

Abstract
The proinflammatory cytokine interleukin (IL)-1β plays a pivotal role in the behavioral manifestations (i.e., sickness) of the stress response. Indeed, exposure to acute and chronic stressors induces the expression of IL-1β in stress-sensitive brain regions. Thus, it is typically presumed that exposure to stressors induces the extra-cellular release of IL-1β in the brain parenchyma. However, this stress-evoked neuroimmune phenomenon has not been directly demonstrated nor has the cellular process of IL-1β release into the extracellular milieu been characterized in brain. This cellular process involves a form of inflammatory cell death, termed pyroptosis, which involves: 1) activation of caspase-1, 2) caspase-1 maturation of IL-1β, 3) caspase-1 cleavage of gasdermin D (GSDMD), and 4) GSDMD-induced permeability of the cell membrane through which IL-1β is released into the extracellular space. Thus, the present study examined whether stress induces the extra-cellular release of IL-1β and engages the above cellular process in mediating IL-1β release in the brain. Male Sprague-Dawley rats were exposed to inescapable tailshock (IS). IL-1β extra-cellular release, caspase-1 activity and cleavage of GSDMD were measured in dorsal hippocampus. We found that exposure to IS induced a transient increase in the release of IL-1β into the extracellular space immediately after termination of the stressor. IS also induced a transient increase in caspase-1 activity prior to IL-1β release, while activation of GSDMD was observed immediately after termination of the stressor. IS also increased mRNA and protein expression of the ESCRTIII protein CHMP4B, which is involved in cellular repair. The present results suggest that exposure to an acute stressor induces the hallmarks of pyroptosis in brain, which might serve as a key cellular process involved in the release of IL-1β into the extracellular milieu of the brain parenchyma.
Keywords: stress, neuroinflammation, pyroptosis, IL-1β, caspase-1, gasdermin
1. Introduction
Exposure to stressors induces an array of neuroimmune changes (Frank et al., 2019) including the induction of proinflammatory cytokines (Johnson et al., 2019). Of these proinflammatory mediators, interleukin (IL)-1β has garnered considerable interest given its role as a “gate keeper” of neuroinflammation (Basu et al., 2004) as well as its pivotal role in the behavioral effects of stress (Goshen and Yirmiya, 2009). Numerous studies have reported stress-induced increases in IL-1β protein in discrete brain regions, typically measured by ELISA applied to brain homogenates. It is often implicitly assumed that this measured increase indicates that IL-1β has been released from its producing cells and is thus available to bind to extracellular receptors. It is also often implicit that this IL-1β represents newly synthesized IL-1β. However, the typical methods cannot distinguish between extracellular and intracellular IL-1β (but see below discussion of Iwata et al., 2016), and so stress-induced increases cannot be taken to mean stress-induced release. Moreover, pro-IL-1β, from which the mature biologically active IL-1β is cleaved, is present in brain constitutively, and so a measured increase could reflect cleavage of existing pro-IL-1β, rather than newly synthesized IL-1β. Notably, the preponderance of studies of IL-1β in the context of stress have examined causal roles for IL-1β using pharmacological and genetic approaches to manipulate IL-1β signaling. While these studies suggest that stress induces the extracellular release of IL-1β in the brain parenchyma, actual IL-1β release and the cellular process involved in release have not been measured or characterized, forming the basis of the present study.
IL-1β lacks an N-terminal secretory signal sequence and thus cannot be released from the cell through classical endoplasmic reticulum-golgi transport mechanisms (Sitia and Rubartelli, 2018). As a result, there has been considerable controversy concerning if and how IL-1β is released from cells. The preponderance of current evidence suggests that release of IL-1β into the extracellular milieu typically involves a cellular process termed pyroptosis or proinflammatory programmed cell death (Cookson and Brennan, 2001). The initial step of pyroptosis requires the processing of pro-caspase-1 into the catalytically active form of caspase-1, formerly known as interleukin-1β converting enzyme (Thornberry et al., 1992). Caspase-1, or cysteine-dependent aspartate-specific protease-1, is activated by a number of inflammasomes, which are multiprotein complexes that form in response to a diverse array of inflammatory stimuli (Martinon et al., 2009; Netea et al., 2015). Recent evidence suggests that the inflammasome-caspase-1 complex functions as a holoenzyme and that caspase-1 activation is a rapid and transient event in macrophages (Boucher et al., 2018). Upon activation, caspase-1 cleaves the 31 kDa pro-IL-1β protein into the mature 17 kDa form, which is the biologically active form of IL-1β (Dinarello, 1997). As mentioned, the 17 kDa mature form of IL-1β lacks an N-terminal signal sequence and thus can only be released into the extracellular milieu via unconventional secretory pathways (Sitia and Rubartelli, 2018).
Of these pathways, IL-1β release via pyroptosis has received renewed interest with the discovery and characterization of gasdermin proteins (Orning et al., 2019b). In particular, gasdermin D (GSDMD) is a substrate for caspase-1 and consists of a C-terminal domain linked to an N-terminal domain, with a linker region containing a caspase-1 sensitive cleavage site (Shi et al., 2015). Once cleaved and released, N-terminal protomers oligomerize to form a ring-shaped β-barrel, which inserts into the plasma membrane to form pores 10–20 nm in diameter (Aglietti et al., 2016; Mulvihill et al., 2018). It is through these pores that IL-1β is then released into the extracellular space. It had been thought that the cell then inevitably dies as a result of entry of extracellular materials through the pores. However, very recent work suggests that pore formation does not necessarily lead to cell death (Orning et al., 2019b). Rather, several studies suggest that pore formation can be a transient cellular state such that pores in the cell membrane are resealed (Chen et al., 2014; Evavold et al., 2018; Heilig et al., 2018; Zanoni et al., 2016), thereby allowing IL-1β release without requiring the cell to die. It has been proposed that this alternate cellular fate, termed “hyperactivation”, constrains IL-1β release and induces a moderate inflammatory response compared to pyroptotic cell fate consequences (Evavold et al., 2018). The ESCRT (endosomal sorting complex required for transport) pathway, in particular ESCRT-III, is considered a key pathway involved in resealing of the plasmalemma after caspase-1 activation (Ruhl et al., 2018), thereby constraining IL-1β release.
Towards examining a potential role for pyroptosis or hyperactivation in stress-induced neuroinflammatory processes, here we investigate in the dorsal hippocampus the effects of an acute stressor on 1) IL-1β extracellular release measured with a novel method employing an optical fiber to which an IL-1β capture antibody is conjugated, and inserted into hippocampus through a perforated guide cannula (IL-1β can only diffuse across the guide cannula if it is extracellular) 2) caspase-1 activity, 3) GSDMD cleavage, and 4) expression of ESCRT-III proteins, which mediate plasma membrane repair.
2. Methods
2.1. Subjects
Male Sprague-Dawley rats (225–250 g; Envigo, Indianapolis, IN) were pair housed on a 12-h light–dark cycle (lights on at 0600 h). Food (standard laboratory chow) and water were available ad libitum. Rats were allowed to acclimate to colony conditions for at least one week prior to experimentation. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Colorado Boulder in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2. Chemicals and materials
Recombinant rat IL-1β protein (501-RL), recombinant rat tumor necrosis factor (TNF) protein (510-RT), recombinant rat IL-6 protein (506-RL), rat IL-1β biotinylated polyclonal goat IgG (capture antibody, BAF501), and rat IL-1β antibody polyclonal goat IgG (detection antibody, AF-501-NA) were obtained from R&D Systems (Minneapolis, MN). Recombinant rat IL-4 (4139) and IL-2 protein (OPSD00011) were purchased from Sapphire Bioscience (Redfern, Australia). The anti-IL-1β detection antibody was conjugated to fluorescent-labeled superparamagnetic iron oxide (SPIO; excitation, 480 nm; emission, 520 nm) microspheres (Bangs Laboratories, Fishers, IN). EZ-Link sulfo-NHS-biotin was obtained from ThermoFisher Scientific. Sulfuric acid (H2SO4; 95.0–98.0%), hydrogen peroxide (H2O2; 30%), (3-aminopropyl)triethoxysilane (APTES), toluene, N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC), 2-(N-morpholino)ethanesulfonic acid hydrate (MES), 1x phosphate buffered saline (PBS), Tween-20, and bovine serum albumin (BSA) were purchased from Sigma-Aldrich. Silica core, glass clad multimode optical fiber (200-μm diameter core) was obtained from Thorlabs (Newton, NJ).
2.3. Fabrication of the fiber sensing interface
The construction of the immunosensing probe was similar to our previous reports (Liu et al., 2017; Zhang et al., 2018a; Zhang et al., 2018b). To prepare the IL-1β sensing interface, the multimode optical fiber was first cut into ~1.2 cm sections and then immersed in piranha solution (H2SO4:H2O2; 7:3) for 12 h in order to clean the glass and to form hydroxyl groups on its surface. The fiber sections were washed with Milli-Q water and ethanol and then air-dried. The cleaned optical fibers were then immersed in APTES (5% v/v) in toluene for 8 h to form an amine-terminated polymer layer. Next, sulfo-NHS-biotin was introduced (20 min) to react with the amine group in order to form a biotin layer on the fiber surface. After a series of PBS washes, the biotin-modified fiber sections were incubated in a streptavidin solution (50 μg/ml in 1x PBS) for 40 min to form a streptavidin-coated layer. Fibers were then subsequently incubated with anti-IL-1β biotinylated capture antibody (50 μg/ml) in 1x PBS for 1 h at room temperature (RT). The capture antibody modified fiber surface was then blocked in a 0.5% BSA solution (1x PBS with 0.1% Tween-20) for 1 h at RT to complete the preparation of the sensing interface. Fibers were stored in blocking buffer (0.5% BSA) until use.
2.4. Preparation of SPIO-IL-1β detection antibody conjugates
Conjugation of the anti-IL-1β detection antibody to fluorescent-labeled SPIO microspheres was achieved using an EDC-NHS coupling procedure. First, 25 μL SPIO stock solution (10 mg/ml) was pipetted into a 0.65 ml microcentrifuge tube, followed by two washes with MES buffer (25 mM, pH 5.5–6.0), and then collected with a magnetic separator. In order to activate the carboxylic acid groups on the bead surface, SPIO microspheres were resuspended in 250 μl freshly prepared EDC (10 mg/ml in MES buffer)/NHS (10 mg/ml in MES buffer) solution for 30 min with occasional brief vortexing. The SPIO were then thoroughly washed with MES buffer to remove excess EDC/NHS reagent. The anti-IL-1β detection antibody (50 μl/ml) was then added to the activated SPIO beads and the mixture was incubated for 2 h at RT with constant rotation in order to facilitate formation of the SPIO-IL-1β conjugate. The newly formed conjugates were subsequently washed three times with MES buffer and then blocked in 0.5% BSA buffer (1x PBS with 0.1% Tween-20) for 2 h at RT. Finally, the obtained SPIO-IL-1β conjugates were re-dispersed in 1x PBS and stored at 4°C until further use.
2.5. Perforation and implantation of guide cannula
Stainless-steel guide cannulae cut 4mm below pedestal (Plastics One Inc.) were perforated by a laser system to create windows such that the fiber-based sensing interface could access the extracellular space once introduced into the cannula. The inner diameter of the cannula (24 gauge) was sufficiently larger than that of the immunocapture fiber so that the immunosensing interface is not damaged upon insertion. Cannula were treated with 85% orthophosphoric acid to etch any laser affected metal oxides and to passivate the stainless steel. They were then thoroughly rinsed and ultrasonic cleaned before implantation. Stereotactic surgeries were carried out under isoflurane (5% induction, 2% maintenance in 2.5 L/min O2; Piramal Critical Care) anesthesia. Rats were implanted with a single perforated cannula targeted to the dorsal hippocampus (A/P: −3.5; M/L: 2.0; D/V: −3.0 mm from skull surface) and secured to the skull with stainless steel screws and dental cement. Dorsal hippocampus was selected as a region of interest given our prior findings that challenge with LPS induces IL-1β release in this hippocampal sub-region (Zhang et al., 2018a). A dummy cannula was inserted into the cannula and held in place with a fitted dust cap (Plastics One). Following surgery, subjects were placed on a heating pad and kept in a recovery box until ambulatory before returning to the colony. Rats were given four weeks to recover from surgery before experimentation. We chose this period of recovery to minimize the effects of cannulation on stress-induced IL-1β release; we have previously demonstrated that cannulation potentiates the IL-1β protein response (i.e. total protein) to immune challenge in dorsal hippocampus at 2 weeks, but not 4 weeks post-cannulation (Holguin et al., 2007).
2.6. Analytical performance of the IL-1β immunosensing probe in vitro
2.6.1. Determination of the optimal incubation of time for IL-1β binding equilibrium
The IL-1β immunosensing probe was exposed to recombinant rat IL-1β (10 pg/ml) in buffer (1x PBS + 0.1% BSA) for various incubation times (0, 10, 20, 30, 60 and 120 min). One fiber per time point was collected. Fibers were subsequently washed 5x in 1x PBS to remove unbound IL-1β, and then exposed to the SPIO-IL-1β conjugate (1 h) to form a sandwich immunocomplex. Fibers were washed 5x to remove unbound SPIO-IL-1β conjugate. The resulting fluorescent signal for each fiber per time point was imaged with a Nikon A1 Laser Scanning Confocal Microscope. The imaging method used here was similar to the method described in our previous report (Zhang et al., 2018a). Fluorescence signal of the immunocomplex on the fiber surface was collected by a z-stack method with the separation of around 5 μm between planes. Three separate images, each along a 1.5 mm length of the sensing fiber, were collected and the fluorescence signal of each image was quantified by integrating over a certain spatial (450 μm) window using ImageJ and Matlab software.
2.6.2. Determination of the detection range of IL-1β protein
The immunosensing probe was incubated with varying concentrations of recombinant rat IL-1β (0, 3.9. 15.6, 62.5, 125, 250 and 500 pg/ml) for 1 h in 1x PBS + 0.1% BSA. One fiber per concentration of IL-1β was collected. Fibers were subsequently washed 5x in PBS to remove unbound IL-1β, and then exposed to the SPIO-IL-1β conjugate (1 h) to form a sandwich immunocomplex. Fibers were washed 5x to remove unbound SPIO-IL-1β conjugate. The immunosensing fibers were then imaged and fluorescence quantified as described in section 2.6.1.
2.6.3. Assessment of the specificity of IL-1β immunosensing fibers
Fibers were exposed to recombinant rat IL-1β (10 pg/ml) in the absence or presence of excess concentrations of alternate cytokines including IL-2 (200 pg/ml), IL-4 (200 pg/ml), IL-6 (200 pg/ml) or TNF (200 pg/ml). Fibers were co-incubated with cytokines for 1h in 1x PBS + 0.1% BSA. One fiber per cytokine condition was collected. Fibers were washed 5x to remove unbound cytokines, and then exposed to the SPIO-IL-1β conjugate (1 h) to form a sandwich immunocomplex. Fibers were washed 5x to remove unbound SPIO-IL-1β conjugate. The immunosensing fibers were then imaged and fluorescence quantified as described in section 2.6.1.
2.7. In vivo detection of hippocampal extracellular IL-1β protein
IL-1β sensing fibers were inserted into the guide cannula for collection of baseline samples (60 and 30 min prior to stressor onset) as well as collection of samples immediately (0 h) and 4 h after termination of tailshock. Fiber insertion and IL-1β collection was taken serially, one fiber per time point per subject. Once inserted into the guide cannula, fibers were left in place for 20 min. Following the 20 min IL-1β collection period, fibers were removed and immediately placed in buffer (1x PBS + 0.1% BSA) until assayed. For home cage control (HCC) rats, fiber insertion occurred at the same time of day as fiber insertion for stress treated rats.
2.8. Inescapable tailshock (IS)
Details of the stressor protocol have been published previously and this protocol reliably potentiates pro-inflammatory cytokine responses in the hippocampus after peripheral immune challenge (Johnson et al., 2003) as well as in isolated hippocampal microglia to LPS ex vivo (Frank et al., 2007). Briefly, rats were placed in Plexiglas tubes (23.4 cm in length × 7 cm in diameter) and exposed to either 10 (50 s total duration), 25 (125 s total duration) or 100 (500 s total duration) −1.6 mA, 5 s tail-shocks with a variable inter-trial interval (ITI) ranging from 30 – 90 s (average ITI = 60 s). All IS treatments occurred between 09:00 and 11:00 h. HCC rats remained undisturbed in their home cages except during fiber insertion and removal.
2.9. Tissue dissection of dorsal hippocampus
Rats were given a lethal dose of sodium pentobarbital. Rats were fully anesthetized and transcardially perfused with ice-cold saline (0.9%) for 3 min to remove peripheral immune leukocytes from the CNS vasculature. Brain was rapidly extracted, placed on ice and dorsal hippocampus adjacent to cannulation site was dissected. Hippocampus was flash frozen in liquid nitrogen for whole tissue analysis. All tissue samples were stored at −80°C.
2.10. Tissue processing for protein assays
Hippocampal samples were sonicated on ice using a tissue extraction reagent (Invitrogen, FNN0071) supplemented with a protease inhibitor cocktail (Sigma-Aldrich, P2714). Homogenates were centrifuged (14,000 × g for 10 min at 4°C) and supernatants collected and stored at −80°C. Total protein was quantified using a Bradford assay.
2.11. Enzyme-linked immunosorbent assay (ELISA)
An ELISA for rat IL-1β (R&D Systems, RLB00) was run according to the manufacturer’s instructions and IL-1β protein levels normalized to total protein (IL-1β pg/mg total protein). Specificity of the ELISA for either mature (17 kDa) or pro (31 kDa) IL-1β was determined by assaying in quadruplicate known concentrations of recombinant rat mature (300 pg/ml; R&D Systems) and pro (300 pg/ml; Sino Biological; 80023-R07E) IL-1β. An ELISA for rat pro-IL-1β (Abkine, KTE100419) was run according to the manufacturer’s instructions and protein levels normalized to total protein (pro-IL-1β pg/mg total protein).
2.12. Caspase-1 activity assay
Caspase-1 activity was measured using the Caspase-Glo® Inflammasome Assay (Promega, G9951). According to the manufacturer’s instructions, the Z-WEHD-aminoluciferin substrate plus the proteasome inhibitor (MG-132), which blocks non-specific cleavage of the Z-WEHD-aminoluciferin substrate, was added to Caspase-Glo® 1 buffer. WEHD (Trp-Glu-His-Asp) substrates are tetrapeptides that exhibit the highest sensitivity to the proteolytic activity of a number of caspases including caspases 1, 11 and 14 (Poreba et al., 2013). The caspase-1 assay used here incorporates a specific caspase-1 inhibitor (Ac-YVAD-CHO)(Thornberry et al., 1992), which is used to determine caspase-1 specific activity apart from total caspase activity. Thus, Ac-YVAD-CHO was added to the Z-WEHD-aminoluciferin substrate buffer to yield 2 substrates: Z-WEHD-aminoluciferin without Ac-YVAD-CHO and Z-WEHD-aminoluciferin including Ac-YVAD-CHO.
For each sample, protein homogenate (25 μl) was added to two wells of a 96 well white-coated plate (Costar, 3917). One well received Z-WEHD-aminoluciferin substrate (25 μl) buffer minus Ac-YVAD-CHO, while the second well received Z-WEHD-aminoluciferin substrate (25 μl) buffer plus Ac-YVAD-CHO resulting in a Z-WEHD concentration of 20 μM and an Ac-YVAD-CHO concentration of 1 μM. Luminescence of the Z-WEHD-aminoluciferin substrate was measured at 15, 30 and 45 min of incubation at 37°C using a Tecan Infinite M200 Pro plate reader (Männedorf, Switzerland). Caspase-1 activity was quantified according to the following formula: total relative luminescence units (RLU)(-Ac-YVAD-CHO) - residual RLU (+Ac-YVAD-CHO) = caspase-1 specific RLU. Data are expressed as RLU/total protein.
2.13. Western Blot
Samples were heated to 70°C for 10 min and 30 μg total protein loaded into a standard polyacrylamide 4–12% Bis-Tris gel (Invitrogen). Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was performed in NuPAGE MES (2-(N-morpholino)-ethane sulfonic acid) SDS running buffer (Invitrogen, NP0002) at 200 V for 30 min. Protein was transferred onto a nitrocellulose membrane using an iBlot dry transfer system (Invitrogen). The membrane was then blocked with Odyssey blocking buffer (LI-COR Biosciences, 927–40000) for 1h and incubated overnight at 4°C in blocking buffer with the following primary antibodies: rabbit anti-rat polyclonal antibody to gasdermin D (1:1000 dilution, Cell Signaling Technology, 93709), rabbit anti-rat polyclonal antibody to CHMP4B (1:1000 dilution, Abcam, ab105767) and mouse anti-rat monoclonal antibody to β-actin (1:100,000 dilution, Sigma-Aldrich, A5316). The membrane was washed 4x in 1x PBS + 0.1% Tween and then incubated in blocking buffer containing either goat anti-rabbit (LI-COR, 925–32211) or goat anti-mouse (LI-COR, 925–32210) IRDye 800CW secondary antibody at a concentration of 1:10,000 for 1 h at room temperature and the membrane was washed 4x in 1x PBS + 0.1% Tween. Protein expression was quantified using an Odyssey Infrared Imager (LI-COR Biosciences) and expressed relative to the housekeeping protein β-actin.
2.14. Real time RT-PCR measurement of gene expression
Total RNA was isolated from dorsal hippocampal using TRI Reagent (MilliPore Sigma, 93289) and a standard method of phenol:chloroform extraction (Chomczynski and Sacchi, 1987). Total RNA was quantified using a NanoDrop 2000 spectrophotometer (ThermoFisher). cDNA synthesis was performed using the SuperScript II Reverse Transcriptase kit (ThermoFisher, 18064014). A detailed description of the PCR amplification protocol has been published previously (Frank et al., 2006). cDNA sequences were obtained from Genbank at the National Center for Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov). Primer sequences were designed using the Operon Oligo Analysis Tool (http://www.operon.com/tools/oligo-analysis-tool.aspx) and tested for sequence specificity using the Basic Local Alignment Search Tool at NCBI (Altschul et al., 1997). Primers were obtained from ThermoFisher. Primer specificity was verified by melt curve analyses. All primers were designed to span exon/exon boundaries and thus exclude amplification of genomic DNA. Primer sequences: Interleukin-1β (Il1b), F: CCTTGTGCAAGTGTCTGAAG, R: GGGCTTGGAAGCAATCCTTA; caspase-1 (Casp1), F: ATGCCGTGGAGAGAAACAAG, R: CCAGGACACATTATCTGGTG; charged multivesicular body protein (Chmp)3, F: AAATCACAGCAGGAGCCTTG, R: CATCTTCCTCATCTCCTTCC; Chmp4b, F: TTGACGAGGATGAGCTCATG, R: CTACAGAGGGGACATTTGGT; gasdermin D (Gsdmd), F: AGCTAGAGTTGGTGAAGCAC, R: GGCCACATTCTTCTAGCAAG; Vacuolar protein sorting 4 homolog A (Vps4a), F: CCATCAGGAGGAGGTTTGAA, R: TCTGTGAGGTTGTGAGGTGT; β-actin (Actb), F: TTCCTTCCTGGGTATGGAAT, R: GAGGAGCAATGATCTTGATC. PCR amplification of cDNA was performed using the Quantitect SYBR Green PCR Kit (Qiagen, 204145). Formation of PCR product was monitored in real time using the CFX96 Touch Real-Time PCR Detection System (BioRad). Relative gene expression was determined using Actb as the housekeeping gene and the 2−ΔΔCT method (Livak and Schmittgen, 2001).
2.15. Juvenile Social Exploration (JSE)
Stress exposure (IS) produces robust decrements in JSE (Christianson et al., 2008), which is a widely used and validated measure of social avoidance and anxiety (File and Seth, 2003) and is sensitive to the neuroinflammatory effects of stress (Goshen and Yirmiya, 2009). Here, JSE was measured 24 h prior to (baseline) and 24 h after IS (test). Each experimental rat was transferred to a novel cage with shaved wood bedding. After a 60-min habituation period, a 28–32 day-old juvenile male rat was introduced to the subject’s cage for 3 min. Exploratory behaviors of the adult (sniffing, pinning, licking and allo-grooming of the juvenile) were timed by an observer blind to treatment condition. After the test, the juvenile was removed and the experimental adult rat was returned to its homecage. Although juvenile stimulus rats were reused for multiple tests, the adult was never re-tested with the same juvenile. For each rat, JSE test data were quantified as a percent of baseline JSE.
2.16. Statistical analysis
All data are presented as mean ± sem. Statistical analyses consisted of t-test or ANOVA followed by post-hoc tests (Tukey’s HSD) using Prism 8 (GraphPad Software). Threshold for statistical significance was set at α = 0.05. Sample sizes are provided in figure captions.
3. Results
3.1. Effect of IS on IL-1β protein release in dorsal hippocampus
3.1.1. Analysis and validation of IL-1β fiber sensors in vitro
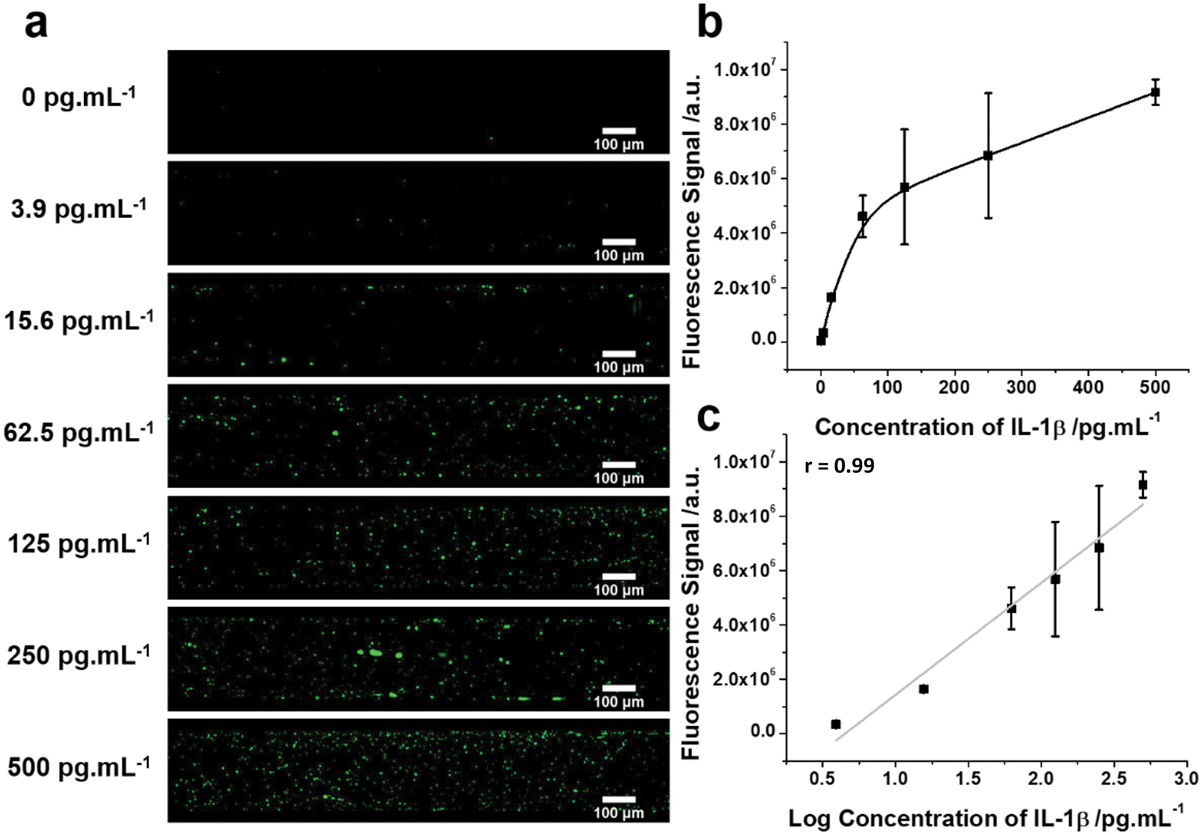
Prior to conducting in vivo studies of the effect of stress on IL-1β release in hippocampus, several in vitro studies were conducted on a batch of fiber sensors to evaluate the analytical performance of the immunosensor. These studies evaluated 1) the kinetics of the formation of the IL-1β capture antibody-antigen immunocomplex, 2) the linear relationship between IL-1β concentrations and fluorescence intensity and 3) the selectivity of the fiber sensor for binding of IL-1β. We found that exposure of the fiber sensor to rat recombinant IL-1β (10 pg/ml) resulted in maximal fluorescence after 60 min of incubation, indicating that the formation of the IL-1β antibody-antigen immunocomplex reaches equilibrium in the range of 30–60 min of incubation (Suppl. Fig. S1). In light of these kinetics of antibody-antigen immunocomplex formation, the fiber sensor was exposed to various concentrations of IL-1β protein for 30 min. Representative confocal z-stack images of fiber fluorescence at various concentrations of IL-1β are presented in Fig. 1A. We found that fluorescence signal increased with increasing concentrations of IL-1β (Fig. 1B) and a linear correlation was obtained for log concentration (3.9 pg/ml to 500 pg/ml) and fluorescence (r = 0.99)(Fig. 1C). The lowest detection limit for IL-1β was calculated to be 1.07 pg/ml. In addition, the precision of the immunosensor was tested using 4 batches (3 fiber samples per batch) of freshly prepared fibers measuring a given concentration of IL-1β (25 pg/ml). The coefficient of variation of the measurements for intra-assay and inter-assay precision was 8.9% and 9.6% respectively, indicating high within-batch and between-batch precision of the fiber sensors. Finally, fiber sensors were exposed to IL-1β (10 pg/ml) in the presence of excess concentrations of various cytokines including IL-2 (200 pg/ml), IL-4 (200 pg/ml), IL-6 (200 pg/ml) and TNF (200 pg/ml). We found that IL-1β fluorescence failed to decrease in the presence of each cytokine suggesting that these alternate cytokines failed to block IL-1β binding to the sensor, thus indicating that the fiber sensor selectively bound IL-1β (Suppl. Fig. S2).
Fig. 1. In vitro analysis of fiber sensor performance.
A. Fiber sensors were exposed to various concentrations of IL-1β protein for 30 min and fiber fluorescence captured. Representative confocal z-stack images of fiber fluorescence are shown after exposure to IL-1β protein. B. Asymptotic relationship between IL-1β concentration and fiber fluorescence. C. Depicted is the linear relationship between log IL-1β concentration and fiber fluorescence. a.u. = arbitrary units.
3.1.2. In vivo detection of stress-induced extracellular IL-1β release in dorsal hippocampus
Given the analytical performance of this batch of sensor fibers tested in 3.1.1, we proceeded to use fibers from this same batch to determine the effect of IS on IL-1β release in dorsal hippocampus immediately after and 4 h after termination of the stressor. The time × stress interaction was significant (df = 1, 6; F = 12.17, p = 0.013) and post-hoc comparisons show IS increased IL-1β release immediately after (p = 0.002), but not 4 h after (p = 0.58) termination of the stressor compared to HCCs (Fig. 2A). Stress as well as IL-1β induce anxiety-like behaviors such as decrements in juvenile social exploration (JSE)(Goshen and Yirmiya, 2009). Therefore, we measured JSE in these same rats, in which IL-1β extracellular release was measured. Consistent with our prior findings (Christianson et al., 2008), IS induced a robust reduction in JSE compared to HCCs measured 24h after stress exposure (Fig. 2B; df = 6, t = 4.42, p = 0.004), which was inversely correlated with IL-1β extracellular levels in dorsal hippocampus (r = −0.65).
Fig. 2. Effect of stress on IL-1β protein release in dorsal hippocampus and anxiety-like behavior.
Rats were exposed to IS or served as home cage controls (HCCs). A. IL-1β specific fiber sensors were inserted via indwelling cannula at 0H and 4H post-stress and extra-cellular IL-1β protein was captured over a 20 min period. Fluorescence of fiber sensors was then measured ex vivo. B. Juvenile social exploration (JSE) was measured 24H after termination of IS and expressed as a percentage of baseline JSE collected 24H prior to IS. Data are presented as the mean+sem. N = 4 per group. RFU = relative fluorescent units. ** p < 0.01, IS vs HCC.
3.2. Effect of cannulation on total IL-1β protein
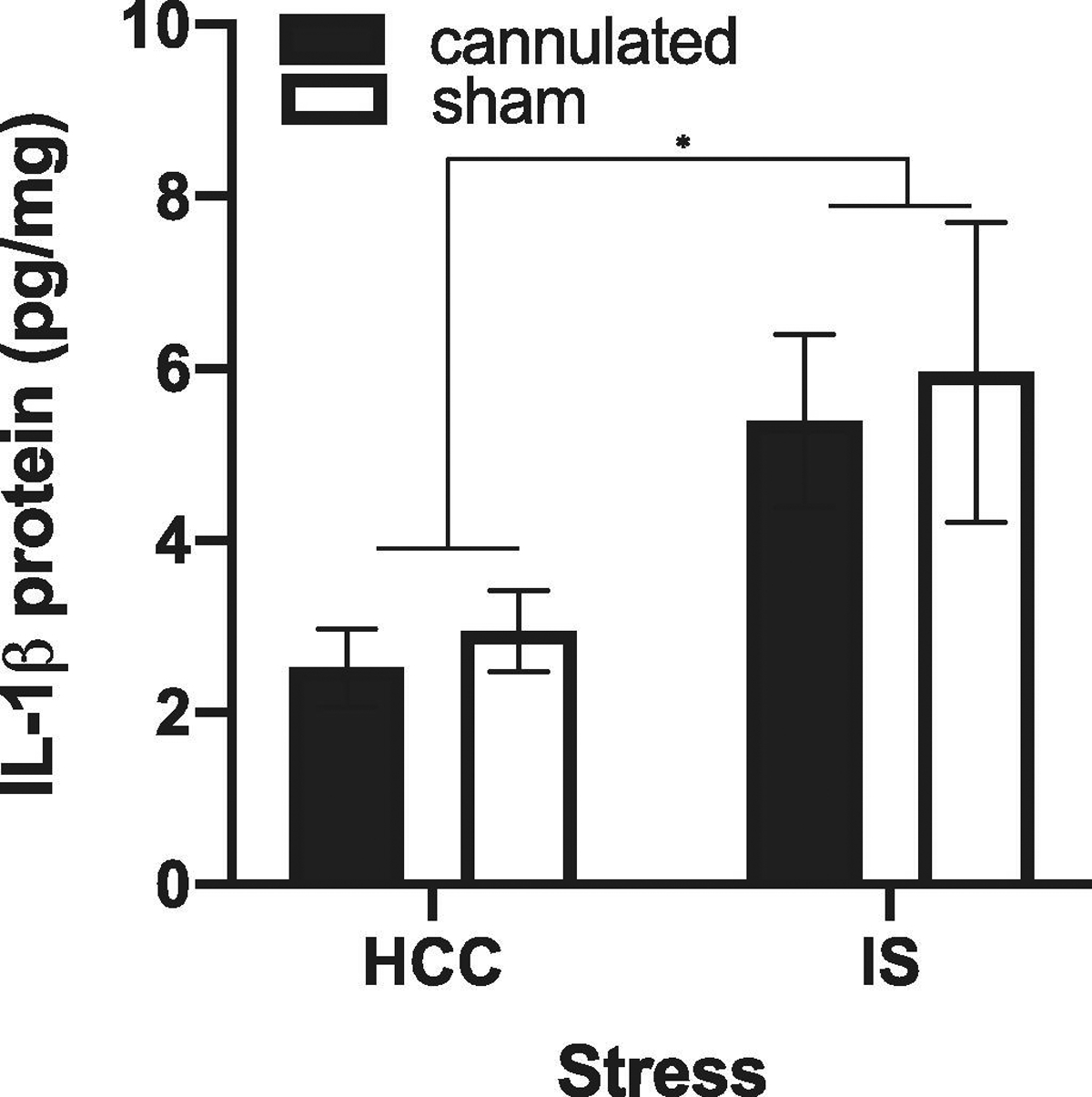
Total IL-1β protein as measured in whole tissue is comprised of both the intracellular and extracellular fraction of total protein. Use of an intra-hippocampal indwelling cannula was necessary to detect the IS-induced extracellular fraction of IL-1β protein. However, a concern regarding this experimental approach is that cannulation per se might: 1) induce a neuroinflammatory response and/or 2) sensitize the neuroinflammatory response to stress exposure. Thus, the effect of stress on IL-1β release might simply reflect the cannulation-induced sensitization of the IL-1β response to IS. Indeed, we have previously demonstrated that intra-hippocampal cannulation potentiates the IL-1β response to a peripheral lipopolysaccharide (LPS) challenge in dorsal hippocampus (Holguin et al., 2007). This cannulation-induced sensitization of the neuroinflammatory response to immune challenge was observed 2 wk, but not 4 wk after cannulation. As noted in section 2.5., stress experiments were conducted 4 wk after intra-hippocampal implantation of cannula to minimize these sensitization effects of cannulation. Nevertheless, to exclude the possibility that cannulation sensitizes the IL-1β protein response to IS, we examined the effect of cannulation (4 wk post-surgery) on total IL-1β protein in dorsal hippocampus of HCCs and stress-exposed rats. While cannulation failed to potentiate the IL-1β response to IS (interaction effect; df = 1, 20, F = 0.005, p = 0.95), stress exposure independent of cannulation increased IL-1β (main effect; df = 1, 20, F = 7.74, p = 0.01). Furthermore, cannulation per se failed to significantly increase IL-1β (main effect; df = 1, 20, F = 1.48, p = 0.64)(Fig. 3).
Fig. 3. Effect of cannulation on total IL-1β protein in dorsal hippocampus.
Rats received sham surgery or were implanted with an indwelling cannula. Four weeks after surgery, rats were then exposed to IS or served as HCCs. At 0H post-stress, total IL-1β protein was measured in dorsal hippocampus. Data are presented as the mean+sem. N = 6 per group. * p < 0.05, IS vs HCC.
3.3. Effect of IS on IL-1β mRNA
In light of the effect of IS on total IL-1β protein, we examined whether IS increased IL-1β mRNA in dorsal hippocampus. Interestingly, we found that immediately after stressor exposure, IS reduced IL-1β mRNA levels compared to HCCs (df = 14, t = 2.51, p = 0.02)(Fig. 4A). This suppressive effect of IS on IL-1β mRNA levels could reflect negative feedback upon de novo synthesis of IL-1β due to increases in IL-1β transcription and translation that might have occurred at an earlier time point. To explore this possibility, we measured IL-1β mRNA levels more proximal to the onset of IS. As a first approximation, we chose to assess IL-1β mRNA levels immediately after 25 shocks or ~ 25 minutes of IS, which is equivalent to 25% of the total stress exposure (100 shocks) typically administered to rats. Notably, the suppressive effect of IS on IL-1β mRNA was even more pronounced after only 25 shocks (df = 10, t = 4.27, p = 0.002)(Fig. 4B). Further, total IL-1β protein levels were not affected by 25 shocks (df = 10, t = 0.32, p = 0.75)(data not shown). These data suggest that the increase in total IL-1β protein observed in Fig. 3 was not due to de novo production of IL-1β, but rather, reflects an increase in the processing of constitutive pro-IL-1β protein into its mature form. Of course, this presumes that the ELISA used here to measure total IL-1β protein preferentially detects the mature form of IL-1β protein (17 kDa) over the pro-form of IL-1β (31 kDa). Indeed, we found that the ELISA used here largely detects mature IL-1β and highly underestimates concentrations of pro-IL-1β (Suppl. Fig. S3A). Further, these findings suggest that IS exposure should reduce levels of pro-IL-1β protein in hippocampus, since it will have become cleaved. Indeed, we found that IS (100 shocks) massively reduced pro-IL-1β protein compared to HCCs (t (10) = 2.21, p = 0.026)(Fig. 4C) using an ELISA kit that preferentially detects pro-IL-1β. We validated that this pro-IL-1β ELISA does detect pro-IL-1β using recombinant pro-IL-1β. However, this ELISA also detects, though underestimates mature IL-1β levels (Suppl. Fig. S3B).
Fig. 4. Effect of stress on IL-1β mRNA in dorsal hippocampus.
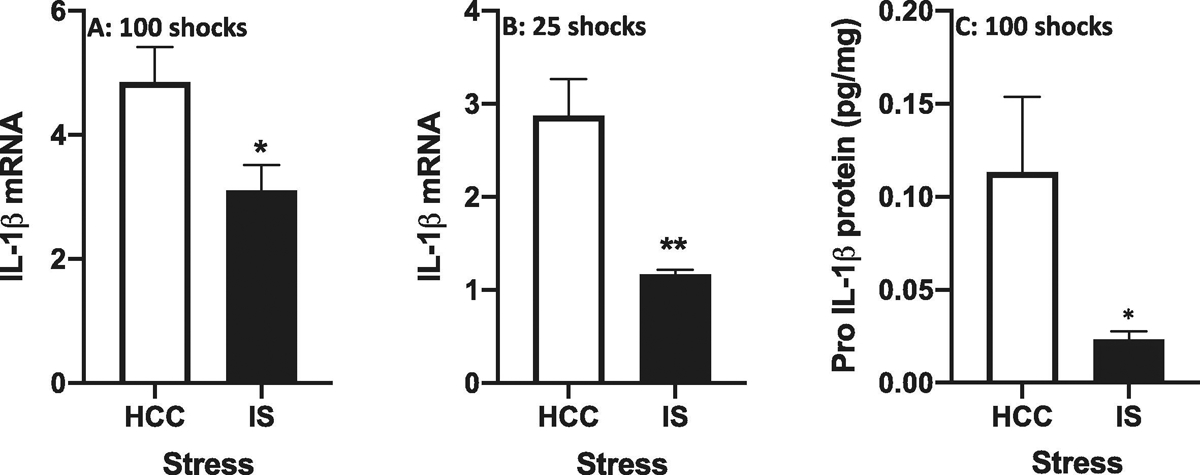
Rats were exposed to IS or served as HCCs. After exposure to (A) 100 shocks and (B) 25 shocks, IL-1β mRNA was measured. After exposure to (C) 100 shocks, pro-IL-1β protein was measured. Data are presented as the mean+sem. N = 6 – 8 per group. * p < 0.05, ** p < 0.01, IS vs HCC.
3.4. Effect of IS on caspase-1 activity
The process of extracellular IL-1β release by pyroptosis requires a number of critical steps (Orning et al., 2019b). These include: 1) inflammasome formation, 2) activation of caspase-1, 3) caspase-1 cleavage of pro-IL-1β into mature IL-1β, and 4) caspase-1 cleavage of gasdermin D, which forms pores in cellular membranes, thereby permitting the extracellular release of mature IL-1β. Thus, given the effect of IS found on extracellular release of IL-1β, we explored the possibility that exposure to IS induces caspase-1 activation. Towards addressing this potential outcome, we utilized a caspase-1 activity assay to measure activity in protein homogenates of dorsal hippocampus. It is important to note that this caspase-1 activity assay has been previously validated in our laboratory (Frank et al., 2020).
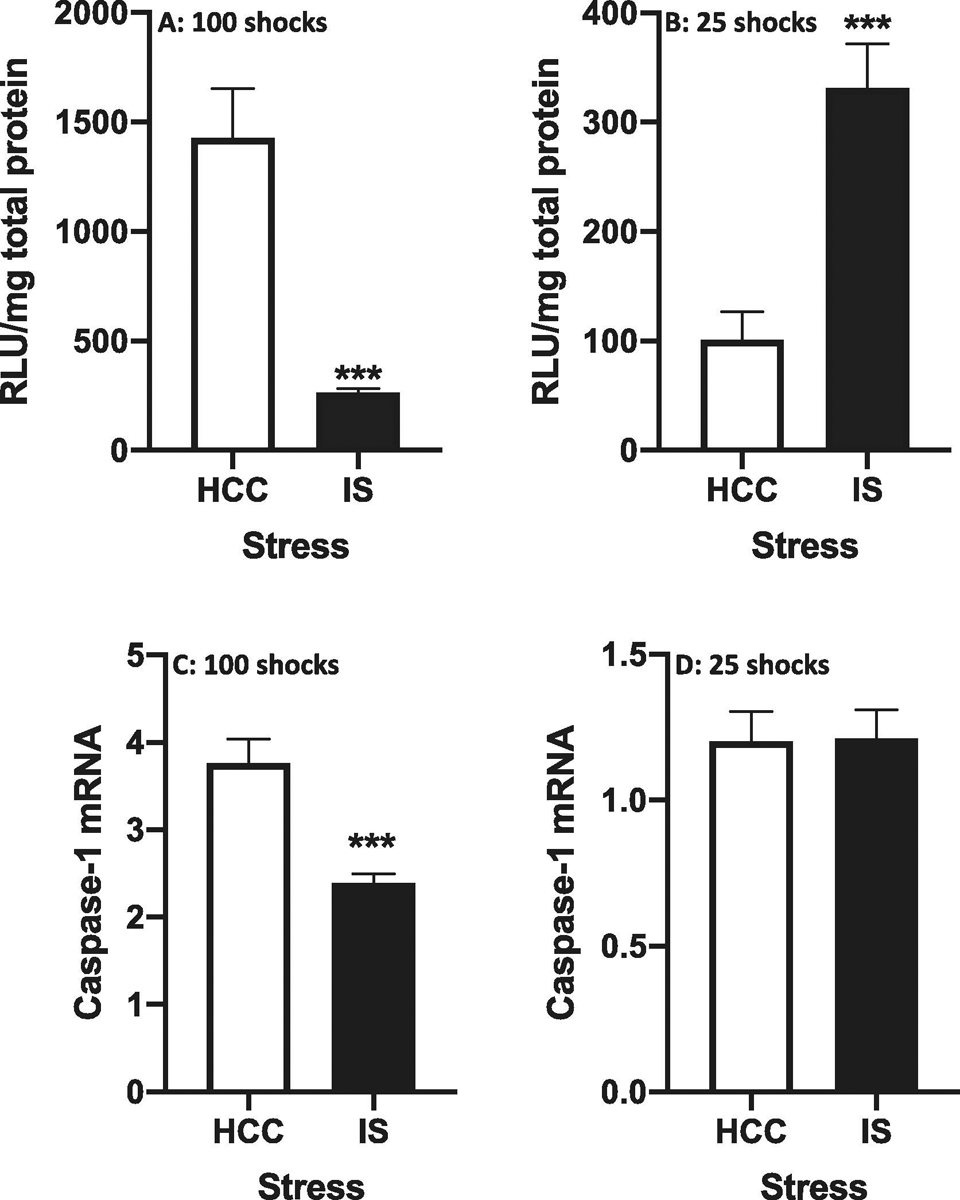
Given that IS induced IL-1β release in dorsal hippocampus immediately after termination of stress exposure (100 shocks), we chose to measure caspase-1 activity at this time point post-stress. Interestingly, we found that IS suppressed caspase-1 activity compared to HCCs (df = 10, t = 5.18, p = 0.0004) (Fig. 5A). In a separate cohort of rats, a similar effect of IS was found for caspase-1 mRNA (df = 10, t = 4.67, p = 0.001)(Fig. 5C). As noted, caspase-1 activation and cleavage of pro-IL-1β precedes the extracellular release of mature IL-1β. Interestingly, a recent study demonstrated that caspase-1 induction in macrophages is a rapid and highly transient event (Boucher et al., 2018). Therefore, we explored the possibility that IS transiently induced caspase-1 activity at a timepoint prior to the detection of IS-induced IL-1β release. Thus, we measured caspase-1 activity after exposure to 25 shocks. After this number of shocks, we found that IS increased caspase-1 activity compared to HCCs (df = 10, t = 4.48, p = 0.0007)(Fig. 5B). However IS failed to alter caspase-1 mRNA levels (df = 10, t = 0.06, p = 0.95)(Fig. 5D). Further, we assessed caspase-1 activity after only 10 shocks and found that IS failed to alter activity (data not shown).
Fig. 5. Effect of stress on caspase-1 activity and mRNA in dorsal hippocampus.
Rats were exposed to IS or served as HCCs. Caspase-1 activity (A and B) or caspase-1 mRNA (C and D) was measured after 100 shocks (A and C) or 25 shocks (B and D). Data are presented as the mean+sem. N = 6 per group. *** p < 0.001, IS vs HCC. RLU = relative luminescence units.
3.5. Effect of IS on cleavage of gasdermin D (GSDMD) in dorsal hippocampus
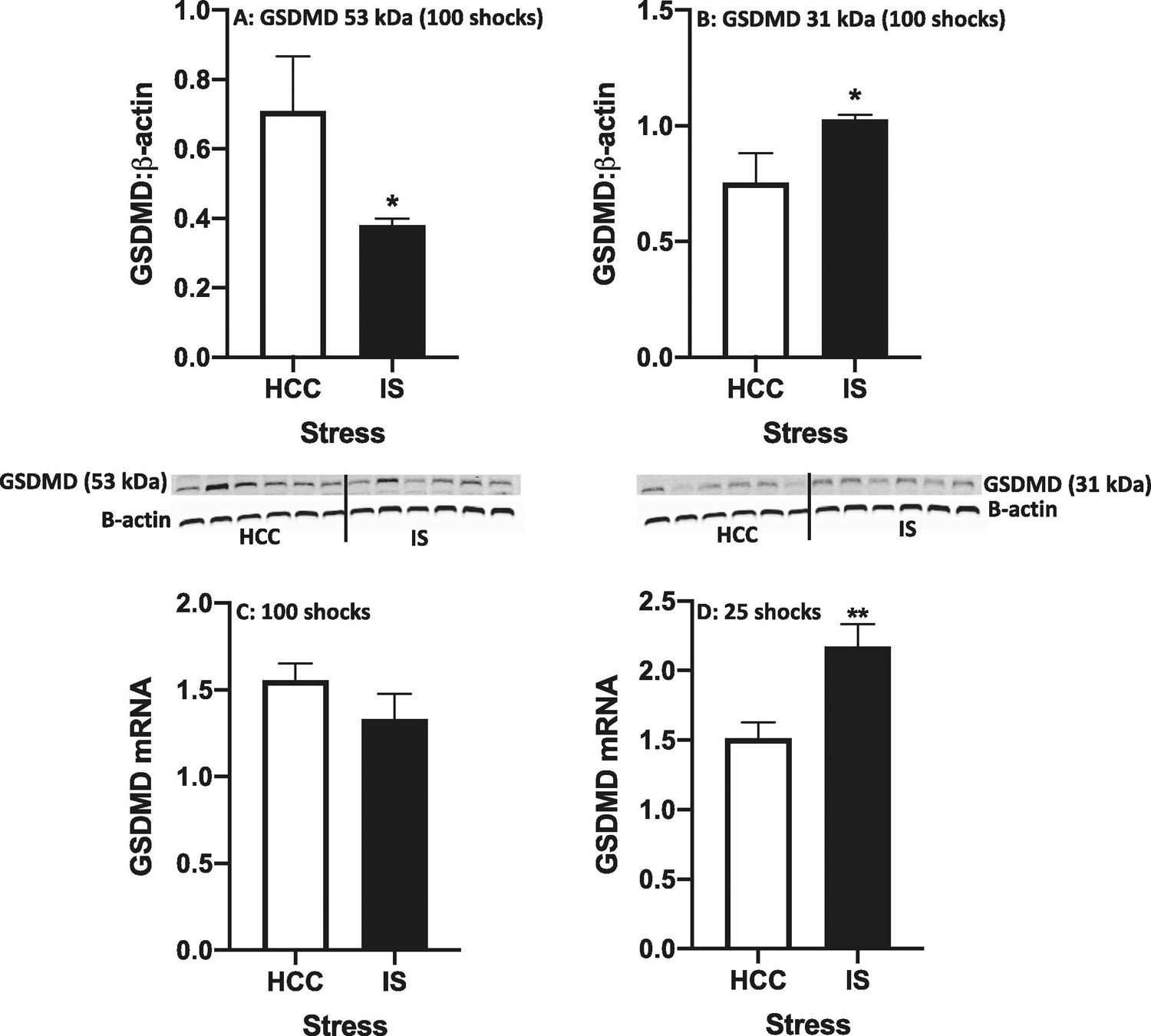
As noted in the Introduction, the GSDMD protein is a substrate for caspase-1 and consists of a C-terminal domain linked to an N-terminal domain, with a linker region containing a caspase-1 sensitive cleavage site (Liu et al., 2016). The C-terminal domain is an inhibitory domain that interferes with the pore-forming N-terminal domain. Upon caspase-1 cleavage, the N-terminal domain oligomerizes and interacts with lipid aspects of the membrane to form 10–20 nm pores in the cell membrane, which permit release of mature IL-1β protein into the extracellular space (Orning et al., 2019b). Given the effects of IS on IL-1β protein release and activation of caspase-1, we examined the effect of IS on GSDMD cleavage. Immediately after termination of stress exposure (100 shocks), we found that IS decreased expression of full-length (53 kDa) GSDMD (df = 9, t = 2.3, p = 0.04) (Fig. 6A), but increased expression of the N-terminal fragment (31 kDa)(df = 9, t = 2.36, p = 0.04) (Fig. 6B). After exposure to 25 shocks, IS failed to affect expression of full-length GSDMD and the N-terminal fragment (data not shown). Given these effects, we examined the effect of IS on GSDMD mRNA levels and found that IS increased GSDMD mRNA after 25 shocks (df = 10, t = 3.31, p = 0.008)(Fig. 6C) and failed to alter GSDMD mRNA after 100 shocks (df = 10, t = 0.30, p = 0.22)(Fig. 6D).
Fig. 6. Effect of stress on cleavage of gasdermin D (GSDMD) protein and mRNA in dorsal hippocampus.
Rats were exposed to IS or served as HCCs. Full length (53 kDa)(A) and cleaved (31 kDa)(B) GSDMD were measured using Western blot (shown below figures) after 100 shocks. GSDMD mRNA was measured after 100 shocks (C) or 25 shocks (D). Data are presented as the mean+sem. N = 5 – 6 per group. * p < 0.05, ** p < 0.01 IS vs HCC.
3.6. Effect of IS on expression of ESCRTIII proteins in dorsal hippocampus
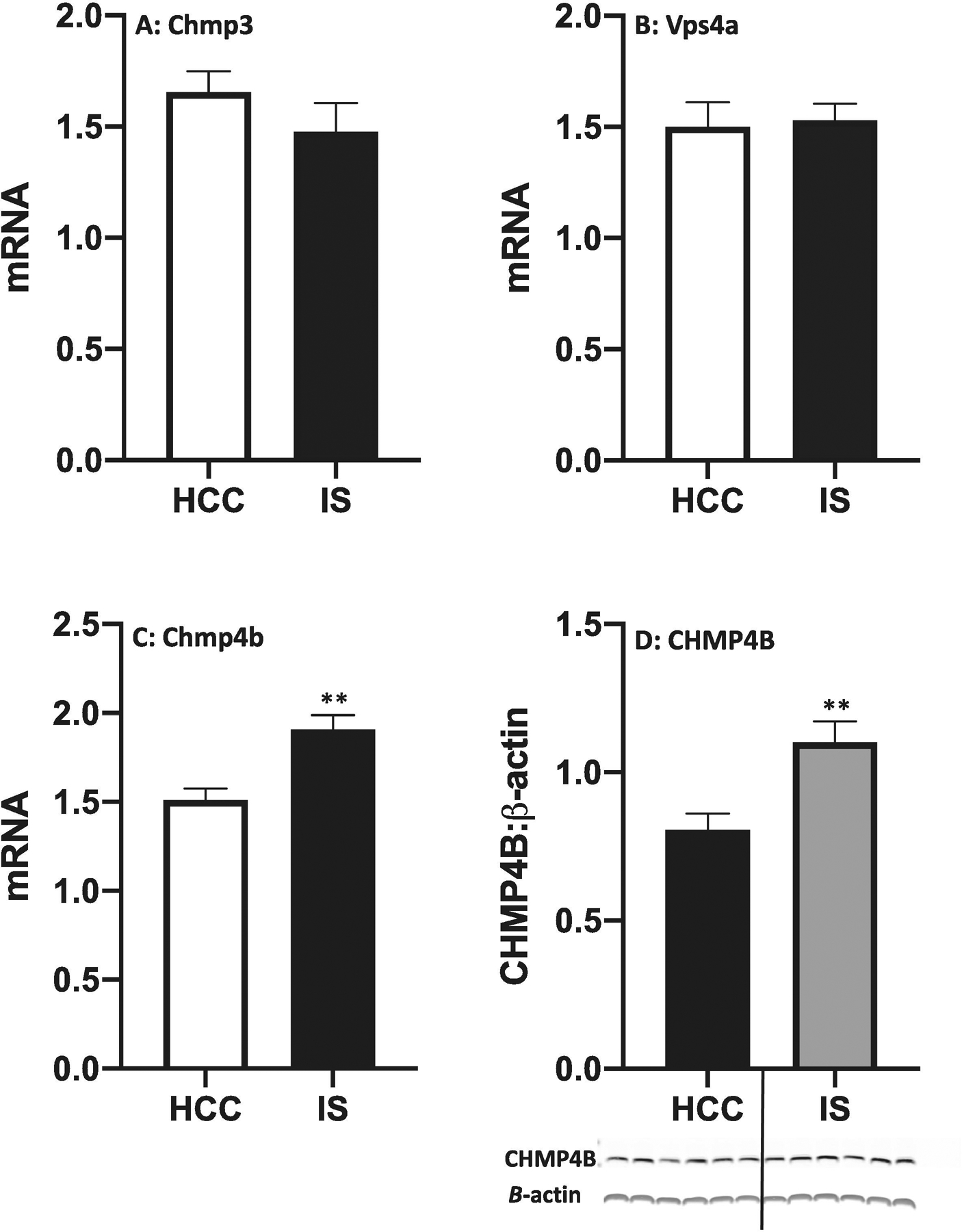
The effect of IS on IL-1β protein release and GSDMD processing suggests that IS might induce pores in cellular membranes to allow release of IL-1β protein. As noted in the Introduction, pore formation can be a transient event, which limits release of IL-1β protein into the extracellular milieu and prevents pyroptotic death from occurring. The ESCRT (endosomal sorting complex required for transport) III proteins are involved in plasma membrane repair (Gatta and Carlton, 2019). Indeed, Ruhl and colleagues have recently demonstrated that several ESCRTIII proteins (CHMP3, CHMP4B and VPS4A) are necessary for cell membrane repair during pyroptotic events and that inhibition of these proteins exacerbates pyroptosis and IL-1β release (Ruhl et al., 2018). In particular, the ESCRTIII protein CHMP4B appears to play a pivotal role in the repair of cell membranes in response to a diverse array of cellular threats (Jimenez et al., 2014). Therefore, we examined the effects of IS on the gene expression of these proteins immediately after termination of the stressor in hippocampus. We found that IS failed to significantly alter expression of Chmp3 (Fig. 7A, df = 10, t = 1.13, p = 0.28) and Vps4a (Fig. 7B, df = 10, t = 0.22, p = 0.83). However, IS induced an increase in the expression of Chmp4b compared to HCCs (Fig. 7C, df = 10, t = 3.9, p = 0.003). In a separate cohort of rats, we found that IS increased protein levels of CHMP4B immediately after termination of the stressor in hippocampus (Fig. 7D, df = 10, t = 3.33, p = 0.008).
Fig. 7. Effect of IS on expression of ESCRTIII proteins in dorsal hippocampus.
Rats were exposed to IS or served as HCCs. Immediately after termination of the stressor (100 shocks), gene expression of (A) Chmp3, (B) Chmp4b and (C) Vps4A was measured. (D) CHMP4B protein was measured using Western blot (shown below figure). Data are presented as the mean+sem. N = 6 per group. ** p < 0.01, IS vs HCC.
4. Discussion
Exposure to stressors can induce a sickness response in rodents, which include behavioral changes such as anhedonia and reduced social investigation as well as physiological changes resulting in fever, aphagia, and weight loss (Dantzer et al., 2008). A considerable number of studies have demonstrated that stress-induced IL-1β in the brain plays a causal role in the sickness response and depressive-like behavior (Goshen and Yirmiya, 2009). Further, exogenous IL-1β recapitulates a number of these stress-induced behavioral and physiological phenotypes (Dantzer, 2009). While these studies clearly implicate extra-cellular release of IL-1β in the brain parenchyma, stress-induced release in the brain has not been demonstrated. Moreover, the cellular mechanism of stress-induced IL-1β release has not been characterized in brain. Here, we demonstrated that exposure to an acute stressor does result in the increased extracellular release of IL-1β because only extracellular IL-1β could access the optical fiber sensor. Increased release of IL-1β in response to stress appears to be a highly transient phenomenon, in that the IL-1β increase was detected immediately after, but not 4 h after termination of the stressor. Interestingly, in a separate cohort of rats, we found that stress induced a profound decrease in IL-1β mRNA immediately after stress exposure and at a time point more proximal to the onset of stress (25 shocks). In addition, we found that pro-IL-1β protein levels were decreased immediately after stress exposure, while mature IL-1β protein levels were increased at this same time point post-stress. Taken together, these effects of stress on IL-1β mRNA, pro-IL-1β protein and mature IL-1β protein suggest that stress exposure induced the maturation of pre-existing pro-IL-1β protein resulting in the release of constitutive IL-1β protein into the extracellular space of the dorsal hippocampus.
In further support of this conclusion, we found that stress increased caspase-1 activity (see below for detailed discussion), which converts pro-IL-1β protein into the mature form of IL-1β protein. Also, we found that stress-induced increases in levels of extracellular IL-1β were inversely correlated with decrements in JSE. A number of studies have demonstrated that IL-1β plays a causal role in the effects of stress on JSE (Goshen and Yirmiya, 2009) and exogenous IL-1β produces profound reductions in JSE (Kent et al., 1992). However, due to the correlative nature of the present study, it is unclear whether IL-1β mediates the effects of stress on JSE. Of note, it is unclear which cell type(s) in the dorsal hippocampus serves as the source of stress-induced IL-1β extra-cellular release. While microglia are considered a key source of IL-1β, astrocytes, oligodendrocytes, endothelial cells and neurons are all capable of producing IL-1β (Basu et al., 2004). Mast cells have also been found in the CNS and could be a source of IL-1β (Mittal et al., 2019). Further, pyroptosis has been observed in microglia, neurons, astrocytes and oligodendrocytes (McKenzie et al., 2020). Pan et al. have demonstrated that chronic stress induces NLRP3 expression in microglia, but not in neurons (Pan et al., 2014). As the NLRP3 inflammasome drives IL-1β production (Swanson et al., 2019), this finding suggests that in the present study microglia might be a key source of stress-induced IL-1β production/release in dorsal hippocampus.
The in vivo detection method used here to capture and quantify IL-1β extra-cellular release has been used previously to detect IL-1β release in dorsal hippocampus in response to an immune challenge (i.e., LPS) (Zhang et al., 2018a). This methodological approach necessitates the placement of a chronic indwelling cannula in dorsal hippocampus. A concern regarding this experimental approach is that cannulation per se might have induced sufficient tissue damage and trauma, which then might have elicited a chronic neuroinflammatory response and/or sensitized the neuroinflammatory response to subsequent stressor exposure. Essentially, cannulation is a stab wound that results in traumatic brain injury and the release of damage-associated molecular patterns (e.g. HMGB1) (Paudel et al., 2018), which are not only neuroinflammatory, but also sensitize neuroinflammatory responses (Frank et al., 2015). Towards mitigating the neuroinflammatory/sensitization effects of cannulation, rats were exposed to stress 4 wk after surgery. We chose this recovery time from surgery based on our prior study demonstrating that cannulation potentiates the IL-1β protein response (i.e. total protein) to immune challenge (LPS) in dorsal hippocampus at 2 wk, but not 4 wk post-cannulation (Holguin et al., 2007). To further explore the possibility that cannulation per se might underpin the observed effect of stress on IL-1β release, we examined the effect of cannulation on hippocampal total IL-1β protein levels in response to stress. Consistent with our prior findings (Holguin et al., 2007), we found that 4 wk after surgery, cannulation failed to increase total IL-1β protein in dorsal hippocampus compared to surgery controls. In addition, cannulation failed to potentiate the stress-induced increase in total IL-1β protein. It is important to emphasize that this control study measured total IL-1β protein in tissue lysates and not release of IL-1β. Therefore, we cannot exclude the possibility, though remote, that cannulation sensitizes the release of IL-1β in response to subsequent stress exposure. It should be noted here that a prior study (Iwata et al., 2016) measured extracellular IL-1β following a stressor using in vivo microdialysis. We did not discuss this study earlier because only 1 wk was allowed for recovery from the guide cannula implantation, and no assessments were made as to whether this procedure did or did not produce sensitization to the stressor. Moreover, potential mechanisms of IL-1β release were not investigated. Here, it is important to consider the clinical relevance of the effect of stress on the extracellular release of IL-1β in this sub-region of the hippocampus. A considerable number of studies have demonstrated that the dorsal hippocampus plays a pivotal role in spatial/contextual memory processes (Fanselow and Dong, 2010). Further, elevations in hippocampal IL-1β, due to aging or immune challenge, play a causal role in spatial/contextual memory impairments (Barrientos et al., 2010). Indeed, we have found that injection of IL-1β protein into the dorsal hippocampus impairs contextual memory formation (Barrientos et al., 2002). Thus, stress-induced IL-1β extracellular release in dorsal hippocampus might play a role in stress-induced spatial/contextual memory impairments (Cazakoff et al., 2010).
Release of the mature (17 kDa), biologically active form of IL-1β into the extra-cellular milieu is a tightly regulated cellular process, which is understandable given its role as a “gatekeeper” of inflammation (Dinarello, 2011). In addition, IL-1β signaling through the type I IL-1 receptor is also tightly regulated via its own receptor antagonist (IL-1RA) as well as the neutralizing action of a decoy receptor (IL-1RII). The initial step in this cellular process of IL-1β maturation and release typically involves the formation and activation of inflammasomes such as NLRP3 (Broz and Dixit, 2016), which are intra-cellular multiprotein scaffolds that form in response to inflammatory stimuli that signal through a number of innate pattern recognition receptors (e.g TLR4)(Latz et al., 2013). Once formed and activated, inflammasomes recruit pro-caspase-1, which undergoes autocatalytic self-cleavage into mature caspase-1 (Boucher et al., 2018), although, it is important to note that pro-caspase-1 does exhibit some enzymatic activity. Boucher and colleagues further demonstrated that active caspase-1 remains bound to the inflammasome and functions as a holoenzyme. Caspase-1 activation is then terminated via a subsequent self-cleavage step. Interestingly, Boucher and colleagues also found that induction of caspase-1 activity was a rapid and transient event, which lasted only 30 minutes after cell stimulation in macrophages. Interestingly, in neutrophils, the kinetics of caspase-1 activation are quite different with induction of caspase-1 activity lasting upwards of 6 hours. Once activated, caspase-1 cleaves pro-IL-1β into its mature form. It is important to note that a number of inflammasome-independent pathways can activate alternate proteases, which process pro-IL-1β into its mature form (Netea et al., 2015). However, these alternate pathways appear to be restricted to neutrophils. In the present study, we examined whether stress induces caspase-1 activation in dorsal hippocampus, given the effect of stress on IL-1β extra-cellular release. Interestingly, we found that exposure to stress suppressed caspase-1 activity immediately after termination of the stressor, which involved our standard protocol of 100 shocks. Of course, caspase-1 activation must precede the processing and release of IL-1β, presuming that stress-induced processing of IL-1β is, at least in part, caspase-1 dependent. Therefore, we examined the effects of stress on caspase-1 activity at a time point preceding the increased release of IL-1β. In this case, we examined caspase-1 activity after 25 shocks and found that activity was increased compared to home cage controls. Clearly, caspase-1 activity must have terminated at a time point between 25 and 100 shocks suggesting that stress-induced caspase-1 activation is a rapid and transient cellular event. This finding is consistent with the findings of Boucher and colleagues who characterized similar caspase-1 kinetics in macrophages (Boucher et al., 2018). While the present results suggest that stress-induced IL-1β extracellular release is caspase-1 dependent, a causal role for caspase-1 in this process is unclear given the correlative nature of these studies.
In addition to performing the catalytic function of processing IL-1β into its mature form, caspase-1 activation is also critical in mediating the cellular process of IL-1β extra-cellular release. Notably, the mature form of IL-1β lacks an N-terminal secretory signal sequence and can thus only be released into the extra-cellular milieu via unconventional secretory pathways (Sitia and Rubartelli, 2018). These pathways include exocytosis via secretory lysosomes, shedding of microvesicles from the plasma membrane, release of exosomes and of particular interest here, passive efflux through pores in the plasma membrane during pyroptotic cell death. It is important to note that signaling of the alarmin ATP through the P2X7 receptor is considered a key step in each of these pathways of IL-1β release (Giuliani et al., 2017). The pyroptotic cell death pathway involves the caspase-1 cleavage of full length gasdermin D (GSDMD; 53 kDa), which is a substrate for caspase-1 and consists of a C-terminal domain linked to an N-terminal domain, with a linker region containing a caspase-1 sensitive cleavage site (Shi et al., 2015). Once cleaved and released, the 31 kDa N-terminal protomers oligomerize to form a ring-shaped β-barrel, which inserts into the plasma membrane to form pores 10–20 nm in diameter (Aglietti et al., 2016; Mulvihill et al., 2018). It is through these pores that IL-1β is then released into the extracellular space. Of note, the molecular diameter of mature IL-1β is approximately 4–5 nm, which readily allows mature IL-1β to pass through GSDMD-formed pores in the cell membrane (Ding et al., 2016; Finzel et al., 1989). If the cellular membrane does not undergo repair and remains porous, cellular swelling and rupture occurs resulting in cell death. This mode of cell death has been termed pyroptosis or inflammatory programmed cell death (Cookson and Brennan, 2001). A number of studies have demonstrated that pyroptosis is critical in controlling viral and bacterial infections (Orning et al., 2019b). Interestingly, GSDMD has also been found to play a causal role in the release of IL-1β-containing microparticles into the extracellular space. These microparticles also contained the P2X7 receptor, which mediate release of IL-1β from microparticles upon binding ATP (Mitra and Sarkar, 2019). It is important to note that alternate gasdermins as well as caspases (e.g. caspase-3) have been implicated in pyroptotic cell death (Orning et al., 2019a). Further, our findings cannot exclude the possibility that stress-induced IL-1β extracellular release involved cellular apoptosis. A number of studies have found that activation of the inflammasome and caspase-1 plays a role in apoptotic cell death in which caspase-3 plays a key role (Tsuchiya, 2020).
Here, stress resulted in the increased extracellular release of IL-1β as well as activation of caspase-1 suggesting that stress might also induce the processing of full-length GSDMD into its pore-forming active N-terminal fragment. Indeed, we found that stress exposure resulted in the reduction of full-length GSDMD protein levels, while increasing protein levels of the N-terminal fragment immediately after termination of the stressor (100 shocks). In addition, we found that stress increased mRNA levels of GSDMD. Taken together, these findings suggest that stress induced the processing of GSDMD into its active pore-forming state. These cellular processes that constitute pyroptosis are induced by a number of inflammasomes including NLRP1, NLRP3, AIM2 and NLRC4 (Netea et al., 2015). Prior studies have implicated NLRP3 in stress-induced priming of neuroinflammatory processes (Alcocer-Gomez et al., 2016; Feng et al., 2019; Iwata et al., 2016; Pan et al., 2014; Weber et al., 2015). However, the present study examined the immediate neuroinflammatory effects of stress per se. Therefore, it is unclear which inflammasome(s) mediates the effects of stress in the present study.
Interestingly, pore formation does not necessarily result in pyroptosis (Orning et al., 2019b). Rather, several studies suggest that pore formation might be a transient cellular state such that pores in the cell membrane are resealed (Chen et al., 2014; Evavold et al., 2018; Heilig et al., 2018; Zanoni et al., 2016), thereby allowing IL-1β release without requiring the cell to die. It has been proposed that this alternate cellular fate, termed “hyperactivation”, constrains IL-1β release and induces a moderate sub-lytic inflammatory response compared to pyroptotic cell fate consequences (Evavold et al., 2018). The ESCRT (endosomal sorting complex required for transport) pathway, in particular ESCRT-III, is considered a key pathway involved in resealing of the plasmalemma after caspase-1 activation (Ruhl et al., 2018), thereby constraining IL-1β release. Here, stress induced a transient release of IL-1β, which suggested to us the possibility that stress might induce only a transient permeability of IL-1β-producing cells. Towards addressing this possibility, we examined the effects of stress on the expression of a number of ESCRT-III proteins including Chmp3, Chmp4b and Vps4a, which have been implicated in cellular membrane repair (Ruhl et al., 2018). Indeed, we found that stress induced both mRNA and protein expression of Chmp4b in dorsal hippocampus. Interestingly, CHMP4B has been found to play a causal role in the repair of cell membranes in response to a diverse array of cellular threats (Jimenez et al., 2014). This finding raises the intriguing possibility that exposure to acute stress might induce transient pore formation in cells and thus serve as a mechanism to limit IL-1β release in the brain.
Taken together, the present set of findings demonstrate that acute stress induces the hallmarks of pyroptosis including 1) IL-1β extracellular release, 2) caspase-1 activity, and 3) GSDMD cleavage. Of course, pyroptosis is a form of inflammatory cell death, which we did not directly measure in the present study. Therefore, the present findings are only suggestive of such a cellular process. In addition, the stress induction of CHMP4B suggests that IL-1β-producing cells might be transiently permeable and thus fail to undergo pyroptosis. Clearly, the correlative nature of the present study precludes definitive conclusions regarding the role of pyroptosis in stress-induced IL-1β release in the brain. Nevertheless, the present findings demonstrate that indeed exposure to stress induces the extracellular release of IL-1β protein and that this process of release exhibits many of the hallmarks of pyroptosis.
Supplementary Material
Fig. S1. Kinetics of IL-1β antigen-antibody immunocomplex formation. Fiber sensors were exposed to recombinant rat IL-β] protein (10 pg/ml) for various times in vitro and fiber fluorescence quantified. One fiber per time point was imaged and 3 separate images were collected along a 1.5 mm length of each fiber. Data are presented as the mean ± sem. a.u. = arbitrary units.
Fig. S2. Assessment of the selectivity of IL-1β immunosensing fibers. Fibers were exposed to recombinant rat IL-1β (10 pg/ml) in the absence or presence of excess concentrations of alternate cytokines including IL-2 (200 pg/ml), IL-4 (200 pg/ml), IL-6 (200 pg/ml) or TNF (200 pg/ml). Fibers were co-incubated with cytokines for 1h. One fiber per cytokine condition was collected and 3 separate images were collected along a 1.5 mm length of each fiber. The immunosensing fibers were then imaged and fluorescence quantified. F0 represents the fluorescence signal of fibers exposed to IL-1β in the absence of competing cytokines. F represents the fluorescence signal of fibers exposed to IL-1β in the presence of competing cytokines. F/ F0 represents the degree of 44 interference of IL-1β binding to the fiber sensor in the presence of alternate cytokines. F/ F0 = 1 indicating no interference and F/ F0 = 0 indicating complete interference. Data are presented as the mean + sem.
Fig. S3. ELISA detection of mature (17 kDa) and pro (31 kDa) rat IL-1β protein.(A) Mature (300 pg/ml) and pro (300 pg/ml) recombinant rat IL-1β protein were assayed using a standard rat IL-1β protein ELISA kit to determine specificity of the ELISA for either form of rat IL-1β. (B) Mature (25 pg/ml) and pro (25 pg/ml) recombinant rat IL-1β protein were assayed using a rat pro-IL-1β protein ELISA kit to determine specificity of the ELISA for either form of rat IL-1β. Data are presented as the mean + sem.
Acute stress induces the extracellular release of IL-1β in dorsal hippocampus.
Acute stress induces transient activation of caspase-1 in dorsal hippocampus.
Acute stress induces the cleavage of gasdermin D.
Acute stress induces the hallmarks of pyroptosis.
Acknowledgments
The authors would like to thank Dr. James Orth at the University of Colorado Boulder Light Microscopy Core Facility and the OptoFab Node of the Australian National Fabrication Facility for their support. M.G.F. and S.F.M. were supported by a grant from the National Institutes of Health (R01MH108523). M.V.B. was supported by an American Australian Association Fellowship. G.L. was supported by funding from the Australia Research Council Future Fellowship (FT160100039), and the National Natural Science Foundation of China (Grant 21575045), University of New South Wales (UNSW) Biomedical Engineering Seed Fund, the UNSW-CAS Collaborative Research Seed Fund (172644KYSB20190059), and UNSW Digital Grid Futures Institute Seed Fund. M.R.H was supported by an Australian Research Council Future Fellowship (FT180100565) and is Director of the Australian Research Council Centre of Excellence for Nanoscale BioPhotonics (CE140100003). E.M.G. was supported by the Australian Research Council Centre of Excellence for Nanoscale BioPhotonics CE140100003.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC, 2016. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcocer-Gomez E, Ulecia-Moron C, Marin-Aguilar F, Rybkina T, Casas-Barquero N, Ruiz-Cabello J, Ryffel B, Apetoh L, Ghiringhelli F, Bullon P, Sanchez-Alcazar JA, Carrion AM, Cordero MD, 2016. Stress-Induced Depressive Behaviors Require a Functional NLRP3 Inflammasome. Molecular neurobiology 53, 4874–4882. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ, 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Watkins LR, Maier SF, 2010. Memory impairments in healthy aging: Role of aging-induced microglial sensitization. Aging Dis 1, 212–231. [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF, 2002. Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res 134, 291–298. [DOI] [PubMed] [Google Scholar]
- Basu A, Krady JK, Levison SW, 2004. Interleukin-1: a master regulator of neuroinflammation. J Neurosci Res 78, 151–156. [DOI] [PubMed] [Google Scholar]
- Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJ, Yap AS, Bezbradica JS, Schroder K, 2018. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. The Journal of experimental medicine 215, 827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Dixit VM, 2016. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Cazakoff BN, Johnson KJ, Howland JG, 2010. Converging effects of acute stress on spatial and recognition memory in rodents: a review of recent behavioural and pharmacological findings. Prog Neuropsychopharmacol Biol Psychiatry 34, 733–741. [DOI] [PubMed] [Google Scholar]
- Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, Schroder K, 2014. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell reports 8, 570–582. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N, 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Christianson JP, Paul ED, Irani M, Thompson BM, Kubala KH, Yirmiya R, Watkins LR, Maier SF, 2008. The role of prior stressor controllability and the dorsal raphe nucleus in sucrose preference and social exploration. Behav Brain Res 193, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson BT, Brennan MA, 2001. Pro-inflammatory programmed cell death. Trends Microbiol 9, 113–114. [DOI] [PubMed] [Google Scholar]
- Dantzer R, 2009. Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am 29, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW, 2008. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, 1997. Interleukin-1. Cytokine Growth Factor Rev 8, 253–265. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, 2011. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol 41, 1203–1217. [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F, 2016. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC, 2018. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Dong HW, 2010. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65, 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Zhao Y, Yang T, Song M, Wang C, Yao Y, Fan H, 2019. Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors. Frontiers in molecular neuroscience 12, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- File SE, Seth P, 2003. A review of 25 years of the social interaction test. Eur J Pharmacol 463, 35–53. [DOI] [PubMed] [Google Scholar]
- Finzel BC, Clancy LL, Holland DR, Muchmore SW, Watenpaugh KD, Einspahr HM, 1989. Crystal structure of recombinant human interleukin-1 beta at 2.0 A resolution. J Mol Biol 209, 779–791. [DOI] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF, 2007. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun 21, 47–59. [DOI] [PubMed] [Google Scholar]
- Frank MG, Fonken LK, Watkins LR, Maier SF, 2019. Microglia: Neuroimmune-sensors of stress. Seminars in cell & developmental biology 94, 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Fonken LK, Watkins LR, Maier SF, 2020. Acute stress induces chronic neuroinflammatory, microglial and behavioral priming: A role for potentiated NLRP3 inflammasome activation. Brain Behav Immun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Weber MD, Watkins LR, Maier SF, 2015. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav Immun 48, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Wieseler-Frank JL, Watkins LR, Maier SF, 2006. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: immunophenotypic and functional characteristics. J Neurosci Methods 151, 121–130. [DOI] [PubMed] [Google Scholar]
- Gatta AT, Carlton JG, 2019. The ESCRT-machinery: closing holes and expanding roles. Curr Opin Cell Biol 59, 121–132. [DOI] [PubMed] [Google Scholar]
- Giuliani AL, Sarti AC, Falzoni S, Di Virgilio F, 2017. The P2X7 Receptor-Interleukin-1 Liaison. Front Pharmacol 8, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshen I, Yirmiya R, 2009. Interleukin-1 (IL-1): a central regulator of stress responses. Front Neuroendocrinol 30, 30–45. [DOI] [PubMed] [Google Scholar]
- Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P, 2018. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol 48, 584–592. [DOI] [PubMed] [Google Scholar]
- Holguin A, Frank MG, Biedenkapp JC, Nelson K, Lippert D, Watkins LR, Rudy JW, Maier SF, 2007. Characterization of the temporo-spatial effects of chronic bilateral intrahippocampal cannulae on interleukin-1beta. J Neurosci Methods 161, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S, Banasr M, Duric V, Yamanashi T, Kaneko K, Rasmussen K, Glasebrook A, Koester A, Song D, Jones KA, Zorn S, Smagin G, Duman RS, 2016. Psychological Stress Activates the Inflammasome via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2X7 Receptor. Biol Psychiatry 80, 12–22. [DOI] [PubMed] [Google Scholar]
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F, 2014. ESCRT machinery is required for plasma membrane repair. Science 343, 1247136. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Barnard DF, Kulp AC, Mehta DM, 2019. Neuroendocrine Regulation of Brain Cytokines After Psychological Stress. J Endocr Soc 3, 1302–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, O’Connor KA, Hansen MK, Watkins LR, Maier SF, 2003. Effects of prior stress on LPS-induced cytokine and sickness responses. Am J Physiol Regul Integr Comp Physiol 284, R422–432. [DOI] [PubMed] [Google Scholar]
- Kent S, Bluthe RM, Dantzer R, Hardwick AJ, Kelley KW, Rothwell NJ, Vannice JL, 1992. Different receptor mechanisms mediate the pyrogenic and behavioral effects of interleukin 1. Proc Natl Acad Sci U S A 89, 9117–9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A, 2013. Activation and regulation of the inflammasomes. Nat Rev Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhang K, Nadort A, Hutchinson MR, Goldys EM, 2017. Sensitive cytokine assay based on optical fiber allowing localized and spatially resolved detection of interleukin-6. ACS Sens 2, 218–226. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J, 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J, 2009. The inflammasomes: guardians of the body. Annual review of immunology 27, 229–265. [DOI] [PubMed] [Google Scholar]
- McKenzie BA, Dixit VM, Power C, 2020. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends in neurosciences 43, 55–73. [DOI] [PubMed] [Google Scholar]
- Mitra S, Sarkar A, 2019. Microparticulate P2X7 and GSDM-D mediated regulation of functional IL-1beta release. Purinergic Signal 15, 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A, Sagi V, Gupta M, Gupta K, 2019. Mast Cell Neural Interactions in Health and Disease. Front Cell Neurosci 13, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Muller DJ, 2018. Mechanism of membrane pore formation by human gasdermin-D. The EMBO journal 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA, 2015. Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol 33, 49–77. [DOI] [PubMed] [Google Scholar]
- Orning P, Lien E, Fitzgerald KA, 2019a. Gasdermins and their role in immunity and inflammation. The Journal of experimental medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Lien E, Fitzgerald KA, 2019b. Gasdermins and their role in immunity and inflammation. The Journal of experimental medicine 216, 2453–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Chen XY, Zhang QY, Kong LD, 2014. Microglial NLRP3 inflammasome activation mediates IL-1beta-related inflammation in prefrontal cortex of depressive rats. Brain Behav Immun 41, 90–100. [DOI] [PubMed] [Google Scholar]
- Paudel YN, Shaikh MF, Chakraborti A, Kumari Y, Aledo-Serrano A, Aleksovska K, Alvim MKM, Othman I, 2018. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Frontiers in neuroscience 12, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poreba M, Strozyk A, Salvesen GS, Drag M, 2013. Caspase substrates and inhibitors. Cold Spring Harbor perspectives in biology 5, a008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P, 2018. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Sitia R, Rubartelli A, 2018. The unconventional secretion of IL-1beta: Handling a dangerous weapon to optimize inflammatory responses. Seminars in cell & developmental biology 83, 12–21. [DOI] [PubMed] [Google Scholar]
- Swanson KV, Deng M, Ting JP, 2019. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. , 1992. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774. [DOI] [PubMed] [Google Scholar]
- Tsuchiya K, 2020. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol Immunol 64, 252–269. [DOI] [PubMed] [Google Scholar]
- Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF, 2015. Stress Induces the Danger-Associated Molecular Pattern HMGB-1 in the Hippocampus of Male Sprague Dawley Rats: A Priming Stimulus of Microglia and the NLRP3 Inflammasome. J Neurosci 35, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, Kagan JC, 2016. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Baratta MV, Liu G, Frank MG, Leslie NR, Watkins LR, Maier SF, Hutchinson MR, Goldys EM, 2018a. A novel platform for in vivo detection of cytokine release within discrete brain regions. Brain Behav Immun 71, 18–22. [DOI] [PubMed] [Google Scholar]
- Zhang K, Liu G, Goldys EM, 2018b. Robust immunosensing system based on biotin-streptavidin coupling for spatially localized femtogram mL(−1) level detection of interleukin-6. Biosens Bioelectron 102, 80–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Kinetics of IL-1β antigen-antibody immunocomplex formation. Fiber sensors were exposed to recombinant rat IL-β] protein (10 pg/ml) for various times in vitro and fiber fluorescence quantified. One fiber per time point was imaged and 3 separate images were collected along a 1.5 mm length of each fiber. Data are presented as the mean ± sem. a.u. = arbitrary units.
Fig. S2. Assessment of the selectivity of IL-1β immunosensing fibers. Fibers were exposed to recombinant rat IL-1β (10 pg/ml) in the absence or presence of excess concentrations of alternate cytokines including IL-2 (200 pg/ml), IL-4 (200 pg/ml), IL-6 (200 pg/ml) or TNF (200 pg/ml). Fibers were co-incubated with cytokines for 1h. One fiber per cytokine condition was collected and 3 separate images were collected along a 1.5 mm length of each fiber. The immunosensing fibers were then imaged and fluorescence quantified. F0 represents the fluorescence signal of fibers exposed to IL-1β in the absence of competing cytokines. F represents the fluorescence signal of fibers exposed to IL-1β in the presence of competing cytokines. F/ F0 represents the degree of 44 interference of IL-1β binding to the fiber sensor in the presence of alternate cytokines. F/ F0 = 1 indicating no interference and F/ F0 = 0 indicating complete interference. Data are presented as the mean + sem.
Fig. S3. ELISA detection of mature (17 kDa) and pro (31 kDa) rat IL-1β protein.(A) Mature (300 pg/ml) and pro (300 pg/ml) recombinant rat IL-1β protein were assayed using a standard rat IL-1β protein ELISA kit to determine specificity of the ELISA for either form of rat IL-1β. (B) Mature (25 pg/ml) and pro (25 pg/ml) recombinant rat IL-1β protein were assayed using a rat pro-IL-1β protein ELISA kit to determine specificity of the ELISA for either form of rat IL-1β. Data are presented as the mean + sem.