

Abstract
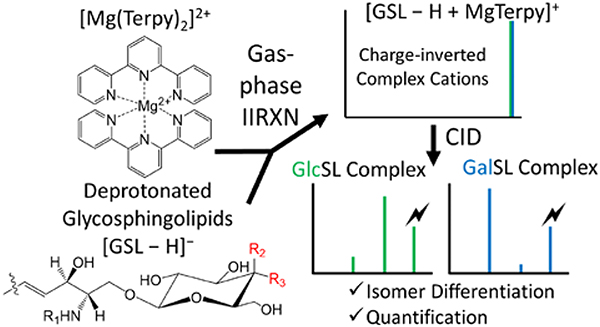
Glycosphingolipids (GSLs), including lyso-glycosphingolipids (lyso-GSLs) and cerebrosides (HexCer), constitute a sphingolipid subclass. The diastereomerism between their monosaccharide head groups, glucose and galactose in mammalian cells, gives rise to an analytical challenge in the differentiation of their biological roles in healthy and disease states. Shotgun tandem mass spectrometry has been demonstrated to be a powerful tool in lipidomics analysis, in which the differentiation of diastereomeric pairs of GSLs could be achieved with offline chemical modifications. However, the limited number of standards, as well as the lack in comprehensive coverage of the GSLs, complicates the qualitative and quantitative analysis of GSLs. In this work, we describe a novel strategy that couples shotgun tandem mass spectrometry with gas-phase ion chemistry to achieve both differentiation and quantification of the diastereomeric pairs of GSLs. In brief, deprotonated GSL anions, [GSL − H]−, and terpyridine-magnesium complex dications, [Mg(Terpy)2]2+, are sequentially injected and mutually stored in a linear ion trap to form charge-inverted complex cations, [GSL – H + MgTerpy]+. Collision-induced dissociation of the charge-inverted complex cations leads to significant spectral differences between the diastereomeric pairs of GSLs, which permits their distinction. Moreover, we describe a relative quantification strategy with the normalized %Area extracted from selected diagnostic ions in binary mixtures. Analytical performance with the selected pure-component pairs, lyso-GSLs and HexCer(d18:1/18:0), were also evaluated in terms of accuracy, repeatability, and inter-day precision. The pure-components could be extended to different fatty acyl chains on cerebrosides with limited error, which allows for the relative quantitation of the diastereomeric pairs without all standards. We successfully applied the presented method to identify and quantify, on a relative basis, the GSLs in commercially available total cerebroside extracts from the porcine brain.
Keywords: glycosphingolipids, tandem mass spectrometry, charge inversion, ion/ion reactions
Graphical Abstract
Introduction
Glycosphingolipids (GSLs), including lyso-glycosphingolipids (lyso-GSLs) and cerebrosides (HexCer), constitute a sphingolipid subclass. In general, lyso-GSL structures consist of a sphingosine moiety linked to a glycan head group while cerebrosides have a single sugar residue and an esterified fatty acyl side chain linked to the sphingoid base, as depicted in Figure 1.1 In mammalian cells, glucose or galactose is the monosaccharide head group linked to the sphingosine backbone via a beta-glycosidic bond.2 The only difference between the diastereomeric pairs of GSLs is the orientation of the hydroxyl group (OH) at the C4’-position on the monosaccharide. The GSL subclass glucosylsphingolipid (GlcSL) has an equatorial OH at the C4’ position whereas galactosylsphingolipid (GalSL) has an axial OH at the C4’ position (Figure 1). As many analytical approaches do not achieve isomeric resolution, this isomeric pair is usually reported as a single species.3,4 However, failing to unambiguously identify these isomers results not only in an underestimation of lipid diversity, but also precludes understanding of their individual roles in biological functions and disease progression.
Figure 1.
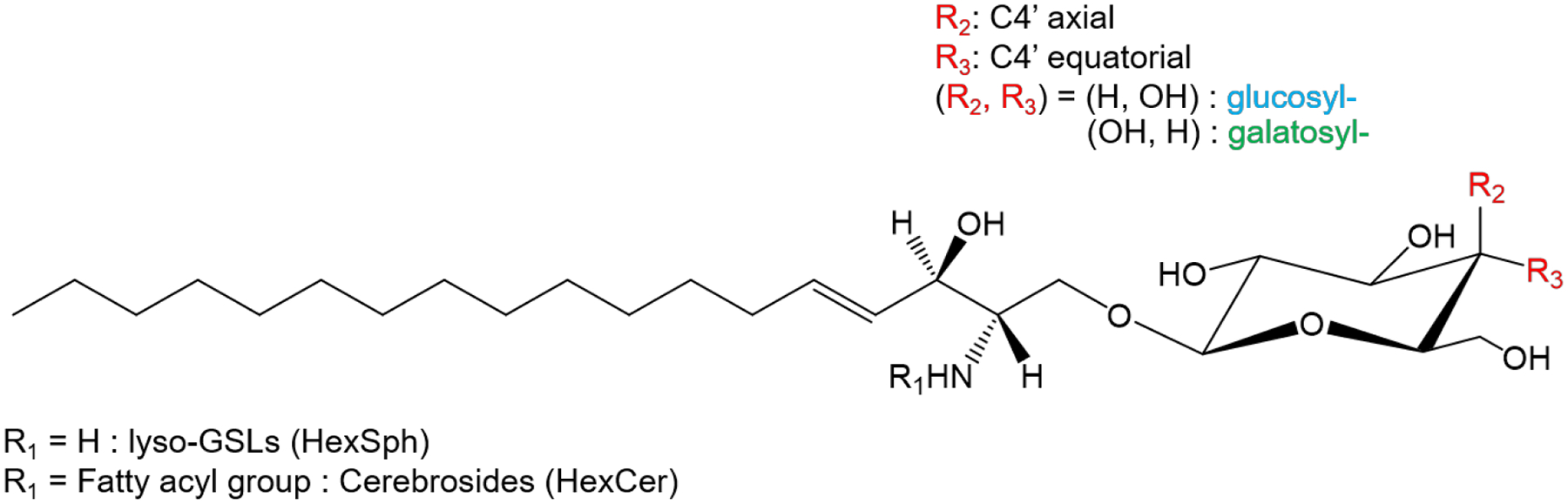
The structure of glycosphingolipids (GSLs).
Gaucher disease is a disorder arising from a mutation in the human GBA gene that causes a deficiency of the glucocerebrosidase (or glucosylceramidase, GCase) enzyme leading to abnormal glucosylceramide (GlcCer) accumulation in the brain.5 In another example, Parkinson’s disease (PD) is also associated with the metabolic pathway of GlcCers in which higher levels of GlcCers in the substantia nigra have been found in PD patients.6 These examples correlate GlcCer levels with neuron dysfunction. However, cerebrosides are also reported to have neuroprotective activity such that increasing cerebroside levels by altering the de novo synthesis pathway of sphingolipids leads to deactivation of downstream apoptosis signaling pathways.7,8 These results of different cerebroside levels reflect an incomplete understanding of the physiological roles of cerebrosides. A more complete understanding of this sphingolipid subclass will likely require both differentiation and quantification of the diastereomers in biological milieu.
Currently, the primary strategy to analyze GSLs utilizes liquid chromatography coupled with mass spectrometry (LC-MS). Normal phase chromatography (NPLC)9 or hydrophilic interaction chromatography (HILIC)10 can separate GlcSL and GalSL diastereomeric pairs. Li et al. developed a platform using 2D NP/RP-LC-MS to analyze GlcSLs and GalSLs that is able to separate both the monosaccharide headgroups and the length of fatty acyl chains.11 However, the 2D separation strategy involves lengthy analysis times (up to hours), pointing to the need for more rapid analytical tools for GSL analysis.
Shotgun analysis via direct electrospray ionization tandem mass spectrometry (ESI-MS/MS) has become a popular tool for lipid analysis due to ease of use, lower sample volume, and decreased analysis time requirements.3 Despite many successes, shotgun approaches often struggle to provide detailed structural information, including the differentiation of GlcSLs and GalSLs. Recently, offline chemical derivatization strategies have helped overcome these difficulties. Leary et al., for example, used metal-ligand complexes to coordinate with the monosaccharides to differentiate the diastereomers.12 Coupled with ion trap tandem mass spectrometry, N-acetylglucosamine and N-acetylgalactosamine could be quantitated by virtue of the distinct product ion relative abundances formed from the isomeric complexes upon activation.13,14 Julian and co-workers reported a strategy using photodissociation (PD) to initiate radical chemistry to differentiate diastereomeric pairs of lyso-GSLs. They used 4-iodobenzoyl 18-crown-6 (IB18C6) to form complexes with the lyso-GSLs followed by PD to generate a radical cation. With further collisional activation, differences in the ratios of relative abundances from the diagnostic product ions derived from the diastereomers were reported.15 Moreover, they also reported a derivatization method using 4-iodophenyl boronic acid (IPBA), which selectively binds to GalCer because of the C4’ axial OH, to separate the diastereomers in solution. Both methods were effective in distinguishing the diastereomers within the context of shotgun analysis. However, offline derivatization steps, as well as post-derivatization sample treatment steps, are time consuming and can lead to low abundances of derivatized products.
Chloride or lithium adduction are alternative approaches to achieve lipid structural elucidation by doping salts into lipid ESI solutions. For example, Han and co-workers developed a strategy in which chloride adducted cerebrosides anions were exploited to differentiate the diastereomeric pairs of cerebrosides (HexCer).16 Briefly, collision-induced dissociation (CID) of the [HexCer + Cl]‾ adduct ion generated different ratios of product ions from GlcCer and GalCer, in which the inner-sugar fragmentation was slightly greater from CID of the [GlcCer + Cl]− ion. The same group also proposed a multidimensional strategy to quantify GalCer in the sample using internal standard addition.16,17 However, chloride adduction can only differentiate between GlcCer and GalCer, but not lyso-GSLs. Furthermore, the method relies on multidimensional operation, which involves multiple salt additions, and can quantify only GalCer, but not GlcCer.
Gas-phase ion/ion chemistry has been shown to be effective in modifying lipid analytes directly within the mass spectrometer. Due to the attractive Coulombic potential associated with the reactions of oppositely charged ions, reaction rates of 10–100 s−1 are typical under readily accessible conditions in quadrupole ion traps.18,19 Recently, our group has described charge inversion ion/ion strategies to facilitate lipid identification and relative quantitation.20,21,22,23 For example, fatty acid (FA) anions derived from non-esterified FAs or released from complex lipid precursors, are reacted in the gas-phase with tris-phenanthroline magnesium complex dications to generate [FA – H + MgPhen]⁺ product ions. Ensuing dissociation of the charge inverted FA complex cation gives rise to spectral patterns that facilitate unambiguous isomeric discrimination and FA identification.20 Additionally, this charge inversion strategy, in combination with a multiple linear regression approach, enables relative quantitation of FA isomers.21 In this study, we describe a shotgun mass spectrometry strategy using ion chemistry to chemically modify GSLs in the gas phase to differentiate and quantify the diastereomeric pairs of GSLs in the samples.
Experimental Section
Materials.
All lipid standards and total cerebrosides extract (porcine brain) were purchased from Avanti Polar Lipids, Inc (Alabaster, AL). Magnesium chloride (MgCl2), 2,2’;6’,2”-terpyridine (Terpy), and 4’-chloro-2,2’;6’,2”-terpyridine (Terpy-Cl) were purchased from Sigma-Aldrich (St. Louis, MO). MS-grade water and methanol (MeOH) were purchased from Fisher Scientific (Pittsburgh, PA).
Sample Preparation.
Solutions of GSL standards were prepared in methanol to a final concentration of 0.01 mg mL−1. Magnesium chloride and 2,2’;6’,2”-terpyridine (Terpy) were mixed in methanolic solution with 1:1 (molar ratio) to a final concentration of ~50 μM for the metal-ligand complex.20 For relative quantification, different ratios of GlcSL/GalSL were prepared, holding the final GSL concentration at 0.01 mg mL−1. For total cerebrosides extract analysis, 1 mg of purified extract powder was dissolved in 1 mL of methanol as the stock solution and stored at −20°C before use. Prior to analysis, the lipid extract was diluted to a final concentration of 0.01 mg mL−1.
Mass Spectrometry.
All experiments were performed on a TripleTOF 5600 quadrupole time-of-flight mass spectrometer (SCIEX, Concord, ON, Canada) that has been modified for ion/ion reactions.24 Alternately pulsed dual nano-electrospray ionization (nESI) allows for the sequential injection of anions and cations.25 GSL anions, denoted [GSL − H]−, were ionized and isolated in Q1, and then accumulated in the high-pressure collision cell q2. Next, metal-ligand reagent dications, referred to as [Mg(Terpy)2]2+ were generated via direct positive nESI, mass-selected in Q1, and transferred to q2 to for storage. Once in q2, the lipid anions and reagent dications were mutually stored for 10–30 ms, undergoing a charge inversion ion/ion reaction. Charge-inverted complex cations were subjected to MSn experiments. Mass analysis was performed via orthogonal acceleration time-of-flight (TOF).
Analytical Performance Evaluation.
The analytical performance was evaluated for accuracy, repeatability, and inter-day precision. Three samples each differing in the ratios of concentrations from both the GlcSph/GalSph and GlcCer(d18:1/18:0)/GalCer(d18:1/18:0) pairs, (i.e., 90/10, 50/50, and 10/90) with a total GSL concentration of 0.01 mg mL−1 were prepared. Note that for each ratio, three individual sample replicates were prepared in order to evaluate repeatability and accuracy. The accuracy was calculated based on the following equation 1:
| (eq. 1) |
The repeatability was expressed by the standard deviation of the calculated accuracy (n = 3). For inter-day precision, the individually prepared samples at different concentration ratios (n = 3) were analyzed on three non-consecutive days. Inter-day precision was calculated using equation 2:
| (eq.2) |
Absolute Quantification.
To achieve absolute quantification, the two different amounts of GlcCer (d18:1/18:0) were spiked into separate aliquots of the total cerebrosides solution. For the high concentration spike, 1μL of 0.01 mg mL−1 of GlcCer(d18:1/18:0) was added to a 99 μL aliquot of the cerebrosides solution. For the low concentration spike, a total of 1μL of 0.001 mg mL−1 of GlcCer(d18:1/18:0) was added to a 49 μL aliquot of the cerebrosides solution. Three replicates were used to calculate the percentage of the GSL pairs in the samples, and further back-calculated the absolute concentration in the total cerebrosides extracts using the known spiked amount of GlcCer(d18:1/18:0) by the following equation 3 and 4:
| (eq. 3) |
| (eq. 4) |
Where the GalCer and GlcCer were expressed in μg mL−1, and %GalCer and %GlcCer were the calculated percentages from the relative quantification.
Results and Discussion
Differentiation of lyso-GSLs via gas-phase ion chemistry.
As described above, diastereomeric GSL pairs differ only in the orientation of the hydroxyl (OH) group at the C4’-position of the monosaccharide. Specifically, glycosphingolipids (GlcSLs) exhibit equatorial OH orientation while galactosylsphingolipids (GalSLs) display axial orientation of the OH moiety (Figure 1). We first examined a diastereomeric pair of lyso-GSLs, (hexosylsphingosine, HexSph) and illustrate with glucosylsphinogsine (GlcSph) and galactosysphingosine (GalSph). As is commonly observed, the diastereomeric pair of lyso-GSLs show only limited differences in their fragmentation patterns from either protonated or deprotonated ions upon collision-induced dissociation (CID) (Figure S1).
Figure 2(a) shows the positive ion spectrum obtained following the reaction between [GlcSph − H]− analyte anions and [Mg(Terpy)2]2+ reagent dications, which yielded the charge-inverted complex cations, [GlcSph – H + MgTerpy]+(m/z 717.4). The reaction between [GalSph – H]− anion and [Mg(Terpy)2]2+ also yielded the charge-inverted complex cation, [GalSph – H + MgTerpy]+. The reactions showed no differences between the two diastereomers, so we labeled the charge-inverted complex cations as the generic label, [HexSph – H + MgTerpy]+ in Figure 2(a). Next, the charge-inverted lyso-GSL complex cation was mass-selected and subjected to ion trap collisional activation. Figures 2(b) and 2(c) show the CID spectra from the charge-inverted GlcSph and GalSph complex cations, respectively. Importantly, significant spectral differences were observed following interrogation of the two charge-inverted lyso-GSL complex cations. Specifically, interrogation of the [GlcSph – H + MgTerpy]+ ion resulted in a dominant neutral loss (NL) of the sugar moiety (m/z 555.3, NL = 162 Da), generating a Y-type ion using the widely used carbohydrate fragment nomenclature.26 A Z-type ion is also observed at much lower abundance corresponding to a NL = 180 Da. In contrast, the CID spectrum of [GalSph – H + MgTerpy]+ shows a dominant product ion corresponding to a neutral loss of a Terpy ligand, NL = 233 Da (m/z 484.3), along with an ion at an apparent NL = 215 Da (m/z 502.3) that arises from the attachment of a water molecule in the collision cell to the NL = 233 Da loss product. Previous work with divalent alkaline earth metal complexes has shown water attachment to occur in the gas phase subsequent to ligand loss.20 Ultimately, these spectral differences permit distinction between the GalSph and GlcSph components of the diastereomeric pair. (We note here that the product ions show the expected isotope distributions and that all of our CID experiments for quantitation (discussed below) involved activation of all isotopes to completion (i.e., the precursor population was fully depleted). Only the monoisotopic product ions, however, were used for quantification.)
Figure 2.
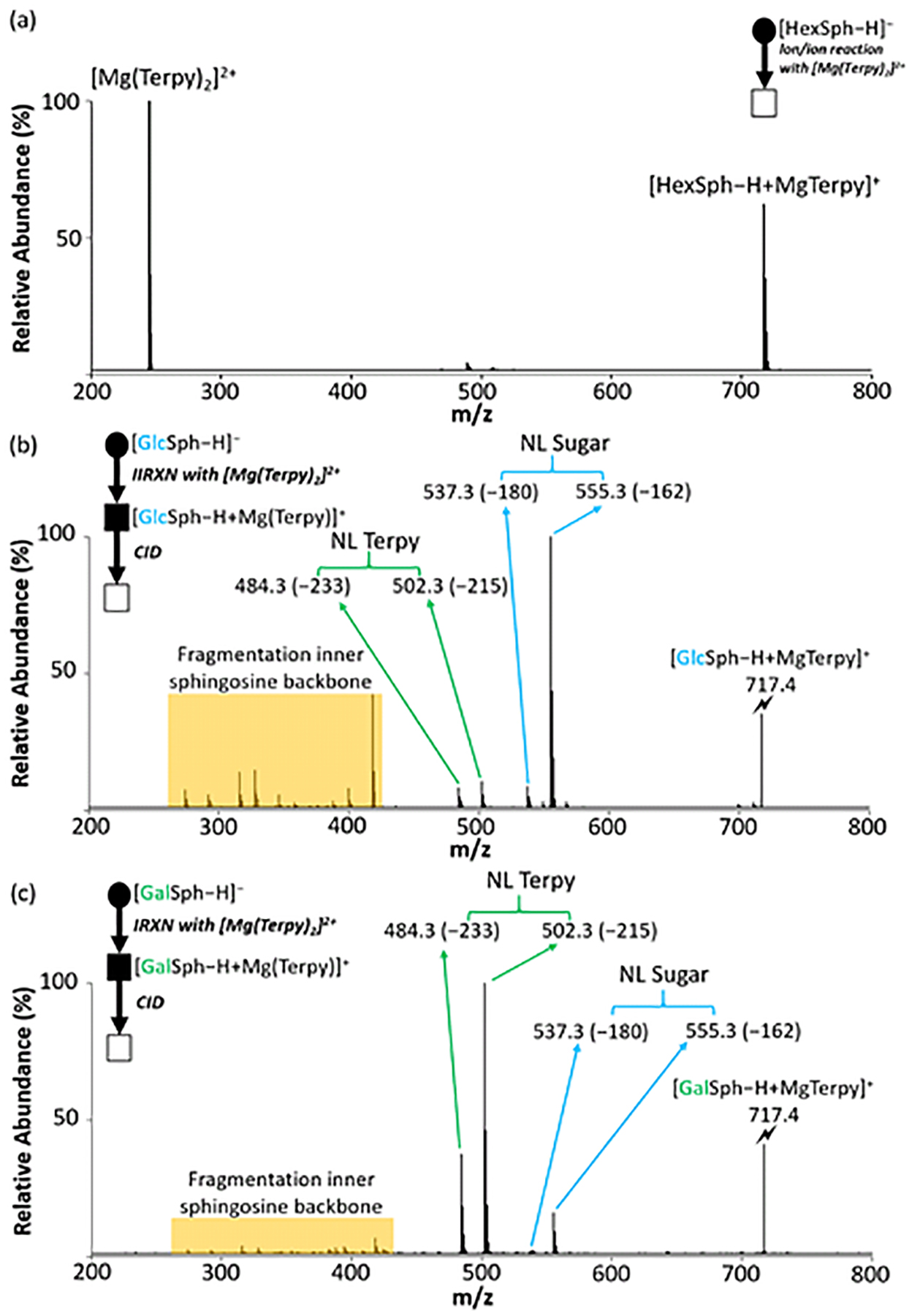
The comparison of the CID spectra between lyso-GSLs after gas-phase ion/ion reaction. (a) The post-ion/ion reaction spectrum of lyso-GSLs anion with Mg(Terpy)2 cation. (b) The CID spectrum of the [GlcSph – H + MgTerpy]+ (m/z 717.4). (c) The CID spectrum of the [GalSph – H + MgTerpy]+ (m/z 717.4). The values inside the parenthesis indicate the neutral loss. The lightning bolt (⚡) signifies collisionally activated precursor ion. The solid circle (●) indicates the mass selection in the negative ion mode analysis and the black and white squares (■/□) indicate the positive ion mode analysis with and without mass selection, respectively.
We have not performed a detailed mechanistic study to determine the origin(s) of the different dissociation behaviors of the [HexSph – H + MgTerpy]+ ions. However, the differences in fragmentation patterns observed following CID of the charge-inverted lyso-GSL complex cations presumably reflect the nature of the interactions of the [Mg(Terpy)2]2+ cation with the lipids. In both cases, upon attachment of the [Mg(Terpy)2]2+ reagent a single Terpy ligand is lost, presumably due to displacement of one of the Terpy ligands by the lipid.20 On the other hand, the loss of the second Terpy ligand is highly dependent on the stereochemistry of the sugar. The [GalSph – H + MgTerpy]+ ion, for example, predominantly loses the second Terpy ligand upon CID, which likely reflects a greater degree of stabilization of the Mg2+ ion than is afforded by the GlcSph lipid. We speculate that the additional stabilization of the Mg2+ ion is afforded by deprotonation of one of the sugar hydroxyl groups at the C4’ or C3’ positions with additional coordination by the hydroxyl group at the C4’ or C3’ site that is not deprotonated (see Scheme 1).27,28 This additional interaction with the Mg2+ ion can facilitate the loss of the second Terpy ligand. In contrast, the glucose head group in GlcSph apparently does not interact with MgTerpy2+ in the same way as the galactose head group in GalSph such that an alternate pathway (i.e., sugar loss) dominates. The facile loss of the neutral sugar indicates that the deprotonation site either originates or can migrate to the sphingosine chain as part of the sugar loss channel.
Scheme 1.
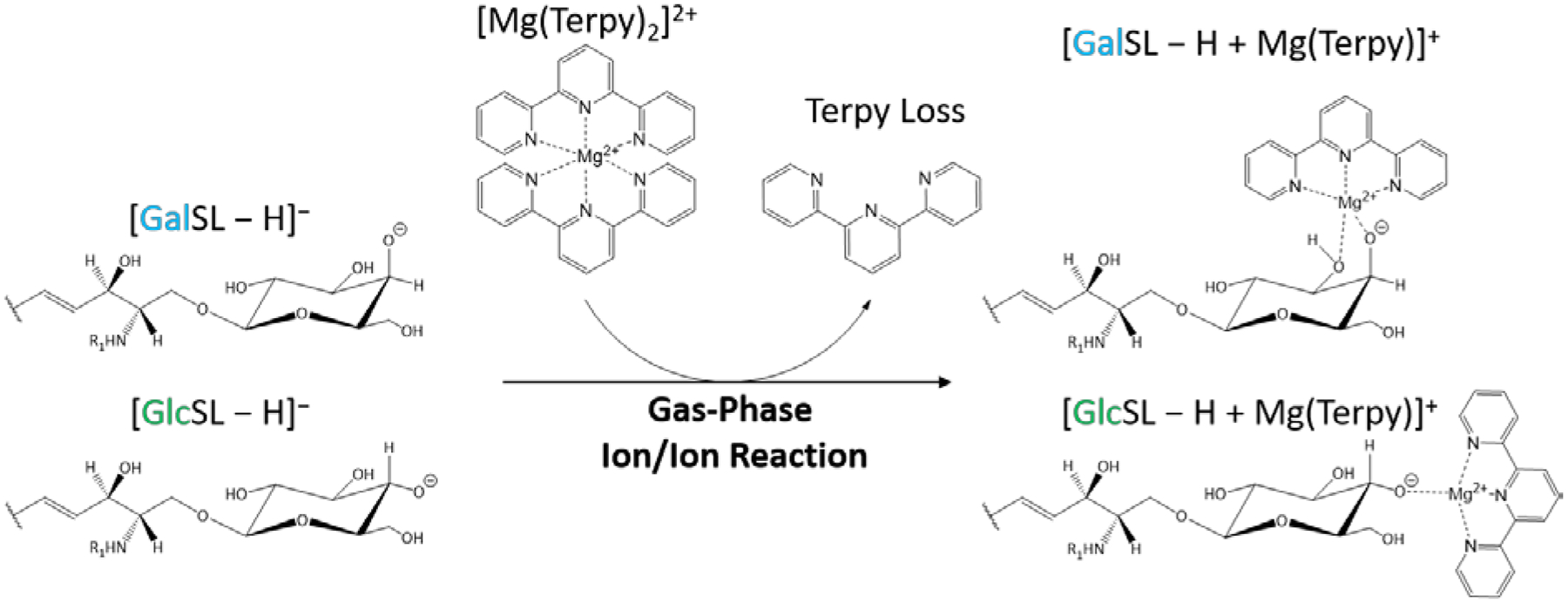
The gas-phase ion/ion reaction between deprotonated GSLs and [Mg(Terpy)2]2+. Note that the multiple structures that solvate the Mg2+ ion can contribute. However, the mixture of structures differs for the two diastereomers.
Differentiation of cerebrosides via gas-phase ion chemistry.
To test the ability of the gas-phase ion/ion reaction in differentiation of cerebrosides (HexCer), we performed the same reaction described above with deprotonated HexCer, [HexCer − H]−. Again, the CID spectra of protonated and deprotonated diastereomeric ions show little or no differences in product ion masses and relative abundances (Figure S2). After the reactions between [GlcCer(d18:1/18:0) − H]− or [GalCer(d18:1/18:0) − H]− with [Mg(Terpy)2]2+, charge-inverted complex cations [GlcCer(d18:1/18:0) – H + MgTerpy]+ or [GalCer(d18:1/18:0) – H + MgTerpy]+ were observed (m/z 983.6). The fragmentation patterns of charge-inverted HexCer complex cations generated by CID are similar but not identical, to those of the analogous charge-inverted lyso-GSLs complex cations (Figure 3). The CID spectrum of [GalCer(d18:1/18:0) – H + MgTerpy]+ (m/z 983.6) is dominated by the neutral loss of Terpy (m/z 750.6, NL = 233 Da) (Figure 3(b)), which is similar to the behavior of the [GalSph − H + MgTerpy]+, with the exception that the addition of a water molecule to the Terpy loss product is far more prominent in the latter case (Figure 2(b)). The [GalCer(d18:1/18:0) – H + Mg]+ product ion clearly solvates the magnesium dication more effectively than the corresponding [GalSph − H + MgTerpy]+ product ion. The CID spectrum of [GlcCer(d18:1/18:0) – H + MgTerpy]+, on the other hand, not only shows a neutral loss of the sugar moiety (m/z 821.6, NL =162 Da), but also shows major neutral losses of water (m/z 965.6, NL = 18 Da) and 443 Da (m/z 540.3).
Figure 3.
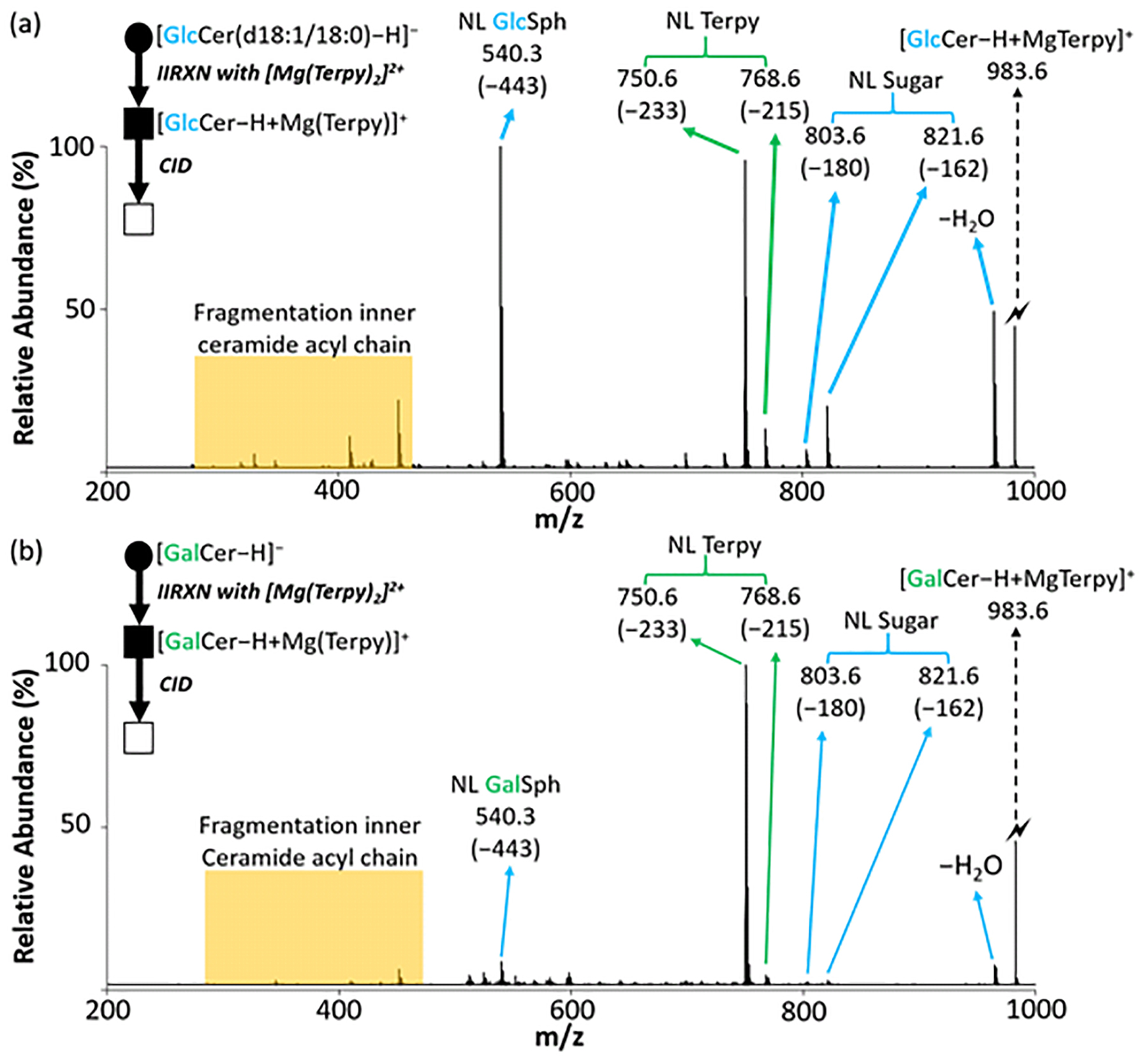
The comparison of the CID spectra between cerebrosides after gas-phase ion/ion reaction. (a) The CID spectrum of the [GlcCer(d18:1/18:0) – H + MgTerpy]+ complex (m/z 983.6) generated via gas-phase ion/ion reaction between singly deprotonated GlcCer anion and [Mg(Terpy)2]2+. (b) The CID spectrum of the [GalCer(d18:1/18:0) – H + MgTerpy]+ complex (m/z 983.6) generated via gas-phase ion/ion reaction between singly deprotonated GalCer anion and [Mg(Terpy)2]2+. The values inside the parenthesis indicate the neutral loss. The lightning bolt (⚡) signifies collisionally activated precursor ion. The solid circle (●) indicates the mass selection in the negative ion mode analysis and the black and white squares (■/□) indicate the positive ion mode analysis with and without mass selection, respectively.
To clarify the origin of the 443 Da loss from the charge-inverted complex after CID, we further applied the same reaction to GlcCer(d18:1/16:0) and a soy-based cerebroside, GlcCer(d18:2/h16:0) (Figure S3(a)), and compared the results. Figures S3(b) and S3(c) show the CID spectra when we subjected the charge-inverted GSL complex cations to CID. A 443 Da loss is observed in the CID spectrum from the [GlcCer(d18:1/16:0) – H + Mg(Terpy)]2+ complex (m/z 955.6), whereas a 441 Da loss is observed from the [GlcCer(d18:2/h16:0) – H + Mg(Terpy)]2+ complex (m/z 969.5). This result suggests that the 443 Da loss from the charge-inverted GSL complex cations described above includes the sphingosine backbone.
We further used MgTerpy-4’-Cl as the ligand for the charge inversion reagent ([Mg(Terpy-Cl)2]2+ (Figure S4 (a)) in reaction with all three glycosylceramides, GlcCer(d18:1/18:0), GlcCer(d18:1/16:0) and GlcCer(d18:2/h16:0) followed by CID of the charge-inverted complex cations. The results show that 443 Da losses from both [GlcCer(d18:1/18:0) – H + Mg(Terpy-Cl)]2+ and [GlcCer(d18:1/16:0) – H + Mg(Terpy-Cl)]2+ are still observed as well as a 441 Da loss from [GlcCer(d18:2/h16:0) – H + Mg(Terpy-Cl)]2+ (Figure S4). This result strongly suggests that the loss of 443 Da does not include the loss of the ligand and that MgTerpy is retained in the product ion. Additionally, we performed MS3 of the 233 Da loss product (the product ion at m/z 750.6 in Figure 3(a)), and it did not show evidence for the loss of 210 Da (Figure S5), which further supports the conclusion that the 443 Da loss described above does not arise from losses of the Terpy ligand and another fragment of mass 210 Da. A previous report suggested a mechanism for losing a hydrocarbon chain from a deprotonated ceramide by various bond cleavages, including cleavage of the N-C bond.29 Therefore, weperformed an MS3 experiment by subjecting the sugar loss ion (m/z 821.6 in Figure 3(a)) to CID, and the m/z 540.3 ion is observed (Figure S6). Hence, the 443 Da loss is comprised of sugar loss (162 Da) and a loss of 281 Da. Furthermore, a 443 Da loss was observed in the CID spectra from both deprotonated cerebrosides (Figure S2(b) and Figure S2(d)). Therefore, the 443 Da loss is most likely to be the sequential cleavages of the glycosidic bond and the N-C bond, with the MgTerpy coordinated with the amide. However, further studies into the detailed mechanism for the net loss of 443 Da from [HexCer(d18:1/18:0) – H + MgTerpy]+ are needed to confirm the product ion structure.
The relative quantification of the diastereomeric pairs of GSLs.
Due to extensive lipid structural diversity, quantification has remained a significant challenge in shotgun lipidomics. The most common strategy to quantify lipids involves preparation of a calibration curve. However, this approach requires the preparation of external calibration standards, which are not always commercially available. Furthermore, calibration curve construction is sample-, time-, and labor-intensive. We developed a quantification strategy exploiting pure component analysis, in which only limited standards are required to achieve relative quantification.
To do so, we first generated the charge-inverted lyso-GSL complex cations in an identical fashion as described above (Figure 2(a)). Next, the charged-inverted complex cations were subjected to ion-trap CID, noting that the entire precursor cation population was fully dissociated. We chose categories of diagnostic product ions mentioned in the previous section, viz. the two major fragments associated with Terpy loss (NL = 233 Da and NL = 215 Da, NL 233 + NL 215) and the ion associated with the loss of the sugar (NL = 162 Da, NL 162). Various molar ratios of the diastereomeric pair of lyso-GSLs were prepared and analyzed using the outlined approach. Specifically, we used GlcSph/GalSph binary mixtures with the ratios 90/10, 80/20, 60/40, 50/50, 40/60, 20/80, and 10/90. The CID spectra of the fully dissociated precursor cation mixtures are provided in Figure S7 and show monotonic changes in relative abundances of the diagnostic product ions as the fractions of GlcSph and GalSph decrease and increase, respectively.
We further extracted areas of the monoisotopic peak from the diagnostic product ions from the pure-component CID results (100% GlcSph and 100% GalSph) for relative quantification. The areas were normalized to the total area from the extracted peaks and expressed as %Area (%A). Table 1 shows the normalized %A of the two groups of product ions from nine replicates (three replicates per day for three days). The %A are placed in the following equations to calculate the percentage of lyso-GSLs (%GlcSph and %GalSph) in unknown samples:
| (eq. 5) |
| (eq. 6) |
Table 1.
The normalized %area for quantifying lyso-GSLs and the analytical performance of lyso-GSLs quantification.
| Normalized %Area for lyso-Glycosphingolipids (HexSph) (N=9) | |||
|---|---|---|---|
| 100% of lyso-GSLs | NL of Terpy (%, NL 215 + NL 233) | NL of sugar (%, NL 162) | SD |
| GlcSph | 15.7 | 84.3 | 1.4 |
| GalSph | 90.5 | 9.5 | 0.6 |
| Analytical Performance | ||||||
|---|---|---|---|---|---|---|
| %GlcSph/%GlcSph | Avg_Cal %GlcSph | Accuracy (%)* | Inter-day Precision (RSD%)** | Avg_Cal %GalSph | Accuracy (%)* | Inter-day Precision (RSD%)** |
| 90/10 | 90.1 | 100.0±0.0 | 0.7 | 9.9 | 98.9±4.7 | 6.3 |
| 50/50 | 52.1 | 104.1±1.2 | 2.7 | 48.4 | 96.8±1.1 | 2.9 |
| 10/90 | 9.8 | 97.7±11.7 | 12.5 | 90.2 | 100.3±1.3 | 1.4 |
Mean±SD, N=3
Relative standard deviation, calculated from three different days with 9 different samples.
Three different molar ratios of GlcSph/GalSph at 90/10, 50/50, and 10/90, were chosen to demonstrate the approach as well as to evaluate the analytical performance in terms of accuracy, repeatability, and inter-day precision. The normalized %A of the product ions from different NL groups were obtained as detected %A and was input into eq. 5 and eq. 6. The bottom panel from Table 1 summarizes the relative quantification results with the analytical performance. The accuracies for relative quantification of the GlcSph and GalSph diastereomeric pair ranged from 96.8 to 104.1% from different molar ratios, with the highest SD around 11.7%. The results suggest that relative quantification is achieved applying the strategy. Moreover, the inter-day precisions of the lyso-GSLs results are all below 12.5%RSD, which suggests that the relative quantification results obtained from different days were still comparable. Correlation curves between the calculated % and the spiked % are also shown in Figure S8. We note that the inter-day precision suggests that the use of the sum of the abundances of the NL = 233 Da and NL = 215 Da signals minimizes sensitivity of this approach to quantification to changes in water levels in the collision cell.
A similar strategy was applied to the cerebroside diastereomeric pair GlcCer(d18:1/18:0) and GalCer(d18:1/18:0). The diagnostic product ions were chosen based on the previous section, and also divided into two groups, NL of Terpy (NL 233 + NL 215) versus NL of water (NL =18 Da, NL 18), NL of sugar (NL 162), and NL of 443 Da (NL 443). The different molar ratios of GlcCer(d18:1/18:0)/GalCer(d18:1/18:0) were analyzed, and the changes in relative abundances of the diagnostic product ions from samples with different molar ratios were also observed (Figure S9). Again, the extracted areas of the monoisotopic peak from the diagnostic product ions were normalized to the total area from the extracted peaks and expressed as %A in the top panel from Table 2. Another set of equations to quantify the percentage of the cerebroside diastereomers are shown as follows:
| (eq. 7) |
| (eq. 8) |
Table 2.
The normalized %area for quantifying cerebrosides and the analytical performance of GSLs quantification
| Normalized %Area for cerebrosides (N=9) | |||
|---|---|---|---|
| 100% of GSLs | NL of Terpy (%, NL215 + NL233) | NL of others* (%, NL18 +N L162 + NL443) | SD |
| GlcCer(d18:1/18:0) | 39.4 | 60.6 | 1.2 |
| GalCer(d18:1/18:0) | 92.5 | 7.5 | 0.2 |
| Analytical Performance | ||||||
|---|---|---|---|---|---|---|
| %GlcCer/%GalCer | Avg_Cal %GlcCer | Accuracy (%)** | Inter-day Precision (RSD%)*** | Avg_Cal %GalCer | Accuracy (%)* | Inter-day Precision (RSD%)** |
| 90/10 | 89.9 | 99.8±0.4 | 1.0 | 10.1 | 101.4±3.9 | 6.8 |
| 50/50 | 49.3 | 98.6±2.1 | 2.2 | 50.7 | 101.4±0.9 | 1.4 |
| 10/90 | 9.3 | 92.6±4.3 | 6.7 | 90.7 | 100.8±0.5 | 0.6 |
NL of water, sugar, and Terpy with sphingosine backbone
Mean±SD, N=3
Relative standard deviation, calculated from three different days with 9 different samples
Three different molar ratios of GlcCer/GalCer were chosen to demonstrate the approach as well as to evaluate the analytical performance. The bottom panel from Table 2 summarizes the relative quantification results with the analytical performances. The accuracies for relatively quantifying the cerebroside diastereomeric pair were ranged from 92.6 to 101.4% with the highest SD around 6.7%, and the inter-day precisions are all below 6.8%RSD. Also, correlation curves between the calculated % and the spiked % are shown in Figure S10. All of the above results show the applicability of the strategy to achieve relative quantification of both lyso-GSLs and the cerebroside diastereomeric pairs, which only require the analysis of the two pure components prior to the quantification.
Lack of commercially available calibration standards is often a challenge in lipidomics analysis, especially for quantification. Therefore, we evaluated the use of the same pure components from a single pair of cerebroside diastereomers to perform the relative quantification for other cerebrosides, which vary only in the fatty acyl chain. This assumes that all charge-inverted cerebroside complex cations have similar fragmentation patterns after activation since none of the selected diagnostic product ions arise from the fatty acyl chain (R1 group in Figure 1). Two different pairs of cerebroside diastereomers, HexCer(d18:1/16:0) and HexCer(d18:1/18:1), at five different molar ratios between the diastereomers (GlcCer/GalCer), including 100/0, 80/20, 50/50, 20/80, and 0/100, were analyzed. The CID spectra of the fully dissociated precursor cations are shown in Figure S11, showing the same monotonic changes in the relative abundances of the diagnostic product ions as a function of molar ratio. The relative quantification is achieved by using the same %A obtained from pure components of HexCer(d18:1/18:0), and eq.7 and eq. 8 The relative quantification results are summarized in Table 3. The accuracies ranged from 96.5 to 113.0%, with the highest SD around 3.2% for the HexCer(d18:1/16:0) pair, and the accuracies ranged from 89.4 to 106.6%, with the highest SD around 8.4%, for HexCer(d18:1/18:1). To validate the results, we also performed the same approach using the pure-component %A obtaining from their own calibration standards. The relative quantification results are also reported (Table S1 and Table S2), and less than 6% error were observed when comparing the two data sets. However, it is noticeable that there is a greater difference in the normalized %A from the pure GlcCer(d18:1/18:1) relative to GlcCer(d18:1/18:0) (i.e., 35.9% (Table S2) vs. 39.4% (Table 1)) than GlcCer(d18:1/16:0) relative to GlcCer(d18:1/18:0) (i.e., 39.9% (Table S1) vs. 39.4% (Table 1)). This suggests that unsaturation in the fatty acyl chain could give rise to slight differences in product ion abundances relative to a saturated fatty acyl chain.30 More studies would be required to establish a firm correlation, however. Overall, the relative quantitation for different fatty acyl chain GSLs using the %area from HexCer(d18:1/18:0) pair is demonstrated.
Table 3.
The relative quantification results of different acyl chains on GSLs with the constants from HexCer(d18:1/18:0).
| HexCer(d18:1/16:0) | ||||
|---|---|---|---|---|
| %GlcCer/%GalCer | Avg_Cal %GlcCer | Accuracy (%)* | Avg_Cal %GalCer | Accuracy (%)* |
| 100/0 | 98.9 | 98.9±0.2 | 1.1 | Not applicable |
| 80/20 | 80.1 | 100.2±0.6 | 19.7 | 98.7±0.8 |
| 50/50 | 49.6 | 99.1±0.4 | 50.4 | 100.9±0.4 |
| 20/80 | 22.6 | 113.0±3.2 | 77.2 | 96.5±2.1 |
| 0/100 | 1.8 | Not applicable | 98.2 | 98.1±0.5 |
| HexCer(d18:1/18:1) | ||||
| 100/0 | 106.4 | 106.4±0.1 | < 0 | Not applicable |
| 80/20 | 82.1 | 102.7±2.1 | 17.9 | 89.4±1.4 |
| 50/50 | 53.3 | 106.6±1.9 | 46.5 | 93.0±1.9 |
| 20/80 | 20.5 | 102.3±5.8 | 79.5 | 99.4±8.4 |
| 0/100 | 2.5 | Not applicable | 97.5 | 97.5±0.2 |
Mean±SD
Analysis of total cerebrosides extracts.
We examined commercially available total cerebroside extracts from porcine brain using the approaches described above. We identified a total of 11 m/z values that correspond to different sizes of cerebrosides common in mammalian systems.31,32 To differentiate the diastereomeric pairs that might be present in the putative cerebrosides, we exploited the strategy described above. Due to the lack of commercially available standards mentioned above, we were able to perform relative quantification on the diastereomeric pairs of cerebrosides using only our evaluated quantification %Area from HexCer(d18:1/18:0) pair. The CID spectra of the fully dissociated precursor ions after all reactions are shown in Figure S12. After the relative quantification, a total of 14 cerebrosides were identified and relatively quantified from the 11 m/z values expected to arise from cerebrosides. The results are reported in Table 4. Most of the cerebrosides that we detected in the porcine brain are GalCers with only three minor components within the diastereomeric pairs, including GlcCer(d18:1/16:0), GlcCer(d18:1/20:0), and GlcCer(d18:1/22:0). The results agree with previous reports showing that the majority of the cerebrosides in mammalian brain are galactosylsphingolipid.5,16,33,34
Table 4.
The relative quantitation results of the profiled cerebrosides from porcine brain extract. (N=3)
| HexCer | Avg Cal_Glc (%) | Avg Cal_Gal (%) | SD |
|---|---|---|---|
| d18:1/14:0 | ND | 99.7 | 0.9 |
| d18:1/16:0 | 10.9 | 89.1 | 0.8 |
| d18:1/18:1 | ND | 106.7 | 2.0 |
| d18:1/18:0 | ND | 99.5 | 1.9 |
| d18:1/20:0 | 1.3 | 98.7 | 0.5 |
| d18:1/22:0 | 0.9 | 99.1 | 0.2 |
| d18:1/23:0 | ND | 100.3 | 0.0 |
| d18:1/24:1 | ND | 102.0 | 0.5 |
| d18:1/24:0 | ND | 100.8 | 1.5 |
| d18:1/26:1 | ND | 100.7 | 0.5 |
| d18:1/26:0 | ND | 101.9 | 0.5 |
ND: non-detectable, indicated the calculated percentages ≤ 0, or ≤ SD.
In the event that a standard is available, such as the case for GlcCer(d18:1/18:0), a measurement of absolute concentration is possible. We spiked two aliquots of the sample with different known amounts of GlcCer(d18:1/18:0). Figure 4 shows the CID spectra of the pre- and post-spiked samples. The changes in the relative abundances of the diagnostic product ions after spiking the standard were observed. The different percentages of GlcCer(d18:1/18:0) and GalCer(d18:1/18:0) from the different spiked samples were obtained using the relative quantitation described above. Due to the fact that we did not detect any GlcCer(d18:1/18:0) in the total cerebrosides extract, we can modify eq. 3 and eq. 4 to yield the absolute amount of GalCer(d18:1/18:0) in the sample by equations 9 and 10:
| (eq. 9) |
| (eq. 10) |
Figure 4.
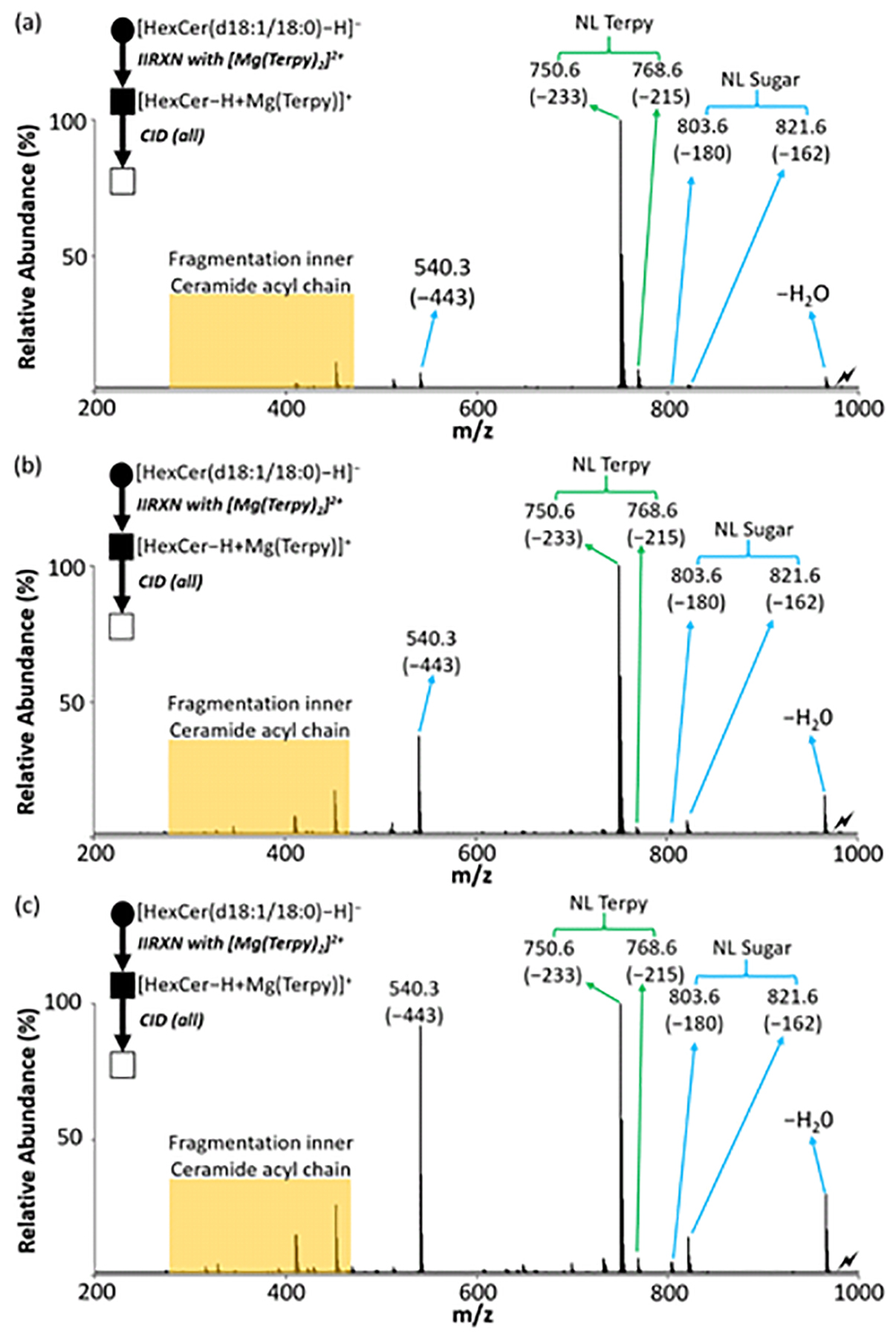
The single standard spiking strategy for absolute quantitation of GSLs in total brain extract. (a) The CID spectrum of the [HexCer(d18:1/18:0) – H + MgTerpy]+ complex (m/z 983.6) without spiking any standard. (b) The CID spectrum of the [HexCer(d18:1/18:0) – H + MgTerpy]+ complex with spiking low concentration of GlcCer(d18:1/18:0). (c) The CID spectrum of the [HexCer(d18:1/18:0) – H + MgTerpy]+ complex with spiking high concentration of GlcCer(d18:1/18:0). The values insides the parenthesis indicate the neutral loss. The lightning bolt (⚡) signifies the location of the fully depleted precursor ion. The solid circle (●) indicates the mass selection in the negative ion mode analysis and the black and white squares (■/□) indicate the positive ion mode analysis with and without mass selection, respectively.
The absolute quantitation results from both high concentration and low concentration spiked samples are shown in Table 5. Normally, spiking two different concentrations in the same sample is needed for applying eq. 3 and eq. 4 if both the cerebroside diastereomers exist in the sample. The mean calculated concentration of GalCer(d18:1/18:0) is 1.90±0.13 μg per mg extract powder. Here we also compared the two results from both high-spiked and low-spiked samples, which are each within one standard deviation of the mean. The primary benefit of this approach is that the absolute quantification of cerebroside diastereomers is achieved using addition of a single calibration standard comprised of only one of the diastereomers. Moreover, no external calibration curve is required, leading to reduced analysis time and cost. However, as with many lipidomics approaches, the lack of commercially available standards presents several challenges, including absolute quantification of GSLs in complex mixtures using the developed platform.
Table 5.
The summary table of the quantitation results from both low concentration and high concentration spiking test (N=3).
| Avg_Cal GalCer(d18:1/18:0) (μg mg−1) | SD | |
|---|---|---|
| High spiked | 1.98 | 0.10 |
| Low spiked | 1.82 | 0.08 |
| Avg (N=6) | 1.90 | 0.13 |
Conclusions
The differentiation of the diastereomeric glycosphingolipids is often a challenge, but essential for defining their biological roles in various diseases. We present a shotgun tandem mass spectrometry strategy using gas-phase ion chemistry to achieve the identification in both lyso-GSLs and cerebrosides without offline chemical derivatization. The gas-phase ion/ion reaction between the deprotonated GSLs ([GSL − H]−) and [Mg(Terpy)2]2+ forming charge-inverted cations, [GSL – H + Terpy]+, followed by collision-induced dissociation yield distinctive product ion spectra for the diastereomers. Moreover, relative quantification is achieved by analyzing the normalized %Area from the diagnostic product ions. The analytical performance of the relative quantification of both lyso-GSL and cerebroside diastereomeric pairs are also evaluated in terms of accuracy, repeatability, and inter-day precision. We also extended the strategy using the %Area from the validated cerebroside diastereomers, GlcCer(d18:1/18:0) and GalCer(d18:1/18:0), to provide relative quantification of other cerebrosides with different fatty acyl chains.
We extended the strategy to the analysis of total cerebroside extracts. A total of 14 cerebrosides were identified and quantified based on of their percentages within the diastereomeric pairs. This work also demonstrated an absolute quantification strategy for a cerebroside component in the total cerebroside extract from the porcine brain. By spiking two different sample solutions with known amounts of GlcCer(d18:1/18:0), the quantity of GalCer(d18:1/18:0) was obtained. The proposed absolute quantification method requires a calibration standard for only one of the diastereomers within the diastereomeric pair, and also with only two spiked concentrations, which reduces the required of numbers of calibration standards and obviates the generation of a calibration curve. However, the lack of a calibration standard from each diastereomeric pair prevents the absolute quantification of all cerebrosides.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NIH) under Grants GM R37–45372 and GM R01-118484. Dr. Elissia T. Franklin, Ms. Caitlin E. Randolph, and Mr. John T. Lawler are acknowledged for helpful discussions.
Footnotes
References
- 1.Merrill AH, Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chem. Rev 2011, 111 (10), 6387–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schnaar RL; Kinoshita T, Glycosphingolipids In Essentials of Glycobiology [Internet], 3rd edition ed.; Varki A, Ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, 2017. [Google Scholar]
- 3.Han X; Gross RW, Shotgun lipidomics: Electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom. Rev 2005, 24 (3), 367–412. [DOI] [PubMed] [Google Scholar]
- 4.Hakomori S-I, Structure and function of glycosphingolipids and sphingolipids: Recollections and future trends. Biochimica et Biophysica Acta (BBA) - General Subjects 2008, 1780 (3), 325–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stirnemann J; Belmatoug N; Camou F; Serratrice C; Froissart R; Caillaud C; Levade T; Astudillo L; Serratrice J; Brassier A; Rose C; Billette de Villemeur T; Berger MG, A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci 2017, 18 (2), 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huebecker M; Moloney EB; van der Spoel AC; Priestman DA; Isacson O; Hallett PJ; Platt FM, Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener 2019, 14 (1), 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bleicher RJ; Cabot MC, Glucosylceramide synthase and apoptosis. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2002, 1585 (2), 172–178. [DOI] [PubMed] [Google Scholar]
- 8.Jin Y; Fan J-T; Gu X-L; Zhang L-Y; Han J; Du S-H; Zhang A-X, Neuroprotective Activity of Cerebrosides from Typhonium giganteum by Regulating Caspase-3 and Bax/Bcl-2 Signaling Pathways in PC12 Cells. J. Nat. Prod 2017, 80 (6), 1734–1741. [DOI] [PubMed] [Google Scholar]
- 9.Shaner RL; Allegood JC; Park H; Wang E; Kelly S; Haynes CA; Sullards MC; Merrill AH, Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res 2009, 50 (8), 1692–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scherer M; Leuthäuser-Jaschinski K; Ecker J; Schmitz G; Liebisch G, A rapid and quantitative LC-MS/MS method to profile sphingolipids. J. Lipid Res 2010, 51 (7), 2001–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li M; Tong X; Lv P; Feng B; Yang L; Wu Z; Cui X; Bai Y; Huang Y; Liu H, A not-stop-flow online normal-/reversed-phase two-dimensional liquid chromatography–quadrupole time-of-flight mass spectrometry method for comprehensive lipid profiling of human plasma from atherosclerosis patients. J. Chromatogr. A 2014, 1372, 110–119. [DOI] [PubMed] [Google Scholar]
- 12.Gaucher SP; Leary JA, Stereochemical Differentiation of Mannose, Glucose, Galactose, and Talose Using Zinc(II) Diethylenetriamine and ESI-Ion Trap Mass Spectrometry. Anal. Chem 1998, 70 (14), 3009–3014. [DOI] [PubMed] [Google Scholar]
- 13.Desaire H; Leary JA, Differentiation of Diastereomeric N-Acetylhexosamine Monosaccharides Using Ion Trap Tandem Mass Spectrometry. Anal. Chem 1999, 71 (10), 1997–2002. [DOI] [PubMed] [Google Scholar]
- 14.Desaire H; Leary JA, Multicomponent Quantification of Diastereomeric Hexosamine Monosaccharides Using Ion Trap Tandem Mass Spectrometry. Anal. Chem 1999, 71 (19), 4142–4147. [DOI] [PubMed] [Google Scholar]
- 15.Pham HT; Julian RR, Characterization of glycosphingolipid epimers by radical-directed dissociation mass spectrometry. Analyst 2016, 141 (4), 1273–1278. [DOI] [PubMed] [Google Scholar]
- 16.Han X; Cheng H, Characterization and direct quantitation of cerebroside molecular species from lipid extracts by shotgun lipidomics. J. Lipid Res 2005, 46 (1), 163–175. [DOI] [PubMed] [Google Scholar]
- 17.Han X; Yang K; Gross RW, Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass. Spectrom. Rev 2012, 31 (1), 134–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLuckey SA; Huang T-Y, Ion/Ion Reactions: New Chemistry for Analytical MS. Anal. Chem 2009, 81 (21), 8669–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He M; McLuckey SA, Two Ion/Ion Charge Inversion Steps To Form a Doubly Protonated Peptide from a Singly Protonated Peptide in the Gas Phase. J. Am. Chem. Soc 2003, 125 (26), 7756–7757. [DOI] [PubMed] [Google Scholar]
- 20.Randolph CE; Foreman DJ; Betancourt SK; Blanksby SJ; McLuckey SA, Gas-Phase Ion/Ion Reactions Involving Tris-Phenanthroline Alkaline Earth Metal Complexes as Charge Inversion Reagents for the Identification of Fatty Acids. Anal. Chem 2018, 90 (21), 12861–12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Randolph CE; Foreman DJ; Blanksby SJ; McLuckey SA, Generating Fatty Acid Profiles in the Gas Phase: Fatty Acid Identification and Relative Quantitation Using Ion/Ion Charge Inversion Chemistry. Anal. Chem 2019, 91 (14), 9032–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Randolph CE; Blanksby SJ; McLuckey SA, Toward Complete Structure Elucidation of Glycerophospholipids in the Gas Phase through Charge Inversion Ion/Ion Chemistry. Anal. Chem 2020, 92 (1), 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franklin ET; Betancourt SK; Randolph CE; McLuckey SA; Xia Y, In-depth structural characterization of phospholipids by pairing solution photochemical reaction with charge inversion ion/ion chemistry. Anal. Bioanal. Chem 2019, 411 (19), 4739–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia Y; Chrisman PA; Erickson DE; Liu J; Liang X; Londry FA; Yang MJ; McLuckey SA, Implementation of Ion/Ion Reactions in a Quadrupole/Time-of-Flight Tandem Mass Spectrometer. Anal. Chem 2006, 78 (12), 4146–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia Y; Liang X; McLuckey SA, Pulsed Dual Electrospray Ionization for Ion/Ion Reactions. J. Am. Soc. Mass Spectrom 2005, 16 (11), 1750–1756. [DOI] [PubMed] [Google Scholar]
- 26.Domon B; Costello CE A Systematic Nomenclature for Carbohydrate Fragmentations in FAB-MS/MS Spectra of Glycoconjugates. Glycconjugate Journal 1988, 5 (4), 397–409. [Google Scholar]
- 27.Salpin J-Y; Tortajada J, Gas-phase acidity of D-glucose. A density functional theory study. J. Mass Spectrom 2004, 39 (8), 930–941. [DOI] [PubMed] [Google Scholar]
- 28.Bythell BJ; Abutokaikah MT; Wagoner AR; Guan S; Rabus JM, Cationized Carbohydrate Gas-Phase Fragmentation Chemistry. J. Am. Soc. Mass Spectrom 2017, 28 (4), 688–703. [DOI] [PubMed] [Google Scholar]
- 29.Hsu F-F, Complete structural characterization of ceramides as [M-H]- ions by multi-stage linear ion trap mass spectrometry. Biochemie 2016, 130, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang K; Zhao Z; Gross RW; Han X, Identification and Quantitation of Unsaturated Fatty Acid Isomers by Electrospray Ionization Tandem Mass Spectrometry: A Shotgun Lipidomics Approach. Anal. Chem 2011, 83 (11), 4243–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fahy E; Sud M; Cotter D; Subramaniam S, LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35 (Web Server issue), W606–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fahy E; Subramaniam S; Murphy RC; Nishijima M; Raetz CRH; Shimizu T; Spener F; van Meer G; Wakelam MJO; Dennis EA, Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res 2009, 50 Suppl (Suppl), S9–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Gerichten J; Schlosser K; Lamprecht D; Morace I; Eckhardt M; Wachten D; Jennemann R; Gröne H-J; Mack M; Sandhoff R, Diastereomer-specific quantification of bioactive hexosylceramides from bacteria and mammals. J. Lipid Res 2017, 58 (6), 1247–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dandana A; Ben Khelifa S; Chahed H; Miled A; Ferchichi S, Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology 2016, 83 (1), 13–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.