro1380Lys) allele in trans.
The observed differences in the sex data in two large ABCA4-associated retinopathy cohorts (with substantial statistical power) suggest further in-depth analysis of this phenomenon across populations. Of additional interest is the fact that differences between the two centers exist in both the overall sex balance in ABCA4-associated retinopathy and separately in the hypomorph subgroups.
In summary, these data strongly suggest our incomplete understanding of this, very important, issue, solving of which requires much more data and analysis. Specifically, this part of the ABCA4 genetic studies requires better understanding of both cis- and trans-modifier alleles. We cannot exclude that there are thus far missed variants, especially in deep-intronic sequences, on the allele carrying the p.(Asn1868Ile) in ABCA4-associated retinopathy cases, which would render these alleles fully penetrant. A primary example for this scenario are the complex alleles harboring both p.(Asn1868Ile) and p.[Gly863Ala,Gly863del] variants, which are fully penetrant (Zernant et al., 2017). However, they would not explain the large differences of expression of ABCA4-associated retinopathy within families, in which trans-modifiers may play a larger role. Finding these, non-ABCA4, modifiers, genetic or non-genetic in nature, which influence the expression of ABCA4-associated retinopathy in a subset of the cases, remains an important task.
10. Animal models
The first animal model for ABCA4-associated retinopathy, the Abca4 knockout (KO) mouse (Abca4−/−), was introduced in 1999 (Weng et al., 1999). Since then this mouse, which has also been generated independently by other groups (Kong et al., 2008), has been the main and only animal model for studying ABCA4-associated retinopathy. It is obvious that the mouse is not the best animal model for macular disease as it lacks the macula and has very few cones, which are the earliest target of ABCA4-associated retinopathy. Therefore, while the Abca4 KO mouse model does not fully replicate the human condition, its most prominent pathological feature, the extensive accumulation of lipofuscin/A2E, faithfully mimics the disease in humans and allows for precise quantification of the disease status, progression, and the therapeutic effect in pre-clinical studies (Charbel Issa et al., 2013; Sparrow et al., 2013). The rest of the disease features, photoreceptor degeneration (Bok, 2005), delayed dark adaptation (Mata et al., 2001; Weng et al., 1999) and ffERG defects are less robust and not always replicated from study to study.
More recently, two Abca4 knock-in (KI) animals have been generated and used to study specific protein defects and progression of the disease (Molday et al., 2018; Zhang et al., 2015). In addition to documenting the influence of several ABCA4 variants on the protein expression, misfolding, trafficking, ATPase activity, etc., these studies also defined the specific ABCA4 variants, p.(Asn965Ser) and p.[Leu541Pro;Ala1038Val], as mostly deleterious. The resulting phenotype did not differ from the KO mouse, i.e. the protein function was absent regardless of the specific mechanism and the phenotype outcome was the same.
In addition to the KO and KI animals, several other strains have been generated which represent dual or triple KOs, where other genes, in addition to Abca4, are knocked out. These include Abca4−/−/Rdh8−/−, Abca4−/−/Rdh8−/−/Nrl−/− and some others. Some of these, especially the Abca4−/−/Rdh8−/− strain, have been extensively used for modeling ABCA4-associated retinopathy and even AMD, although they do not, strictly speaking, represent models for these diseases. The Abca4−/−/Rdh8−/− strain presents with much more advanced phenotype, which is not surprising since the two consecutive proteins in the visual cycle are eliminated, thereby causing an earlier onset and fast progressing retinal degeneration (Maeda et al., 2008).
Anatomical differences between the mouse and human retina, namely the absence of a macula due to the spatial distribution of rods and cones in the former has been a long standing challenge in recapitulating disease features found in human patients. As such, extensive efforts over the last 20 years have been devoted to identify a more suitable model system, such as certain breeds of dogs, which have a macula-like “visual streak” that have been efficiently used to study severe retinal degenerations resulting from mutations in the RPE65 gene (Acland et al., 2001; Jacobson et al., 2005) and among others. While the existence of a dog with ABCA4-associated retinopathy was very likely due to the extensive genetic variability not only in the human ABCA4 locus but also in other mammals, the search was successful only very recently, when when several Labrador retrievers were identified and characterized as a KO for ABCA4 (Makelainen et al., 2019). The affected animals, homozygous for the deleterious, c.4176insC, p.(Phe1393Leufs*2), allele presented with a phenotype more closely resembling the ABCA4-associated retinopathy in humans (Makelainen et al., 2019), including visual impairment at 10 years of age, abnormal fundus images, a complete loss of ABCA4 protein, profound reduction of cone outer segments, and an approximately 50% reduction of photoreceptor nuclei in the affected retinae. The RPE autofluorescence in the affected animal, indicating lipofuscin accumulation, was ~7X higher compared to the unaffected dogs and a clear functional defect was detected by flash-electroretinography (Makelainen et al., 2019). In summary, the dogs with no functional ABCA4 closely resembled the human phenotype, although the disease severity was still less profound than in patients lacking ABCA4 (Tanaka et al., 2018).
11. Therapeutic intervention
11.1. Clinical trials
Given its relatively high prevalence as well as its disease course, ABCA4-associated retinopathy is an attractive target for therapeutic intervention, yet no approved therapy exists. For several reasons, the eye (or retina) is an extremely suitable organ for the development and implementation of novel therapies, i.e. its easy accessibility, compartmentalized and immune-privileged nature, and the possibilities to measure potential therapeutic outcome non-invasively. Like for every other subtype of IRD, the chosen therapeutic strategy for ABCA4-associated retinopathy can range from mutation-specific approaches to more generally applicable cell replacement, mainly depending on the primary genetic defect, and the disease stage at the time of treatment (Vazquez-Dominguez et al., 2019). However, in particular for ABCA4-associated retinopathy, one of the most challenging aspects of implementing new treatments is the ability to measure therapeutic benefit. Below, we outline the various therapeutic strategies that are currently in clinical trials or in preclinical development, and discuss how disease progression can be accurately and quantitatively measured, with the ultimate goal to define useful clinical outcome parameters to prove therapeutic benefit.
As shown in Table 6, currently there are 16 clinical trials registered at http://clinicaltrials.gov describing therapeutic intervention for ABCA4-associated retinopathy (combining the results of search queries ‘ABCA4’ and ‘Stargardt’ and selecting those containing a therapeutic intervention). One additional trial was only registered at https://www.clinicaltrialsregister.eu/. These trials can roughly be divided into three categories, i.e. cell replacement, compound administration and gene augmentation (Fig. 9a).
Table 6.
Registered clinical trials for ABCA4-associated retinopathy.
| Cell transplantation | ||||
|---|---|---|---|---|
| Reg. number | Intervention | Phase | Status | References |
| NCT01920867 | Bone marrow-derived stem cells | n.a. | Enrolling by invitation | |
| NCT03011541 | Bone marrow-derived stem cells | n.a. | Recruiting | |
| NCT02903576 | hESC-derived RPE cells | Phase 1/2 | Unknown | |
| NCT01345006 | hESC-derived RPE cells (MA09-hRPE) | Phase 1/2 | Completed | Schwartz et al. (2012) |
| NCT01469832 | hESC-derived RPE cells (MA09-hRPE) | Phase 1/2 | Completed | Mehat et al. (2018) |
| NCT03772938 | Stem/progenitor cells | Phase 1/2 | Enrolling by invitation | |
| Compound administration | ||||
| Reg. number | Intervention | Phase | Status | References |
| NCT00346853 | 4-Methylpyrazole (alcohol dehydrogenase inhibitor) | Phase 1 | Completed | |
| NCT02402660 | ALK-001 (chemically modified vitamin A) | Phase 2 | Recruiting | |
| NCT00060749 | DHA (omega-3 fatty acid) | Phase 1 | Completed | MacDonald and Sieving (2018) |
| NCT03033108 | Emixustat (inhibitor of RPE65) | Phase 2 | Completed | |
| NCT03772665 | Emixustat (inhibitor of RPE65) | Phase 3 | Recruiting | |
| NCT03297515 | Madeos (omega-3 fatty acid) | n.a. | Recruiting | |
| NCT01278277 | Saffron (neuroprotectant) | Phase 1/2 | Unknown | Piccardi et al. (2019) |
| 2018-001496-20 | Soraprazan (H+,K+-ATPase inhibitor) | Phase 2 | Active | |
| NCT03364153 | Zimura (inhibitor of complement factor C5) | Phase 2 | Active, not recruiting | |
| Gene augmentation | ||||
| Reg. number | Intervention | Phase | Status | References |
| NCT01367444 | SAR422459 (lentiviral delivery ABCA4 cDNA) | Phase 1/2 | Terminated | Parker et al. (2016) |
| NCT01736592 | SAR422459 (lentiviral delivery ABCA4 cDNA) | Phase 1/2 | Active, not recruiting |
Trials are subdivided into three categories. Trials are retrieved from http://www.clinicaltrials.gov and https://www.clinicaltrialsregister.eu/.
Fig. 9.

Therapeutic interventions for ABCA4-associated retinopathy. (A)Overview of all therapeutic strategies currently in clinical trials; left panel: cell replacement therapy: cells, either in a stem cell state or pre-differentiated ex vivo towards a retinal fate are directly injected into the retina; middle panel: structure formulas of compounds currently in clinical trials; right panel: gene augmentation therapy, in which wild-type ABCA4 cDNA is packaged into a lentiviral vector which is injected into the retina of subjects with ABCA4-associated retinopathy. (B) Examples of therapeutic strategies currently in preclinical development. Left panel: dual AAV-based gene augmentation, in which ABCA4 cDNA is split into two halves that are aimed to recombine once inside the target cell; right panel: AON-based modulation of pre-mRNA splicing to correct aberrant splicing processes. The figure was created with the aid of BioRender and Illustrator software. Image of the eye was adapted from pixabay.com.
11.1.1. Cell replacement therapy
In ABCA4-associated retinopathy, lipofuscin accumulation exerts a toxic effect causing cell death of neuroretinal and/or RPE cells, mainly in and around the macula. An obvious therapeutic strategy to combat the degeneration of these cells is cell replacement therapy. So far, cell replacement strategies have focused either on the delivery of stem cells, destined to differentiate towards the desired cell once inside the human body, or the delivery of cells that were first differentiated to RPE cells in vitro prior to administration. The exact delivery of cells differs per study, including (a combination of) retrobulbar, subtenon, intravitreal, subretinal or intravenous injection of these cells. In addition, the source of the stem cells varies, and includes bone marrow-derived cells as well as human embryonic stem cells (hESCs). The first study was started in 2011, with initial results reported in 2012, demonstrating interim safety and moderate efficacy in a single subject with ABCA4-associated retinopathy (Schwartz et al., 2012). Later, results of nine subjects in a dose-escalation study were published, again with no major safety issues, although some complications related to vitreoretinal surgery or immunosuppression were reported. In the majority of subjects, visual function appeared to improve in the treated compared to the contralateral eye (Schwartz et al., 2015). Delivery of hESC-derived RPE cells to the retina of ABCA4-associated retinopathy cases also was safe, yet no improvement in visual function could be measured (Mehat et al., 2018). For the other clinical trials aiming to assess the safety and efficacy of cell transplantation, no results have been published yet. In addition to the official clinical trial studies reported at http://www.clinicaltrials.gov, a number of other studies using cell replacement therapy for ABCA4-associated retinopathy have been published. In one study, four subjects received a graft of adipose tissue-derived mesenchymal stem cells that was bilaterally delivered between the choroid and the sclera (Oner et al., 2018). Improvements were found in visual performance as well as responses in multifocal ERG analyses, without any safety complications. Another study described the delivery of hESC-derived RPE cells, with safety and moderate efficacy reported up to one year after treatment (Song et al., 2015b). Overall, despite some moderate efficacy reported in a few studies, one should carefully take the pathophysiological mechanism of ABCA4-associated retinopathy into consideration when applying cell replacement. With ABCA4 being (mainly) expressed in photoreceptors, providing RPE cells (or cells destined to become RPE) will likely not have a long-term beneficial effect. Future research should thus be more directed towards the transplantation of cell sheets that contain both RPE and photoreceptor cells. Thus, although in essence, cell replacement therapy may hold great promise for the treatment of ABCA4-associated retinopathy, there are many variables that still need to be optimized, including selection of the optimal origin of stem cells, whether or not to differentiate cells ex vivo prior to transplantation and if so, until what stage, whether to deliver individual cells or cell sheets, and how to surgically deliver these cells. Finally, also the disease stage of the subject to be treated needs to be taken into account, and can have a major influence on the therapeutic outcome.
11.1.2. Compound administration therapy
An attractive alternative to cell replacement therapy is the administration of compounds, each of which aims to modify either the physiological or the pathological pathways that are affected in ABCA4-associated retinopathy. In total, nine of these compounds have been or are currently being tested in human subjects (Table 6). Only for two of these, initial results have been published (MacDonald and Sieving, 2018; Piccardi et al., 2019). In one study, saffron was used, a compound harbouring carotenoid constituents that are able to counteract oxidative stress. Oral administration of either saffron or placebo (in a crossover trial design) to a total of 31 cases with ABCA4-associated retinopathy revealed that the supplement was well tolerated but on the short term did not seem to give any measurable improvement in visual function (Piccardi et al., 2019). Long-term studies are needed to investigate the potential therapeutic efficacy of this drug. The other published study also made use of oral delivery of a compound, namely docosahexaenoic acid (DHA), in a cross-over trial design with 11 subjects with ABCA4-associated retinopathy. DHA is a major polyunsaturated fatty acid present in high concentrations in the retina (Fliesler and Anderson, 1983) and important for retinal structure and function. It is believed that in ABCA4-associated retinopathy cases, DHA metabolism is altered. Overall, the administration of DHA did not lead to visual improvements in this relatively small group. Small adverse events were noted but it was concluded that they were unrelated to the drug (MacDonald and Sieving, 2018). MADEOS is the abbreviation for Macular Degeneration Omega-3 Study in which the efficacy of omega-3 fatty acids is tested in subjects with ABCA4-associated retinopathy or AMD. This trial however is still in its recruiting phase.
Another potential drug used for the treatment of ABCA4-associated retinopathy is 4-methylpyrazole (4-MP), an alcohol dehydrogenase inhibitor that can delay dark adaptation, at least in laboratory animals. In healthy individuals, intravenous administration did not seem to have an effect on dark adaptation and thus the potential therapeutic efficacy was questioned (Jurgensmeier et al., 2007). However, 4-MP could potentially halt or delay the processing of vitamin A derivatives and thereby prevent the formation of toxic lipofuscin, and was therefore also tested in ten subjects with ABCA4-associated retinopathy. No trial results however were published despite the fact that the study was completed more than a decade ago. ALK-001 is also a molecule that aims to prevent the formation of lipofuscin, yet by a slightly different mechanism. In fact, ALK-001 is a deuterated form of vitamin A that is less capable of forming vitamin A dimers, and thereby toxic lipofuscin. The therapeutic potential of ALK-001 was demonstrated in a murine model of ABCA4-associated retinopathy, by showing a reduced A2E dimer formation and lipofuscin accumulation compared to age-matched wild-type mice (Charbel Issa et al., 2015). A phase 1 study, in which ALK-001 was administered to 40 healthy adult volunteers, has been completed (NCT02230228), after which the phase II study, where 50 ABCA4-associated retinopathy cases were administered with either ALK-001 or placebo at a daily basis, was initiated. No results have been published to date. The last two compounds that are currently being tested in clinical trials both act at the level of the RPE. Emixustat hydrochloride (also known as ACU-4429) is a nonretinoid compound that can exert an inhibitory effect on one of the enzymes involved in the visual cycle, the RPE-specific 65 kDa protein isomerase, encoded by the RPE65 gene. A phase 1 placebo-controlled study exploring the safety of Emixustat that was administered to healthy individuals at a daily basis (NCT00942240) did not reveal any systemic adverse events, although ocular side effects were observed in the majority of participants (Kubota et al., 2014). However, these effects were considered mild and transient, disappearing after completion of the trial, and warranted further testing in subjects with impaired visual function. Initially, Emixustat was tested in subjects with geographic atrophy associated with dry AMD, with results that are supportive of the anticipated mode of action, yet not having led to spectacular results in terms of visual improvement (Dugel et al., 2015). As illustrated in Table 6, Emixustat is now also being evaluated as a drug for ABCA4-associated retinopathy, in two independent trials. The phase 2 trial that aimed to look at the potential short-term benefit has just been completed; however, no results have been reported. The multicentre phase 3 trial in which 162 subjects are planned to be enrolled is currently ongoing, with the aim to monitor the long-term effect of this new drug. One compound that is planned to be studied in human subjects is Zimura, an aptamer that can inhibit the activity of complement factor C5 (Drolet et al., 2016). Zimura was first tested in subjects with age-related macular degeneration but now also for cases with ABCA4-associated retinopathy. Finally, a compound called Soraprazan, a fast-acting inhibitor of H+,K+-ATPase (Simon et al., 2007), is currently being investigated in a multi-national, multicenter, double-masked, placebo-controlled proof of concept trial. Previously, Soraprazan was used for the treatment of patients with gastroesophageal reflux disease. After the discovery that this compound could remove lipofuscin from the RPE in monkeys (Julien and Schraermeyer, 2012), its potential for the treatment of ABCA4-associated retinopathy is now also being studied, with no data reported so far.
Together, the various compounds that have been tested so far in subjects with ABCA4-associated retinopathy overall can be considered safe, yet none of them succeeded to demonstrate a high therapeutic effect. The advantage of many of these compounds, i.e. the fact that they can be administered orally, also can be considered a disadvantage, since this delivery route does not always allow the active compound to reach a sufficient concentration within the retina. Further research to identify the optimal route of administration, the concentrations needed to exert their effect without causing adverse events, and the ideal treatment regime, is needed to reveal the true therapeutic potential of the aforementioned compounds, as well as those that are currently in preclinical development.
11.1.3. Gene augmentation therapy
The approval of gene augmentation therapy for another subtype of inherited retinal disease that is caused by bi-allelic mutations in RPE65, has provided hope for many visually impaired individuals, and is paralleled by the development of similar therapeutic strategies for other genes underlying these disorders (Vazquez-Dominguez et al., 2019). The vast majority of trials assessing gene augmentation for retinal diseases employs adeno-associated viruses (AAVs) to deliver the wild type cDNA of the gene that is mutated. The one and only exception is gene augmentation therapy for ABCA4 disease, foremost because the size of wild-type ABCA4 cDNA (6.8 kb) surpasses AAV’s cargo capacity. A phase 1/2 clinical study employing lentiviral delivery of ABCA4 cDNA (therapeutic molecule SAR422459) has commenced in 2012, but recently has been terminated, without any data of efficacy published. However, in 2016, Parker et al. reported on test-retest variability for a number of clinical outcomes in subjects participating in this trial (Parker et al., 2016). The exact reasons for terminating this trial are unknown; a second trial employing the same therapeutic molecule however is still active and recruiting (Table 6).
Summarizing the clinical trials so far, one can conclude that there are several different treatments for ABCA4-associated retinopathy under development, yet none of them so far revealed itself to be the ideal therapeutic intervention. Not only the therapeutic drug itself needs to be further optimized, also the high degree of variability observed in different clinical tests (between and within a subject) warrants an improved trial design. This is further illustrated by the many pre-clinical studies that are currently ongoing, to identify novel therapeutic strategies as well as improved clinical diagnostics, as further outlined below.
11.2. Preclinical studies
Besides the clinical studies mentioned above, numerous therapeutic strategies are currently being assessed in preclinical models (examples are illustrated in Fig. 9b). Many of these studies focus on the replacement of stem cell-derived RPE cells, as summarized by Sachdeva et al. (Sachdeva and Eliott, 2016). In addition, novel compounds that e.g. interfere with lipofuscin accumulation, or affect intracellular trafficking of mutant ABCA4 protein are constantly being investigated, a few examples of which are provided below. Administration of BPN-14136, a non-retinoid antagonist of the retinol-binding protein 3 involved in the visual cycle, to Abca4−/− mice inhibited bisretinoid synthesis while not altering the rate of the visual cycle, demonstrating its potential for the treatment of ABCA4-associated retinopathy (Racz et al., 2018). A systems pharmacological approach identified a number of G-protein coupled receptors that showed improved photoreceptor cell survival and function in Abca4−/− mice (Sabirzhanova et al., 2015). VX-809, a molecule previously found to be efficacious for rescuing trafficking of the CFTR protein in cystic fibrosis, also increased the membrane localization of some mutant ABCA4 proteins in cultured HEK293T cells (Liu et al., 2019), although care is warranted when deciding which allele can be amenable for which type of therapy, as the pathogenic mechanism for many variants is still not entirely understood.
As no data on potential efficacy have been reported for lentiviral gene augmentation, alternative strategies to deliver ABCA4 cDNA to the retinal cells were developed. One delivery strategy uses nanoparticles, whereas others make use of a dual AAV approach. Han and colleagues developed non-viral nanoparticles, and demonstrated that upon sub-retinal delivery in Abca4−/− mice, Abca4 transgene expression persisted up to eight months after injection, and resulted in reduced lipofuscin accumulation in the treated animals (Han et al., 2012). With dual AAV technology, cDNA fragments that exceed the cargo capacity of a single AAV can be split into two halves and each packaged into a separate AAV, with additional sequences that allow reconstitution of the complete cDNA once inside the target cell (Trapani, 2019). Dual AAVs have been used to deliver ABCA4 cDNA to the (cone-enriched) porcine retina (Trapani et al., 2014, 2015), as well as the retina of Abca4−/− mice, with reduced lipofuscin accumulation and/or correction of the autofluorescent phenotype measured in a number of separate studies (Dyka et al., 2019; McClements et al., 2019; Trapani et al., 2015). Although no clinical trials exploring the safety and efficacy of these gene augmentation strategies have been reported, it is expected that these will soon commence. Yet, it is important to realize that the transduction efficiency of both dual AAV vectors and non-viral vectors generally is lower when compared to classical AAV vectors.
Finally, there are also several mutation-specific therapies under development for ABCA4-associated retinopathy. These strategies mainly employ antisense oligonucleotides (AONs) and are focussed on those variants that affect pre-mRNA splicing of ABCA4. AONs are relatively small and versatile RNA molecules that can be synthesized in such a way that their sequence is complementary to their target pre-mRNA. So far, AONs have mainly been used to block PE inclusions caused by deep-intronic mutations. The first AON described for a retinal disease targets a recurrent deep-intronic mutation in CEP290, and was initially tested in lymphoblastoid and fibroblast cells derived from patients homozygously harbouring this mutation (Collin et al., 2012; Gerard et al., 2012). Following further demonstration of efficacy in a humanized mouse model (Garanto et al., 2016) and in iPSC-derived retinal organoids (Dulla et al., 2018; Parfitt et al., 2016), a clinical trial was initiated, with recently reported positive interim results achieved by intravitreal delivery of AONs in subjects with CEP290-associated LCA (Cideciyan et al., 2019). As stated in section 6.4, an increasing number of deep-intronic ABCA4 variants that result in PE inclusion have been identified over the last few years. Using midigene splice assays and patient-derived cells, the ability of AONs to prevents the aberrant PE inclusions caused by several different ABCA4 variants has been demonstrated (Albert et al., 2018; Bauwens et al., 2019; Garanto et al., 2019; Sangermano et al., 2019), although the therapeutic efficacy so far was only assessed at the RNA level. Besides deep-intronic variants, there are also several ABCA4 variants reported that are located in or near exons and result in altered pre-mRNA splicing, i.e. exon skipping or exon elongation. Also for these variants, AONs could be employed, e.g. by blocking splice silencer motifs that are created by such variants, or alternative cryptic splice sites that are used. Overall, the versatile nature of AONs render these attractive therapeutic molecules, yet the fact that some of the variants targeted by AONs are rare, or sometimes even ultra-rare, prevent a broad applicability of this therapeutic strategy for a large group of ABCA4-associated retinopathy cases. In addition to AON-based splicing correction, genome editing (or RNA editing) approaches are booming, and have the potential to correct mutations regardless of the size of the gene, or the position within the genome. By employing CRISPR/Cas9 technology with the homology-directed repair (HDR) pathway, one can replace e.g. single basepair substitutions with the wild-type nucleotide and in that way repair mutations (Hsu et al., 2014; Yanik et al., 2017). Other recently described strategies include e.g. base pair-editing systems (Billon et al., 2017; Rees and Liu, 2018). It is expected that these strategies will soon also be applied for the mutation-specific correction of ABCA4 variants.
12. Clinical outcome measures
The selection of appropriate clinical outcome measures for ABCA4-associated retinopathy should be predicated on a deep understanding of the factors underlying progression. The natural history of ABCA4-associated retinopathy consists of multiple trajectories that is largely determined by an individual’s genotype. Effective outcome measures should be generalizable across patients but at the same time, account for their respective differences.
12.1. Understanding the parameters of atrophy growth
Monitoring the size of the atrophic lesion has been the primary outcome measure of most ABCA4-associated retinopathy clinical trials to date. At face value, atrophy progression appears to be a logical endpoint as it is the most discernible anatomic manifestation of cellular degeneration and historically, the primary endpoint in clinical trials for AMD (Klein et al., 2010; Knudtson et al., 2004; Lindblad et al., 2009). Although convenient, following the AMD model may not be an effective strategy for several reasons: (1) the two conditions share very little pathophysiological and demographic overlap, and (2) studies have already reported extensive differences in their respective rates and spatial correlation with the functional scotoma (Bernstein et al., 2016; Lindner et al., 2017; Sunness and Steiner, 2008). Furthermore, the atrophic process in ABCA4-associated retinopathy is difficult to uniformly define, as it is an evolving entity that progressively spans multiple layers of the retina over time across the natural history of the disease (Fig. 1). The sensitivity of SW-AF imaging provides a reasonable assessment of cellular level involvement (Sunness et al., 2006) and as such, the investigators of ProgStar have proposed distinguishing lesions decreased autofluorescence (DDAF) or questionably decreased autofluorescence (QDAF) (ProgStar Report No. 9) (Strauss et al., 2017a, 2017b). Nevertheless, an analysis of reported rates of “DDAF” across the several prominent articles still uncovered a range of variability— 0.94 ± 0.87) mm2/year (range 0.2–2.13 mm2/year (Chen et al., 2010) 1.58 ± 1.25 (standard deviations) mm2/year (range 0.13–5.27 mm2/year) (McBain et al., 2012) and 2.5 ± 2.9 mm2 (range, 0.02–16.03 mm2) (Strauss et al., 2016)— indicating the presence of other unaccounted factors. Fujinami et al. stratified patients according to “AF type”, or the background heterogeneity of flecks, and found that the rate of atrophy enlargement (RAE) (or “DDAF”) in a more severe background exhibit significantly increased rates of growth (Fujinami et al., 2013a). More recent studies have looked to different modes of optical coherence tomography (OCT) to monitor the rate of lesion progression or even the delineation of new lesion types such as “dark atrophy” on OCT angiography (Pellegrini et al., 2016) allowing for a more restrictive definition of atrophy by observable changes in the anatomical layers visible on OCT scans (Arepalli et al., 2018; Cai et al., 2018; Kong et al., 2019; Park et al., 2015; Tanna et al., 2019). Increasing interest is being garnered in this area particularly with advancements in scan resolution, wide-field capture and analytical capabilities such as en face analysis (Alabduljalil et al., 2019; Greenstein et al., 2017; Melillo et al., 2016; Sodi et al., 2016).
12.2. Targeting the lipofuscin biomarker
Most ABCA4-associated retinopathy patients exhibit a spatially homogenous and localized increase in RPE lipofuscin because of ABCA4 dysfunction (Cideciyan et al., 2004). Augmented RPE lipofuscin confers a Vermillion hue to the fundus under white light imaging and can obstruct fluorescence emanating from the underlying choroid in during fluorescein angiograms (FA) giving rise to the distinct “dark” or “silent” choroid in up to 62% of patients (Anmarkrud, 1979; Ernest and Krill, 1966; Fish et al., 1981). Despite the historical utility of FA, its role in the evaluation of STGD1 has been increasingly limited as it is no longer performed on a routine basis in favor of newer, less invasive imaging modalities. Further development of the confocal scanning laser ophthalmoscope (cSLO) introduced various modes of imaging that allow for the capture of an inherent autofluorescence emitted by photoreceptor and RPE fluorophores belonging to the family of bisretinoids that includes N-retinyl-N-retinylidene ethanolamine (A2E), its cis isomers and other related compounds (Fishkin et al., 2005; Kim and Sparrow, 2018; Parish et al., 1998; Wu et al., 2009; Yamamoto et al., 2011). This autofluorescence signal is emitted at wavelengths between 520 and 800 nm and can be captured by the standard SW-AF (448-nm excitation) and the more recently developed ultra-wide field AF (532-nm excitation). The components of photoreceptor/RPE lipofuscin contribute most predominantly to the 488-nm excitation wavelength (SW-AF) where its distribution and pattern in the normal retina has been well described (Delori et al., 1995; Delori et al., 2001; von Ruckmann et al., 1995, 1997). Deviations in SW-AF intensity and texture can be observed in nearly all inherited retinal degenerative diseases where a decrease or absence of SW-AF is generally indicative of a disruption in the tissue architecture in regions not obstructed by retinal vessels or luteal macular pigment in the fovea.
Early attempts at measuring SW-AF (Cideciyan et al., 2004; Lois et al., 2004) lead to the development of quantitative autofluorescence (qAF) by Delori and colleagues (Delori et al., 2011) wherein the SW-AF intensities in non-normalized images (acquired without histogram stretching) were calibrated to the fluorescence intensities of an internal reference mounted within the cSLO and captured simultaneously to compensate for variations in laser power and detector gain. Using this method, Burke et al. verified the increase in autofluorescence and additionally, reported differences amongst genotypes (Burke et al., 2014; Sparrow et al., 2020). In a similar study, the use of qAF was shown to be effective in differentiating ABCA4-associated retinopathy from non-ABCA4-associated retinopathy (masquerading) bull’s eye maculopathy phenotypes (Duncker et al., 2015b). Near-infrared autofluorescence imaging (NIR-AF), which employs an excitation of signal at wavelength 787 nm, generates a signal corresponding to RPE and choroidal melanin (Keilhauer and Delori, 2006). This has also recently emerged as an effective modality for inherited retinal diseases and ABCA4-associated retinopathy (Cideciyan et al., 2007; Cukras et al., 2012; Duncker et al., 2013, 2014; Kellner et al., 2009; Sparrow et al., 2015). Interestingly, Paavo et al. reported quantitative increases in the NIR-AF (787-nm) signal in ABCA4-associated retinopathy as well as Abca4−/− mice, corroborating results from an earlier study (Charbel Issa et al., 2013) prompting further revisions to either the interpretation of the anatomical origins of the NIR-AF signal or ABCA4-associated pathophysiology (e.g. the role of melano-lipofuscin) (Paavo et al., 2018). Additionally, NIR-AF has the practical advantage of being low luminance for ease in acquisition and possible safety consideration.
Effort should be allocated towards tracking other dynamic lipofuscin-related features such as the evolution of flecks patterns and the “leading disease front” (Cideciyan et al., 2015). However, as is the case with qAF, data acquisition and analysis may require a high-level expertise and computational proficiency that can impede its adoption as a routine method in the clinic or treatment trials. Nevertheless, the quantitation of autofluorescence holds promise as an effective outcome measure in ABCA4-associated retinopathy, particularly for monitoring the effects of lipofuscin-targeted therapies. Further studies evaluating the longitudinal sensitivity of qAF and a more precise understanding of the cellular origins of autofluorescence may further support its adoption as a primary outcome measure for clinical trials.
12.3. Mapping the range of functional loss
Impairment of visual function is the predominant symptom of ABCA4-associated retinopathy and all inherited retinal diseases and as such, developing effective ways to track it over time is essential to assessing the disease natural history and response to therapies. The most direct approach is measurement of best-corrected visual acuity (BCVA). Numerous systematic protocols have been developed and this is the standard used by all clinical trials (Table 6). Many studies have documented broad trends in BCVA progression and its relationship to other clinical features in ABCA4-associated retinopathy (Birch et al., 2001; Collison and Fishman, 2018; Ergun et al., 2005; Kong et al., 2016; Parodi et al., 2015; Querques et al., 2008; Testa et al., 2012, 2014), however as a method, it is perhaps most susceptible to technical limitations such as measurement bias, repeatability as well as confounders that are specific to ABCA4-associated retinopathy such as the variable status of the fovea (Bax et al., 2019b; Collison et al., 2019; Nakao et al., 2012; van Huet et al., 2014) and shifting of the preferred retinal locus (PRL) into consideration (Bethlehem et al., 2014; Greenstein et al., 2008; Krishnan and Bedell, 2018; Schonbach et al., 2017a, 2018).
The full-field electroretinogram (ffERG) is a powerful tool in the diagnostic repertoire of a retinal disease clinic. By measuring the summation of electrical activity generated by cones and rods across the entire retina (separately and combined), its application has been highly effective in broadly classifying severity and predicting the prognosis of individuals with ABCA4-associated retinopathy (Lois et al., 2001). The drawback to this generalized approach however, is an insensitivity to subtle changes in the macula, which disproportionately contributes to the aggregate electrophysiological response of the entire retina. The multifocal electroretinogram (mfERG) is more precise as local ERG responses can be recorded simultaneously from many defined regions of the retina. Few studies to date have examined its suitability for monitoring ABCA4-associated retinopathy progression (Kuniyoshi et al., 2014; Sisk and Leng, 2014; Tosha et al., 2010), which may be due to the requirement of fixation stability in a disease where most patients have profound central vision loss. The same issue is encountered with conventional methods of static and kinetic perimetry in the mapping of visual fields as they are performed under free-viewing conditions (fixation is not tracked) required but not monitored increasing the likelihood of obtaining false positives (Acton and Greenstein, 2013).
Methods circumventing the fixation requirement include full-field stimulus testing (FST) which provides a psychophysical measure of luminance. Although FST elicits a full-field response, Collison et al. found that cone-mediated thresholds in ABCA4-associated retinopathy correlate well with locally defined changes such as visual acuity and macular thickness (Collison et al., 2014). Furthermore, the improvements on FST were instrumental in demonstrating the efficacy of voretigene neparvovec (AAV2-hRPE65v2, Luxturna) in RPE65-associated LCA (NCT00999609) supporting its suitability as an endpoint for aggressive therapies in patients with advanced phenotypes (Jacobson et al., 2009; Maguire et al., 2019). For a more localized assessment, microperimetry (MP) allows for the quantification of mesopic and scotopic visual sensitivity at user-defined points on the retina and fundus tracking which addresses unstable fixation. Studies assessing the scotoma of ABCA4-associated retinopathy conclude that it is reliable in tracking longitudinal changes and is highly correlated with changes in BCVA (Cideciyan et al., 2012; Schonbach et al., 2017b).
13. Future research
Despite significant advances in deciphering all aspects of ABCA4-associated retinopathy, there are still several areas requiring attention in the future. Despite its worldwide occurrence, the prevalence of ABCA4-associated retinopathy is not yet known. The 1:8000–10,000 estimate, which everybody cites, comes from the textbook chapter by Blacharski in 1988 (Blacharski, 1988), which states: “We have seen this condition much more commonly than retinoblastoma, which has been estimated at 1 in 15,000 live births. Fundus flavimaculatus is not as common as retinitis pigmentosa, which has a prevalence of no more than 1 in 5000. We have roughly estimated the incidence to be between 1 in 8000 and 1 in 10,000.” It is obviously not a scientific way to determine a disease prevalence. The actual prevalence of ABCA4 -associated retinopathy is very difficult to estimate due to enormous clinical and genetic heterogeneity, variable age of onset, and (still) incomplete genetic data. Most of the genetic information for ABCA4-associated retinopathy thus far has been collected from individuals of European descent. To fully appreciate the allelic heterogeneity for ABCA4-associated retinopathy and to better understand the differences in genetic background that may influence the expression of this disease, more emphasis should be put on the sequence analysis of ABCA4-associated retinopathy cases from non-Caucasian populations.
The wide functional spectrum of ABCA4 variants, from extremely hypomorphic to deleterious, leaves us with many ABCA4 variants for which the penetrance is still not known. This has a profound effect on genetic testing and genetic counseling of patients. For example, the disease causality for the p.(Asn1868Ile) allele is still not widely accepted, and this is complicated by the discussion about its penetrance. At least 10% of the entire ABCA4-associated retinopathy remains not genetically confirmed if the genetic testing laboratories do not detect and/or report the p.(Asn1868Ile) variant. Considering the p.(Asn1868Ile) allele “benign” has also a detrimental effect on family planning. Screening of large cohorts of familial cases of ABCA4-associated retinopathy will continue helping to decipher the penetrance of selected ABCA4 alleles on a specific genetic background. We estimate that ~10% of ABCA4-associated retinopathy probands carries a causal deep-intronic variant and that ~2% of the cases carries a SV. Routine diagnostics therefore cannot only rely on the sequence analysis of the coding regions but also needs to sequence the entire ABCA4 genomic locus. To establish pathogenicity of novel deep-intronic variants, NCSS variants and rare synonymous coding variants should be tested using in vitro splice assays. The accuracy of these assays in the future can be improved by using cell lines that mimic the splicing processes in the normal retina. Another emerging important area is the concept of genetic modifiers in the ABCA4 locus and in the genome; i.e., both cis- and trans-modifiers for ABCA4-associated retinopathy. We have started to determine some of the cis-modifiers (see the example of the p.[Gly863Ala,Gly863del] allele above), but most of these remain obscure. Trans-modifiers will be even harder to identify, but with the complete sequencing of the ABCA4 locus (Bauwens et al., 2019; Sangermano et al., 2019; Zernant et al., 2014b) and the entire exome and genome, these are likely to be found through the analysis of very large familial cohorts, which are, fortunately, possible to obtain even for a relatively rare Mendelian disease. More than 50% of ABCA4 variants are missense mutations. For frequent missense mutations, we can predict their severity quite accurately based on genotype-phenotype correlations. Most of the rare missense variants however are classified as ‘variants of unknown significance’. If a crystal structure would be determined at high resolution for the ABCA4 protein, proper functional studies could be conducted. Currently, functional studies relying on animal models and in vitro data, can reach correct conclusions in some cases; however, in many cases these do not correlate with genetic and clinical studies. One of the best examples is the c.2588G>C, p.[Gly863Ala,Gly863del] variant, which has been shown to result in a dual effect and also in significant defects in in vitro assays (Maugeri et al., 1999; Sangermano et al., 2018), but is not penetrant based on clinical/genetic studies unless it is in cis with the p.(Asn1868Ile) allele. Determining the high-resolution protein structure would allow investigating the predicted effect of this and many other missense ABCA4 variants with great precision.
The relatively high prevalence of ABCA4-associated retinopathy as well as its progressive nature have led to an enormous attention from academia as well as industry with regard to the development of molecular and cellular therapies. Although ABCA4 is expressed in photoreceptors, its pathogenic effects are manifest primarily in the RPE (Cideciyan et al., 2004), where accumulation of toxic bisretinoids (Sparrow et al., 2012) occur from the perpetual shedding and subsequent phagocytosis of outer segments (Steinberg et al., 1977; Young, 1967; Young and Bok, 1969). This prevailing disease model, while in many ways consistent with general mechanism of ABCA4 dysfunction, may not be that straightforward. Rod discs are enclosed, self-containing structures and thus bisretinoids generated within would indeed eventually be phagocytosed, along with the rest of the shed outer segment, by RPE. Cone lamellae, however, are contiguous with its plasma membrane and therefore, accumulating bisretinoids may be retained in other cellular compartments, where their toxic effects can be expressed. Consistent with this model is the histopathological observation of lipofuscin-like autofluorescence in the cone inner segments of the retina of a patient with fundus flavimaculatus (ABCA4-associated retinopathy) (Birnbach et al., 1994). Clinical imaging studies in patients have largely demonstrated the trend that RPE cell death precedes photoreceptor cell death (Cideciyan et al., 2007; Duncker et al., 2014; Greenstein et al., 2017; Kellner et al., 2009); however most, if not all of these studies, report notable exceptions. For instance, in a study of 24 patients (45 eyes), Duncker et al. reported that zones of RPE atrophy (reduced AF) are statistically larger in NIR-AF compared to SW-AF in the same eye, although a subgroup of younger patients with relatively severe, early-onset disease exhibited larger areas of photoreceptor-attributable EZ loss compared to reduced NIR-AF (Duncker et al., 2014). Similarly, AO-SLO imaging of two STGD1 patients revealed abnormal spacing of rods and cones in otherwise unaffected areas (Song et al., 2015a). Whether photoreceptor cell degeneration precedes or follows RPE loss in ABCA4-associated retinopathy remains unresolved. Given the complex role of ABCA4 dysfunction in both cell types, it is likely that the sequence of incapacitation varies in accordance with disease stage, location and overall severity of progression. There are still many challenges ahead, both in terms of identifying and optimizing the right therapeutic drug and finding the right clinical parameters to accurately measure disease progression as well as therapeutic benefit. In the end, there will likely not be a ‘one-strategy-fits-all’ treatment; rather every individual case may need its own tailor-made therapeutic intervention, based on their genetic profile as well as the stage of their disease.
In conclusion, clinical, molecular genetics and therapeutic studies have resulted in a large body of knowledge regarding the fascinating complex ABCA4-associated retinopathy. At the same time, new research avenues have opened to unravel the unexplained differences in disease expression between ABCA4-associated retinopathy cases, both within and between families. Long-read sequencing technologies will allow phasing of different genomic variants to establish the role of cis-modifiers and enable us to appreciate in great detail normal and abnormal RNA splicing. A full comprehension of the molecular genetic causes and molecular mechanisms of ABCA4-associated retinopathy will allow us to develop ‘personalized’ therapies to slow down or stop disease progression.
Acknowledgements
FPMC and RWJC wish to acknowledge long-standing collaborations with Drs. Carel B. Hoyng and L. Ingeborgh van den Born and the contributions of Drs. Claire-Marie Dhaenens, Alex Garanto, Riccardo Sangermano and Irene Vazquez-Dominguez, and of Ms. Mubeen Khan, and Ms. Esmee H. Runhart. RA and WL wish to acknowledge the patients and their families, long-standing collaborations with Drs. Stephen H. Tsang, Janet R. Sparrow and Gerald A. Fishman and, especially, Dr. Jana Zernant for almost 20 years of work in ABCA4 genetics and help with data analysis for this manuscript. This work was supported in part by the Retina UK grants no. GR591 (to FPMC), and GR596 (to RWJC and FPMC), a Fighting Blindness Ireland grant no. FB18CRE (to FPMC), a Horizon 2020, Marie Sklodowska-Curie Innovative Training Network entitled European Training Network to Diagnose, Understand and Treat Stargardt Disease, a Frequent Inherited Blinding Disorder-StarT (813490) (to FPMC, RWJC), the Foundation Fighting Blindness USA, grant no. PPA-0517-0717-RAD (to FPMC and RWJC), the Foundation Fighting Blindness USA, grant no. BR-GE-1018-0738-RAD (to FPMC), the Rotterdamse Stichting Blindenbelangen, the Stichting Blindenhulp, and the Stichting tot Verbetering van het Lot der Blinden (to FPMC), and by the Landelijke Stichting voor Blinden en Slechtzienden, Macula Degeneratie fonds and the Stichting Blinden-Penning that contributed through Uitzicht 2016-12 (to FPMC). This work was supported, in part, by the National Eye Institute, NIH grants R01 EY028203, R01 EY028954, R01 EY029315, R01 EY024091, P30 19007 (Core Grant for Vision Research), the Foundation Fighting Blindness USA, grant no. PPA-1218-0751-COLU, and the unrestricted grant to the Department of Ophthalmology, Columbia University, from Research to Prevent Blindness (to RA and WL).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J, 2001. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet 28, 92–95. [DOI] [PubMed] [Google Scholar]
- Acton JH, Greenstein VC, 2013. Fundus-driven perimetry (microperimetry) compared to conventional static automated perimetry: similarities, differences, and clinical applications. Can. J. Ophthalmol 48, 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agbaga MP, Brush RS, Mandal MN, Henry K, Elliott MH, Anderson RE, 2008. Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc. Natl. Acad. Sci. U. S. A 105, 12843–12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Lamban J, Gonzalez-Aguilera JJ, Riveiro-Alvarez R, Cantalapiedra D, Avila-Fernandez A, Villaverde-Montero C, Corton M, Bianco-Kelly F, Garcia-Sandoval B, Ayuso C, 2011. Further associations between mutations and polymorphisms in the ABCA4 gene: clinical implication of allelic variants and their role as protector/risk factors. Invest. Ophthalmol. Vis. Sci 52, 6206–6212. [DOI] [PubMed] [Google Scholar]
- Ahn J, Molday RS, 2000. Purification and characterization of ABCR from bovine rod outer segments. Methods Enzymol. 315, 864–879. [DOI] [PubMed] [Google Scholar]
- Alabduljalil T, Patel RC, Alqahtani AA, Gao SS, Gale MJ, Zhang M, Jia Y, Huang D, Chiang PW, Chen R, Wang J, Weleber RG, Pennesi ME, Yang P, 2019. Correlation of outer retinal degeneration and choriocapillaris loss in stargardt disease using en face optical coherence tomography and optical coherence tomography angiography. Am. J. Ophthalmol 202, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert S, Garanto A, Sangermano R, Khan M, Bax NM, Hoyng CB, Zernant J, Lee W, Allikmets R, Collin RWJ, Cremers FPM, 2018. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying stargardt disease. Am. J. Hum. Genet 102, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldahmesh MA, Mohamed JY, Alkuraya HS, Verma IC, Puri RD, Alaiya AA, Rizzo WB, Alkuraya FS, 2011. Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am. J. Hum. Genet 89, 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, 2000. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am. J. Hum. Genet 67, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, 2007. Stargardt disease: from gene discovery to therapy. In: Tombran-Tink J, Barnstable CJ (Eds.), Biology. Diagnostics and Therapeutics Humana Press, Totowa, NJ, pp. 105–118 Retinal Degenerations. [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewus RA, Nathans J, Leppert M, Dean M, Lupski JR, 1997. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet 15, 236–246. [DOI] [PubMed] [Google Scholar]
- Allikmets R, Zernant J, Lee W, 2018. Penetrance of the ABCA4 p.Asn1868Ile allele in stargardt disease. Invest. Ophthalmol. Vis. Sci 59, 5564–5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KL, Baird L, Lewds RA, Chinault AC, Otterud B, Leppert M, Lupski JR, 1995. A YAC contig encompassing the recessive Stargardt disease gene (STGD) on chromosome 1p. Am. J. Hum. Genet 57, 1351–1363. [PMC free article] [PubMed] [Google Scholar]
- Anmarkrud N, 1979. Fundus fluorescein angiography in fundus flavimaculatus and Stargardts disease. Acta Ophthalmol. 57, 172–182. [DOI] [PubMed] [Google Scholar]
- Arepalli S, Traboulsi EI, Ehlers JP, 2018. Ellipsoid zone mapping and outer retinal assessment in stargardt disease. Retina 38, 1427–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauwens M, De Zaeytijd J, Weisschuh N, Kohl S, Meire F, Dahan K, Depasse F, De Jaegere S, De Ravel T, De Rademaeker M, Loeys B, Coppieters F, Leroy BP, De Baere E, 2015. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum. Mutat 36, 39–42. [DOI] [PubMed] [Google Scholar]
- Bauwens M, Garanto A, Sangermano R, Naessens S, Weisschuh N, De Zaeytijd J, Khan M, Sadler F, Balikova I, Van Cauwenbergh C, Rosseel T, Bauwens J, De Leeneer K, De Jaegere S, Van Laethem T, De Vries M, Carss K, Arno G, Fakin A, Webster AR, de Ravel de l’Argentiere TJL, Sznajer Y, Vuylsteke M, Kohl S, Wissinger B, Cherry T, Collin RWJ, Cremers FPM, Leroy BP, De Baere E, 2019. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med 21, 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax NM, Sangermano R, Roosing S, Thiadens AA, Hoefsloot LH, van den Born LI, Phan M, Klevering BJ, Westeneng-van Haaften C, Braun TA, Zonneveld-Vrieling MN, de Wijs I, Mutlu M, Stone EM, den Hollander AI, Klaver CC, Hoyng CB, Cremers FP, 2015. Heterozygous deep-intronic variants and deletions in ABCA4 in persons with retinal dystrophies and one exonic ABCA4 variant. Hum. Mutat 36, 43–47. [DOI] [PubMed] [Google Scholar]
- Bax NM, Lambertus S, Cremers FPM, Klevering BJ, Hoyng CB, 2019a. The absence of fundus abnormalities in Stargardt disease. Graefes Arch. Clin. Exp. Ophthalmol 257, 1147–1157. [DOI] [PubMed] [Google Scholar]
- Bax NM, Valkenburg D, Lambertus S, Klevering BJ, Boon CJF, Holz FG, Cremers FPM, Fleckenstein M, Hoyng CB, Lindner M, for the Foveal Sparing Atrophy Study, 2019b. Foveal sparing in central retinal dystrophies. T. Invest. Ophthalmol. Vis. Sci 60, 3456–3467. [DOI] [PubMed] [Google Scholar]
- Beharry S, Zhong M, Molday RS, 2004. N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J. Biol. Chem 279, 53972–53979. [DOI] [PubMed] [Google Scholar]
- Beit-Ya’acov A, Mizrahi-Meissonnier L, Obolensky A, Landau C, Blumenfeld A, Rosenmann A, Banin E, Sharon D, 2007. Homozygosity for a novel ABCA4 founder splicing mutation is associated with progressive and severe Stargardt-like disease. Invest. Ophthalmol. Vis. Sci 48, 4308–4314. [DOI] [PubMed] [Google Scholar]
- Bernstein A, Sunness JS, Applegate CA, Tegins EO, 2016. Mapping the dense scotoma and its enlargement in stargardt disease. Retina 36, 1741–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen M, Zernant J, Larsen M, Duno M, Allikmets R, Rosenberg T, 2014. Generalized choriocapillaris dystrophy, a distinct phenotype in the spectrum of ABCA4-associated retinopathies. Invest. Ophthalmol. Vis. Sci 55, 2766–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethlehem RA, Dumoulin SO, Dalmaijer ES, Smit M, Berendschot TT, Nijboer TC, Van der Stigchel S, 2014. Decreased fixation stability of the preferred retinal location in juvenile macular degeneration. PloS One 9, e100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billon P, Bryant EE, Joseph SA, Nambiar TS, Hayward SB, Rothstein R, Ciccia A, 2017. CRISPR-mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP codons. Mol. Cell. 67, 1068–1079 e1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch DG, Peters AY, Locke KL, Spencer R, Megarity CF, Travis GH, 2001. Visual function in patients with cone-rod dystrophy (CRD) associated with mutations in the ABCA4(ABCR) gene. Exp. Eye Res 73, 877–886. [DOI] [PubMed] [Google Scholar]
- Birnbach CD, Jarvelainen M, Possin DE, Milam AH, 1994. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology 101, 1211–1219. [DOI] [PubMed] [Google Scholar]
- Birtel J, Eisenberger T, Gliem M, Muller PL, Herrmann P, Betz C, Zahnleiter D, Neuhaus C, Lenzner S, Holz FG, Mangold E, Bolz HJ, Charbel Issa P, 2018. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep 8, 4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacharski P, 1988. Retinal Dystrophies and Degenerations. Raven Press, New York. [Google Scholar]
- Bok D, 2005. Cellular mechanisms of retinal degenerations: RPE65, ABCA4, RDS, and bicarbonate transporter genes as examples. Retina 25, S18–S20. [DOI] [PubMed] [Google Scholar]
- Braun TA, Mullins RF, Wagner AH, Andorf JL, Johnston RM, Bakall BB, Deluca AP, Fishman GA, Lam BL, Weleber RG, Cideciyan AV, Jacobson SG, Sheffield VC, Tucker BA, Stone EM, 2013. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet 22, 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TR, Fishman GA, Zernant J, Schubert C, Tsang SH, Smith RT, Ayyagari R, Koenekoop RK, Umfress A, Ciccarelli ML, Baldi A, Iannaccone A, Cremers FP, Klaver CC, Allikmets R, 2012. Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci 53, 4458–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TR, Yzer S, Zernant J, Smith RT, Tsang SH, Allikmets R, 2013. Abnormality in the external limiting membrane in early Stargardt disease. Ophthalmic Genet. 34, 75–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TR, Duncker T, Woods RL, Greenberg JP, Zernant J, Tsang SH, Smith RT, Allikmets R, Sparrow JR, Delori FC, 2014. Quantitative fundus autofluorescence in recessive Stargardt disease. Invest. Ophthalmol. Vis. Sci 55, 2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai CX, Light JG, Handa JT, 2018. Quantifying the rate of ellipsoid zone loss in stargardt disease. Am. J. Ophthalmol 186, 1–9. [DOI] [PubMed] [Google Scholar]
- Carss KJ, Arno G, Erwood M, Stephens J, Sanchis-Juan A, Hull S, Megy K, Grozeva D, Dewhurst E, Malka S, Plagnol V, Penkett C, Stirrups K, Rizzo R, Wright G, Josifova D, Bitner-Glindzicz M, Scott RH, Clement E, Allen L, Armstrong R, Brady AF, Carmichael J, Chitre M, Henderson RHH, Hurst J, MacLaren RE, Murphy E, Paterson J, Rosser E, Thompson DA, Wakeling E, Ouwehand WH, Michaelides M, Moore AT, Consortium NI-BRD, Webster AR, Raymond FL, 2017. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet 100, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celia W, Greenstein VC, Zernant-Rajang J, Smith TR, Barile G, Allikmets R, Tsang SH, 2009. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp. Eye Res 89, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon-Camacho OF, Granillo-Alvarez M, Ayala-Ramirez R, Zenteno JC, 2013. ABCA4 mutational spectrum in Mexican patients with Stargardt disease: identification of 12 novel mutations and evidence of a founder effect for the common p.A1773V mutation. Exp. Eye Res 109, 77–82. [DOI] [PubMed] [Google Scholar]
- Chapi M, Sabbaghi H, Suri F, Alehabib E, Rahimi-Aliabadi S, Jamali F, Jamshidi J, Emamalizadeh B, Darvish H, Mirrahimi M, Ahmadieh H, Daftarian N, 2019. Incomplete penetrance of CRX gene for autosomal dominant form of cone-rod dystrophy. Ophthalmic Genet. 40, 259–266. [DOI] [PubMed] [Google Scholar]
- Charbel Issa P, Barnard AR, Singh MS, Carter E, Jiang Z, Radu RA, Schraermeyer U, MacLaren RE, 2013. Fundus autofluorescence in the Abca4(-/-) mouse model of Stargardt disease-correlation with accumulation of A2E, retinal function, and histology. Invest. Ophthalmol. Vis. Sci 54, 5602–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbel Issa P, Barnard AR, Herrmann P, Washington I, MacLaren RE, 2015. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc. Natl. Acad. Sci. U. S. A 112, 8415–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Tosha C, Gorin MB, Nusinowitz S, 2010. Analysis of autofluorescent retinal images and measurement of atrophic lesion growth in Stargardt disease. Exp. Eye Res 91, 143–152. [DOI] [PubMed] [Google Scholar]
- Chen L, Lee W, de Carvalho JRL Jr., Chang S, Tsang SH, Allikmets R, Sparrow JR, 2019. Multi-platform imaging in ABCA4-associated disease. Sci. Rep 9, 6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry TJ, Yang MG, Harmin DA, Tao P, Timms AE, Bauwens M, Allikmets R, Jones EM, Chen R, De Baere E, Greenberg ME, 2020. Mapping the cfs-regulatory architecture of the human retina reveals noncoding genetic variation in disease. Proc. Natl. Acad. Sci. U. S. A 117, 9001–9012. https://www.pnas.org/content/117/16/9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Swider M, Schwartz SB, Steinberg JD, Brucker AJ, Maguire AM, Bennett J, Stone EM, Jacobson SG, 2004. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum. Mol. Genet 13, 525–534. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Swider M, Aleman TS, Sumaroka A, Schwartz SB, Roman MI, Milam AH, Bennett J, Stone EM, Jacobson SG, 2005. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest. Ophthalmol. Vis. Sci 46, 4739–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Swider M, Aleman TS, Roman MI, Sumaroka A, Schwartz SB, Stone EM, Jacobson SG, 2007. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J Opt Soc Am A Opt Image Sci Vis 24, 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Swider M, Aleman TS, Feuer WJ, Schwartz SB, Russell RC, Steinberg JD, Stone EM, Jacobson SG, 2012. Macular function in macular degenerations: repeatability of microperimetry as a potential outcome measure for ABCA4-associated retinopathy trials. Invest. Ophthalmol. Vis. Sci 53, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Swider M, Schwartz SB, Stone EM, Jacobson SG, 2015. Predicting progression of ABCA4-associated retinal degenerations based on longitudinal measurements of the leading disease front. Invest. Ophthalmol. Vis. Sci 56, 5946–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, Roman AJ, Sumaroka A, Han IC, Hochstedler MD, Pfeifer WL, Sohn EH, Taiel M, Schwartz MR, Biasutto P, Wit W, Cheetham ME, Adamson P, Rodman DM, Platenburg G, Tome MD, Balikova I, Nerinckx F, Zaeytijd J, Van Cauwenbergh B, Leroy BP, Russell SR, 2019. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med 25, 225–228. [DOI] [PubMed] [Google Scholar]
- Collin RW, den Hollander AI, van der Velde-Visser SD, Bennicelli J, Bennett J, Cremers FP, 2012. Antisense oligonucleotide (AON)-based therapy for leber congenital amaurosis caused by a frequent mutation in CEP290. Mol. Ther. Nucleic Acids 1, el4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison FT, Fishman GA, 2018. Visual acuity in patients with stargardt disease after age 40. Retina 38, 2387–2394. [DOI] [PubMed] [Google Scholar]
- Collison FT, Fishman GA, McAnany JJ, Zernant J, Allikmets R, 2014. Psychophysical measurement of rod and cone thresholds in stargardt disease with full-field stimuli. Retina 34, 1888–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison FT, Lee W, Fishman GA, Park JC, Zernant J, McAnany JJ, Allikmets R, 2019. Clinical characterization of Stargardt disease patients with the p.N1868I ABCA4 mutation. Retina 39, 2311–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornells SS, Bax NM, Zernant J, Allikmets R, Fritsche LG, den Dunnen JT, Ajmal M, Hoyng CB, Cremers FP, 2017. Silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum. Mutat 38, 400–408. [DOI] [PubMed] [Google Scholar]
- Cremers FP, van de Pol DJ, van Driel M, den Hollander AI, van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Deutman AF, Hoyng CB, 1998. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardťs disease gene ABCR. Hum. Mol. Genet 7, 355–362. [DOI] [PubMed] [Google Scholar]
- Cremers FP, Maugeri A, den Hollander AI, Hoyng CB, 2004. The expanding roles of ABCA4 and CRB1 In inherited blindness. Novartis Found Symp 255,68–79; discussion 79–84, 177–178. [DOI] [PubMed] [Google Scholar]
- Cremers FPM, Cornells SS, Runhart EH, Astuti GDN, 2018. Author response: penetrance of the ABCA4 p.Asn1868Ile allele in stargardt disease. Invest. Ophthalmol. Vis. Sci 59, 5566–5568. [DOI] [PubMed] [Google Scholar]
- Cukras CA, Wong WT, Caruso R, Cunningham D, Zein W, Sieving PA, 2012. Centrifugal expansion of fundus autofluorescence patterns in Stargardt disease over time. Arch. Ophthalmol 130, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl SG, Sylte I, Ravna AW, 2004. Structures and models of transporter proteins. J. Pharmacol. Exp. Therapeut 309, 853–860. [DOI] [PubMed] [Google Scholar]
- Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Welter JJ, 1995. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest. Ophthalmol. Vis. Sci 36, 718–729. [PubMed] [Google Scholar]
- Delori FC, Goger DG, Dorey CK, 2001. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest. Ophthalmol. Vis. Sci 42, 1855–1866. [PubMed] [Google Scholar]
- Delori F, Greenberg JP, Woods RL, Fischer J, Duncker T, Sparrow J, Smith RT, 2011. Quantitative measurements of autofluorescence with the scanning laser ophthalmoscope. Invest. Ophthalmol. Vis. Sci 52, 9379–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutman AF, 1971. The hereditary dystrophies of the posterior pole of the eye van Gorcum & Comp. N.V. [Google Scholar]
- Drolet DW, Green LS, Gold L, Janjic N, 2016. Fit for the eye: aptamers in ocular disorders. Nucleic Acid Therapeut 26, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducroq D, Shalev S, Habib A, Munnich A, Kaplan J, Rozet JM, 2006. Three different ABCA4 mutations in the same large family with several consanguineous loops affected with autosomal recessive cone-rod dystrophy. Eur. J. Hum. Genet 14, 1269–1273. [DOI] [PubMed] [Google Scholar]
- Dugel PU, Novack RL, Csaky KG, Richmond PP, Birch DG, Kubota R, 2015. Phase ii, randomized, placebo-controlled, 90-day study of emixustat hydrochloride in geographic atrophy associated with dry age-related macular degeneration. Retina 35, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulla K, Aguila M, Lane A, Jovanovic K, Parfitt DA, Schulkens I, Chan HL, Schmidt I, Beumer W, Vorthoren L, Collin RWJ, Garanto A, Duijkers L, Brugulat-Panes A, Semo M, Vugler AA, Biasutto P, Adamson P, Cheetham ME, 2018. Splice-modulating oligonucleotide QR-110 restores CEP290 mRNA and function in human c.2991 + 1655A>G LCA10 models. Mol. Ther. Nucleic Acids 12, 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker T, Lee W, Tsang SH, Greenberg JP, Zernant J, Allikmets R, Sparrow JR, 2013. Distinct characteristics of inferonasal fundus autofluorescence patterns in stargardt disease and retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci 54, 6820–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker T, Marsiglia M, Lee W, Zernant J, Tsang SH, Allikmets R, Greenstein VC, Sparrow JR, 2014. Correlations among near-infrared and short-wavelength autofluorescence and spectral-domain optical coherence tomography in recessive Stargardt disease. Invest. Ophthalmol. Vis. Sci 55, 8134–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker T, Stein GE, Lee W, Tsang SH, Zernant J, Bearelly S, Hood DC, Greenstein VC, Delori FC, Allikmets R, Sparrow JR, 2015a. Quantitative fundus autofluorescence and optical coherence tomography in ABCA4 carriers. Invest. Ophthalmol. Vis. Sci 56, 7274–7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker T, Tsang SH, Lee W, Zernant J, Allikmets R, Delori FC, Sparrow JR, 2015b. Quantitative fundus autofluorescence distinguishes ABCA4-associated and non-ABCA4-associated bull’s-eye maculopathy. Ophthalmology 122, 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker T, Tsang SH, Woods RL, Lee W, Zernant J, Allikmets R, Delori FC, Sparrow JR, 2015c. Quantitative fundus autofluorescence and optical coherence tomography in PRPH2/RDS- and ABCA4-associated disease exhibiting phenotypic overlap. Invest. Ophthalmol. Vis. Sci 56, 3159–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyka FM, Molday LL, Chiodo VA, Molday RS, Hauswirth WW, 2019. Dual ABCA4-AAV vector treatment reduces pathogenic retinal A2E accumulation in a mouse model of autosomal recessive stargardt disease. Hum. Gene Ther 30, 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle RC Jr., Lucier AC, Bernardino VB Jr., Yanoff M, 1980. Retinal pigment epithelial abnormalities in fundus flavimaculatus: a light and electron microscopic study. Ophthalmology 87, 1189–1200. [DOI] [PubMed] [Google Scholar]
- Ergun E, Hermann B, Wirtitsch M, Unterhuber A, Ko TH, Sattmann H, Scholda C, Fujimoto JG, Stur M, Drexler W, 2005. Assessment of central visual function in Stargardťs disease/fundus flavimaculatus with ultrahigh-resolution optical coherence tomography. Invest. Ophthalmol. Vis. Sci 46, 310–316. [DOI] [PubMed] [Google Scholar]
- Ernest JT, Krill AE, 1966. Fluorescein studies in fundus flavimaculatus and drusen. Am. J. Ophthalmol 62, 1–6. [DOI] [PubMed] [Google Scholar]
- Fadaie Z, Khan M, Del Pozo-Valero M, Cornells SS, Ayuso C, Cremers FPM, Roosing S, The ABCA4 Study Group, 2019. Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4. Hum. Mutat 40, 2365–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falfoul Y, Habibi I, Turki A, Chebil A, Hassairi A, Schorderet DF, El Matri L, 2018. Phenotypic progression of stargardt disease in a large consanguineous Tunisian family harboring new ABCA4 mutations. J Ophthalmol 2018, 1030184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingert JH, Eliason DA, Phillips NC, Lotery AJ, Sheffield VC, Stone EM, 2006. Case of Stargardt disease caused by uniparental isodisomy. Arch. Ophthalmol 124, 744–745. [DOI] [PubMed] [Google Scholar]
- Fish G, Grey R, Sehmi KS, Bird AC, 1981. The dark choroid in posterior retinal dystrophies. Br. J. Ophthalmol 65, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishkin NE, Sparrow JR, Allikmets R, Nakanishi K, 2005. Isolation and characterization of a retinal pigment epithelial cell fluorophore: an all-trans-retinal dimer conjugate. Proc. Natl. Acad. Sci. U. S. A 102, 7091–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman GA, 1976. Fundus flavimaculatus. A clinical classification. Arch. Ophthalmol 94, 2061–2067. [DOI] [PubMed] [Google Scholar]
- Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR, 1999. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch. Ophthalmol 117, 504–510. [DOI] [PubMed] [Google Scholar]
- Fliesler SJ, Anderson RE, 1983. Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid Res 22, 79–131. [DOI] [PubMed] [Google Scholar]
- Franceschetti A, 1965. A special form of tapetoretinal degeneration: fundus flavimaculatus. Trans. Am. Acad. Ophthalmol. Otolaryngol 69, 1048–1053. [PubMed] [Google Scholar]
- Franceschetti A, François J, 1965. [Fundus flavimaculatus]. Arch. Ophtalmol. Rev Gen. Ophtalmol 25, 505–530. [PubMed] [Google Scholar]
- François P, Turut P, Puech B, Hache JC, 1975. [Stargardťs disease and fundus flavimaculatus]. Arch. Ophtalmol. Rev Gen. Ophtalmol 35, 817–846. [PubMed] [Google Scholar]
- Fujinami K, Lois N, Mukherjee R, McBain VA, Tsunoda K, Tsubota K, Stone EM, Fitzke FW, Bunce C, Moore AT, Webster AR, Michaelides M, 2013a. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest. Ophthalmol. Vis. Sci 54, 8181–8190. [DOI] [PubMed] [Google Scholar]
- Fujinami K, Sergouniotis PI, Davidson AE, Wright G, Chana RK, Tsunoda K, Tsubota K, Egan CA, Robson AG, Moore AT, Holder GE, Michaelides M, Webster AR, 2013b. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am. J. Ophthalmol 156, 487–501 e481. [DOI] [PubMed] [Google Scholar]
- Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, Tsubota K, Robson AG, Holder GE, Allikmets R, Michaelides M, Moore AT, 2015. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 122, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinami K, Strauss RW, Chiang JP, Audo IS, Bernstein PS, Birch DG, Bomotti SM, Cideciyan AV, Ervin AM, Marino MJ, Sahel JA, Mohand-Said S, Sunness JS, Traboulsi EI, West S, Wojciechowski R, Zrenner E, Michaelides M, Scholl HPN, ProgStar Study G, ProgStar Study G, 2019. Detailed genetic characteristics of an international large cohort of patients with Stargardt disease: ProgStar study report 8. Br. J. Ophthalmol 103, 390–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui T, Yamamoto S, Nakano K, Tsujikawa M, Morimura H, Nishida K, Ohguro N, Fujikado T, Irifune M, Kuniyoshi K, Okada AA, Hirakata A, Miyake Y, Tano Y, 2002. ABCA4 gene mutations in Japanese patients with Stargardt disease and retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci 43, 2819–2824. [PubMed] [Google Scholar]
- Garanto A, Chung DC, Duijkers L, Corral-Serrano JC, Messchaert M, Xiao R, Bennett J, Vandenberghe LH, Collin RW, 2016. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum. Mol. Genet 25, 2552–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garanto A, Duijkers L, Tomkiewicz TZ, Collin RWJ, 2019. Antisense Oligonucleotide Screening to Optimize the Rescue of the Splicing Defect Caused by the Recurrent Deep-intronic ABCA4 Variant C.4539 + 2001 G> A in Stargardt Disease. Genes, Basel, pp. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Lee W, Sengillo JD, Allikmets R, Garg K, Tsang SH, 2017. Peripapillary sparing in RDH12-associated Leber congenital amaurosis. Ophthalmic Genet 38, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard X, Perrault I, Hanein S, Silva E, Bigot K, Defoort-Delhemmes S, Rio M, Munnich A, Scherman D, Kaplan J, Kichler A, Rozet JM, 2012. AON-mediated exon skipping restores ciliation in fibroblasts harboring the common leber congenital amaurosis CEP290 mutation. Mol. Ther. Nucleic Acids 1, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber S, Rozet JM, Bonneau D, Souied E, Camuzat A, Dufier JL, Amalric P, Weissenbach J, Munnich A, Kaplan J, 1995. A gene for late-onset fundus flavimaculatus with macular dystrophy maps to chromosome 1p13. Am. J. Hum. Genet 56, 396–399. [PMC free article] [PubMed] [Google Scholar]
- Giroux JM, Barbeau A, 1972. Erythrokeratodermia with ataxia. Arch. Dermatol 106, 183–188. [PubMed] [Google Scholar]
- Greenstein VC, Santos RA, Tsang SH, Smith RT, Barile GR, Seiple W, 2008. Preferred retinal locus in macular disease: characteristics and clinical implications. Retina 28, 1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein VC, Nunez J, Lee W, Schuerch K, Fortune B, Tsang SH, Allikmets R, Sparrow JR, Hood DC, 2017. A comparison of en face optical coherence tomography and fundus autofluorescence in stargardt disease. Invest. Ophthalmol. Vis. Sci 58, 5227–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guymer RH, Heon E, Lotery AJ, Munier FL, Schorderet DF, Baird PN, McNeil RJ, Haines H, Sheffield VC, Stone EM, 2001. Variation of codons 1961 and 2177 of the Stargardt disease gene is not associated with age-related macular degeneration. Arch. Ophthalmol 119, 745–751. [DOI] [PubMed] [Google Scholar]
- Westeneng-van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, den Hollander AI, Hoyng CB, 2012. Clinical and genetic characteristics of late-onset Stargardťs disease. Ophthalmology 119, 1199–1210. [DOI] [PubMed] [Google Scholar]
- Han Z, Conley SM, Makkia RS, Cooper MJ, Naash MI, 2012. DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J. Clin. Invest 122, 3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F, 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu FY, Li JK, Gao FJ, Qi YH, Xu P, Zhang YJ, Wang DD, Wang LS, Li W, Wang M, Chen F, Shen SM, Xu GZ, Zhang SH, Chang Q, Wu JH, 2019. ABCA4 gene screening in a Chinese cohort with stargardt disease: identification of 37 novel variants. Front. Genet 10, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckfeldt RM, East JS, Stone EM, Sohn EH, 2016. Phenotypic variation in a family with pseudodominant stargardt disease. JAMA Ophthalmol 134, 580–583. [DOI] [PubMed] [Google Scholar]
- Jaakson K, Zernant J, Kulm M, Hutchinson A, Tonisson N, Glavac D, Ravnik-Glavac M, Hawlina M, Meltzer MR, Caruso RC, Testa F, Maugeri A, Hoyng CB, Gouras P, Simonelli F, Lewis RA, Lupski JR, Cremers FP, Allikmets R, 2003. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat 22, 395–403. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Windsor EA, Traboulsi EI, Heon E, Pittler SJ, Milam AH, Maguire AM, Palczewski K, Stone EM, Bennett J, 2005. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc. Natl. Acad. Sci. U. S. A 102, 6177–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Aleman TS, Cideciyan AV, Roman AJ, Sumaroka A, Windsor EA, Schwartz SB, Heon E, Stone EM, 2009. Defining the residual vision in leber congenital amaurosis caused by RPE65 mutations. Invest. Ophthalmol. Vis. Sci 50, 2368–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersgaard C, Fang M, Bertelsen M, Dang X, Jensen H, Chen Y, Bech N, Dai L, Rosenberg T, Zhang J, Moller LB, Tumer Z, Brondum-Nielsen K, Gronskov K, 2019. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep 9, 1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Pan Z, Xu K, Tian L, Xie Y, Zhang X, Chen J, Dong B, Li Y, 2016. Screening of ABCA4 gene in a Chinese cohort with stargardt disease or cone-rod dystrophy with a report on 85 novel mutations. Invest. Ophthalmol. Vis. Sci 57, 145–152. [DOI] [PubMed] [Google Scholar]
- Julien S, Schraermeyer U, 2012. Lipofuscin can be eliminated from the retinal pigment epithelium of monkeys. Neurobiol. Aging 33, 2390–2397. [DOI] [PubMed] [Google Scholar]
- Jurgensmeier C, Bhosale P, Bernstein PS, 2007. Evaluation of 4-methylpyrazole as a potential therapeutic dark adaptation inhibitor. Curr. Eye Res 32, 911–915. [DOI] [PubMed] [Google Scholar]
- Kang Derwent JJ, Derlacki DJ, Hetling JR, Fishman GA, Birch DG, Grover S, Stone EM, Pepperberg DR, 2004. Dark adaptation of rod photoreceptors in normal subjects, and in patients with Stargardt disease and an ABCA4 mutation. Invest. Ophthalmol. Vis. Sci 45, 2447–2456. [DOI] [PubMed] [Google Scholar]
- Kaplan J, Gerber S, Larget-Piet D, Rozet JM, Dollfus H, Dufier JL, Odent S, Postel-Vinay A, Janin N, Briard ML, et al. , 1993. A gene for Stargardťs disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat. Genet 5, 308–311. [DOI] [PubMed] [Google Scholar]
- Keilhauer CN, Delori FC, 2006. Near-infrared autofluorescence imaging of the fundus: visualization of ocular melanin. Invest. Ophthalmol. Vis. Sci 47, 3556–3564. [DOI] [PubMed] [Google Scholar]
- Kellner S, Kellner U, Weber BH, Fiebig B, Weinitz S, Ruether K, 2009. Lipofuscin- and melanin-related fundus autofluorescence in patients with ABCA4-associated retinal dystrophies. Am. J. Ophthalmol 147, 895–902 902, e891. [DOI] [PubMed] [Google Scholar]
- Khan KN, Kasilian M, Mahroo OAR, Tanna P, Kalitzeos A, Robson AG, Tsunoda K, Iwata T, Moore AT, Fujinami K, Michaelides M, 2018. Early patterns of macular degeneration in ABCA4-associated retinopathy. Ophthalmology 125, 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Cornells SS, Khan MI, Elmelik D, Manders E, Bakker S, Derks R, Neveling K, van de Vorst M, Gilissen C, Meunier I, Defoort S, Puech B, Devos A, Schulz HL, Stohr H, Grassmann F, Weber BHF, Dhaenens CM, Cremers FPM, 2019. Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease. Hum. Mutat 40, 1749–1759. [DOI] [PubMed] [Google Scholar]
- Khan M, Cornells SS, del Pozo-Valero M, Whelan L, Runhart EH, Mishra K, Bults F, AlSwaiti Y, AlTabishi A, De Baere E, Banff S, Banin E, Bauwens M, Ben-Yosef T, Boon CJF, van den Born LI, Defoort S, Devos A, Dockery A, Dudakova L, Fakin A, Farrar GJ, Ferraz Sallum JM, Fujinami K, Gilissen C, Glavac D, Gorin MB, Greenberg J, Hayashi T, Hettinga Y, Hoischen A, Hoyng CB, Hufendiek K, Jägle H, Kamakari S, Karali M, Kellner U, Klaver CCW, Kousal B, Lamey T, MacDonald IM, Matynia A, McLaren T, Mena MD, Meunier I, Miller R, Newman H, Ntozini B, Oldak M, Pieterse M, Podhajcer OL, Puech B, Ramesar R, Rüther K, Salameh M, Vallim Salles M, Sharon D, Simonelli F, Spital G, Steehouwer M, Szaflik JP, Thompson JA, Thuillier C, Tracewska AM, van Zweeden M, Vincent AL, Zanlonghi X, Liskova P, Stöhr H, De Roach J, Ayuso C, Roberts L, Weber BHF, Dhaenens C-M, Cremers FPM, 2020. Resolving the dark matter of ABCA4 for 1,054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med https://www.nature.com/articles/s41436-020-0787-4#article-info. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Sparrow JR, 2018. Novel bisretinoids of human retina are lyso alkyl ether glycerophosphoethanolamine-bearing A2PE species. J. Lipid Res 59, 1620–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellstrom U, 2015. Reduced macular function in ABCA4 carriers. Mol. Vis 21, 767–782. [PMC free article] [PubMed] [Google Scholar]
- Klein ML, Ferris FL 3rd, Francis PJ, Lindblad AS, Chew EY, Hamon SC, Ott J, 2010. Progression of geographic atrophy and genotype in age-related macular degeneration. Ophthalmology 117, 1554–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevering BJ, Blankenagel A, Maugeri A, Cremers FP, Hoyng CB, Rohrschneider K, 2002. Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest. Ophthalmol. Vis. Sci 43, 1980–1985. [PubMed] [Google Scholar]
- Klien BA, Krill AE, 1967. Fundus flavimaculatus. Clinical, functional and histopathologic observations. Am. J. Ophthalmol 64, 3–23. [PubMed] [Google Scholar]
- Kniazeva M, Chiang MF, Morgan B, Anduze AL, Zack DJ, Han M, Zhang K, 1999. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. Am. J. Hum. Genet 64, 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudtson MD, Klein R, Klein BE, Lee KE, Meuer SM, Tomany SC, 2004. Location of lesions associated with age-related maculopathy over a 10-year period: the Beaver Dam Eye Study. Invest. Ophthalmol. Vis. Sci 45, 2135–2142. [DOI] [PubMed] [Google Scholar]
- Kong J, Kim SR, Binley K, Pata I, Doi K, Mannik J, Zernant-Rajang J, Kan O, Iqball S, Naylor S, Sparrow JR, Gouras P, Allikmets R, 2008. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther 15, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Strauss RW, Michaelides M, Cideciyan AV, Sahel JA, Munoz B, West S, Scholl HP, ProgStar Study G, 2016. Visual acuity loss and associated risk factors in the retrospective progression of stargardt disease study (ProgStar report No. 2). Ophthalmology 123, 1887–1897. [DOI] [PubMed] [Google Scholar]
- Kong X, Ho A, Munoz B, West S, Strauss RW, Jha A, Ervin A, Buzas J, Singh M, Hu Z, Cheetham J, Ip M, Scholl HPN, 2019. Reproducibility of measurements of retinal structural parameters using optical coherence tomography in stargardt disease. Transl Vis Sci Technol 8, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan AK, Bedell HE, 2018. Functional changes at the preferred retinal locus in subjects with bilateral central vision loss. Graefes Arch. Clin. Exp. Ophthalmol 256, 29–37. [DOI] [PubMed] [Google Scholar]
- Kubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, Chandler JW, 2014. Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina 34, 603–609. [DOI] [PubMed] [Google Scholar]
- Kuniyoshi K, Terasaki H, Arai M, Hirose T, 2014. Multifocal electroretinograms in Stargardťs disease/fundus flavimaculatus. Ophthalmologica 232, 118–125. [DOI] [PubMed] [Google Scholar]
- Lambertus S, van Huet RA, Bax NM, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, Hoyng CB, 2015. Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology 122, 335–344. [DOI] [PubMed] [Google Scholar]
- Lambertus S, Lindner M, Bax NM, Mauschitz MM, Nadal J, Schmid M, Schmitz-Valckenberg S, den Hollander AI, Weber BH, Holz FG, van der Wilt GJ, Fleckenstein M, Hoyng CB, Foveal sparing Atrophy Study, 2016. Progression of late-onset stargardt disease. T.. Invest. Ophthalmol. Vis. Sci 57, 5186–5191. [DOI] [PubMed] [Google Scholar]
- Lang W, 1885. Central choroiditis with disseminated patches in remainder of fundus. Trans. Ophthalmol. Soc. U. K 5, 140–141. [Google Scholar]
- Lee W, Noupuu K, Oll M, Duncker T, Burke T, Zernant J, Bearelly S, Tsang SH, Sparrow JR, Allikmets R, 2014. The external limiting membrane in early-onset Stargardt disease. Invest. Ophthalmol. Vis. Sci 55, 6139–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Xie Y, Zernant J, Yuan B, Bearelly S, Tsang SH, Lupski JR, Allikmets R, 2016. Complex inheritance of ABCA4 disease: four mutations in a family with multiple macular phenotypes. Hum. Genet 135, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Schuerch K, Zernant J, Collison FT, Bearelly S, Fishman GA, Tsang SH, Sparrow JR, Allikmets R, 2017. Genotypic spectrum and phenotype correlations of ABCA4-associated disease in patients of south Asian descent. Eur. J. Hum. Genet 25, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Zernant J, Nagasaki T, Tsang SH, Allikmets R, 2018. Deep scleral exposure: a degenerative outcome of end-stage stargardt disease. Am. J. Ophthalmol 195, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Paavo M, Zernant J, Stong N, Laurente Z, Bearelly S, Nagasaki T, Tsang SH, Goldstein DB, Allikmets R, 2019. Modification of the PROM1 disease phenotype by a mutation in ABCA4. Ophthalmic Genet 40, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenis TL, Hu J, Ng SY, Jiang Z, Sarfare S, Lloyd MB, Esposito NJ, Samuel W, Jaworski C, Bok D, Finnemann SC, Radeke MJ, Redmond TM, Travis GH, Radu RA, 2018. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc. Natl. Acad. Sci. U. S. A 115, El1120–El1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, Lupski JR, Leppert M, Dean M, 1999. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am. J. Hum. Genet 64, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad AS, Lloyd PC, Clemons TE, Gensler GR, Ferris FL 3rd, Klein ML, Armstrong JR, Age-Related Eye Disease Study Research, 2009. Change in area of geographic atrophy in the Age-Related Eye Disease Study: AREDS report number 26. G. Arch. Ophthalmol 127, 1168–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner M, Lambertus S, Mauschitz MM, Bax NM, Kersten E, Luning A, Nadal J, Schmitz-Valckenberg S, Schmid M, Holz FG, Hoyng CB, Fleckenstein M, Foveal sparing Atrophy Study, 2017. Differential disease progression in atrophic age-related macular degeneration and late-onset stargardt disease. T.. Invest. Ophthalmol. Vis. Sci 58, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Liu Q, Sabirzhanova I, Bergbower EAS, Yanda M, Guggino WG, Cebotaru L, 2019. The CFTR corrector, VX-809 (lumacaftor), rescues ABCA4 trafficking mutants: a potential treatment for stargardt disease. Cell. Physiol. Biochem 53, 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC, 2001. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch. Ophthalmol 119, 359–369. [DOI] [PubMed] [Google Scholar]
- Lois N, Halfyard AS, Bird AC, Holder GE, Fitzke FW, 2004. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am. J. Ophthalmol 138, 55–63. [DOI] [PubMed] [Google Scholar]
- Lopez PF, Maumenee IH, de la Cruz Z, Green WR, 1990. Autosomal-dominant fundus flavimaculatus. Clinicopathologic correlation. Ophthalmology 97, 798–809. [DOI] [PubMed] [Google Scholar]
- Lopez-Rubio S, Chacon-Camacho OF, Matsui R, Guadarrama-Vallejo D, Astiazaran MC, Zenteno JC, 2018. Retinal phenotypic characterization of patients with ABCA4 retinopathydue to the homozygous p.Alal773Val mutation. Mol. Vis 24, 105–114. [PMC free article] [PubMed] [Google Scholar]
- Ma CJ, Lee W, Stong N, Zernant J, Chang S, Goldstein D, Nagasaki T, Allikmets R, 2019. Late-onset pattern macular dystrophy mimicking ABGA4 and PRPH2 disease is caused by a homozygous frameshift mutation in ROM1. Cold Spring Harb Mol Case Stud 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald IM, Sieving PA, 2018. Investigation of the effect of dietary docosahexaenoic acid (DHA) supplementation on macular function in subjects with autosomal recessive Stargardt macular dystrophy. Ophthalmic Genet 39, 477–486. [DOI] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Golczak M, Palczewski K, 2008. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem 283, 26684–26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, Marshall KA, McCague S, Reichert H, Davis M, Simonelli F, Leroy BP, Wright JF, High KA, Bennett J, 2019. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: results of phase 1 and 3 trials. Ophthalmology 126, 1273–1285. [DOI] [PubMed] [Google Scholar]
- Maia-Lopes S, Silva ED, Silva MF, Reis A, Faria P, Castelo-Branco M, 2008. Evidence of widespread retinal dysfunction in patients with stargardt disease and morphologically unaffected carrier relatives. Invest. Ophthalmol. Vis. Sci 49, 1191–1199. [DOI] [PubMed] [Google Scholar]
- Makelainen S, Godia M, Hellsand M, Viluma A, Hahn D, Makdoumi K, Zeiss CJ, Mellersh C, Ricketts SL, Narfstrom K, Hallbook F, Ekesten B, Andersson G, Bergstrom TF, 2019. An ABCA4 loss-of-function mutation causes a canine form of Stargardt disease. PLoS Genet 15, e1007873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Mir A, Paloma E, Allikmets R, Ayuso C, del Rio T, Dean M, Vilageliu L, Gonzalez-Duarte R, Balcells S, 1998. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat. Genet 18, 11–12. [DOI] [PubMed] [Google Scholar]
- Mata NL, Tzekov RT, Liu X, Weng J, Birch DG, Travis GH, 2001. Delayed dark-adaptation and lipofuscin accumulation in abcr + /− mice: implications for involvement of ABCR In age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 42, 1685–1690. [PubMed] [Google Scholar]
- Maugeri A, van Driel MA, van de Pol DJ, Klevering BJ, van Haren FJ, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Dahl N, Brunner HG, Deutman AF, Hoyng CB, Cremers FP, 1999. The 2588G->C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am. J. Hum. Genet 64, 1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri A, Klevering BJ, Rohrschneider K, Blankenagel A, Brunner HG, Deutman AF, Hoyng CB, Cremers FP, 2000. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet 67, 960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri A, Flothmann K, Hemmrich N, Ingvast S, Jorge P, Paloma E, Patel R, Rozet JM, Tammur J, Testa F, Balcells S, Bird AC, Brunner HG, Hoyng CB, Metspalu A, Simonelli F, Allikmets R, Bhattacharya SS, D’Urso M, Gonzalez-Duarte R, Kaplan J, te Meerman GJ, Santos R, Schwartz M, Van Camp G, Wadelius C, Weber BH, Cremers FP, 2002. The ABCA4 2588G>C Stargardt mutation: single origin and increasing frequency from South-West to North-East Europe. Eur. J. Hum. Genet 10, 197–203. [DOI] [PubMed] [Google Scholar]
- Maw MA, Corbeil D, Koch J, Hellwig A, Wilson-Wheeler JC, Bridges RJ, Kumaramanickavel G, John S, Nancarrow D, Roper K, Weigmann A, Huttner WB, Denton MJ, 2000. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum. Mol. Genet 9, 27–34. [DOI] [PubMed] [Google Scholar]
- McBain VA, Townend J, Lois N, 2012. Progression of retinal pigment epithelial atrophy in stargardt disease. Am. J. Ophthalmol 154, 146–154. [DOI] [PubMed] [Google Scholar]
- McClements ME, Barnard AR, Singh MS, Charbel Issa P, Jiang Z, Radu RA, MacLaren RE, 2019. An AAV dual vector strategy ameliorates the stargardt phenotype in adult Abca4(-/-) mice. Hum. Gene Ther 30, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehat MS, Sundaram V, Ripamonti C, Robson AG, Smith AJ, Borooah S, Robinson M, Rosenthal AN, Innes W, Weleber RG, Lee RWJ, Crossland M, Rubin GS, Dhillon B, Steel DHW, Anglade E, Lanza RP, Ali RR, Michaelides M, Bainbridge JWB, 2018. Transplantation of human embryonic stem cell-derived retinal pigment epithelial cells in macular degeneration. Ophthalmology 125, 1765–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo P, Testa F, Rossi S, Di Iorio V, Orrico A, Auricchio A, Simonelli F, 2016. En face spectral-domain optical coherence tomography for the monitoring of lesion area progression in stargardt disease. Invest. Ophthalmol. Vis. Sci 57, OCT247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelides M, Holder GE, Bradshaw K, Hunt DM, Moore AT, 2005. Cone-rod dystrophy, intrafamilial variability, and incomplete penetrance associated with the R172W mutation in the peripherin/RDS gene. Ophthalmology 112, 1592–1598. [DOI] [PubMed] [Google Scholar]
- Molday RS, 2015. Insights into the molecular properties of ABCA4 and its role in the visual cycle and stargardt disease. Prog Mol Biol Transl Sci 134, 415–431. [DOI] [PubMed] [Google Scholar]
- Molday LL, Molday Rs AR, 2016. Localization and Functional Analysis of ABCA4 Variants Associated with Stargardt Disease, Association for Research in Vision and Ophthalmology. Seattle, WA. [Google Scholar]
- Molday LL, Wahl D, Sarunic MV, Molday RS, 2018. Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum. Mol. Genet 27, 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PL, Gliem M, Mangold E, Bolz HJ, Finger RP, McGuinness M, Betz C, Jiang Z, Weber BH, MacLaren RE, Holz FG, Radu RA, Charbel Issa P, 2015. Monoallelic ABGA4 mutations appear insufficient to cause retinopathy: a quantitative auto fluorescence study. Invest. Ophthalmol. Vis. Sci 56, 8179–8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PL, Fimmers R, Gliem M, Holz FG, Charbel Issa P, 2017. Choroidal alterations in abca4-related retinopathy. Retina 37, 359–367. [DOI] [PubMed] [Google Scholar]
- Nakao T, Tsujikawa M, Sawa M, Gomi F, Nishida K, 2012. Foveal sparing in patients with Japanese Stargard’s disease and good visual acuity. Jpn. J. Ophthalmol 56, 584–588. [DOI] [PubMed] [Google Scholar]
- Nassisi M, Mohand-Said S, Andrieu C, Antonio A, Condroyer C, Mejecase C, Varin J, Wohlschlegel J, Dhaenens CM, Sahel JA, Zeitz C, Audo I, 2019. Prevalence of ABCA4 deep-intronic variants and related phenotype in an unsolved “One-Hit” cohort with stargardt disease. Int. J. Mol. Sci 20, E5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noupuu K, Lee W, Zernant J, Tsang SH, Allikmets R, 2014. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Invest. Ophthalmol. Vis. Sci 55, 7217–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noupuu K, Lee W, Zernant J, Greenstein VC, Tsang S, Allikmets R, 2016. Recessive Stargardt disease phenocopying hydroxychloroquine retinopathy. Graefes Arch. Clin. Exp. Ophthalmol 254, 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oner A, Gonen ZB, Sevim DG, Smim Kahraman N, Unlu M, 2018. Suprachoroidal adipose tissue-derived mesenchymal stem cell implantation in patients with dry-type Age-related macular degeneration and stargardťs macular dystrophy: 6-month follow-up results of a phase 2 study. Cell. Reprogr 20, 329–336. [DOI] [PubMed] [Google Scholar]
- Paavo M, Zhao J, Kim HJ, Lee W, Zernant J, Cai C, Allikmets R, Tsang SH, Sparrow JR, 2018. Mutations in gpr143/OA1 and ABCA4 inform interpretations of short-wavelength and near-infrared fundus auto fluorescence. Invest. Ophthalmol. Vis. Sci 59, 2459–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paavo M, Lee W, Allikmets R, Tsang S, Sparrow JR, 2019. Photoreceptor cells as a source of fundus autofluorescence in recessive Stargardt disease. J. Neurosci. Res 97, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang CE, Suqin Y, Sherman J, Freund KB, 2015. New insights into Stargardt disease with multimodal imaging. Ophthalmic Surg Lasers Imaging Retina 46, 257–261. [DOI] [PubMed] [Google Scholar]
- Papermaster DS, Converse CA, Zorn M, 1976. Biosynthetic and immunochemical characterization of large protein in frog and cattle rod outer segment membranes. Exp. Eye Res 23, 105–115. [DOI] [PubMed] [Google Scholar]
- Parfitt DA, Lane A, Ramsden CM, Carr AJ, Munro PM, Jovanovic K, Schwarz N, Kanuga N, Muthiah MN, Hull S, Gallo JM, da Cruz L, Moore AT, Hardcastle AJ, Coffey PJ, Cheetham ME, 2016. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell 18, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J, 1998. Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc. Natl. Acad. Sci. U. S. A 95, 14609–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JC, Collison FT, Fishman GA, Allikmets R, Zernant J, Liu M, McAnany JJ, 2015. Objective analysis of hyperreflective outer retinal bands imaged by optical coherence tomography in patients with stargardt disease. Invest. Ophthalmol. Vis. Sci 56, 4662–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MA, Choi D, Erker LR, Pennesi ME, Yang P, Chegarnov EN, Steinkamp PN, Schlechter CL, Dhaenens CM, Mohand-Said S, Audo I, Sahel J, Weleber RG, Wilson DJ, 2016. Test-retest variability of functional and structural parameters in patients with stargardt disease participating in the SAR422459 gene therapy trial. Transl Vis Sci Technol 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi MB, Iacono P, Triolo G, La Spina C, Zucchiatti I, Cicinelli MV, Borrelli E, Manitto MP, Martina E, Bandello F, 2015. Morpho-functional correlation of fundus autofluorescence in Stargardt disease. Br. J. Ophthalmol 99, 1354–1359. [DOI] [PubMed] [Google Scholar]
- Passerini I, Sodi A, Giambene B, Mariottini A, Menchini U, Torricelli F, 2010. Novel mutations in of the ABCR gene in Italian patients with Stargardt disease. Eye 24, 158–164. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Acquistapace A, Oldani M, Cereda MG, Giani A, Cozzi M, Staurenghi G, 2016. Dark atrophy: an optical coherence tomography angiography study. Ophthalmology 123, 1879–1886. [DOI] [PubMed] [Google Scholar]
- Piccardi M, Fadda A, Martelli F, Marangoni D, Magli A, Minnella AM, Bertelli M, Di Marco S, Bisti S, Falsini B, 2019. Antioxidant saffron and central retinal function in ABCA4-related stargardt macular dystrophy. Nutrients 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, Molday RS, 2013. Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J. Biol. Chem 288, 34414–34426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, Molday RS, 2014. ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc. Natl. Acad. Sci. U. S. A Ill, 5024–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, Lenevich S, Molday RS, 2012. ABCA4 is an N-retinylidene-phosphatidy-lethanolamine and phosphatidylethanolamine importer. Nat. Commun 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querques G, Leveziel N, Benhamou N, Voigt M, Soubrane G, Souied EH, 2006. Analysis of retinal flecks in fundus flavimaculatus using optical coherence tomography. Br. J. Ophthalmol 90, 1157–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querques G, Prato R, Iaculli C, Voigt M, Delle Noci N, Coscas G, Soubrane G, Souied EH, 2008. Correlation of visual function impairment and OCT findings in patients with Stargardt disease and fundus flavimaculatus. Eur. J. Ophthalmol 18, 239–247. [DOI] [PubMed] [Google Scholar]
- Racz B, Varadi A, Kong J, Allikmets R, Pearson PG, Johnson G, Cioffi CL, Petrukhin K, 2018. A non-retinoid antagonist of retinol-binding protein 4 rescues phenotype in a model of Stargardt disease without inhibiting the visual cycle. J. Biol. Chem 293, 11574–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees HA, Liu DR, 2018. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet 19, 770–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveiro-Alvarez R, Valverde D, Lorda-Sanchez I, Trujillo-Tiebas MJ, Cantalapiedra D, Vallespin E, Aguirre-Lamban J, Ramos C, Ayuso C, 2007. Partial paternal uniparental disomy (UPD) of chromosome 1 in a patient with Stargardt disease. Mol. Vis 13, 96–101. [PMC free article] [PubMed] [Google Scholar]
- Riveiro-Alvarez R, Lopez-Martinez MA, Zernant J, Aguirre-Lamban J, Cantalapiedra D, Avila-Fernandez A, Gimenez A, Lopez-Molina MI, Garcia-Sandoval B, Bianco-Kelly F, Corton M, Tatu S, Fernandez-San Jose P, Trujillo-Tiebas MJ, Ramos C, Allikmets R, Ayuso C, 2013. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology 120, 2332–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera A, White K, Stohr H, Steiner K, Hemmrich N, Grimm T, Jurklies B, Lorenz B, Scholl HP, Apfelstedt-Sylla E, Weber BH, 2000. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet 67, 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg T, Klie F, Garred P, Schwartz M, 2007. N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol. Vis 13, 1962–1969. [PubMed] [Google Scholar]
- Rotenstreich Y, Fishman GA, Anderson RJ, 2003. Visual acuity loss and clinical observations in a large series of patients with Stargardt disease. Ophthalmology 110, 1151–1158. [DOI] [PubMed] [Google Scholar]
- Rozet JM, Gerber S, Ghazi I, Perrault I, Ducroq D, Souied E, Cabot A, Dufier JL, Munnich A, Kaplan J, 1999. Mutations of the retinal specific ATP binding transporter gene (ABCR) in a single family segregating both autosomal recessive retinitis pigmentosa RP19 and Stargardt disease: evidence of clinical heterogeneity at this locus. J. Med. Genet 36, 447–451. [PMC free article] [PubMed] [Google Scholar]
- Runhart EH, Sangermano R, Cornells SS, Verheij J, Plomp AS, Boon CJF, Lugtenberg D, Roosing S, Bax NM, Blokland EAW, Jacobs-Camps MHM, van der Velde-Visser SD, Pott JR, Rohrschneider K, Thiadens A, Klaver CCW, van den Born LI, Hoyng CB, Cremers FPM, 2018. The common ABCA4 variant p.Asnl868Ile shows nonpenetrance and variable expression of stargardt disease when present in trans with severe variants. Invest. Ophthalmol. Vis. Sci 59, 3220–3231. [DOI] [PubMed] [Google Scholar]
- Runhart EH, Valkenburg D, Cornells SS, Khan M, Sangermano R, Albert S, Bax NM, Astuti GDN, Gilissen C, Pott JR, Verheij J, Blokland EAW, Cremers FPM, van den Born LI, Hoyng CB, 2019. Late-onset stargardt disease due to mild, deep-intronic ABCA4 alleles. Invest. Ophthalmol. Vis. Sci 60, 4249–4256. [DOI] [PubMed] [Google Scholar]
- Sabirzhanova I, Lopes Pacheco M, Rapino D, Grover R, Handa JT, Guggino WB, Cebotaru L, 2015. Rescuing trafficking mutants of the ATP-binding cassette protein, ABCA4, with small molecule correctors as a treatment for stargardt eye disease. J. Biol. Chem 290, 19743–19755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva MM, Eliott D, 2016. Stem cell-based therapy for diseases of the retinal pigment epithelium: from bench to bedside. Semin. Ophthalmol 31, 25–29. [DOI] [PubMed] [Google Scholar]
- Salvatore S, Fishman GA, McAnany JJ, Genead MA, 2014. Association of dark-adapted visual function with retinal structural changes in patients with Stargardt disease. Retina 34, 989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangermano R, Bax NM, Bauwens M, van den Born LI, De Baere E, Garanto A, Collin RW, Goercharn-Ramlal AS, den Engelsman-van Dijk AH, Rohrschneider K, Hoyng CB, Cremers FP, Albert S, 2016. Photoreceptor progenitor mRNA analysis reveals exon skipping resulting from the ABCA4 c.5461–10T> C mutation in Stargardt disease. Ophthalmology 123, 1375–1385. [DOI] [PubMed] [Google Scholar]
- Sangermano R, Khan M, Cornells SS, Richelle V, Albert S, Garanto A, Elmelik D, Qamar R, Lugtenberg D, van den Born LI, Collin RWJ, Cremers FPM, 2018. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res 28, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangermano R, Garanto A, Khan M, Runhart EH, Bauwens M, Bax NM, van den Born LI, Khan MI, Cornells SS, Verheij J, Pott JR, Thiadens A, Klaver CCW, Puech B, Meunier I, Naessens S, Arno G, Fakin A, Carss KJ, Raymond FL, Webster AR, Dhaenens CM, Stohr H, Grassmann F, Weber BHF, Hoyng CB, De Baere E, Albert S, Collin RWJ, Cremers FPM, 2019. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med 21, 1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl HP, Besch D, Vonthein R, Weber BH, Apfelstedt-Sylla E, 2002. Alterations of slow and fast rod ERG signals in patients with molecularly confirmed Stargardt disease type 1. Invest. Ophthalmol. Vis. Sci 43, 1248–1256. [PubMed] [Google Scholar]
- Schonbach EM, Ibrahim MA, Strauss RW, Birch DG, Cideciyan AV, Hahn GA, Ho A, Kong X, Nasser F, Sunness JS, Zrenner E, Sadda SR, West SK, Scholl HPN, Progression of Stargardt Disease Study, 2017a. Fixation location and stability using the MP-1 microperimeter in stargardt disease: ProgStar report No. 3. G. Ophthalmol Retina 1, 68–76. [DOI] [PubMed] [Google Scholar]
- Schonbach EM, Wolfson Y, Strauss RW, Ibrahim MA, Kong X, Munoz B, Birch DG, Cideciyan AV, Hahn GA, Nittala M, Sunness JS, Sadda SR, West SK, Scholl HPN, ProgStar Study G, 2017b. Macular Sensitivity Measured with Microperimetry in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 7. JAMA Ophthalmol 135. pp. 696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonbach EM, Strauss RW, Kong X, Munoz B, Ibrahim MA, Sunness JS, Birch DG, Hahn GA, Nasser F, Zrenner E, Sadda SR, West SK, Scholl HPN, ProgStar Study G, 2018. Longitudinal changes of fixation location and stability within 12 Months in stargardt disease: ProgStar report No. 12. Am. J. Ophthalmol 193, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorderet DF, Iouranova A, Favez T, Tiab L, Escher P, 2013. IROme, a new high-throughput molecular tool for the diagnosis of inherited retinal dystrophies. BioMed Res. Int 2013, 198089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz HL, Grassmann F, Kellner U, Spital G, Ruther K, Jagle H, Hufendiek K, Rating P, Huchzermeyer C, Baier MJ, Weber BH, Stohr H, 2017. Mutation spectrum of the ABCA4 gene in 335 stargardt disease patients from a multicenter German cohort-impact of selected deep intronic variants and common SNPs. Invest. Ophthalmol. Vis. Sci 58, 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, Mickunas E, Gay R, Klimanskaya I, Lanza R, 2012. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379, 713–720. [DOI] [PubMed] [Google Scholar]
- Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, Hubschman JP, Davis JL, Heilwell G, Spirn M, Maguire J, Gay R, Bateman J, Ostrick RM, Morris D, Vincent M, Anglade E, Del Priore LV, Lanza R, 2015. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardťs macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 385, 509–516. [DOI] [PubMed] [Google Scholar]
- Sciezynska A, Ozieblo D, Ambroziak AM, Korwin M, Szulborski K, Krawczynski M, Stawinski P, Szaflik J, Szaflik JP, Ploski R, Oldak M, 2016. Next-generation sequencing of ABCA4: high frequency of complex alleles and novel mutations in patients with retinal dystrophies from Central Europe. Exp. Eye Res 145, 93–99. [DOI] [PubMed] [Google Scholar]
- Shankar SP, Hughbanks-Wheaton DK, Birch DG, Sullivan LS, Conneely KN, Bowne SJ, Stone EM, Daiger SP, 2016. Autosomal dominant retinal dystrophies caused by a founder splice site mutation, C.828 + 3A>T, in PRPH2 and protein haplotypes in trans as modifiers. Invest. Ophthalmol. Vis. Sci 57, 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon D, Ben-Yosef T, Goldenberg-Cohen N, Pras E, Gradstein L, Soudry S, Mezer E, Zur D, Abbasi AH, Zeitz C, Cremers FPM, Khan MI, Levy J, Rotenstreich Y, Birk OS, Ehrenberg M, Leibu R, Newman H, Shomron N, Banin E, Perlman I, 2020. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat 41, 140–149. [DOI] [PubMed] [Google Scholar]
- Shroyer NF, Lewis RA, Lupski JR, 2000. Complex inheritance of ABCR mutations in Stargardt disease: linkage disequilibrium, complex alleles, and pseudodominance. Hum. Genet 106, 244–248. [DOI] [PubMed] [Google Scholar]
- Shroyer NF, Lewis RA, Lupski JR, 2001a. Analysis of the ABCR (ABCA4) gene in 4-aminoquinoline retinopathy: is retinal toxicity by chloroquine and hydroxychloroquine related to Stargardt disease? Am. J. Ophthalmol 131, 761–766. [DOI] [PubMed] [Google Scholar]
- Shroyer NF, Lewis RA, Yatsenko AN, Lupski JR, 2001b. Null missense ABCR (ABCA4) mutations in a family with stargardt disease and retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci 42, 2757–2761. [PubMed] [Google Scholar]
- Simon WA, Herrmann M, Klein T, Shin JM, Huber R, Senn-Bilfinger J, Postius S, 2007. Soraprazan: setting new standards in inhibition of gastric acid secretion. J. Pharmacol. Exp. Therapeut 321, 866–874. [DOI] [PubMed] [Google Scholar]
- Sisk RA, Leng T, 2014. Multimodal imaging and multifocal electroretinography demonstrate autosomal recessive Stargardt disease may present like occult macular dystrophy. Retina 34, 1567–1575. [DOI] [PubMed] [Google Scholar]
- Sodi A, Mucciolo DP, Cipollini F, Murro V, Caporossi O, Virgili G, Rizzo S, 2016. En face OCT in Stargardt disease. Graefes Arch. Clin. Exp. Ophthalmol 254, 1669–1679. [DOI] [PubMed] [Google Scholar]
- Sohocki MM, Sullivan LS, Mintz-Hittner HA, Birch D, Heckenlively JR, Freund CL, McInnes RR, Daiger SP, 1998. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am. J. Hum. Genet 63, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Rossi EA, Latchney L, Bessette A, Stone E, Hunter JJ, Williams DR, Chung M, 2015a. Cone and rod loss in Stargardt disease revealed by adaptive optics scanning light ophthalmoscopy. JAMA Ophthalmol 133, 1198–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WK, Park KM, Kim HJ, Lee JH, Choi J, Chong SY, Shim SH, Del Priore LV, Lanza R, 2015b. Treatment of macular degeneration using embryonic stem cell-derived retinal pigment epithelium: preliminary results in Asian patients. Stem Cell Reports 4, 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souied EH, Ducroq D, Gerber S, Ghazi I, Rozet JM, Perrault I, Munnich A, Dufier JL, Coscas G, Soubrane G, Kaplan J, 1999. Age-related macular degeneration in grandparents of patients with Stargardt disease: genetic study. Am. J. Ophthalmol 128, 173–178. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Boulton M, 2005. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res 80, 595–606. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Yamamoto K, 2012. The bisretinoids of RPE lipofuscin: a complex mixture. Adv. Exp. Med. Biol 723, 761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, Hicks D, Hamel CP, 2010. The retinal pigment epithelium in health and disease. Curr. Mol. Med 10, 802–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, Gregory-Roberts E, Yamamoto K, Blonska A, Ghosh SK, Ueda K, Zhou J, 2012. The bisretinoids of retinal pigment epithelium. Prog. Retin. Eye Res 31, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, Blonska A, Flynn E, Duncker T, Greenberg JP, Secondi R, Ueda K, Delori FC, 2013. Quantitative fundus autofluorescence in mice: correlation with HPLC quantitation of RPE lipofuscin and measurement of retina outer nuclear layer thickness. Invest. Ophthalmol. Vis. Sci 54, 2812–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, Marsiglia M, Allikmets R, Tsang S, Lee W, Duncker T, Zernant J, 2015. Flecks in recessive stargardt disease: short-wavelength Autofluorescence, near-infrared autofluorescence, and optical coherence tomography. Invest. Ophthalmol. Vis. Sci 56, 5029–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, Duncker T, Schuerch K, Paavo M, de Carvalho JRL Jr., 2020. Lessons learned from quantitative fundus autofluorescence. Prog. Retin. Eye Res 74,100774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stargardt K, 1909. Über familiäre, progressive Degeneration in der Maculagegend des Auges. Albrecht von Graefes Arch Klin Ophthalmology 71, 534–550. [Google Scholar]
- Steinberg RH, Wood I, Hogan MJ, 1977. Pigment epithelial ensheathment and phagocytosis of extrafoveal cones in human retina. Philos. Trans. R. Soc. Lond. B Biol. Sci 277, 459–474. [DOI] [PubMed] [Google Scholar]
- Stenirri S, Battistella S, Fermo I, Manitto MP, Martina E, Brancato R, Ferrari M, Cremonesi L, 2006. De novo deletion removes a conserved motif in the C-terminus of ABCA4 and results in cone-rod dystrophy. Clin. Chem. Lab. Med 44, 533–537. [DOI] [PubMed] [Google Scholar]
- Stone EM, Nichols BE, Kimura AE, Weingeist TA, Drack A, Sheffield VC, 1994. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch. Ophthalmol 112, 765–772. [DOI] [PubMed] [Google Scholar]
- Strauss RW, Ho A, Munoz B, Cideciyan AV, Sahel JA, Sunness JS, Birch DG, Bernstein PS, Michaelides M, Traboulsi EI, Zrenner E, Sadda S, Ervin AM, West S, Scholl HP, Progression of Stargardt Disease Study, G., 2016. The natural history of the progression of atrophy secondary to stargardt disease (ProgStar) studies: design and baseline characteristics: ProgStar report No. 1. Ophthalmology 123, 817–828. [DOI] [PubMed] [Google Scholar]
- Strauss RW, Munoz B, Ho A, Jha A, Michaelides M, Cideciyan AV, Audo I, Birch DG, Hariri AH, Nittala MG, Sadda S, West S, Scholl HPN, ProgStar Study G, 2017a. Progression of stargardt disease as determined by fundus autofluorescence in the retrospective progression of stargardt disease study (ProgStar report No. 9). JAMA Ophthalmol 135, 1232–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss RW, Munoz B, Ho A, Jha A, Michaelides M, Mohand-Said S, Cideciyan AV, Birch D, Hariri AH, Nittala MG, Sadda S, Scholl HPN, ProgStar Study G, 2017b. Incidence of Atrophic Lesions in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 5. JAMA Ophthalmol 135, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Molday RS, Nathans J, 1999. Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J. Biol. Chem 274, 8269–8281. [DOI] [PubMed] [Google Scholar]
- Sunness JS, Steiner JN, 2008. Retinal function and loss of autofluorescence in stargardt disease. Retina 28, 794–800. [DOI] [PubMed] [Google Scholar]
- Sunness JS, Ziegler MD, Applegate CA, 2006. Issues in quantifying atrophic macular disease using retinal autofluorescence. Retina 26, 666–672. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Lee W, Zernant J, Schuerch K, Ciccone L, Tsang SH, Sparrow JR, Allikmets R, 2018. The rapid-onset chorioretinopathy phenotype of ABCA4 disease. Ophthalmology 125, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanna P, Georgiou M, Strauss RW, Ali N, Kumaran N, Kalitzeos A, Fujinami K, Michaelides M, 2019. Cross-sectional and longitudinal assessment of the ellipsoid zone in childhood-onset stargardt disease. Transl Vis Sci Technol 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa F, Rossi S, Sodi A, Passerini I, Di Iorio V, Della Corte M, Banff S, Surace EM, Menchini U, Auricchio A, Simonelli F, 2012. Correlation between photoreceptor layer integrity and visual function in patients with Stargardt disease: implications for gene therapy. Invest. Ophthalmol. Vis. Sci 53, 4409–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa F, Melillo P, Di Iorio V, Orrico A, Attanasio M, Rossi S, Simonelli F, 2014. Macular function and morphologic features in juvenile stargardt disease: longitudinal study. Ophthalmology 121, 2399–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teussink MM, Lee MD, Smith RT, van Huet RA, Klaver CC, Klevering BJ, Theelen T, Hoyng CB, 2015. The effect of light deprivation in patients with Stargardt disease. Am. J. Ophthalmol 159, 964–972 e962. [DOI] [PubMed] [Google Scholar]
- Tosha C, Gorin MB, Nusinowitz S, 2010. Test-retest reliability and inter-ocular symmetry of multi-focal electroretinography in Stargardt disease. Curr. Eye Res 35, 63–72. [DOI] [PubMed] [Google Scholar]
- Tracewska AM, Kocyla-Karczmarewicz B, Rafalska A, Murawska J, Jakubaszko-Jablonska J, Rydzanicz M, Stawinski P, Ciara E, Khan MI, Henkes A, Hoischen A, Gilissen C, van de Vorst M, Cremers FPM, Ploski R, Chrzanowska KH, 2019. Genetic spectrum of ABCA4-associated retinal degeneration in Poland. Genes (Basel) 10, E959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani I, 2019. Adeno-associated viral vectors as a tool for large gene delivery to the retina. Genes (Basel) 10, E287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani I, Colella P, Sommella A, Iodice C, Cesi G, de Simone S, Marrocco E, Rossi S, Giunti M, Palfi A, Farrar GJ, Polishchuk R, Auricchio A, 2014. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med 6, 194–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani I, Toriello E, de Simone S, Colella P, Iodice C, Polishchuk EV, Sommella A, Colecchi L, Rossi S, Simonelli F, Giunti M, Bacci ML, Polishchuk RS, Auricchio A, 2015. Improved dual AAV vectors with reduced expression of truncated proteins are safe and effective in the retina of a mouse model of Stargardt disease. Hum. Mol. Genet 24, 6811–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsybovsky Y, Palczewski K, 2014. Expression, purification and structural properties of ABC transporter ABCA4 and its individual domains. Protein Expr. Purif 97, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsybovsky Y, Molday RS, Palczewski K, 2010. The ATP-binding cassette transporter ABCA4: structural and functional properties and role in retinal disease. Adv. Exp. Med. Biol 703, 105–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsybovsky Y, Orban T, Molday RS, Taylor D, Palczewski K, 2013. Molecular organization and ATP-induced conformational changes of ABCA4, the photoreceptor-specific ABC transporter. Structure 21, 854–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte Gauthier MT, 2010. Etude clinique et genetique d’une nouvelle forme d’ataxie spinocerebelleuse pure associee a l’erythrokeratodermie, Faculte de Medecine. Univ. Montreal [Google Scholar]
- Valverde D, Riveiro-Alvarez R, Bernal S, Jaakson K, Baiget M, Navarro R, Ayuso C, 2006. Microarray-based mutation analysis of the ABCA4 gene in Spanish patients with Stargardt disease: evidence of a prevalent mutated allele. Mol. Vis 12, 902–908. [PubMed] [Google Scholar]
- van Driel MA, Maugeri A, Klevering BJ, Hoyng CB, Cremers FP, 1998. ABCR unites what ophthalmologists divide(s). Ophthalmic Genet 19, 117–122. [DOI] [PubMed] [Google Scholar]
- van Huet RA, Bax NM, Westeneng-Van Haaften SC, Muhamad M, Zonneveld-Vrieling MN, Hoefsloot LH, Cremers FP, Boon CJ, Klevering BJ, Hoyng CB, 2014. Foveal sparing in Stargardt disease. Invest. Ophthalmol. Vis. Sci 55, 7467–7478. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke T, Buyl R, De Zaeytijd J, Bauwens M, Uvijls A, De Baere E, Leroy BP, 2015. Colour vision in stargardt disease. Ophthalmic Res 54, 181–194. [DOI] [PubMed] [Google Scholar]
- Vazquez-Dominguez I, Garanto A, Collin RWJ, 2019. Molecular Therapies for Inherited Retinal Diseases-Current Standing, Opportunities and Challenges. Genes, Basel 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ, 2018. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res 66, 157–186. [DOI] [PubMed] [Google Scholar]
- Verdina T, Tsang SH, Greenstein VC, Zernant J, Sodi A, Lima LH, Chang S, Allikmets R, Menchini U, 2012. Functional analysis of retinal flecks in stargardt disease. J. Clin. Exp. Ophthalmol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ruckmann A, Fitzke FW, Bird AC, 1995. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br. J. Ophthalmol 79, 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ruckmann A, Fitzke FW, Bird AC, 1997. In vivo fundus autofluorescence in macular dystrophies. Arch. Ophthalmol 115, 609–615. [DOI] [PubMed] [Google Scholar]
- Webster AR, Heon E, Lotery AJ, Vandenburgh K, Casavant TL, Oh KT, Beck G, Fishman GA, Lam BL, Levin A, Heckenlively JR, Jacobson SG, Weleber RG, Sheffield VC, Stone EM, 2001. An analysis of allelic variation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci 42, 1179–1189. [PubMed] [Google Scholar]
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH, 1999. Insights into the function of Rim protein in photoreceptors and etiology of Stargardťs disease from the phenotype in abcr knockout mice. Cell 98, 13–23. [DOI] [PubMed] [Google Scholar]
- Wolock CJ, Stong N, Ma CJ, Nagasaki T, Lee W, Tsang SH, Kamalakaran S, Goldstein DB, Allikmets R, 2019. A case-control collapsing analysis identifies retinal dystrophy genes associated with ophthalmic disease in patients with no pathogenic ABCA4 variants. Genet. Med 21, 2336–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Fishkin NE, Pande A, Pande J, Sparrow JR, 2009. Novel lipofuscin bisretinoids prominent in human retina and in a model of recessive Stargardt disease. J. Biol. Chem 284, 20155–20166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Yoon KD, Ueda K, Hashimoto M, Sparrow JR, 2011. A novel bisretinoid of retina is an adduct on glycerophosphoethanolamine. Invest. Ophthalmol. Vis. Sci 52, 9084–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanik M, Muller B, Song F, Gall J, Wagner F, Wende W, Lorenz B, Stieger K, 2017. In vivo genome editing as a potential treatment strategy for inherited retinal dystrophies. Prog. Retin. Eye Res 56, 1–18. [DOI] [PubMed] [Google Scholar]
- Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR, 2001. Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum. Genet 108, 346–355. [DOI] [PubMed] [Google Scholar]
- Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR, 2003. An ABCA4 genomic deletion in patients with Stargardt disease. Hum. Mutat 21, 636–644. [DOI] [PubMed] [Google Scholar]
- Young RW, 1967. The renewal of photoreceptor cell outer segments. J. Cell Biol 33, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW, Bok D, 1969. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J. Cell Biol 42, 392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernant J, Collison FT, Lee W, Fishman GA, Noupuu K, Yuan B, Cai C, Lupski JR, Yannuzzi LA, Tsang SH, Allikmets R, 2014a. Genetic and clinical analysis of ABCA4-associated disease in African American patients. Hum. Mutat 35,1187–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernant J, Xie YA, Ayuso C, Riveiro-Alvarez R, Lopez-Martinez MA, Simonelli F, Testa F, Gorin MB, Strom SP, Bertelsen M, Rosenberg T, Boone PM, Yuan B, Ayyagari R, Nagy PL, Tsang SH, Gouras P, Collison FT, Lupski JR, Fishman GA, Allikmets R, 2014b. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet 23, 6797–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernant J, Lee W, Collison FT, Fishman GA, Sergeev YV, Schuerch K, Sparrow JR, Tsang SH, Allikmets R, 2017. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet 54, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernant J, Lee W, Nagasaki T, Collison FT, Fishman GA, Bertelsen M, Rosenberg T, Gouras P, Tsang SH, Allikmets R, 2018. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb Mol Case Stud 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA, Ahmad Z, Caruso R, MacDonald I, Sieving P, Riazuddin S, Hejtmancik JF, 2007. Severe retinitis pigmentosa mapped to 4pl5 and associated with a novel mutation in the PROM1 gene. Hum. Genet 122, 293–299. [DOI] [PubMed] [Google Scholar]
- Zhang N, Tsybovsky Y, Kolesnikov AV, Rozanowska M, Swider M, Schwartz SB, Stone EM, Palczewska G, Maeda A, Kefalov VJ, Jacobson SG, Cideciyan AV, Palczewski K, 2015. Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum. Mol. Genet 24, 3220–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolnikova IV, Strelnikov VV, Skvortsova NA, Tanas AS, Barh D, Rogatina EV, Egorova IV, Levina DV, Demenkova ON, Prikaziuk EG, Ivanova ME, 2017. Stargardt disease-associated mutation spectrum of a Russian Federation cohort. Eur. J. Med. Genet 60, 140–147. [DOI] [PubMed] [Google Scholar]