

Abstract
SARS-CoV-2, an emerging coronavirus, has spread rapidly around the world, resulting in over ten million cases and more than half a million deaths as of July 1, 2020. Effective treatments and vaccines for SARS-CoV-2 infection do not currently exist. Previous studies demonstrated that nonstructural protein 16 (nsp16) of coronavirus is an S-adenosyl methionine (SAM)-dependent 2’-O-methyltransferase (2’-O-MTase) that has an important role in viral replication and prevents recognition by the host innate immune system. In the present study, we employed structural analysis, virtual screening, and molecular simulation approaches to identify clinically investigated and approved drugs which can act as promising inhibitors against nsp16 2′-O-MTase of SARS-CoV-2. Comparative analysis of primary amino acid sequences and crystal structures of seven human CoVs defined the key residues for nsp16 2-O’-MTase functions. Virtual screening and docking analysis ranked the potential inhibitors of nsp16 from more than 4,500 clinically investigated and approved drugs. Furthermore, molecular dynamics simulations were carried out on eight top candidates, including Hesperidin, Rimegepant, Gs-9667, and Sonedenoson, to calculate various structural parameters and understand the dynamic behavior of the drug-protein complexes. Our studies provided the foundation to further test and repurpose these candidate drugs experimentally and/or clinically for COVID-19 treatment.
Communicated by Ramaswamy H. Sarma
Keywords: SARS-CoV-2, nsp16, methyltransferase, KDKE motif, virtual screening, molecular dynamics simulation, inhibitor
Introduction
Three coronaviruses (CoVs): severe acute respiratory syndrome coronavirus (SARS-CoV-1), Middle East respiratory syndrome coronavirus (MERS-CoV), and the recently identified SARS-CoV-2 in December 2019, have caused deadly pneumonia in humans since the beginning of the 21st century (Coronaviridae Study Group of the International Committee on Taxonomy of, 2020; Wu, Zhao, et al., 2020; Zhu et al., 2020). The SARS-CoV-2 causes coronavirus disease-19 (COVID-19) with influenza-like symptoms ranging from mild discomfort to severe lung injury and multi-organ failure, eventually leading to death. As of July 1, 2020, more than ten million COVID-19 cases were reported worldwide, and more than half million patients have died (https://www.who.int/emergencies/diseases/novel-coronavirus-2019). Effective treatments and vaccines for SARS-CoV-2 infection do not currently exist. Thus, there is an urgent and unmet need for the discovery and development of antiviral therapeutics for the treatment of COVID-19.
CoVs are positive-sense RNA viruses that replicate in the cytoplasm of infected cells. Replication and transcription of the CoV RNA genome are achieved by a complex RNA replication/transcription machinery, consisting of at least 16 viral nonstructural proteins (nsps) (Coronaviridae Study Group of the International Committee on Taxonomy of, 2020; Perlman & Netland, 2009). Previous studies demonstrated that nsp16 proteins of SARS-CoV-1 and MERS-CoV have methyltransferase (MTase) activities that catalyze methylation of the first transcribed nucleotide at the ribose 2′-O position (2′-O-Me) (Aouadi et al., 2017; Chen et al., 2011; Decroly et al., 2011). The 2′-O-Me of viral cap RNA protects itself from degradation by 5′-3′ exoribonucleases, ensures efficient translation, and helps to prevent recognition by the host innate immune system (Menachery et al., 2014). The importance of 2′-O-MTase activity for CoV infection and pathogenesis was previously documented by in vitro and in vivo studies (Gonzales-van Horn & Sarnow, 2017; Menachery et al., 2014; Sevajol et al., 2014; Subissi et al., 2014). For SARS-CoV-1, the absence of nsp16 2′-O-MTase activity results in significant attenuation characterized by decreased viral replication, reduced weight loss, and limited breathing dysfunction in mice (Menachery et al., 2014). In addition, 2′-O-Me acts as a recognition marker that helps the host cell recognize its own RNA species (Byszewska et al., 2014; Züst et al., 2011). Inhibition of nsp16 2′-O-MTase activities should restrain viral replication and enable recognition by the host innate immune system. This makes the nsp16-MTase a promising target for the identification of new anti-SARS-CoV-2 drugs.
Recent advances in structural bioinformatics and virtual screening approaches have revolutionized the identification and/or repurposing of marketed drugs or bioactive compounds for effective treatment of various human diseases, including infectious diseases (Chang et al., 2016; Gioia et al., 2017; Kitchen et al., 2004; Lasko et al., 2017; Maia et al., 2020; Pinzi & Rastelli, 2019; Slater & Kontoyianni, 2019). Moreover, in silico approaches, including molecular dynamics (MD), have also been widely used to determine the conformational space of the investigated targets, ligands, and ligand-target complexes, and thus better understand the dynamic behavior of ligand-target complexes (Bhardwaj & Purohit, 2020; Pinzi & Rastelli, 2019; Rajendran et al., 2018; Slater & Kontoyianni, 2019; Śledź & Caflisch, 2018). Very recently, a virtual screening approach was used to identify potential drugs to inhibit SARS-CoV-2 proteins, including surface spike glycoprotein, main protease, and nsp16 (Bhardwaj et al., 2020; Panda et al., 2020; Tazikeh-Lemeski et al., 2020; Vijayan et al., 2020). For example, Bhardwaj et al. reported the identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors (Bhardwaj et al., 2020). Molecular docking and virtual screening approaches also have been attempted to identify compounds targeting 2′-O-MTase of SARS-CoV-2 (Encinar & Menendez, 2020; Hijikata et al., 2020; Tazikeh-Lemeski et al., 2020; Vijayan et al., 2020).
In the present study, we employed structural analysis, virtual screening, and molecular simulation approaches to identify potential inhibitors targeting 2′-O-MTase of SARS-CoV-2. We first performed comparative analysis of primary amino acid sequences and crystal structures of seven human CoVs and defined the key residues for nsp16 2′-O-MTase functions. We executed virtual screening and docking analysis to rank the potential inhibitors of nsp16 from more than 4,500 clinically investigated and approved drugs. MD simulations were carried out on eight candidate compounds to calculate various structural parameters and understand the dynamic behavior of the drug-protein complexes. Our studies provided the foundation to further test and repurpose these candidate drugs experimentally and/or clinically for COVID-19 treatment.
Results
Comparative sequence and structure analysis of nsp16 2’-O-MTases
To identify inhibitors targeting nsp16, we first performed comparative analysis of primary amino acid sequences and crystal structures of seven human CoVs. Supplementary Table 1 lists the detailed genome and protein information that were employed in this study. In primary amino acid sequences, nsp16 of SARS-CoV-2 was found to be 93.3% identical to SARS-CoV-1, but only 56.6 − 65.9% identical to five other human CoVs (MERS-CoV, HCoV-OC43, -HKU1, -NL63, and -229E). The nsp16 proteins belong to a class of S-adenosyl methionine (SAM) - dependent 2′-O-MTases present in all life forms, and they all contain the conserved catalytic KDKE motif (K46, D130, K170, and E203 in SARS-CoV-2) (Figure 1A & B) (Bouvet et al., 2010; Chen et al., 2011; Decroly et al., 2011).
Table 1.
Top ten scoring drugs for SARS-CoV-2 nsp16.
| Drug Name | 2D Structure | AutoDock Vina Score (Energy) | Rosetta’s Energy Score (REU) | Clinical Stage | ||
|---|---|---|---|---|---|---|
| Hesperidin | 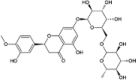 |
−10.30 | −1502.39 | Phase 3/ Approved | ||
| Rebastinib | 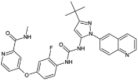 |
−10.20 | −1498.69 | Phase 2 | ||
| Losulazine | 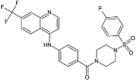 |
−10.27 | −1497.67 | Phase 0 | ||
| Cep-32496 | 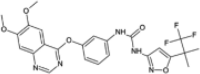 |
−10.20 | −1497.02 | Phase 1 | ||
| R428 | 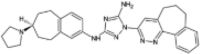 |
−10.40 | −1495.17 | Phase 2 | ||
| Entrectinib | 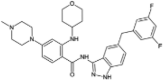 |
−10.57 | −1494.42 | Approved | ||
| Osi-027 | 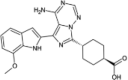 |
−10.50 | −1492.85 | Phase 1 | ||
| MK3207 | 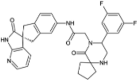 |
−10.97 | −1491.61 | Phase 2 | ||
| Rimegepant | 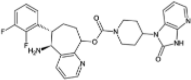 |
−10.77 | −1486.36 | Approved | ||
| Bolazine | 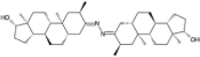 |
−10.47 | −1484.41 | |||
Note: For the drugs with multiple stereoisomers in MTiOpenScreen database, only one stereoisomer with the highest AutoDock Vina Score was listed.
Figure 1.

Comparative analysis of primary amino acid sequences and crystal structures of CoV 2’-O-MTases. (A) Schematic presentation of the SARS-CoV-2 genome organization. Expression of two open reading frames (ORF1a and ORF1b) yields 16 nsps, including 2’-O-MTase nsp16. S, E, M, and N indicate the four structural proteins: spike, envelope, membrane, and nucleocapsid. (B) Sequence alignment of nsp16 proteins derived from genome sequences of the following: SARS-CoV-2, SARS-CoV-1, MERS-CoV, HCoV-OC43, HCoV-HKU1, HCoV-NL63, and HCoV-229E. The secondary structure of SARS-CoV-2 nsp16 is shown above. Residues with 100% conservation are indicated in solid red boxes and those with identity of 70% or higher are depicted in light red color. The red stars indicate the conserved KDKE motif in 2’-O-MTases. (C) Surface representation of the SAM binding pocket and the RNA binding groove in SARS-CoV-2 nsp16 with coloring according to the electrostatic potential. The surface electrostatic potential diagram (±5 kT/e) in SARS-CoV-2 nsp16 (PDB: 6W4H Chain A) was generated by PyMol; the blue areas represent positively charged areas, while the red areas represent negatively charged areas. (D) The KDKE catalytic tetrad motif is located at the bottom of the RNA binding groove of nsp16.
We next analyzed crystal structures of nsp16s obtained from SARS-CoV-2, SARS-CoV-1, and MERS-CoV that are available in RCSB Protein Data Bank (PDB) as of April 30, 2020 (Supplementary Table S1) (Berman et al., 2000; Chen et al., 2011; Decroly et al., 2011). The three-dimensional (3 D) structures of nsp16s were solved in complex with nsp10 that functions as a stimulatory factor to regulate 2′-O-MTase function (Supplementary Figure S1) (Chen et al., 2011; Decroly et al., 2011). Structural alignment was performed using TM-align, an algorithm for sequence-independent protein structure comparisons (Zhang & Skolnick, 2005). We revealed the highest coverage, percent identity, and structural conservation between SARS-CoV-2 and SARS-CoV-1 nsp16 bound to natural ligand SAM [PDB: 6W4H chain A (Resolution: 1.80 Å); and 3R24 chain A (Resolution: 2.00 Å), respectively], with TM-score 0.958 and Root Mean Square Deviation (RMSD) = 0.89. The SARS-CoV-2 nsp16 structure is also highly similar to the MERS-CoV complex bound to SAM and a cap analogue: 7-methyl-GpppA [PDB:5YNM: chain A (Resolution: 1.68 Å)], with TM-score 0.956 and RMSD = 0.80 (Supplementary Figure S2). The superposition of 6W4H and 5YNM revealed that the conserved KDKE motif of SARS-CoV-2 nsp16 is located at the bottom of the RNA binding pocket (Figure 1C & D). Structural analysis also revealed that residues N43, Y47, G71, G81, D99, L100, N101, D114, and M131 coordinate SAM binding in both SARS-CoV-1 and −2 through hydrogen bonds and water-mediated interactions (Aouadi et al., 2017; Viswanathan et al., 2020).
Figure 2.

Comparison of 2’-O-MTase domain of human CMTR1 with nsp16 of SARS-CoV-2. Top: Surface representation with electrostatic potentials showing the RNA binding groove and the SAM binding pocket of human CMTR1 (PDB: 4N48). Bottom: Superposition of the KDKE catalytic tetrad motifs of nsp16 of SARS-CoV-2 (PDB: 6W4H) and 2’-O-MTase domain of human CMTR1 (PDB: 4N48).
In humans, cap methyltransferase 1 (CMTR1) is a specific 2′-O-MTase that catalyzes methylation of the first transcribed nucleotide (N1) in cellular mRNA (Cap 1) (Belanger et al., 2010; Smietanski et al., 2014). We also performed comparative analysis of the crystal structures of CoV-2 nsp16 and human 2′-O-MTase CMTR1. SARS-CoV-2 nsp16 and human CMTR1 MTase domain share little primary amino acid sequence identity (12.1%) based on the Pairwise Sequence Alignment with the Needleman-Wunsch algorithm at EMBL-EBI services (https://www.ebi.ac.uk/Tools/psa/). A search of the PDB using DALI service (http://ekhidna2.biocenter.helsinki.fi/dali/) revealed structural similarity between CoV-2 nsp16 (PDB: 6W4H chain A) and human CMTR1 (PDB: 4N48 and 4N49), which have Dali Z-Scores 13.7 and 13.8, respectively, and RMSD 3.4 (Supplementary Figure S3). However, even though both nsp16 and CMTR1 contain a KDKE catalytic tetrad motif and a conserved folding made of a Rossmann β-sheet surrounded by helices on each side, substantial differences are found in the RNA binding groove as well as the SAM binding pocket (Figures 1C and 2). In CMTR1, Cap 0 binds in a deep pocket, with a triphosphate bridge and N1 phosphate backbone stabilized by a stack of arginine residues along the RNA binding groove (Smietanski et al., 2014). In contrast, the RNA binding groove on SARS-CoV-2 nsp16 appears to be less positively charged and shallower (Figure 1C). Additionally, the SAM binding pocket on SARS-CoV-2 nsp16 is more negatively charged than that of human CMTR1. Also, the SAM binding pocket on SARS-CoV-2 nsp16 is more open and flexible compared to that of CMTR1, which is deeper and longer (Figures 1C and 2). The difference between the sequences and structures of the human 2′-O-MTase CMTR1 and CoV nsp16 could be exploited to develop inhibitors that could specifically block viral MTases.
Figure 3.

(A) Similarity chart of the 1,380 top-scoring compounds for the SARS-CoV-2 nsp16 using the FragFp descriptors in DataWarrior. Compounds with high chemical similarity are connected by lines. Stereoisomers of the same drug such as Hesperidin (38 Stereoisomers) are clustered together. Compounds are colored according to their predicted AutoDock Vina score. (B) Neighbor tree shows seven drugs that have FragFp similarity scores to SAM greater than 0.8. Drugs are colored according to their FragFp similarity score to SAM. (C) Pie charts show target and pathway classes of hit compounds that were queried in the Probes and Drugs Portal.
Virtual screening of drugs against nsp16 of SARS-CoV-2
We hypothesize that the small-molecule antagonists of nsp16 of SARS-CoV-2 will limit viral replication and/or unmask viral RNA to intracellular innate immunity. To identify new drugs that have the potential to inhibit nsp16 functions of SARS-CoV-2, we first examined the druggability of all binding sites of 2′-O-MTase by the DoGSiteScorer tool (Volkamer et al., 2012). With values between zero and one, the SAM pocket has the highest drug score (0.85) and RNA binding pocket has a score of 0.55 in SARS-CoV-2 nsp16 (Supplementary Figure S1). Similar scores were observed in MERS-CoV nsp16 (PDB: 5YNM) that was solved in complex with both SAM and the cap RNA analogue (Supplementary Figure S1).
As the aim of this work was to identify clinically investigated and approved drugs for quickly repurposing to treat COVID-19 infections, we made use of an MTiOpenScreen database Drugs-lib that contains 7,173 stereoisomers corresponding to 4,574 drugs, generated from ChEMBL, DrugBank, DrugCentral, and SuperDrug2 (Lagarde et al., 2018, 2019). Among them, the nsp16 natural ligand SAM would serve as a positive control for virtual screening. Among the five available nsp16 structures of SARS-CoV-2 (April 30, 2020), we selected the PDB 6W4H structure which presents the best resolution (1.8 Å) to screen the Drugs-lib using the MTiOpenScreen service (Lagarde et al., 2019). The computations of MTiOpenScreen were carried out with AutoDock Vina, a gradient optimization algorithm, which provided a list of the 1,500 highest-scoring compounds (Trott & Olson, 2010). We repeated the MTiOpenScreen screening protocol three times using the 6W4H structure to ensure the reliability of the results. We found that four stereoisomers of SAM were ranked in the top 1,500 best compounds in all three runs, with energy equal to −8.10 kcal/mol (Figure 3A & B, Supplementary Table S2). Among the 1,500 top hit compounds in each screening, 1,380 compounds were shared across all three MTiOpenScreen runs with highly similar AutoDock Vina scores. Based on the means of scores, we ranked these 1,380 compounds (stereoisomers) that correspond to 967 drugs (Supplementary Table S2). The top ten drugs with the highest scores were MK3207, Rimegepant, Entrectinib, Osi-027, Bolazine, R428, Hesperidin, Losulazine, Rebastinib, and Cep-32496, with the means of energy around −10.97 ∼ −10.17 kcal/mol (Figure 3A and Table 1). Notably, there are 38 Hesperidin stereoisomers and four MK3207 stereoisomers among the 1,380 hits (Figure 3A and Supplementary Table S2).
Table 2.
Seven drugs chemically similar to natural ligand (SAM).
| Drug Name | 2D Structure | AutoDock Vina Score (Energy) | Rosetta’s Energy Score (REU) | Clinical Stage | |
|---|---|---|---|---|---|
| Gs-9667 |  |
−8.87 | −1499.57 | Phase 1 | |
| Sonedenoson | 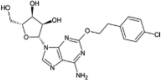 |
−8.30 | −1494.49 | Phase 2 | |
| Regadenoson | 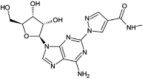 |
−8.33 | −1491.72 | Approved | |
| Metrifudil | 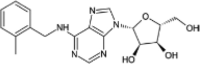 |
−8.20 | −1491.61 | Phase 0 | |
| Binodenoson |  |
−8.57 | −1490.97 | Phase 3 | |
| Trabodenoson |  |
−8.40 | −1489.93 | Phase 3 | |
| Selodenoson | 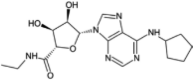 |
−8.10 | −1489.55 | Phase 0 | |
| S-adenosyl-methionine (SAM) | 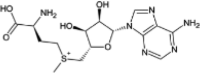 |
−8.1 | −1509.65 | ||
Note: For the drugs with multiple stereoisomers in MTiOpenScreen database, only one stereoisomer with the highest AutoDock Vina Score was listed.
To understand previously reported pharmacological action of hit compounds, we queried the biological target and pathway classes in the Probes and Drugs Portal, in which 713 standardized (693 non-isomeric) drugs were available for these 967 drugs (Skuta et al., 2017). The top target classes of these drugs are G-protein-coupled receptors (241), catalytic receptors (139), and kinases (129). The top pathways are signal transduction (453), immune system (270), and transcription (247) (Figure 3C). For example, MK3207 and Rimegepant are calcitonin gene-related peptide (CGRP) receptor antagonists, which have been clinically investigated and/or approved for migraine treatment (Bell, 2014; Lipton et al., 2019; Salvatore et al., 2010). Hesperidin possesses biological and pharmacological properties as an effective antioxidant, anti-inflammatory, anti-carcinogenic, and anti-hypertensive agent (Aggarwal et al., 2020; Tejada et al., 2018).
Drugs similar to ligand of nsp16
Using the principles of structural similarity searching, analogues of the natural ligand (SAM) in nsp16 might be potent inhibitors against 2′-O-MTase function. Since SAM was among our 1,380 highest-scoring compounds, we next computed the similarity between each pair of compounds using the FragFp descriptors in DataWarrior (Sander et al., 2015). Among them, we found that seven drugs are chemically similar to SAM, namely Gs-9667, Trabodenoson, Binodenoson, Sonedenoson, Regadenoson, Metrifudil, and Selodenoson (Figure 3B and Table 2). All seven of these drugs are adenosine receptor agonists that were clinically investigated for treating various diseases, including cardiac arrhythmias, neuropathic pain, inflammatory diseases, and cancer (Gao & Jacobson, 2011; Jacobson et al., 2019).
Molecular docking simulation of top candidate drugs against nsp16 of SARS-CoV-2
Rigid docking performed using Autodock Vina provided insight into the binding affinity and hydrogen bond formation of the candidate compounds on the nsp16 2′-O-MTase. Next, the top ten drugs and seven drugs structurally similar to SAM were chosen for the ligand flexible docking simulation by using the RosettaLigand program (Davis & Baker, 2009; Lemmon & Meiler, 2012). RosettaLigand employs the Monte Carlo minimization protocol in which the ligand position and orientation are randomly perturbed by a small deviation (0.1 Å and 3°). A scoring function in RosettaLigand includes an electrostatics model, an explicit orientation-dependent hydrogen bonding potential, an implicit solvation model, and van der Walls interactions (Davis & Baker, 2009; Lemmon & Meiler, 2012). Rosetta’s energy scores (REU) of these 17 drugs were shown in Tables 1 and 2. With the RosettaLigand simulation, Hesperidin ranked the highest with the lowest REU among the top ten hits obtained from the Autodock Vina screening. In the seven SAM-like drugs, similar to AutoDock Vina results, Gs-9667 had the lowest REU score (Table 2).
Binding mode analysis and intermolecular interaction of top drugs and nsp16
We next examined the interaction of these 17 drugs with key residues for nsp16 2′-O-MTase function. We found that Hesperidin, Osi-027, Gs-9667, and Sonedenoson have many hydrogen bonds or hydrophobic interactions with key functional residues involved in 2′-O-MTase function, which are represented in Figure 4. Hesperidin forms hydrogen bonds with N43, K46, G71, L100, N101, C115, Y132, and K170 and hydrophobic interactions with Y132, and F149. Osi-027 forms hydrogen bonds with N43, K46, L100, N101, C115, Y132, and K170, a salt bridge with D99, and hydrophobic interactions with L100, D114, M131, and F149. Gs-9667 forms hydrogen bonds with L100, C115, and Y132 and salt bridges with D99 and D114. Sonedenoson forms hydrogen bonds with N43, K46, D75, N101, Y132, and K170, salt bridges with D75 and D99, and hydrophobic interactions with L100, M131, and F149. Notably, Hesperidin, Osi-027 and Sonedenoson form hydrogen bonds with two key residues in the KDKE motif: K46 and K170.
Figure 4.

Interaction of four drugs (Hesperidin, Osi-027, Gs-9667 and Sonedenoson) with SAM binding pocket of SARS-CoV-2 nsp16. Left: Surface representation with charge showing predicted binding of SARS-CoV-2 nsp16 with the four drugs in the SAM binding pocket. The drugs are depicted by sticks. Right: 3 D representation of 2’-O-MTase active site residues interacting with Hesperidin, Osi-027, Gs-9667 and Sonedenoson.
Molecular dynamics simulation of eight candidate compounds
To gain insight into the dynamic behavior of the compounds at the active site of 2′-O-MTase, a set of 100 ns MD simulations were executed for eight selected hits (top hits and/or clinically approved drugs, Table 3) by employing the GROningen MAchine for Chemical Simulations (GROMACS) package (Van Der Spoel et al., 2005). The obtained results were read as RMSD, which is essential to quantify the structural stability of protein-drug complexes within a regular time frame. RMSD analysis of the free form of 2′-O-MTase revealed that the protein molecule achieves stability at around 20 ns, and it mostly maintained its stability until 100 ns. Nsp16-SAM and nsp16-Hesperidin complexes attained stability at around 50 ns and maintained that stability until the end. Based on average RMSD, all eight drugs were very comparable to SAM, and the mean values of RMSD in all compound-protein complexes are lower than those in free protein (Table 3). The lowest mean value of RMSD is observed for the Gs-9667 simulation (Table 3).
Table 3.
Time averaged structural properties obtained from MD simulation of 2′-O-MTase of SARS-CoV-2.
| Analysis | Blank | Hesperidin | Entrectinib | Osi-027 | MK3207 | Rimegepant | Gs-9667 | Sonedenoson | Regadenoson | SAM |
|---|---|---|---|---|---|---|---|---|---|---|
| Average RMSD (nm) | 0.534 | 0.514 | 0.474 | 0.451 | 0.493 | 0.504 | 0.445 | 0.496 | 0.484 | 0.491 |
| Average RMSF for Chain A (nm) | 0.177 | 0.177 | 0.144 | 0.157 | 0.151 | 0.165 | 0.175 | 0.145 | 0.163 | 0.150 |
| Average Rg (nm) | 2.156 | 2.127 | 2.130 | 2.149 | 2.174 | 2.135 | 2.150 | 2.158 | 2.202 | 2.145 |
| PCA (Trace of Covariance Matrix nm2) | 176.151 | 219.597 | 128.648 | 114.024 | 123.147 | 144.829 | 195.177 | 132.557 | 163.854 | 127.930 |
| Average Intermolecular Hydrogen Bonds (Protein-Ligand) | N/A | 5 | 3 | 1 | 2 | 2 | 2 | 2 | 5 | 3 |
| Average SASA (nm2) | 176.111 | 172.096 | 173.087 | 177.480 | 175.793 | 173.861 | 176.027 | 174.493 | 175.085 | 170.143 |
Root Mean Square Fluctuation (RMSF) is used to estimate the average atomic mobility of backbone atoms (N, Cα, and C) during MD simulation. The 2′-O-MTase nsp16 of SARS-CoV-2 has an extensively looped structure, and we observed the fluctuations throughout the entire nsp16 protein (Figure 5). Even though the average fluctuations of all eight protein-drug complexes were very similar, the nsp16-Hersperidin complex was found to have the highest, while nsp16-Entrectinib complex had the lowest average RMSF value (Table 3).
Figure 5.

Plot of MD simulation trajectories of free 2’-O-MTase and drug-protein complexes during 100 ns simulation. RMSD: Root Mean Square Deviation; RMSF: Root Mean Square Fluctuation; SASA: Solvent Accessible Surface Area; H-Bond: hydrogen bond.
We also calculated additional structural parameters from the MD simulations, like Radius of Gyration (Rg), Solvent-Accessible Surface Area (SASA), Inter-molecular Hydrogen Bonding (H-bonding), and Principal Component Analysis (PCA) based on essential dynamics (ED) approach (Figures 5 and 6). Based on the Rg analysis that estimates the compactness of protein and protein-drug complexes, the 2′-O-MTase was undergoing some compression in its 3 D conformation (Figure S4). Among the protein-drug complexes, the most compressed complex was that of Hesperidin (Table 3). These results are consistent with the SASA analysis, which determined that the amount of overall surface area is reduced for solvent accessibility due to compression in the protein structure (Figure 5). Additionally, our MD simulation analysis showed an average of five hydrogen bonds present between nsp16 protein and two drugs (Hesperidin and Regadenoson), while others form an average of one to three hydrogen bonds (Figure 5 and Table 3).
Figure 6.

Principal Component Analysis (PCA) of free 2’-O-MTase, along with eight drug- and SAM-protein complexes. The bottom shows a superimposed plot of all ten PCA analyses.
The PCA was employed to reveal relevant motions from the global trajectories of unbound proteins as well as the protein-drug complexes using the ED approach. ED projects the combined fluctuations of the most unsteady regions of protein molecules into two variables, namely Principal Component 1 (PC1) and Principal Component 2 (PC2). The projections of trajectories for free protein as well as complexes are illuminated in Figure 6. Among them, the Hesperidin-protein complex was found to be occupying the most, while the Osi-027 complex had the least conformational space (Table 3).
MM-PBSA analysis of eight candidate compounds
Next, we performed the MM-PBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) analysis to estimate the protein-drug binding affinity in dynamic state. As shown in Table 4, we found that three compounds, Hesperidin, Rimegepant, and Entrectinib, had the highest binding affinities with protein in dynamic state. Among three SAM-like drugs with MD simulation analysis, Sonedenoson and Gs-9667 had relatively higher binding affinities, compared to the SAM with 2′-O-MTase in dynamic state. To determine the energy contributions of individual residues to the total binding free energy, energy decomposition analysis was conducted on a per-residue basis and the result is presented in Supplementary Table S3. As illuminated in Figure 7, the calculations of residue-based free energy decomposition identified the hot interaction spots of 2′-O-MTase with four compounds. For example, four residues (D75, L100, Y132, and F149) of nsp16 were notable hotspots (< −10.0 kcal/mol) that contribute favorably to the binding of Hesperidin. In the Gs-9667 complex, two residues, D130 and Y132, of nsp16 were dramatically favorable toward binding (-10.6 and −20.2 kcal/mol). As seen from MM-PBSA results and docking studies, drugs including Hesperidin, Osi-027, Rimegepant, Sonedenoson, and Gs-9667 had higher binding affinities than SAM with the 2′-O-MTase of SARS-CoV-2.
Table 4.
MM-PBSA calculations of binding affinity for eight selected drugs and SAM.
| Complexes | Binding (kj / mol) | SASA (kj / mol) | Polar Solvation (kj / mol) | Electrostatic (kj / mol) | Van der Waals (kj / mol) |
|---|---|---|---|---|---|
| Hesperidin | −166.759 +/- 2.938 | −23.518 +/- 0.072 | 327.379 +/- 2.124 | −215.678 +/- 2.658 | −254.942 +/- 1.784 |
| Entrectinib | −171.784 +/- 1.635 | −23.523 +/- 0.053 | 180.869 +/- 1.036 | −78.331 +/- 1.580 | −250.800 +/- 1.155 |
| Osi-027 | −110.927 +/- 1.676 | −19.411 +/- 0.038 | 183.292 +/- 1.146 | −60.872 +/- 1.354 | −213.936 +/- 1.025 |
| Mk3207 | −134.973 +/- 1.814 | −21.603 +/- 0.126 | 158.480 +/- 2.169 | −38.778 +/- 1.657 | −233.071 +/- 2.064 |
| Rimegepant | −209.740 +/- 1.690 | −22.181 +/- 0.068 | 99.705 +/- 0.639 | −5.437 +/- 1.340 | −281.828 +/- 1.373 |
| Gs-9667 | −128.897 +/- 2.006 | −18.201 +/- 0.071 | 132.429 +/- 1.512 | −62.692 +/- 1.491 | −180.433 +/- 1.472 |
| Sonedenoson | −129.205 +/- 1.845 | −17.312 +/- 0.057 | 127.457 +/- 1.833 | −67.639 +/- 1.670 | −171.711 +/- 1.194 |
| Regadenoson | −118.419 +/- 2.273 | −17.685 +/- 0.045 | 225.210 +/- 1.721 | −146.241 +/- 1.896 | −179.704 +/- 1.171 |
| SAM | −81.029 +/- 2.157 | −16.564 +/- 0.067 | 172.481 +/- 2.493 | −78.414 +/- 2.029 | −158.531 +/- 1.326 |
Figure 7.

The per-residue free energy contribution spectrums of nsp16 in four compound-protein complexes. Only residues contributing above a ± 10 kcal/mol threshold were colored and labeled.
Discussion
Methylation of RNA, which occurs in all kingdoms of life, plays important roles in RNA metabolism, processing, stability, nuclear export, translation efficiency, and others (Kadumuri & Janga, 2018; Li & Mason, 2014; Menachery et al., 2014; Roundtree et al., 2017; Shi et al., 2019). 2′-O-Me is predominantly found in rRNA and tRNA of bacteria and eukaryotes, as well as in the 5′ mRNA Cap of higher eukaryotes (Ayadi et al., 2019; Boccaletto et al., 2018). In humans, five 2′-O-MTases [FtsJ RNA methyltransferase homolog 1 (FTSJ1), FTSJ2, FTSJ3, CMTR1, and CMTR2] have been reported to catalyze 2′-O-Me of various RNAs. These 2′-O-MTases regulate important cellular functions, and are associated with developmental disorders and cancer (Bügl et al., 2000; Hager et al., 2002; Lee & Bogenhagen, 2014; Li, Wang, et al., 2020; Ringeard et al., 2019). Mutations of FTSJ1, a tRNA 2′-O-MTase, cause autosomal-recessive intellectual disability (Freude et al., 2004; Guy et al., 2015; Li, Wang, et al., 2020). FTSJ2 (also known as MRM2) is a mitochondrial rRNA methyltransferase (Lee et al., 2013). Human FTSJ3, which likely catalyzes 2′-O-Me of both rRNA and internal sites of mRNA, is a potential regulator of breast cancer progression (Manning et al., 2020). Furthermore, FTSJ3 catalyzes 2′-O-Me of HIV RNAs and leads to the inhibition of innate immune sensing and response (Ringeard et al., 2019). Both CMTR1 and CMTR2 catalyze mRNA cap methylation, CMTR1 for the first transcribed nucleotide and CMTR2 for the second transcribed nucleotide (Inesta-Vaquera & Cowling, 2017). Notably, CMTR1 has previously been identified as ISG95, a protein implicated in the response to interferon treatment and viral infection (Geiss et al., 2003; Su et al., 2002). A very recent study in the influenza A virus revealed that loss of CMTR1 in host cells inhibits viral replication and up-regulates anti-viral genes (Li, Clohisey, et al., 2020). Thus, 2′-O-MTases have critical roles in viral replication and anti-viral immune response.
Many RNA viruses, including CoVs, replicate in the cytoplasm using their own viral 2′-O-MTases, which catalyze the formation of cap structures on viral mRNA that mimic those present on host mRNAs (Hyde & Diamond, 2015; Netzband & Pager, 2020). Previous studies on SARS-CoV-1 and MERS-CoV demonstrated that targeting nsp16 2′-O-MTases might interfere with viral replication by inhibiting the replication process and promoting intracellular recognition/immune response to viral RNA species (Menachery et al., 2014; Sevajol et al., 2014; Subissi et al., 2014). Thus, nsp16 is a promising target for identification of drugs for viral infections, including SARS-CoV-2.
In this study, we performed virtual screening and ranked the predicted binding affinities of approximately 1,000 therapeutic drugs that have the potential to inhibit nsp16 2′-O-MTase function. We identified top ten candidates and seven SAM-like compounds. These drugs have been experimentally and clinically investigated for treatment of various diseases. For example, Hesperidin is one of the main components of a Chinese medicine, beniol (Bai et al., 2019). Hesperidin had demonstrated antiviral properties in in vitro models against influenza A virus (H1N1) by stimulating cell-autonomous immunity through interferon gene expression (Ding et al., 2018). Rimegepant recently received FDA approval for the acute treatment of migraine in adults (Scott, 2020). Additionally, Entrectinib and Osi-027 are both anti-cancer drugs (Drilon, 2019; Mateo et al., 2016; Sartore-Bianchi et al., 2020). SAM-like compound Gs-9667 was clinically investigated for treating Type 2 diabetes (Staehr et al., 2013). To additionally compare the features of these candidate compounds in Tables 1 and 2, we also performed pharmacophore analysis using an online PharmaGist server (Schneidman-Duhovny et al., 2008). The number of features and spatial feature set for each compound were summarized in Supplementary Table S4. Multiple aligned structures of the top ten compounds revealed that the highest PharmaGist score is 29.7, which resulted from the alignment of six compounds (Supplementary Figure S5). These six compounds shared four pharmacophoric features: three aromatic rings and one hydrogen bond acceptor. Furthermore, as expected, the seven SAM-like compounds have a relatively higher PharmaGist score. The highest score (50.6) resulted from the alignment of four compounds (Sonedenoson, Regadenoson, Metrifudil, Selodenoson) plus SAM. They shared ten features including two aromatic rings, two hydrogen bond donors, five hydrogen bond acceptors, and one positive charge (Supplementary Figure S6). These four compounds and SAM likely form hydrogen bond with residue N101 of nsp16 protein. In the future, after determining how some of these compounds directly bind nsp16 of SARS-CoV-2 with various biochemical approaches, the pharmacophore model will help to identify highly potent compounds.
To gain insight into the dynamic behavior of the small molecules at the active site of 2′-O-MTase, we performed MD simulation analysis for eight top hits including MK3207, Hesperidin, Osi-027, Entrectinib, Rimegepant, Gs-9667, Sonedenoson, and Regadenoson. The results suggest that all eight compound-protein complexes were overall stable with acceptable deviation (Figure 5). Among the eight complexes, Gs-9667 had the lowest mean value of RMSD. Additionally, RMSF analysis revealed the high fluctuations at the looped regions within the nsp16 structure, particularly around the amino acids 28, 140, and 260. Even though the average fluctuations of the eight protein-drug complexes were very similar, the 2′-O-MTase-Hesperidin complex showed the most significant fluctuations. Furthermore, the conserved K-D-K-E (K46, D130, K170, and E203) motif remained remarkably stable throughout the MD simulation. Our MD simulation analysis also found an average of five hydrogen bonds present between nsp16 protein and Hesperidin. Structural analysis indicated that Hesperidin might form hydrogen bonds with K46 and K170 of the KDKE motif, as well as N43 and L100, which are key residues for SAM binding in nsp16. Recent virtual screening studies also found that Hesperidin may disrupt the interaction of SARS-CoV-2 Spike protein and its host cellular receptor ACE2 (Angiotensin-converting enzyme 2) (Haggag et al., 2020; Wu, Liu, et al., 2020). It will be interesting to determine how effectively Hesperidin binds nsp16 of SARS-CoV-2, and to demonstrate its antiviral activity in cellular and animal models related to COVID-19.
Conclusion
In summary, we have analyzed structural features and performed functional determination of 2′-O-MTase nsp16 of SARS-CoV-2, which causes the devastating COVID-19. The nsp16 has a druggable pocket, which is associated with substrate binding and enzymatic activity. Using high-throughput virtual screening, molecular docking, and MD simulation approaches, we identified 17 therapeutic drugs, including several clinically approved drugs that have the potential to inhibit 2′-O-MTase of SARS-COV-2. The identified top candidates, including Hesperidin, Rimegepant, Gs-9667, and Sonedenoson, merit further testing and repurposing experimentally and/or clinically for COVID-19 treatment.
Material and methods
Sequence and structure analysis
The primary amino acid sequences of seven human CoV nsp16 proteins and human CMTR1 were retrieved from the NCBI (National Center for Biotechnology Information) Database, and Supplementary Table 1 lists the detailed genome and protein ID for these 2′-O-MTases. The protein sequences were aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/), and was presented with the ESPript 3.0 program (http://espript.ibcp.fr/ESPript/ESPript/). Crystal structures of three CoV nsp16s and human CMTR1 were obtained from RCSB Protein Data Bank (Berman et al., 2000), and analyzed with PyMOL, UCSF Chimera and Protein-Ligand Interaction Profiler programs (Pettersen et al., 2004; Salentin et al., 2015). Structural alignment and comparison were performed using TM-align algorithm and Dali server (Holm, 2020; Zhang & Skolnick, 2005).
Virtual screening
Crystal structure (PDB: 6W4H) of SARS-CoV-2 nsp16 with 1.8 Å resolution was prepared with UCSF Chimera programs for virtual screening against a Drugs-lib that contains 7,173 stereoisomers corresponding to 4,574 “approved” drugs in MTiOpenScreen service (Lagarde et al., 2018, 2019). The Drugs-lib is generated from four compound databases of the “drug” subset of the ChEMBL database, the “approved” subset of DrugBank, the DrugCentral database, and the “approved” SuperDrug2 database (Lagarde et al., 2019). For the MTiOpenScreen Vina docking, the (x, y, z) grid center coordinates used for SAM binding pocket of 6W4H are (83.7, 14.8, 27.7), and the size of the search space was set to 20 Å x 20 Å x 20 Å. The MTiOpenScreen screening was repeated three times, and the compounds shared across all three runs were analyzed by DataWarrior, PyMOL, and UCSF Chimera programs. Fragfp descriptors in the DataWarrior were used to compute the similarity between each pair of compounds (Sander et al., 2015). The targets and pathways of identified candidate compounds were analyzed by querying Probes and Drugs Portal (Skuta et al., 2017). The interaction of candidate drugs with key residues for nsp16 2′-O-MTase function were analyzed by PyMOL, LIGPLOT, and PLIP (protein–ligand interaction profiler) (Salentin et al., 2015; Wallace et al., 1995).
Molecular docking simulation using RosettaLigand program
Ligand flexible docking simulation was performed using the RosettaLigand program that employs the Monte Carlo minimization protocol (Davis & Baker, 2009; Lemmon & Meiler, 2012). In the first, low-resolution stage, the ligand was placed near the SAM binding pocket of nsp16, where it was perturbed 50 times and rotated 1,000 times in a random direction. The best-scoring models were filtered by root-mean-square deviation to eliminate near-duplicates and one of the remaining models was selected at random. To ensure that the ligand did not “walk away” from the protein, the ligand was moved towards the protein. The second, high-resolution stage employed the Monte Carlo minimization protocol in which the ligand position and orientation were randomly perturbed by a small deviation (0.1 Å and 3°); receptor side chains were repacked using a rotamer library; the ligand position, orientation, and torsions and protein side-chain torsions were simultaneously optimized using quasi-Newton minimization; and the end result was accepted or rejected based on the Metropolis criterion. Scoring used the full-atom Rosetta energy function with softened van der Waals repulsion (Davis & Baker, 2009; Lemmon & Meiler, 2012). The full repack made 1,000 random rotamer substitutions at random positions and accepted or rejected each on the Metropolis criterion. Rotamer trials chose the single best rotamer at a random position in the context of the current state of the rest of the system, with the positions visited once each in random order. Six cycles of rotamer trials and a full repack after every three cycles were performed. The third and final stage was a more stringent, gradient-based minimization of the ligand position, orientation, and torsions and receptor torsions for both side chains and the backbone. Scoring used the same Rosetta energy function, but with a hard-repulsive van der Waals potential, which created a more rugged energy landscape that was better at discriminating native from non-native binding modes (Davis & Baker, 2009; Lemmon & Meiler, 2012). The entire structure was then scored using the partial covalent interactions energy function, which was developed to score H-bonds more accurately.
Molecular dynamics simulations
To make the docking results mimic the physiological state of protein molecules, we performed MD simulations on PDB 6W4H, which contained both nsp16 and nsp10, with the eight selected inhibitors and natural ligand SAM. The MD simulations were executed by the GROningen MAchine for Chemical Simulations (GROMACS) version 2020.1 with GROMOS96 43a1 force field parameters (Chiu et al., 2009). The topology of the drug molecules was created using PRODRG webserver (Schuttelkopf & van Aalten, 2004). The simulation of all the proteins and protein-drug complexes were run for a period of 100 ns. To make the system electrostatically neutral, counter ions were added to the protein-drug complexes. The complexes were solvated within a 10 nm SPC/E (extended simple point charge) water cube (Khan et al., 2020). The protein-drug complexes were minimized in multiple steps using the steepest descent method, where minimizations of the whole system, water cube, and non-heavy atoms of the complexes were accomplished. The entire systems were then progressively heated up to 300 K on a time scale of 100 ps. The equilibration steps were performed in two different phases, one with constant pressure and temperature (NPT) and the other with steady volume and temperature (NVT). Various structural parameters, like Root Mean Square Deviation (RMSD), Root Mean Square Fluctuations (RMSF), Radius of Gyration (Rg), Principal Component Analysis (PCA) based on essential dynamics (ED) approach, Inter-molecular Hydrogen Bonding (H-bonding), and Solvent-Accessible Surface Area (SASA) were calculated as a function of time to explore the structural behavior of the proteins and protein-drug complexes.
Binding affinity calculations using MM-PBSA
The binding affinity between the protein and drug molecules were calculated using molecular mechanics/Poisson-Boltzmann surface area (MM-PBSA) method [84]. MM-PBSA utilizes the effects of thermal averaging with a force field/continuum solvent model to post-process a series of representative snapshots from MD trajectories (Poli et al., 2020). The method expresses the binding affinity (ΔG bind) as the difference between the free binding energy of the complex and the free binding energy of the receptor plus the ligand. In this study, the binding free energy was calculated as the average of the MM-PBSA obtained from 400 equally distributed snapshots of MD trajectory of each protein–ligand complex after removing the water molecules by using GMXPBSA 2.1 tool available in GROMACS (Paissoni et al., 2015). A g_mmpbsa tool was used calculate the contribution of each residue to the total binding free energy (Kumari et al., 2014).
Supplementary Material
Funding Statement
This work was partially supported by grants from the Department of Defense (DoD) Breast Cancer Program BC161536, Elsa U. Pardee Foundation, DMC Foundation and Molecular Therapeutics Program of Karmanos Cancer Institute to Dr. Zeng-Quan Yang; and by funding from Susan G. Komen GTDR14299438 and Wayne State University Graduate School Dean Mathur Fellowship to Morenci Manning.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Aggarwal, V., Tuli, H. S., Thakral, F., Singhal, P., Aggarwal, D., Srivastava, S., Pandey, A., Sak, K., Varol, M., Khan, M. A., & Sethi, G. (2020). Molecular mechanisms of action of hesperidin in cancer: Recent trends and advancements. Experimental Biology and Medicine (Maywood, N.J.).), 245(5), 486–497. 10.1177/1535370220903671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi, W., Blanjoie, A., Vasseur, J. J., Debart, F., Canard, B., & Decroly, E. (2017). Binding of the methyl donor S-adenosyl-l-methionine to Middle East respiratory syndrome coronavirus 2'-O-methyltransferase nsp16 promotes recruitment of the allosteric activator nsp10. Journal of Virology, 91(5), e02217-16. 10.1128/JVI.02217-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayadi, L., Galvanin, A., Pichot, F., Marchand, V., & Motorin, Y. (2019). RNA ribose methylation (2'-O-methylation): Occurrence, biosynthesis and biological functions. Biochimica et Biophysica Acta Gene Regulatory Mechanisms, 1862(3), 253–269. 10.1016/j.bbagrm.2018.11.009 [DOI] [PubMed] [Google Scholar]
- Bai, L., Li, X., He, L., Zheng, Y., Lu, H., Li, J., Zhong, L., Tong, R., Jiang, Z., Shi, J., & Li, J. (2019). Antidiabetic potential of flavonoids from traditional Chinese medicine: A review. The American Journal of Chinese Medicine, 47(5), 933–957. 10.1142/S0192415X19500496 [DOI] [PubMed] [Google Scholar]
- Belanger, F., Stepinski, J., Darzynkiewicz, E., & Pelletier, J. (2010). Characterization of hMTr1, a human Cap1 2'-O-ribose methyltransferase. The Journal of Biological Chemistry, 285(43), 33037–33044. 10.1074/jbc.M110.155283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, I. M. (2014). Calcitonin gene-related peptide receptor antagonists: New therapeutic agents for migraine. Journal of Medicinal Chemistry, 57(19), 7838–7858. 10.1021/jm500364u [DOI] [PubMed] [Google Scholar]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The protein data bank. Nucleic Acids Research, 28(1), 235–242. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj, V. K., & Purohit, R. (2020). A new insight into protein-protein interactions and the effect of conformational alterations in PCNA. International Journal of Biological Macromolecules, 148, 999–1009. 10.1016/j.ijbiomac.2020.01.212 [DOI] [PubMed] [Google Scholar]
- Bhardwaj, V. K., Singh, R., Sharma, J., Rajendran, V., Purohit, R., & Kumar, S. (2020). Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1766572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., Wirecki, T. K., de Crécy-Lagard, V., Ross, R., Limbach, P. A., Kotter, A., Helm, M., & Bujnicki, J. M. (2018). Modomics: A database of RNA modification pathways. 2017 update. Nucleic Acids Research, 46(D1), D303–D307. 10.1093/nar/gkx1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet, M., Debarnot, C., Imbert, I., Selisko, B., Snijder, E. J., Canard, B., & Decroly, E. (2010). In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathogens, 6(4), e1000863. 10.1371/journal.ppat.1000863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bügl, H., Fauman, E. B., Staker, B. L., Zheng, F., Kushner, S. R., Saper, M. A., Bardwell, J. C., & Jakob, U. (2000). RNA methylation under heat shock control. Molecular Cell, 6(2), 349–360. 10.1016/S1097-2765(00)00035-6 [DOI] [PubMed] [Google Scholar]
- Byszewska, M., Śmietański, M., Purta, E., & Bujnicki, J. M. (2014). RNA methyltransferases involved in 5' cap biosynthesis. RNA Biology, 11(12), 1597–1607. 10.1080/15476286.2015.1004955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C-k., Jeyachandran, S., Hu, N.-J., Liu, C.-L., Lin, S.-Y., Wang, Y.-S., Chang, Y.-M., & Hou, M.-H. (2016). Structure-based virtual screening and experimental validation of the discovery of inhibitors targeted towards the human coronavirus nucleocapsid protein. Molecular Biosystems, 12(1), 59–66. 10.1039/c5mb00582e [DOI] [PubMed] [Google Scholar]
- Chen, Y., Su, C., Ke, M., Jin, X., Xu, L., Zhang, Z., Wu, A., Sun, Y., Yang, Z., Tien, P., Ahola, T., Liang, Y., Liu, X., & Guo, D. (2011). Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2'-O-methylation by nsp16/nsp10 protein complex. PLoS Pathogens, 7(10), e1002294. 10.1371/journal.ppat.1002294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, S. W., Pandit, S. A., Scott, H. L., & Jakobsson, E. (2009). An improved united atom force field for simulation of mixed lipid bilayers. The Journal of Physical Chemistry B, 113(9), 2748–2763. 10.1021/jp807056c [DOI] [PubMed] [Google Scholar]
- Coronaviridae Study Group of the International Committee on Taxonomy of, V. (2020). The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology, 5(4), 536–544. 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, I. W., & Baker, D. (2009). RosettaLigand docking with full ligand and receptor flexibility. Journal of Molecular Biology, 385(2), 381–392. 10.1016/j.jmb.2008.11.010 [DOI] [PubMed] [Google Scholar]
- Decroly, E., Debarnot, C., Ferron, F., Bouvet, M., Coutard, B., Imbert, I., Gluais, L., Papageorgiou, N., Sharff, A., Bricogne, G., Ortiz-Lombardia, M., Lescar, J., & Canard, B. (2011). Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2'-O-methyltransferase nsp10/nsp16 complex. PLoS Pathogens, 7(5), e1002059. 10.1371/journal.ppat.1002059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Z., Sun, G., & Zhu, Z. (2018). Hesperidin attenuates influenza A virus (H1N1) induced lung injury in rats through its anti-inflammatory effect. Antiviral Therapy, 23(7), 611–615. 10.3851/IMP3235 [DOI] [PubMed] [Google Scholar]
- Drilon, A. (2019). TRK inhibitors in TRK fusion-positive cancers. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 30(Suppl 8), viii23–viii30. 10.1093/annonc/mdz282 [DOI] [PubMed] [Google Scholar]
- Encinar, J. A., & Menendez, J. A. (2020). Potential drugs targeting early innate immune evasion of SARS-coronavirus 2 via 2'-O-methylation of viral RNA. Viruses, 12(5), 525. 10.3390/v12050525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freude, K., Hoffmann, K., Jensen, L.-R., Delatycki, M. B., Des Portes, V., Moser, B., Hamel, B., van Bokhoven, H., Moraine, C., Fryns, J.-P., Chelly, J., Gécz, J., Lenzner, S., Kalscheuer, V. M., & Ropers, H.-H. (2004). Mutations in the FTSJ1 gene coding for a novel S-adenosylmethionine-binding protein cause nonsyndromic X-linked mental retardation. American Journal of Human Genetics, 75(2), 305–309. 10.1086/422507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Z. G., & Jacobson, K. A. (2011). Emerging adenosine receptor agonists: An update. Expert Opinion on Emerging Drugs, 16(4), 597–602. 10.1517/14728214.2011.644786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss, G. K., Carter, V. S., He, Y., Kwieciszewski, B. K., Holzman, T., Korth, M. J., Lazaro, C. A., Fausto, N., Bumgarner, R. E., & Katze, M. G. (2003). Gene expression profiling of the cellular transcriptional network regulated by alpha/beta interferon and its partial attenuation by the hepatitis C virus nonstructural 5A protein. Journal of Virology, 77(11), 6367–6375. 10.1128/jvi.77.11.6367-6375.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioia, D., Bertazzo, M., Recanatini, M., Masetti, M., & Cavalli, A. (2017). Dynamic docking: A paradigm shift in computational drug discovery. Molecules, 22(11), 2029. 10.3390/molecules22112029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales-van Horn, S. R., & Sarnow, P. (2017). Making the mark: The role of adenosine modifications in the life cycle of RNA viruses. Cell Host & Microbe, 21(6), 661–669. 10.1016/j.chom.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy, M. P., Shaw, M., Weiner, C. L., Hobson, L., Stark, Z., Rose, K., Kalscheuer, V. M., Gecz, J., & Phizicky, E. M. (2015). Defects in tRNA anticodon loop 2'-O-methylation are implicated in nonsyndromic X-linked intellectual disability due to mutations in FTSJ1. Human Mutation, 36(12), 1176–1187. 10.1002/humu.22897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager, J., Staker, B. L., Bugl, H., & Jakob, U. (2002). Active site in RrmJ, a heat shock-induced methyltransferase. The Journal of Biological Chemistry, 277(44), 41978–41986. 10.1074/jbc.M205423200 [DOI] [PubMed] [Google Scholar]
- Haggag, Y. A., El-Ashmawy, N. E., & Okasha, K. M. (2020). Is hesperidin essential for prophylaxis and treatment of COVID-19 Infection? Medical Hypotheses, 144, 109957. 10.1016/j.mehy.2020.109957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata, A., Shionyu-Mitsuyama, C., Nakae, S., Shionyu, M., Ota, M., Kanaya, S., & Shirai, T. (2020). Knowledge-based structural models of SARS-CoV-2 proteins and their complexes with potential drugs. FEBS Letters, 594(12), 1960–1973. 10.1002/1873-3468.13806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. (2020). Using Dali for protein structure comparison. Methods Mol. Biol.), 2112, 29–42. 10.1007/978-1-0716-0270-6_3 [DOI] [PubMed] [Google Scholar]
- Hyde, J. L., & Diamond, M. S. (2015). Innate immune restriction and antagonism of viral RNA lacking 2׳-O methylation. Virology, 479–480, 66–74. 10.1016/j.virol.2015.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inesta-Vaquera, F., & Cowling, V. H. (2017). Regulation and function of CMTR1-dependent mRNA cap methylation. Wiley Interdisciplinary Reviews: RNA, 8(6). 10.1002/wrna.1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, K. A., Tosh, D. K., Jain, S., & Gao, Z. G. (2019). Historical and current adenosine receptor agonists in preclinical and clinical development. Frontiers in Cellular Neuroscience, 13, 124. 10.3389/fncel.2019.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadumuri, R. V., & Janga, S. C. (2018). Epitranscriptomic code and its alterations in human disease. Trends in Molecular Medicine, 24(10), 886–903. 10.1016/j.molmed.2018.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, R. J., Jha, R. K., Amera, G. M., Jain, M., Singh, E., Pathak, A., Singh, R. P., Muthukumaran, J., & Singh, A. K. (2020). Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2'-O-ribose methyltransferase. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen, D. B., Decornez, H., Furr, J. R., & Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews Drug Discovery, 3(11), 935–949. 10.1038/nrd1549 [DOI] [PubMed] [Google Scholar]
- Kumari, R., Kumar, R., Open Source Drug Discovery Consortium , & Lynn, A. (2014). g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations . Journal of Chemical Information and Modeling, 54(7), 1951–1962. 10.1021/ci500020m [DOI] [PubMed] [Google Scholar]
- Lagarde, N., Goldwaser, E., Pencheva, T., Jereva, D., Pajeva, I., Rey, J., Tuffery, P., Villoutreix, B. O., & Miteva, M. A. (2019). A free web-based protocol to assist structure-based virtual screening experiments. International Journal of Molecular Sciences, 20(18), 4648. 10.3390/ijms20184648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde, N., Rey, J., Gyulkhandanyan, A., Tuffery, P., Miteva, M. A., & Villoutreix, B. O. (2018). Online structure-based screening of purchasable approved drugs and natural compounds: Retrospective examples of drug repositioning on cancer targets. Oncotarget, 9(64), 32346–32361. 10.18632/oncotarget.25966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko, L. M., Jakob, C. G., Edalji, R. P., Qiu, W., Montgomery, D., Digiammarino, E. L., Hansen, T. M., Risi, R. M., Frey, R., Manaves, V., Shaw, B., Algire, M., Hessler, P., Lam, L. T., Uziel, T., Faivre, E., Ferguson, D., Buchanan, F. G., Martin, R. L., … Bromberg, K. D. (2017). Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature, 550(7674), 128–132. 10.1038/nature24028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. W., & Bogenhagen, D. F. (2014). Assignment of 2'-O-methyltransferases to modification sites on the mammalian mitochondrial large subunit 16 S ribosomal RNA (rRNA). The Journal of Biological Chemistry, 289(36), 24936–24942. 10.1074/jbc.C114.581868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. W., Okot-Kotber, C., LaComb, J. F., & Bogenhagen, D. F. (2013). Mitochondrial ribosomal RNA (rRNA) methyltransferase family members are positioned to modify nascent rRNA in foci near the mitochondrial DNA nucleoid. The Journal of Biological Chemistry, 288(43), 31386–31399. 10.1074/jbc.M113.515692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, G., & Meiler, J. (2012). Rosetta Ligand docking with flexible XML protocols. Methods in Molecular Biology (Clifton, N.J.).), 819, 143–155. 10.1007/978-1-61779-465-0_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B., Clohisey, S. M., Chia, B. S., Wang, B., Cui, A., Eisenhaure, T., Schweitzer, L. D., Hoover, P., Parkinson, N. J., Nachshon, A., Smith, N., Regan, T., Farr, D., Gutmann, M. U., Bukhari, S. I., Law, A., Sangesland, M., Gat-Viks, I., Digard, P., … Hacohen, N. (2020). Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nature Communications, 11(1), 164. 10.1038/s41467-019-13965-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S., & Mason, C. E. (2014). The pivotal regulatory landscape of RNA modifications. Annual Review of Genomics and Human Genetics, 15, 127–150. 10.1146/annurev-genom-090413-025405 [DOI] [PubMed] [Google Scholar]
- Lipton, R. B., Croop, R., Stock, E. G., Stock, D. A., Morris, B. A., Frost, M., Dubowchik, G. M., Conway, C. M., Coric, V., & Goadsby, P. J. (2019). Rimegepant, an oral calcitonin gene-related peptide receptor antagonist, for migraine. New England Journal of Medicine, 381(2), 142–149. 10.1056/NEJMoa1811090 [DOI] [PubMed] [Google Scholar]
- Li, J., Wang, Y. ‐N., Xu, B. ‐S., Liu, Y. ‐P., Zhou, M., Long, T., Li, H., Dong, H., Nie, Y., Chen, P. R., Wang, E. ‐D., & Liu, R. ‐J. (2020). Intellectual disability-associated gene ftsj1 is responsible for 2'-O-methylation of specific tRNAs. EMBO Reports, 21(8), e50095. 10.15252/embr.202050095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia, E. H. B., Assis, L. C., de Oliveira, T. A., da Silva, A. M., & Taranto, A. G. (2020). Structure-based virtual screening: From classical to artificial intelligence. Frontiers in Chemistry, 8, 343. 10.3389/fchem.2020.00343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning, M., Jiang, Y., Wang, R., Liu, L., Rode, S., Bonahoom, M., Kim, S., & Yang, Z.-Q. (2020). Pan-cancer analysis of RNA methyltransferases identifies FTSJ3 as a potential regulator of breast cancer progression. RNA Biology, 17(4), 474–486. 10.1080/15476286.2019.1708549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo, J., Olmos, D., Dumez, H., Poondru, S., Samberg, N. L., Barr, S., Van Tornout, J. M., Jie, F., Sandhu, S., Tan, D. S., Moreno, V., LoRusso, P. M., Kaye, S. B., & Schöffski, P. (2016). A first in man, dose-finding study of the mTORC1/mTORC2 inhibitor OSI-027 in patients with advanced solid malignancies. British Journal of Cancer, 114(8), 889–896. 10.1038/bjc.2016.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery, V. D., Debbink, K., & Baric, R. S. (2014). Coronavirus non-structural protein 16: Evasion, attenuation, and possible treatments. Virus Research, 194, 191–199. 10.1016/j.virusres.2014.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery, V. D., Yount, B. L., Josset, L., Gralinski, L. E., Scobey, T., Agnihothram, S., Katze, M. G., & Baric, R. S. (2014). Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2'-o-methyltransferase activity. Journal of Virology, 88(8), 4251–4264. 10.1128/JVI.03571-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzband, R., & Pager, C. T. (2020). Epitranscriptomic marks: Emerging modulators of RNA virus gene expression. Wiley Interdisciplinary Reviews RNA, 11(3), e1576. 10.1002/wrna.1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paissoni, C., Spiliotopoulos, D., Musco, G., & Spitaleri, A. (2015). GMXPBSA 2.1: A GROMACS tool to perform MM/PBSA and computational alanine scanning. Computer Physics Communications, 186, 105–107. 10.1016/j.cpc.2014.09.010 [DOI] [Google Scholar]
- Panda, P. K., Arul, M. N., Patel, P., Verma, S. K., Luo, W., Rubahn, H.-G., Mishra, Y. K., Suar, M., & Ahuja, R. (2020). Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Science Advances, 6(28), eabb8097. 10.1126/sciadv.abb8097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman, S., & Netland, J. (2009). Coronaviruses post-SARS: Update on replication and pathogenesis. Nature Reviews Microbiology, 7(6), 439–450. 10.1038/nrmicro2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pinzi, L., & Rastelli, G. (2019). Molecular docking: Shifting paradigms in drug discovery. International Journal of Molecular Sciences, 20(18), 4331. 10.3390/ijms20184331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli, G., Granchi, C., Rizzolio, F., & Tuccinardi, T. (2020). Application of MM-PBSA methods in virtual screening. Molecules, 25(8), 1971. 10.3390/molecules25081971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran, V., Gopalakrishnan, C., & Sethumadhavan, R. (2018). Pathological role of a point mutation (T315I) in BCR-ABL1 protein-A computational insight. Journal of Cellular Biochemistry, 119(1), 918–925. 10.1002/jcb.26257 [DOI] [PubMed] [Google Scholar]
- Ringeard, M., Marchand, V., Decroly, E., Motorin, Y., & Bennasser, Y. (2019). FTSJ3 is an RNA 2'-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature, 565(7740), 500–504. 10.1038/s41586-018-0841-4 [DOI] [PubMed] [Google Scholar]
- Roundtree, I. A., Evans, M. E., Pan, T., & He, C. (2017). Dynamic RNA modifications in gene expression regulation. Cell, 169(7), 1187–1200. 10.1016/j.cell.2017.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F., & Schroeder, M. (2015). PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Research, 43(W1), W443–447. 10.1093/nar/gkv315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore, C. A., Moore, E. L., Calamari, A., Cook, J. J., Michener, M. S., O'Malley, S., Miller, P. J., Sur, C., Williams, D. L., Zeng, Z., Danziger, A., Lynch, J. J., Regan, C. P., Fay, J. F., Tang, Y. S., Li, C.-C., Pudvah, N. T., White, R. B., Bell, I. M., … Kane, S. A. (2010). Pharmacological properties of MK-3207, a potent and orally active calcitonin gene-related peptide receptor antagonist. The Journal of Pharmacology and Experimental Therapeutics, 333(1), 152–160. 10.1124/jpet.109.163816 [DOI] [PubMed] [Google Scholar]
- Sander, T., Freyss, J., von Korff, M., & Rufener, C. (2015). DataWarrior: An open-source program for chemistry aware data visualization and analysis. Journal of Chemical Information and Modeling, 55(2), 460–473. 10.1021/ci500588j [DOI] [PubMed] [Google Scholar]
- Sartore-Bianchi, A., Pizzutilo, E. G., Marrapese, G., Tosi, F., Cerea, G., & Siena, S. (2020). Entrectinib for the treatment of metastatic NSCLC: Safety and efficacy. Expert Review of Anticancer Therapy, 20(5), 333–341. 10.1080/14737140.2020.1747439 [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny, D., Dror, O., Inbar, Y., Nussinov, R., & Wolfson, H. J. (2008). PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Research, 36(Web Server issue), W223–228. (Web Server issue), 10.1093/nar/gkn187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttelkopf, A. W., & van Aalten, D. M. (2004). PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica Section D, Biological Crystallography, 60(Pt 8), 1355–1363. 10.1107/S0907444904011679 [DOI] [PubMed] [Google Scholar]
- Scott, L. J. (2020). Rimegepant: First Approval. Drugs, 80(7), 741–746. 10.1007/s40265-020-01301-3 [DOI] [PubMed] [Google Scholar]
- Sevajol, M., Subissi, L., Decroly, E., Canard, B., & Imbert, I. (2014). Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Research, 194, 90–99. 10.1016/j.virusres.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H., Wei, J., & He, C. (2019). Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Molecular Cell, 74(4), 640–650. 10.1016/j.molcel.2019.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuta, C., Popr, M., Muller, T., Jindrich, J., Kahle, M., Sedlak, D., Svozil, D., & Bartunek, P. (2017). Probes &Drugs portal: An interactive, open data resource for chemical biology. Nature Methods, 14(8), 759–760. 10.1038/nmeth.4365 [DOI] [PubMed] [Google Scholar]
- Slater, O., & Kontoyianni, M. (2019). The compromise of virtual screening and its impact on drug discovery. Expert Opinion on Drug Discovery, 14(7), 619–637. 10.1080/17460441.2019.1604677 [DOI] [PubMed] [Google Scholar]
- Śledź, P., & Caflisch, A. (2018). Protein structure-based drug design: From docking to molecular dynamics. Current Opinion in Structural Biology, 48, 93–102. 10.1016/j.sbi.2017.10.010 [DOI] [PubMed] [Google Scholar]
- Smietanski, M., Werner, M., Purta, E., Kaminska, K. H., Stepinski, J., Darzynkiewicz, E., Nowotny, M., & Bujnicki, J. M. (2014). Structural analysis of human 2'-O-ribose methyltransferases involved in mRNA cap structure formation. Nature Communications, 5, 3004. 10.1038/ncomms4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehr, P. M., Dhalla, A. K., Zack, J., Wang, X., Ho, Y. L., Bingham, J., & Belardinelli, L. (2013). Reduction of free fatty acids, safety, and pharmacokinetics of oral GS-9667, an A(1) adenosine receptor partial agonist. The Journal of Clinical Pharmacology, 53(4), 385–392. 10.1002/jcph.9 [DOI] [PubMed] [Google Scholar]
- Su, A. I., Pezacki, J. P., Wodicka, L., Brideau, A. D., Supekova, L., Thimme, R., Wieland, S., Bukh, J., Purcell, R. H., Schultz, P. G., & Chisari, F. V. (2002). Genomic analysis of the host response to hepatitis C virus infection. Proceedings of the National Academy of Sciences of the United States of America, 99(24), 15669–15674. 10.1073/pnas.202608199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subissi, L., Imbert, I., Ferron, F., Collet, A., Coutard, B., Decroly, E., & Canard, B. (2014). SARS-CoV ORF1b-encoded nonstructural proteins 12-16: Replicative enzymes as antiviral targets. Antiviral Research, 101, 122–130. 10.1016/j.antiviral.2013.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazikeh-Lemeski, E., Moradi, S., Raoufi, R., Shahlaei, M., Janlou, M. A. M., & Zolghadri, S. (2020). Targeting SARS-COV-2 non-structural protein 16: A virtual drug repurposing study. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1779133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejada, S., Pinya, S., Martorell, M., Capo, X., Tur, J. A., Pons, A., & Sureda, A. (2018). Potential anti-inflammatory effects of hesperidin from the genus citrus. Current Medicinal Chemistry, 25(37), 4929–4945. 10.2174/0929867324666170718104412 [DOI] [PubMed] [Google Scholar]
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., & Berendsen, H. J. (2005). GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 26(16), 1701–1718. 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- Vijayan, V., Pant, P., Vikram, N., Kaur, P., Singh, T. P., Sharma, S., & Sharma, P. (2020). Identification of promising drug candidates against NSP16 of SARS-CoV-2 through computational drug repurposing study. Journal of Biomolecular Structure and Dynamics, 1–15. 10.1080/07391102.2020.1802349 [DOI] [PubMed] [Google Scholar]
- Viswanathan, T., Arya, S., Chan, S.-H., Qi, S., Dai, N., Misra, A., Park, J.-G., Oladunni, F., Kovalskyy, D., Hromas, R. A., Martinez-Sobrido, L., & Gupta, Y. K. (2020). Structural basis of RNA cap modification by SARS-CoV-2. Nature Communications, 11(1), 3718. 10.1038/s41467-020-17496-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkamer, A., Kuhn, D., Rippmann, F., & Rarey, M. (2012). DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics (Oxford, England)), 28(15), 2074–2075. 10.1093/bioinformatics/bts310 [DOI] [PubMed] [Google Scholar]
- Wallace, A. C., Laskowski, R. A., & Thornton, J. M. (1995). LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Engineering, 8(2), 127–134. 10.1093/protein/8.2.127 [DOI] [PubMed] [Google Scholar]
- Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., Wang, Y., Wang, Q., Xu, Y., Li, M., Li, X., Zheng, M., Chen, L., & Li, H. (2020). Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B, 10(5), 766–788. 10.1016/j.apsb.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., Hu, Y., Tao, Z.-W., Tian, J.-H., Pei, Y.-Y., Yuan, M.-L., Zhang, Y.-L., Dai, F.-H., Liu, Y., Wang, Q.-M., Zheng, J.-J., Xu, L., Holmes, E. C., & Zhang, Y.-Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., & Skolnick, J. (2005). TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Research, 33(7), 2302–2309. 10.1093/nar/gki524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G. F., & Tan, W. (2020). A novel coronavirus from patients with pneumonia in China, 2019. The New England Journal of Medicine, 382(8), 727–733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züst, R., Cervantes-Barragan, L., Habjan, M., Maier, R., Neuman, B. W., Ziebuhr, J., Szretter, K. J., Baker, S. C., Barchet, W., Diamond, M. S., Siddell, S. G., Ludewig, B., & Thiel, V. (2011). Ribose 2'-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nature Immunology, 12(2), 137–143. 10.1038/ni.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.