

Abstract
COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) primarily appeared in Wuhan, China, in December 2019. At present, no proper therapy and vaccinations are available for the disease, and it is increasing day by day with a high mortality rate. Pharmacophore based virtual screening of the selected natural product databases followed by Glide molecular docking and dynamics studies against SARS-CoV-2 main protease was investigated to identify potential ligands that may act as inhibitors. The molecules SN00293542 and SN00382835 revealed the highest docking score of −14.57 and −12.42 kcal/mol, respectively, when compared with the co-crystal ligands of PDB-6Y2F (O6K) and 6W63 (X77) of the SARS-CoV-2 Mpro. To further validate the interactions of top scored molecules SN00293542 and SN00382835, molecular dynamics study of 100 ns was carried out. This indicated that the protein-ligand complex was stable throughout the simulation period, and minimal backbone fluctuations have ensued in the system. Post-MM-GBSA analysis of molecular dynamics data showed free binding energy-71.7004 +/− 7.98, −56.81+/− 7.54 kcal/mol, respectively. The computational study identified several ligands that may act as potential inhibitors of SARS-CoV-2 Mpro. The top-ranked molecules SN00293542, and SN00382835 occupied the active site of the target, the main protease like that of the co-crystal ligand. These molecules may emerge as a promising ligands against SARS-CoV-2 and thus needs further detailed investigations.
Communicated by Ramaswamy H. Sarma
Keywords: Covid-19, main protease, pharmacophore, virtual screening, molecular docking, molecular dynamics, MM-GBSA
Graphical Abstract

Introduction
Coronaviruses are a group of viruses that generally affect the respiratory system of mammals, including humans, and can cause severe acute respiratory tract infections. After the last two attacks by Severe Acute Respiratory Syndrome Coronavirus-1 (SARS-CoV-1) and the Middle East Respiratory Syndrome Coronavirus (MERS-CoV), which caused high morbidity and mortality in some parts of the world, its third new strain known as SARS-CoV-2 is causing havoc across the entire globe. Initially, the virus was originated from Wuhan city, Hubei province of China, in late December 2019 and then later spread across the entire globe affecting more than 200 countries (Adhikari et al., 2020). WHO declared coronavirus disease 2019 (COVID-19), a pandemic on March 11, 2020, and as of June 2, 2020, is responsible for 376 320 deaths globally. The symptoms associated with COVID-19 include pyrexia, cough, hemoptysis, muscle soreness, diarrhea, lymphopenia, dyssomnia, dyspnea, and dysgeusia. Certain atypical symptoms of coronavirus infections have also been reported, like gastrointestinal distress and lower respiratory tract infections (Keyhan et al., 2020; Rothan & Byrareddy, 2020).
Currently, no drug or vaccine has been approved for the treatment or prevention of COVID-19. Considerable efforts are being taken to repurpose or develop novel molecules for the treatment of COVID-19. The targets that are currently being explored for the development of novel inhibitors include SARS-CoV-2 Spike (S) protein, Angiotensin-converting enzyme-2 (ACE-2), human proteases such as Transmembrane protease, serine 2 (TMPRSS2), Furin, viral proteases like RNA-dependent RNA-polymerase (RdRp) and Papain like protease-2 (PLpro) (Andersen et al., 2020; Bestle et al., 2020; Coutard et al., 2020; Hoffmann et al., 2020; Walls et al., 2020; Xia, Liu, et al., 2020; Xia, Zhu, et al., 2020). Of all the targets that are being explored for SARS-CoV-2, the Main protease (Mpro) of SARS-CoV-2, also known as 3-Chymotrypsin like protease (3-CLpro) has gathered much attention from the scientists around the world owing to its crucial role in the life cycle of SARS-CoV-2. Sequence similarity studies have revealed that the Mpro of SARS-CoV-2 is 96% identical to that of Mpro of SARS-CoV-1. SARS-CoV-2 Mpro cleaves the pp1ab at 11 specific sites to release 12 nsps (nsp4, nsp6-16), the recognition sites being Leu-Gln↓(Ser, Ala, Gly) (Wu et al., 2020; Zhang et al., 2020). Structurally Mpro is a dimer, and each monomer consists of three domains, namely domain I, II, and III. The substrate-binding site containing the catalytic dyad (Cys145 and His61) is positioned between domains II and III (Jin et al., 2020a). The inhibitors designed to target Mpro broadly fall into two groups- peptidomimetic inhibitors and small molecule-based inhibitors (Pillaiyar et al., 2016). Peptidomimetic inhibitors were designed by attaching a “warhead” groups like Michael acceptors, ketones, aldehydes, halomethyl ketones, etc. to a peptide that resembles the natural substrate. In general, these inhibitors exhibit their action via two steps- (i) they first bind non-covalently with the enzyme such that the “warhead” is in close vicinity with the catalytic residue. (ii) this is followed by a nucleophilic attack by Cys145 that results in the formation of a covalent bond, thereby inhibiting the enzyme reversibly or irreversibly (Pillaiyar et al., 2016). Recently, Jin et al. reported the irreversible inhibition of SARS-CoV-2 Mpro by Michael acceptor type peptidomimetic inhibitor N3 (Figure 1) (Jin et al., 2020a). The X-ray crystal structure of N3 in the binding pocket of SARS-CoV-2 Mpro (PDB ID- 6LU7) is shown in Figure 2A, B. Dai et al. reported the SARS-CoV-2 Mpro inhibitory potential of two novel peptidomimetic aldehydes 1 and 2 (Figure 1). The peptidomimetic aldehydes 1 and 2 exhibited IC50 values of 0.053 µM and 0.040 µM, respectively, against the Mpro of SARS-CoV-2 (Dai et al., 2020). Zhang et al. reported the optimization of α-ketoamide inhibitors targeting the SARS-CoV-2 Mpro. The α-ketoamides offers an advantage over the Michael acceptor or aldehydic based inhibitors in that its warhead is capable of forming two hydrogen bond interaction with catalytic residues rather than one hydrogen bond interaction as in the case of Michael acceptors or aldehydes (Zhang et al., 2020). The crystal structure of N3 and α-ketoamide inhibitor 3, in the binding site of SARS-CoV-2 Mpro (PDB ID- 6LU7, and PDB ID- 6Y2F) is depicted in Figures 2A, B and 3A, B. A pharmacophore is an ensemble of spatial and electronic features that is necessary for interaction with a macromolecular target that results in a biological response. Pharmit (http://pharmit.csb.pitt.edu/) is an open-source web server that provides a platform for the virtual screening of large compound databases based on pharmacophore, molecular shape, and energy minimization. The users can input a pre-defined pharmacophore query, or Pharmit can elucidate the pharmacophore queries from the receptor and/or ligand structures (Sunseri & Koes, 2016). Molecular docking is a widely used approach for structure-based drug discovery that enables modeling of interaction between a macromolecule and a ligand. It is used to forecast the ideal conformation of the ligand in the macromolecule (binding pose) and, the binding affinity. In structural biology, molecular dynamics (MD) is a powerful computational technique to study the dynamics of macromolecules like proteins and nucleic acids.
Figure 1.

Structure of inhibitors of SARS-CoV-2 Mpro.
Figure 2.

(A) 3D interactions of compound N3 with active site residues of the target protein, color interpretation- yellow hydrogen bond, pink- aromatic hydrogen bond; (B) 3D view of compound N3 inside the binding pocket of SARS-CoV-2 Mpro (B) (PDB ID- 6LU7).
Figure 3.

(A) 3D interactions of compound 3 with active site residues of the target protein, color interpretation- yellow hydrogen bond, pink- aromatic hydrogen bond; (B) 3D view of compound 3 inside the binding pocket of SARS-CoV-2 Mpro (B) (PDB ID- 6Y2F).
In the current study, two crystal structures of SARS-CoV-2 Mpro in complex with a covalent inhibitor O6K (PDB ID- 6Y2F), and a non-covalent inhibitor X77 (PDB ID- 6W63) were used for the generation of two pharmacophore models (Figure 4). A large number of studies have been done in the past to identify anti-CoV natural products (Islam et al., 2020; Lin et al., 2014; Mani et al., 2020). In the past, different classes of natural products like flavonoids and terpenoids have also been reported to inhibit Mpro of SARS-CoV (Pillaiyar et al., 2016). Recently, shikonin, a naturally occurring naphthoquinone, was reported to inhibit SARS CoV-2 Mpro (Jin et al., 2020b). Recent studies have also highlighted the importance of Traditional Chinese Medicine for the treatment or prevention of COVID-19 (Luo et al., 2020; Yang et al., 2020). Therefore, a natural products-based treatment regimen promises to be a useful ally in the raging war against COVID-19. The generated pharmacophore models were used as a query for the virtual screening of natural product databases like Supernatural product (SNP), Zinc natural database, and Marine Natural Products (MNP), with the hope of identifying potential inhibitors of SARS-CoV-2 Mpro. The generated HITS from the virtual screening were then docked into their respective proteins. Finally, the interactions of the top molecule were validated by performing MD simulation study.
Figure 4.

Workflow adopted during the current study. The numbers inside the brackets indicate the total number of molecules.
Materials and method
Pharmacophore modeling and virtual screening
The pharmacophore models were generated by using the Pharmit server (http://pharmit.csb.pitt.edu/) (Sunseri & Koes, 2016). The models were constructed by using the selected PDB codes 6Y2F and 6W63 obtained from the RCSB protein data bank (http://www.rcsb.org/structure/6y2f, https://www.rcsb.org/structure/6w63). The pharmacophoric models were generated by keeping the default parameters in the server. The generated models were used for virtual screening of Supernatural product (SNP) database consisting of 274,363 molecules, Zinc natural database consisting of 120,720 molecules (Sterling & Irwin, 2015) and Marine Natural Products (MNP) database consisting of 14,064 molecules.
Molecular docking studies
Docking studies of the database compounds were performed using the Glide module of Schrodinger software (Schrödinger Release, 2019c) installed on Intel XenonW3565 processor and Ubuntu enterprise version 14.04 as an operating system. The selected target protein structure was retrieved from the RCSB protein data bank.
Ligand preparation
The ligands used as an input for docking study was downloaded from virtual screening hits of the natural databases. Then, ligands were incorporated into the workstation, and the energy was minimized using OPLS3e (Optimized Potentials for Liquid Simulations) force field in the Ligprep module of the software (Schrodinger, 2019). This minimization helps to assign bond orders, the addition of hydrogens to the ligands, and conversion of 2D to 3D structure for the docking studies. The generated output file (Best conformations of the ligands) was further used for docking studies (Schrödinger Release, 2019b).
Protein preparation
Protein preparation wizard (Version 2019-1, Schrodinger) (Schrödinger Release, 2019e) is the primary tool in Schrodinger to prepare and minimize the energy of protein. Hydrogen atom was added to the protein, and charges were assigned. Generated Het states using Epik at pH 7.0 ± 2.0. Pre-process the protein and refine, modify the protein by analyzing the workspace water molecules and others. The critical water molecules remained the same, and the rest of the molecules apart from the heteroatoms was deleted. Finally, the protein was minimized using the OPLS3 force field. A grid was created by considering co-crystal ligand, which was included in the active site of the selected protein target (PDB-6Y2F and 6W63). After the final step of docking with the co-crystal ligand in extra precision(XP) mode, root mean square deviation (RMSD) was checked to validate the protein. (Schrödinger Release, 2019e).
Receptor grid generation
A receptor grid was generated around the proteins (PDB-6Y2F [co-ordinates X-11.0, Y- −0.61, Z-20.83, 10 × 10 × 10] and 6W63 [co-ordinates X- −20.46, Y- 18.17, Z- −26.28, 10 × 10 × 10]) by choosing the inhibitory ligand (X-ray pose of the ligand in the protein). The centroid of the ligand was selected to create a grid box around it, and Vander Waal radius of receptor atoms was scaled to 1.00 Å with a partial atomic charge of 0.25
MM-GBSA analysis
The MM-GBSA (Molecular Mechanics, the Generalized Born model and Solvent Accessibility) analysis was performed to investigate the free binding energies of the protein and ligand complexes. The prime module of Schrödinger software was used to calculate the optimal binding energy of the selected complexes whose docking score was lowest among all. For the analysis, VSGB 2.0 model was exploited, having OPLS-AA force field inclusive of an implicit solvent model in addition to physics-based modifications for π-π interactions, hydrophobic interactions, and hydrogen bonding self-contact interactions (Li et al., 2011)
In silico predicted physico-chemical parameters
The Physico-chemical parameters of the obtained hits after the docking studies were in silico predicted using the Qikprop module of Schrodinger. The diverse parameters predicted were molecular weight (M.Wt.), total solvent accessible surface area (SASA), number of hydrogen bond donor (HBD), number of hydrogen bond acceptor (HBA), octanol/water partition coefficient (log P), aqueous solubility (Log S), predicted apparent Caco-2 cell permeability in nm/sec (P Caco) and number of rotatable bonds (Rot) (QikProp Descriptors and Properties PISA, 2015; Schrödinger Release, 2019d).
Molecular dynamics and post-MM-GBSA analysis
MD study was performed using the Desmond module of Schrodinger software (Schrödinger Release, 2019a) through the system's builder panel; the orthorhombic simulation box was prepared with the simple point-charge (SPC) explicit water model in such a way that the minimum distance between the protein surface and the solvent surface is 10 Å. Protein-ligand docked complexes were solvated using the orthorhombic SPC water model (Mark & Nilsson, 2001). The solvated system was neutralized with counter ions, and physiological salt concentration was limited to 0.15 M. The receptor-ligand complex system was designated with the OPLS3 force field (Jorgensen et al., 1996). The simulation was for a total of 100 ns using NPT (Isothermal-Isobaric ensemble, constant temperature, and constant pressure, constant number of particles) ensemble (Kalibaeva et al., 2003) at a temperature of 300 K and atmospheric pressure (1.013 bars) with the default settings of relaxation before simulation. The MD simulation was run by using the MD simulation tool, the system with 36136 atoms including 10434water molecules loaded, and simulation time setup to 1000 ns. Further, for viewing the trajectories and creating a movie, _out.cms file was imported, and the movie was exported with high resolution (1280 × 1024) with improved quality. During the MD simulation, the trajectory was written with 2002 frames. To understand the stability of the complex during MD simulation, the protein backbone frames were aligned to the backbone of the initial frame. Finally, the analysis of the simulation interaction diagram was achieved after loading the _out.cms file and selected Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) in the analysis type to obtain the mentioned plots. To perform the post-MM-GBSA analysis, the thermal_MMGBSA.py script of the Prime/Desmond module of the Schrodinger suite was used (Masetti et al., 2020). The binding energy calculation was performed on the basis of this parameter- MM-GBSA ΔG Bind: The binding energy of the receptor and ligand as calculated by the Prime Energy, a Molecular Mechanics + Implicit Solvent Energy Function (kcal/mol).
Results and discussion
Pharmacophore modeling and virtual screening
A pharmacophore is an ensemble of spatial and electronic features that is necessary for interaction with a macromolecular target that results in a biological response. In the present study, two structure-based pharmacophore models were developed based on the crystal structure of SARS-CoV-2 co-crystallized with alpha-ketoamide 13b and non-covalent inhibitor X-77 (PDB ID- 6Y2F and PDB ID- 6W63, respectively) using Pharmit server that provides a setting for virtual screening of databases using appropriate pharmacophore models. The initially generated pharmacophore model for PDB 6Y2F is stemmed from the active site which includes the following essential features of ligand-
Four hydrogen bond acceptors (Acc) - F1 for interacting with amino-acid residues Gly143, Cys145; F2 for accepting a hydrogen bond from amino-acid His41, F4 and F5 for interacting with His163 and Glu166 amino-acids, respectively.
Two hydrogen bond donors (Don)- F3 for interacting with amino-acid Phe140, and F6 for interacting with amino-acid Glu166 (Figure 5).
Figure 5.

The pharmacophore model developed using the Pharmit server for the target protein (PDB ID- 6Y2F). Orange spheres- Hydrogen bond acceptors; White spheres- Hydrogen bond donors; Acc- Acceptors; Don- Donors.
To develop a useful pharmacophore query for virtual screening, six essential features were only chosen. Secondly, the essential pharmacophoric features of ligand X77 complexed with SARS-CoV-2 Mpro (PDB ID- 6W63) includes-
Four hydrogen bond acceptors, F1-F4 for interacting with amino-acid residues Glu166, His163, Gly143, Gly143, respectively.
One hydrophobic center (Hyd) F5 for interacting with amino-acid residue His41 (Figure 6).
Figure 6.

The pharmacophore model generated using the Pharmit server for the target protein (PDB ID- 6W63). Orange spheres- Hydrogen bond acceptors; Green sphere- Hydrophobic center; Acc- Acceptor; Hyd- Hydrophobic.
The generated pharmacophore models were used as a filter to screen the natural product databases such as Zinc natural product database consisting of 120,720 molecules, SNP containing 274,363 molecules, and MNP containing 14064 molecules. A total of 124 hits (108 from SNP, 15 from MNP and 1 from Zinc natural product database) were obtained for PDB- 6Y2F and a total of 313 Hits (292 from SNP, 20 from MNP, and 1 from Zinc natural product database) were obtained for PDB- 6W63 which were utilized for further docking study.
Molecular docking studies
The principal objective of the present study was to inspect the natural product databases in order to find out potential inhibitors of SARS-CoV-2 main protease Mpro. The generated pharmacophore-based hits (124 for PDB ID 6y2f, and 313 for PDB ID 6w63) were further selected for the profound molecular docking studies by using Schrodinger's Glide. Prior to screening of all ligands, co-crystal structures of PDB-6Y2F and 6W63 with their inhibitors were chosen and were re-docked back into their active site. The RMSD values between the crystallographic orientation and the best-docked pose were generated. The RMSD values of the selected targets were found to be 1.7 Å and 0.4 Å, respectively (Figures 7 and 8). The lower RMSD value indicates that the docking protocol could be reliable for the final docking studies of the selected compounds against the selected targets.
Figure 7.

Superimposition view of the X-ray native pose of co-crystal ligand and the re-docked pose of the co-crystal ligand in the active site of the target PDB-6Y2F (RMSD- 1.7 Å). Color interpretation: Green- X-ray native pose, Black- Re-docked pose.
Figure 8.

Superimposition view of the X-ray native pose of co-crystal ligand and the re-docked pose of the co-crystal ligand in the active site of the target PDB-6W63 (RMSD- 0.4 Å). Color interpretation: Green- X-ray native pose, Pink- Re-docked pose.
The docking studies revealed the presumed binding modes in the active site of the selected targets and exhibited the maximum docking scores. The results were surprising; the co-crystal ligands of the selected targets revealed less docking scores when compared with the top hit natural product database compounds. The docking scores of the co-crystal ligand and natural product database compounds are depicted in Tables 1 and 2. Top-ranked (Top 3) compounds against both the targets (PDB – 6Y2F, 6W63) were selected for the exhaustive analysis.
Table 1.
Hits identified after extra precision (XP) docking studies of Supernatural product database against the target PDB-6Y2F.
| S.no | Compound code | Glide score (Kcal/mol) | S.no | Compound code | Glide score (Kcal/mol) |
|---|---|---|---|---|---|
| 1 | SN00293542 | −14.57 | 31 | SN00213301 | −9.10 |
| 2 | SN00334894 | −13.78 | 32 | SN00038641 | −9.06 |
| 3 | SN00213037 | −13.32 | 33 | SN00213550 | −8.96 |
| 4 | SN00007464 | −11.53 | 34 | SN00237624 | −8.90 |
| 5 | SN00340755 | −11.48 | 35 | SN00331876 | −8.65 |
| 6 | SN00249174 | −11.30 | 36 | SN00220693 | −8.64 |
| 7 | SN00296151 | −11.18 | 37 | SN00165570 | −8.62 |
| 8 | SN00352807 | −10.98 | 38 | SN00388072 | −8.41 |
| 9 | SN00216715 | −10.66 | 39 | SN00171785 | −8.41 |
| 10 | SN00299979 | −10.61 | 40 | SN00274778 | −8.29 |
| 11 | SN00213181 | −10.60 | 41 | SN00396679 | −8.28 |
| 12 | SN00334175 | −10.53 | 42 | SN00311904 | −8.25 |
| 13 | SN00165563 | −10.46 | 43 | SN00311904 | −8.25 |
| 14 | SN00216711 | −10.39 | 44 | SN00311904 | −8.25 |
| 15 | SN00215944 | −10.27 | 45 | SN00260154 | −8.23 |
| 16 | SN00216710 | −10.19 | 46 | SN00366013 | −7.99 |
| 17 | SN00347461 | −10.07 | 47 | SN00364207 | −7.86 |
| 18 | SN00220722 | −10.07 | 48 | SN00310067 | −7.78 |
| 19 | SN00350811 | −9.90 | 49 | SN00292930 | −7.76 |
| 20 | SN00347999 | −9.73 | 50 | SN00231630 | −7.68 |
| 21 | SN00347999 | −9.73 | 51 | SN00395488 | −7.68 |
| 22 | SN00288247 | −9.63 | 52 | SN00215337 | −7.67 |
| 23 | SN00213547 | −9.61 | 53 | SN00265167 | −7.60 |
| 24 | SN00321138 | −9.59 | 54 | SN00256000 | −7.37 |
| 25 | SN00302560 | −9.59 | 55 | SN00266598 | −7.35 |
| 26 | SN00384731 | −9.58 | 56 | SN00302377 | −7.32 |
| 27 | SN00215773 | −9.36 | 57 | SN00378515 | −7.28 |
| 28 | SN00341530 | −9.30 | 58 | SN00317208 | −7.13 |
| 29 | SN00240768 | −9.29 | 59 | SN00386537 | −7.02 |
| 30 | SN00242594 | −9.18 | 60 | SN00305874 | −7.02 |
Table 2.
Hits identified after extra precision (XP) docking studies of Marine natural product database and Zinc natural database against the target PDB-6Y2F.
| S.no | Compound code | Glide score (Kcal/mol) |
|---|---|---|
| 1 | 96626-37-8 | −12.72 |
| 2 | 865369-05-7 | −12.55 |
| 3 | 474794-52-0 | −12.43 |
| 4 | 105404-83-9 | −11.99 |
| 5 | 32581-42-3 | −10.91 |
| 6 | 113322-01-3 | −9.94 |
| 7 | 134439-73-9 | −9.37 |
| 8 | 106543-01-5 | −9.32 |
| 9 | 157171-93-2 | −7.45 |
| 10 | ZINC70665993 | −11.80 |
The co-crystal ligand of the 6Y2F (alpha-Ketoamide), unveiled seven hydrogen bonds in the active sites of both the selected targets PDB- 6Y2F (Figure 9). Apart from the hydrogen bonds, alpha-Ketoamide revealed an aromatic interaction with THR-26. The alpha-ketoamide displayed the hydrogen bond interactions with the amino-acid residues like GLU-166, HIE- 163, GLY-143, CYS-145, PHE-140 along with one water-mediated interaction as well. All these critical amino-acid residue interactions of the alpha-Ketoamide revealed a comparable docking score of −7.720 kcal/mol in the active site of the target (Table 3).
Figure 9.

Amino-acid residue interactions exhibited by the co-crystal ligand in the active site of the target PDB-6Y2F. (A) 3D interaction diagram of the co-crystal ligand showing the hydrogen bond interaction in black, aromatic bond in blue color. (B) 2D interaction diagram of the co-crystal ligand showing the hydrogen bond interaction in magenta color.
Table 3.
In-depth amino-acid residues interactions exhibited by the co-crystal ligand and Top-3 compounds against the target PDB-6Y2F.
| S.no | Compound Code | Hydrogen bond with distances (Å) | Aromatic bond with distance (Å) | Glide score (Kcal/mol) |
|---|---|---|---|---|
| 1 | SN00293542(1st) | ASN-142 (2.62), GLY-143 (1.75), SER-144 (2.21), HIE-163 (2.09), HIE-164 (2.66), GLU-166 (2.32), THR-45 (2.77), SER-46 (2.40), GLN-189 (2.03), H20 (2.77,2.43, 2.05) | – | −14.565 |
| 2 | SN00334894(2nd) | SER-46 (2.21), ASN-142 (2.22), GLY-143 (2.14), THR-24 (1.94, 2.62), HIE-164 (1.50, 1.95), H20 (1.74,2.44) | – | −13.780 |
| 3 | SN00213037(3rd) | HIE-164(1.74), ASN-142 (1.65), GLY-143(2.32), CYS-145(2.20), H20 (2.40,2.08,1.93,1.63) | – | −13.322 |
| 4 | Co-crystal ligand(PDB-6Y2F) | GLU-166 (1.92,2.15), HIE-163 (1.89), GLY-143 (2.06), CYS-145(2.06), PHE-140 (2.30), H20 (2.21) | THR-26 (2.37) | −7.720 |
SN00293542 is 1F-fructosylnystose and belongs to the class of fructooligosaccharides (FOC) that consists of one molecule of sucrose and three molecules of fructose. These are widely distributed in onions, asparagus, and edible burdock (Mitsuoka et al., 1987). FOC has been traditionally used as a sweetener in Japan. 1F-fructosylnystose is an ingredient in the commercial product Neosugar that consists of a mixture of FOCs (Hidaka et al., 1986). The FOCs have exhibited beneficial effects in humans, such as prebiotic effect, reduced blood glucose levels, decreased blood triglycerides, cholesterols, and phospholipids (Mitsuoka et al., 1987; Sabater-Molina et al., 2009). FOCs have also been isolated from roots of Morinda officinalis, a traditional Chinese medicine (TCM), and have been widely explored for its antidepressant effect (Qiu et al., 2016; Zhang et al., 2002, 2018). This molecule produced the highest docking score of −14.565 kcal/mol in the active site of the target 6Y2F, which is almost double of the docking score of the co-crystal ligand. This molecule also revealed nine hydrogen bond interactions and three water medicated interactions in the active site of the target, which strongly suggested that the molecule occupied the active receptor site and revealed its maximum docking score (Figure 10). Amino-acid residues like THR-45, SER-46, ASN-142, GLY-143, SER-144, HIE-164, GLU-166, and GLN-189 exhibited hydrogen bond interaction with the SN00293542 molecule. No aromatic and Pi-Pi interactions were revealed by the same molecule.
Figure 10.

Amino-acid residue interactions exhibited by the molecule SN00293542 in the active site of the target PDB-6Y2F. (A) 3D interaction diagram of the SN00293542 showing the hydrogen bond interaction in black. (B) 2D interaction diagram of the SN00293542 showing the hydrogen bond interaction in magenta color.
In the same series of Supernatural product database, the second top-ranked molecule is SN00334894. SN00334894 is a glycan produced by Rhizobium meliloti J7017 (Hisamatsu et al., 1985). This molecule revealed less docking scores when compared with the previously discussed top-ranked molecule and significant docking score when compared with the co-crystal ligand. Similarly, like the previous molecule, the SN00334894 revealed nine hydrogen bond communications, including some water-mediated interactions. In both cases, ASN-142, GLY-143, and HIE-164 are the common amino-acid residues (Figure 11). Apart from the common residues, amino-acids SER-46 and THR-24 are involved in the hydrogen bond interaction. The docking score exposed by this compound is −13.780 kcal/mol.
Figure 11.

Amino-acid residue interactions exhibited by the molecule SN00334894 in the active site of the target PDB-6Y2F. (A) 3D interaction diagram of the SN00334894 showing the hydrogen bond interaction in black. (B) 2D interaction diagram of the SN00334894 showing the hydrogen bond interaction in magenta color.
SN00213037, Dihydrostreptomycin 3'alpha,6-bisphosphate is in the third position of the list. Dihydrostreptomycin 3'alpha,6-bisphosphate is produced by the reaction between dihydrostreptomycin-6-phosphate and ATP catalyzed by enzyme Dihydrostreptomycin-6-phosphate 3′-alpha-kinase that is produced by Streptomyces. The biological significance of Dihydrostreptomycin 3'alpha,6-bisphosphate, has not been elucidated yet, but it is speculated to be slowly converted into its active antibiotic in vivo (Walker & Walker, 1970). This molecule also exhibited frequent amino-acid interactions as that of molecule SN00334894. Additionally, this compound also exhibited a hydrogen bond interaction with the catalytic residue of CYS-145 (Figure 12). However, in all the cases of the top-ranked molecules, this molecule exposed the highest water-mediated interactions.
Figure 12.

Amino-acid residue interactions exhibited by the molecule SN00213037 in the active site of the target PDB-6Y2F. (A) 3D interaction diagram of the SN00213037 showing the hydrogen bond interaction in black. (B) 2D interaction diagram of the SN00213037 showing the hydrogen bond interaction in magenta color.
Similarly, a non-covalent bond inhibitor in PDB-6W63 exhibited five-hydrogen bond interactions with the surrounded amino-acid residues. The docking scores of the co-crystal ligand and natural product database compounds are depicted in Tables 4 and 5. Two aromatic bond interactions with HIE-164 and ASN-142 and one pi-pi interaction with HIE-41 was exhibited by the co-crystal ligand. GLU-166, HIE-163, and GLY-143 are the amino-acid residues which contributed a hydrogen bond with the co-crystal ligand (Figure 13). All these interactions made the docking score of −7.20 kcal/mol of the co-crystal ligand (Table 6).
Table 4.
Hits identified after extra precision (XP) docking studies of Supernatural product database against the target PDB-6W63.
| S.no | compound code | Docking score (Kcal/mol) | s.no | compound code | Docking score (Kcal/mol) |
|---|---|---|---|---|---|
| 1 | SN00382835 | −12.425 | 31 | SN00388072 | −8.972 |
| 2 | SN00403420 | −12.402 | 32 | SN00141985 | −8.912 |
| 3 | SN00041592 | −12.142 | 33 | SN00032643 | −8.764 |
| 4 | SN00168969 | −11.966 | 34 | SN00042584 | −8.724 |
| 5 | SN00391842 | −11.751 | 35 | SN00166870 | −8.376 |
| 6 | SN00040401 | −11.621 | 36 | SN00335100 | −8.351 |
| 7 | SN00114482 | −11.538 | 37 | SN00386048 | −8.344 |
| 8 | SN00162335 | −11.512 | 38 | SN00165676 | −8.321 |
| 9 | SN00330810 | −11.082 | 39 | SN00038775 | −8.273 |
| 10 | SN00392377 | −10.899 | 40 | SN00164881 | −8.22 |
| 11 | SN00216726 | −10.822 | 41 | SN00173581 | −8.21 |
| 12 | SN00038781 | −10.736 | 42 | SN00173831 | −8.209 |
| 13 | SN00175930 | −10.628 | 43 | SN00329478 | −8.085 |
| 14 | SN00379716 | −10.557 | 44 | SN00399664 | −7.915 |
| 15 | SN00161123 | −10.444 | 45 | SN00160358 | −7.911 |
| 16 | SN00339099 | −10.286 | 46 | SN00393877 | −7.887 |
| 17 | SN00338961 | −10.22 | 47 | SN00380351 | −7.873 |
| 18 | SN00143458 | −10.068 | 48 | SN00160458 | −7.795 |
| 19 | SN00162745 | −10.052 | 49 | SN00025215 | −7.763 |
| 20 | SN00213304 | −9.936 | 50 | SN00173164 | −7.756 |
| 21 | SN00382588 | −9.851 | 51 | SN00040115 | −7.747 |
| 22 | SN00165557 | −9.782 | 52 | SN00218781 | −7.702 |
| 23 | SN00332128 | −9.743 | 53 | SN00388121 | −7.682 |
| 24 | SN00037946 | −9.499 | 54 | SN00096441 | −7.521 |
| 25 | SN00398348 | −9.27 | 55 | SN00100418 | −7.46 |
| 26 | SN00106829 | −9.2 | 56 | SN00218714 | −7.409 |
| 27 | SN00339802 | −9.108 | 57 | SN00214165 | −7.4 |
| 28 | SN00106842 | −9.067 | 58 | SN00164074 | −7.368 |
| 29 | SN00387960 | −9.008 | 59 | SN00397542 | −7.132 |
| 30 | SN00011125 | −9.007 | 60 | SN00096315 | −7.094 |
Table 5.
Hits identified after extra precision (XP) docking studies of Marine natural product database and Zinc natural database against the target PDB-6W63.
| S.no | compound code | Docking score (Kcal/mol) |
|---|---|---|
| 1 | 20633-84-5 | −12.203 |
| 2 | 139933-53-2 | −12.007 |
| 3 | 102040-09-5 | −9.888 |
| 4 | 476437-86-2 | −8.58 |
| 5 | 103425-21-4 | −8.553 |
| 6 | 23235-67-8 | −8.543 |
| 7 | 98166-57-5 | −7.911 |
| 8 | ZINC02030982 | −8.033 |
Figure 13.

Amino-acid residue interactions exhibited by the co-crystal ligand in the active site of the target PDB-6W63. (A) 3D interaction diagram of the co-crystal ligand showing the hydrogen bond interaction in black, aromatic bond in blue color. (B) 2D interaction diagram of the co-crystal ligand showing the hydrogen bond interaction in magenta color and Pi interaction in green color.
Table 6.
In-depth amino-acid residues interactions exhibited by the co-crystal ligand and Top-3 compounds against the target PDB-6W63.
| S.no | Compound Code | Hydrogen bond with distances (Å) | Aromatic bond with distance (Å) | Pi-Pi interactions with distance (Å) | Glide score (Kcal/mol) |
|---|---|---|---|---|---|
| 1 | SN00382835(1st) | GLU-166 (2.56), ASN-142 (2.06), GLY-143 (1.83), H20 (1.79, 2.25) | TYR-54 (2.77) | – | −12.425 |
| 2 | SN00403420(2nd) | GLU-166 (1.98,2.13), HIE-163 (1.92), HIE-164 (2.41), GLY-143 (2.75), THR-25 (2.15), H20 (1.67) | – | HIE-41 (4.30) | −12.402 |
| 3 | 20633-84-5(3rd) | GLU-166 (1.65,1.66 2.03), HIE-163 (1.79), THR-190 (1.81, 1.84) H20 (1.81) | – | – | −12.203 |
| 4 | Co-crystal ligand(PDB-6W63) | GLU-166 (1.89), GLY-143 (2.09), HIE-163 (2.08), H20 (1.98, 2.25) | HIE-164 (2.48), ASN-142 (2.68) | HIE-41 (4.86) | −7.20 |
Supernatural product database compound SN00382835 is a multi-ester oligosaccharide- 6-O-benzoyl-3′-O-sinapoylsucrose that is isolated from various Polygala species like P. tenulifolia, P. sibirica, P. tricornis, and P. telephioides. These plants are used in Traditional Chinese medicine as a tranquilizer, as an expectorant, tonic, and for the prevention of memory failure (Chang & Tu, 2007; Li et al., 2005; Miyase et al., 1999; Miyase & Ueno, 1993). Molecule SN00382835 unveiled the maximum docking score of −12.425 kcal/mol, and it occupied top rank in the 6W63 target. The amino-acid residues like GLU-166, GLY-143 are common residues involved in the interaction in the case of both the co-crystal ligand and the compound SN00382835. Apart from the common amino-acid residue, ASN-142 exhibited a hydrogen bond interaction with the molecule SN00382835 (Figure 14). Two water-mediated interactions were also observed in the same molecule.
Figure 14.

Amino-acid residue interactions exhibited by the molecule SN00382835 in the active site of the target PDB-6W63. (A) 3D interaction diagram of the molecule SN00382835 showing the hydrogen bond interaction in black and aromatic interaction in blue color. (B) 2D interaction diagram of the molecule SN00382835 showing the hydrogen bond interaction in magenta color.
Molecule SN00403420 is a natural color pigment- Delphinidin 3-(6”-malonyl-glucoside) that belongs to the class of anthocyanidin glycosides. It is responsible for giving a blue color to the plant Clitoria ternatea (Kazuma et al., 2003). It has also been reported to be present in fruits like Mulberry (Morus atropurpurea) and blood oranges Sanguinello and Tarocco (Citrus sinensis) (Cebadera-Miranda et al., 2019; Wu et al., 2011). Delphinidin 3-(6”-malonyl-glucoside), being a flavonoid, is widely explored for its antioxidant activity (Rocchetti et al., 2017; Wu et al., 2011). This molecule also revealed a comparable docking score of −12.402 kcal/mol, which was ranked second in the order. The common amino-acid residues that are involved in the interaction with the molecule SN00382835 are also involved in the interaction with SN00403420. Apart from these common connections, the molecule additionally also connected with the amino-acid residues HIE-163,164 and THR-25 with a hydrogen bond (Figure 15). One water-mediated interaction was also observed in the molecule. HIE-41 exhibited pi-pi interaction with the molecule SN00403420.
Figure 15.

Amino-acid residue interactions exhibited by the molecule SN00403420 in the active site of the target PDB-6W63. (A) 3D interaction diagram of the molecule SN00403420 showing the hydrogen bond interaction in black and Pi- interaction in magenta color. (B) 2D interaction diagram of the molecule SN00403420 showing the hydrogen bond interaction in magenta color and Pi- interaction in green color.
A Zinc natural product database compound 20633-84-5 came in the picture next to the Supernatural product database compounds and occupied the third position in the list. The molecule 20633-84-5 is a flavonoid glycoside- Luteolin-7-rutinoside. It is reported to possess a wide range of biological actions like antioxidant, antigenotoxic, anticancer, antiallergic, and antimicrobial, among others (Inoue et al., 2002; Orhan et al., 2016; Zhu et al., 2004). It is also reported to interact and inhibit enzymes like sentrin-specific protease 1 (SENP1) (a SUMO protease that is involved in the development of prostate cancer), aldose reductase (an enzyme that is responsible for conversion of glucose to sorbitol and has a pathophysiological role in Diabetic retinopathy), and Matrix metalloproteases (MMPs) (an enzyme that is overexpressed in cancer, arthritis, atherosclerosis, etc.) (AID 651697, 2012; Crascì et al., 2017; Jung et al., 2011). This molecule exhibited one common amino-acid residue interaction with GLU-166 (Figure 16). Overall, the compound 20633-84-5 exhibited seven hydrogen bond interactions. Apart from the common amino-acid residue interactions, this molecule also exhibited three hydrogen bond interactions with the amino-acid residues like HIE-163, THR-190, and one water-mediated hydrogen bond interaction as well.
Figure 16.

Amino-acid residue interactions exhibited by the molecule 20633-84-5 in the active site of the target PDB-6W63. (A) 3D interaction diagram of the molecule 20633-84-5 showing the hydrogen bond interaction in black color. (B) 2D interaction diagram of the molecule 20633-84-5 showing the hydrogen bond interaction in Magenta color.
The structures and brief details of the selected molecules are depicted in Table 7. No common molecules were identified among the top-3 ranked compounds after docking studies on PDB 6Y2F and 6W63. This was expected as the hits identified after virtual screening was unique owing to their different pharmacophoric features. The identified molecules briefly belonged to glycans, oligosaccharides and flavonoids. The selected molecules have been explored for their CNS effects, antioxidants effects, among others. Notably, flavonoids have been reported to possess significant activity against coronaviruses (Chiow et al., 2016; Jo et al., 2020). Therefore, it would be interesting to test if 20633-84-5 has any effect on SARS-CoV-2. GLU-166 and GLY-143 are the amino-acid residues which commonly contributed their role in the active hydrogen bond formation with these hit molecules. Water played an essential role in the formation of water-mediated interactions. All these interactions strongly suggested that these top hit molecules may act as potential inhibitor of SARS-CoV-2 Mpro. Upon keen observation of all hydrogen bond distances with the surrounded amino-acid residues and the water molecules, the distances are very close to the molecules, and these connections are also tightly bonded to the molecules, which may further, in turn, contributed to its significant docking score when compared with its co-crystal ligand.
Table 7.
Description of the identified hits after docking studies.
| Compound ID | Compound Name | Structure | Class | Found in / Produced by |
Uses / Activities reported |
|---|---|---|---|---|---|
| SN00293542 | 1F-fructosylnystose | 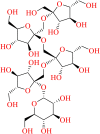 |
Fructooligosaccharides | Onions, asparagus, and edible burdock, Morinda officinalis | Sweetener, prebiotic, antidepressant |
| SN00334894 | – |  |
– | Rhizobium meliloti | – |
| SN00213037 | Dihydrostreptomycin 3′alpha,6-bisphosphate | 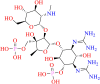 |
Aminoglycosides | Streptomyces | Antibiotic |
| SN00382835 | 6-O-benzoyl-3′-O-sinapoylsucrose | 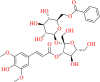 |
Oligosaccharides multi-ester | Polygala | Tranquilizer, Expectorant, tonic, prevention of memory failure |
| SN00403420 | Delphinidin 3-(6"-malonyl-glucoside) |  |
Anthocyanidin glycosides |
Clitoria ternatea,
Morus atropurpurea, Citrus sinensis |
Antioxidant |
| 20633-84-5 | Luteolin 7-rutinoside | 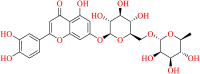 |
Flavonoid glycoside | Cynara scolymus, Mentha piperita, Artemisia montana, Olea europaea | Antioxidant, antigenotoxic, anticancer, antiallergic, antimicrobial |
In silico predicted physico-chemical parameters
In medicinal chemistry, Absorption, Distribution, Metabolism, and Excretion (ADME) plays a significant role. These parameters will decide the drug likeliness of the molecules. In silico Physico-chemical (Lipinski's rule of five and Jorgensen's rule of three), parameters of the top 5 hits from the docking study was predicted using the QikProp module of the Schrodinger. The parameters are depicted in Table 8. The molecules exhibited a higher molecular weight than the prescribed limit. Log Po/w was predicted, and these values are within the prescribed limit of −2.0 to 6 except for the compounds SN00334894, SN00293542 and SN00213037 for the PDB 6Y2F. The violations are considered up to 4. For all the predicted molecules, the maximum violation is 3 only. These results suggested that the molecule may exhibit suitable drug likeliness property. The molecules exhibited good solubility, indicated by the Log S values except for the compounds SN00334894 and SN00213037. The in silico predicted Caco-2 cell permeability for the selected compounds was found to very low, suggesting that it may face some hurdles in absorption process. The number of likely metabolic reactions of the compounds were found to be high. Overall, the results are satisfied with the rules with fewer violations only.
Table 8.
In silico predicted Physico-chemical parameters of the top-5 compounds against each PDB.
| PDB-
6W63 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound code | Lipinski rule
of five |
Jorgensen
rule of three |
|||||||
| MW | donorHB | accptHB | logPo/w | Violation | logS | PCaco | metab | Violation | |
| SN00382835 | 652.6 | 7 | 19.65 | −0.325 | 3 | −2.417 | 10.426 | 9 | 2 |
| SN00041592 | 476.4 | 7 | 19.75 | −2.547 | 2 | −1.186 | 14.907 | 7 | 2 |
| 20633-84-5 | 594.5 | 8 | 19.8 | −2.013 | 3 | −2.436 | 2.518 | 9 | 2 |
| 139933-53-2 | 971.1 | 11 | 29.2 | 0.474 | 3 | −3.687 | 0.939 | 13 | 2 |
| SN00403420 | 550.4 | 7 | 16.05 | −1.525 | 3 | −2.815 | 0.058 | 8 | 2 |
| PDB - 6Y2F | |||||||||
| SN00293542 | 828.7 | 17 | 36.6 | −7.824 | 3 | 1.174 | 0.007 | 17 | 2 |
| SN00213037 | 743.5 | 19 | 31.55 | −8.037 | 3 | −0.067 | 0 | 9 | 2 |
| SN00334894 | 990.8 | 20 | 52.7 | −12.325 | 3 | 2 | 0.003 | 20 | 2 |
| SN00007464 | 358.2 | 8 | 18 | −3.247 | 2 | −0.494 | 1.172 | 8 | 2 |
| SN00340755 | 1138.1 | 18 | 31.4 | −4.464 | 3 | −2.296 | 0 | 17 | 2 |
Description: Lipinski rule- Number of violations of Lipinski's rule of five. The rules are: mol MW < 500, logPo/w < 5, donor HB ≤ 5, accept HB ≤ 10 and maximum 4 violations.
Jorgensen's rule of three - Number of violations of Jorgensen's rule of three. The three rules are: logS > −5.7, PCaco > 22 nm/s, # Primary Metabolites < 7. Compounds with fewer (maximum 3) violations.
Additionally, the Prime MM-GBSA module analyzed the binding energy calculation of selected top two compounds based on their binding affinity towards the active site binding pocket of the target molecule. MM-GBSA score of lead molecule SN00293542 was −82.04, whereas, the second lead molecule SN00382835 has shown significantly lower MM-GBSA score −64.81 (Table 9). With these results, it can be concluded that the free binding energy score of SN00293542 was better in comparison to the SN00382835 compound.
Table 9.
The computed (MM-GBSA) binding free energies (ΔG bind) of the selected compounds against Mpro enzyme.
| S. No | Compound code | MMGBSA score (Kcal/mol) |
|---|---|---|
| Pre dynamics MM-GBSA | ||
| 1 | SN00293542 | −84.04 |
| 2 | SN00382835 | −64.81 |
| 3 | Cocrystal ligand (PDB-6Y2F) | −58.20 |
| 4 | Cocrystal ligand (PDB-6W63) | −75.50 |
| Post dynamics MM-GBSA | ||
| 1 | SN00293542 | −71.70 +/− 7.98 |
| 2 | SN00382835 | −56.81 +/−7.54 |
Molecular dynamics and post-MM-GBSA analysis
To validate the protein-ligand complex of the docked compound and to measure the constancy of the ligand binding in the active site of the selected target, molecular dynamics simulation study was conducted for the top hit molecule from each PDB. The compounds SN00293542 SN00382835 with the protein complex was immersed in an orthorhombic box of SPC water molecules. The solvated system was neutralized by adding one Na+ counterion. To equilibrate the system, the solutes were subjected to NPT ensemble. Finally, the full system was subjected to an MD simulation run for 100 ns at 300 K temperature and 1 bar pressure. The obtained simulation results were analyzed using the backbone RMSD values. All conformations disclosed significant RMSD values in the selected targets. Both the targets revealed the maximum RMSD value of 2 Å, which describes that the protein-ligand complex throughout the simulation period was maintained continuously (Figures 17 and 18).
Figure 17.

Root Mean Square Deviation of the protein-ligand complex of PDB-6Y2F with the lowest binding energy compound SN00293542.
Figure 18.

Root Mean Square Deviation of the protein-ligand complex of PDB-6W63 with the lowest binding energy compound SN00382835.
Protein interactions with the ligand were supervised throughout the simulation. These interactions can be organized by type, reviewed, and represented in Figures 19 and 20. Protein-ligand interactions were categorized into four types: Hydrogen Bonds, Hydrophobic, Ionic and water bridges. The type of aminoacid residues present in the active site of the target protein plays a crucial role in making the protein-ligand complex stable. The MD simulation study indicates stable results of the molecules SN00293542, SN00382835 in the protein-ligand complex. The aminoacid residues GLU-166 and GLY-143 exhibited hydrogen bond contact with the molecule SN00293542, as revealed by docking studies was also existing in MD throughout the trajectory, with 88 and 79% interaction in the stipulated time. The second hit molecule form the other target 6W63 revealed significant hydrogen bond interaction with aminoacid residue GLU-166, with 20–26% interacted during the entire simulation. Apart from these residues, a crucial aminoacid residue CYS-145 was also actively contributed to the hydrogen bond with the target molecule in the active site. The amino acid residues like GLN-189 (89%), GLU-47 (75%) and ASP-48 (56,40%) were involved in the hydrogen bond formation with the molecule SN00293542. Aminoacid residue GLY-192 was involved in the bond formation with the molecule SN00382835 in the active site of 6W63 (Figures 21 and 22). All the aminoacid interactions revealed upon docking studies of the target molecule was also exhibited during the dynamic study. This indicated that the protein-ligand complex was stable throughout the simulation period, and minimal backbone fluctuations have ensued in the system. Overall, the simulation revealed more water-mediated connections with the hit molecules; amino-acid mediated water bridges were also found in the MD simulations study. Specific timeline contacts with the aminoacid residues that were present in the targets are briefly presented in Figure 23. The darker the color represents the number of connections with respective of the aminoacid was high. Apart from this, Pre-MM-GBSA analysis of the same hit compounds revealed the binding free energy of −84.04, −64.81 kcal/mol, respectively. The co-crystal ligands, which were present in the targets also revealed significant binding free energies. Co-crystal ligand presents in the target 6Y2F revealed −58.20 kcal/mol, which was comparatively lesser than that of the molecule SN00293542. Another co-crystal ligand of 6W63 revealed significant binding free energy of −75.50 when compared with analogue SN00382835. The post-MM-GBSA analysis of free binding energy calculation was carried out with the generation of 0-2002 frames having around 10-step sampling size. A total of 201 frames were processed and analyzed throughout the post-MM-GBSA calculation of 100 ns MD data of both the hit ligands revealed by the dynamics studies. The calculated binding free energy, ΔG average of the molecule SN00293542 was found to be −71.70 +/− 7.98 kcal/mol and the second hit molecule SN00382835 found to be −56.81 +/− 7.54 kcal/mol SN00382835. Pre dynamic MM-GBSA results displayed the best binding free energy scores when compared with the post dynamic MM-GBSA calculations.
Figure 19.

Plot (stacked bar charts) of Protein interactions with the ligand supervised throughout the simulation of the compound SN00293542 (PDB-6Y2F).
Figure 20.

Plot (stacked bar charts) of Protein interactions with the ligand supervised throughout the simulation of the compound SN00382835 (PDB-6W63).
Figure 21.

Detailed ligand SN00293542 atomic interactions with the protein residues of PDB-6Y2F.
Figure 22.

Detailed ligand SN00382835 atomic interactions with the protein residues of PDB-6W63.
Figure 23.

Specific contacts made by the protein with the ligand throughout the trajectory. (Darker the color indicates more specific contact with the ligand) A- SN00293542 (PDB-6Y2F), B- SN00382835 (PDB-6W63).
Conclusion
There is an urgent requirement to discover novel therapeutics for treating the early stages of COVID-19 caused by SARS-CoV-2. Mpro is one of the potential targets for antiviral treatment against SARS-CoV-2. The abundant natural resources can be exploited for the discovery of natural products of pharmacological significance. In the present study, a combination of computational approaches, namely- pharmacophore based virtual screening, molecular docking, MD simulation, and MM-GBSA were performed to identify potential inhibitors of SARS-CoV-2 Mpro from available natural products databases.
Two pharmacophore models were generated based on the selected crystal structure of SARS-CoV-2 Mpro with a covalent inhibitor O6K (PDB ID- 6Y2F), and a non-covalent inhibitor X77 (PDB ID- 6W63). Various natural products belonging to different classes have been reported to possess significant anti-CoV activity in the past. Therefore, performing a virtual screening against the natural product databases could result in the identification of putative active compounds against SARS-CoV-2. Hence, the generated pharmacophore models were used as a filter for screening the selected natural product databases - SNP, MNP and Zinc natural product database to identify potential inhibitors of SARS-CoV-2 Mpro. The adopted virtual screening procedure identified a total of 124 hits for the developed pharmacophore model with PDB ID - 6Y2F and a total of 313 hits for the developed pharmacophore model with PDB ID - 6W63, all of which were then subjected to molecular docking study. The detailed interactions of six natural compounds (Top 3 hits for each target) after the docking study have been thoroughly investigated, and they exhibited strong predicted binding affinities for the SARS-CoV-2 Mpro. To further validate the stability of a potential inhibitors SN00293542 and SN00382835, a MD simulation study was performed for 100 ns. The MD simulation result revealed significant stability of test compounds SN00293542 and SN00382835 in the active site of SARS-CoV-2 Mpro and exhibited good interactions with the surrounded amino acid residues. The pre (after docking study) and post (after MD simulation) MM-GBSA analysis of the compounds SN00293542 and SN00382835 showed significant binding affinity with its selected target.
The six identified potential inhibitors belong to different classes of natural compounds like- FOCs, aminoglycosides, anthocyanin glycosides, and flavonoid glycosides. These molecules have been isolated from various plant and bacterial species. These plants have been used as traditional Chinese medicine for different purposes. Structurally, all these compounds possessed at least one sugar molecule: pentose or a hexose and were also characterized by the presence of many hydroxyl groups, which played a crucial role in hydrogen bond formation with the surrounding amino acid residues, thus shaped significant docking scores. Compounds SN00382835, SN00403420 and 20633-84-5 also shared a phenolic nucleus in their structures. Fascinatingly, compounds SN00403420 and 20633-84-5 are flavonoids. Flavonoids have been experimentally proven to inhibit Mpro of corona viruses. Moreover, flavonoids are also reported to possess potent anti-inflammatory activity and thus may prove to be beneficial against the cytokine storm during SARS-CoV-2 infection. Further, in vitro studies of the identified natural products against SARS-CoV-2 Mpro needs to be performed in order to substantiate them as a potential inhibitory ligand of SARS-CoV-2 Mpro.
Supplementary Material
Acknowledgements
Authors Banoth Karan Kumar, Faheem and Sankaranarayanan Murugesan gratefully acknowledge to the BITS-Pilani, Pilani campus for providing the adequate facilities to do this research. Our special thanks to Dr. Pritesh Bhat, Senior scientist, Schrodinger, LLC, Bangalore.
Glossary
Abbreviations
- HTVS
High throughput virtual screening
- SP
Standard precession
- XP
Extra precession
- RMSD
Root mean square deviation
- RMSF
Root mean square fluctuation
- MD
Molecular dynamics
- Å
Angstrom
- SARS-CoV-2 Mpro
Severe Acute Respiratory Syndrome Corona Virus 2 main protease
- MM-GBSA
Molecular Mechanics, the Generalized Born model and Solvent Accessibility
Funding Statement
Authors Banoth Karan Kumar, Faheem and Sankaranarayanan Murugesan gratefully acknowledge to the Department of Biotechnology, Indo-Spain, New Delhi. (Ref. No: BT/IN/Spain/39/SM/2017-2018). Additionally, Banoth Karan Kumar thankful to the Ministry of Tribal affairs, Government of India for providing financial assistance (Award no- 201920-NFST-TEL-01497).
Authors contribution
Banoth Karan Kumar - Conceptualization, Methodology, Software, Validation, Data curation, writing the original draft. Faheem – Conceptualization, writing, review, editing. Rupal Ojha – Scientific discussion, review. Vijay Kumar Prajapati- Scientific discussion. Aravinda Pai - Scientific discussion. Kondapalli Venkata Gowri Chandra Sekhar – Discussion, review, editing. Murugesan Sankaranarayanan – Supervision, Project administration, Funding acquisition, review and editing.
Disclosure statement
The authors have no conflicts of interest to declare.
References
- Adhikari, S. P., Meng, S., Wu, Y.-J., Mao, Y.-P., Ye, R.-X., Wang, Q.-Z., Sun, C., Sylvia, S., Rozelle, S., Raat, H., & Zhou, H. (2020). Epidemiology, causes, clinical manifestation and diagnosis, prevention and control of coronavirus disease (COVID-19) during the early outbreak period: A scoping review. Infectious Diseases of Poverty, 9(1), 29. 10.1186/s40249-020-00646-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- AID 651697 . (2012). Dose response confirmation of small molecule inhibitors of the catalytic domain of the SUMO protease, SENP1 in a kinetic FRET assay - PubChem.
- Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature Medicine, 26(4), 450–452. 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestle, D., Heindl, M. R., Limburg, H., Van, T. V. L., Pilgram, O., Moulton, H., & Böttcher-Friebertshäuser, E. (2020). TMPRSS2 and furin are both essential for proteolytic activation and spread of SARS-CoV-2 in human airway epithelial cells and provide promising drug targets. BioRxiv. 10.1101/2020.04.15.042085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebadera-Miranda, L., Domínguez, L., Dias, M. I., Barros, L., Ferreira, I. C. F. R., Igual, M., Martínez-Navarrete, N., Fernández-Ruiz, V., Morales, P., & Cámara, M. (2019). Sanguinello and Tarocco (Citrus sinensis [L.] Osbeck): Bioactive compounds and colour appearance of blood oranges. Food Chemistry, 270, 395–402. 10.1016/j.foodchem.2018.07.094 [DOI] [PubMed] [Google Scholar]
- Chang, H. T., & Tu, P. F. (2007). New oligosaccharide esters and xanthone C-glucosides from Polygala telephioides. Helvetica Chimica Acta, 90(5), 944–950. 10.1002/hlca.200790095 [DOI] [Google Scholar]
- Chiow, K. H., Phoon, M. C., Putti, T., Tan, B. K. H., & Chow, V. T. (2016). Evaluation of antiviral activities of Houttuynia cordata Thunb. extract, quercetin, quercetrin and cinanserin on murine coronavirus and dengue virus infection. Asian Pacific Journal of Tropical Medicine, 9(1), 1–7. 10.1016/j.apjtm.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard, B., Valle, C., de Lamballerie, X., Canard, B., Seidah, N. G., & Decroly, E. (2020). The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Research, 176, 104742. 10.1016/j.antiviral.2020.104742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crascì, L., Basile, L., Panico, A., Puglia, C., Bonina, F. P., Basile, P. M., Rizza, L., & Guccione, S. (2017). Correlating in vitro target oriented screening and docking: Inhibition of matrix metalloproteinases activities by flavonoids. Planta Medica, 83(11), 901–911. 10.1055/s-0043-104775 [DOI] [PubMed] [Google Scholar]
- Dai, W., Zhang, B., Jiang, X.-M., Su, H., Li, J., Zhao, Y., Xie, X., Jin, Z., Peng, J., Liu, F., Li, C., Li, Y., Bai, F., Wang, H., Cheng, X., Cen, X., Hu, S., Yang, X., Wang, J., … Liu, H. (2020). Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science (New York, N.Y.), 368(6497), 1331–1335. 10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka, H., Eida, T., Takizawa, T., Tokunaga, T., & Tashiro, Y. (1986). Effects of fructooligosaccharides on intestinal flora and human health. Bifidobacteria and Microflora, 5(1), 37–50. 10.12938/bifidus1982.5.1_37 [DOI] [Google Scholar]
- Hisamatsu, M., Yamada, T., Akaki, M., Nashinoki, H., Takai, Y., & Amemura, A. (1985). Structural studies on new, non-reducing oligosaccharides produced by rhizobium meliloti j7017. Agricultural and Biological Chemistry, 49(5), 1447–1451. 10.1080/00021369.1985.10866904 [DOI] [Google Scholar]
- Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., Schiergens, T. S., Herrler, G., Wu, N.-H., Nitsche, A., Müller, M. A., Drosten, C., & Pöhlmann, S. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell, 181(2), 210–271. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, T., Sugimoto, Y., Masuda, H., & Kamei, C. (2002). Antiallergic effect of flavonoid glycosides obtained from Mentha piperita L. Biological & Pharmaceutical Bulletin, 25(2), 256–259. 10.1248/bpb.25.256 [DOI] [PubMed] [Google Scholar]
- Islam, M. T., Sarkar, C., El-Kersh, D. M., Jamaddar, S., Uddin, S. J., Shilpi, J. A., & Mubarak, M. S. (2020). Natural products and their derivatives against coronavirus: A review of the non-clinical and pre-clinical data. Phytotherapy Research. Wiley. 10.1002/ptr.6700 [DOI] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., & Yang, H. (2020. a). Structure of Mpro from COVID-19 virus and discovery of its inhibitors. BioRxiv. 10.1101/2020.02.26.964882 [DOI] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020. b). Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature, 582(7811), 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Jo, S., Kim, S., Shin, D. H., & Kim, M. S. (2020). Inhibition of SARS-CoV 3CL protease by flavonoids. Journal of Enzyme Inhibition and Medicinal Chemistry, 35(1), 145–151. 10.1080/14756366.2019.1690480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, W. L., Maxwell, D. S., & Tirado-Rives, J. (1996). Development and testing of the OPLS all atom force field on conformational energetics and properties of organic liquids. Journal of the American Chemical Society, 118(45), 11225–11236. 10.1021/ja9621760 [DOI] [Google Scholar]
- Jung, H. A., Islam, M. D. N., Kwon, Y. S., Jin, S. E., Son, Y. K., Park, J. J., Sohn, H. S., & Choi, J. S. (2011). Extraction and identification of three major aldose reductase inhibitors from Artemisia montana. Food and Chemical Toxicology: Toxicology, 49(2), 376–384. 10.1016/j.fct.2010.11.012 [DOI] [PubMed] [Google Scholar]
- Kalibaeva, G., Ferrario, M., & Ciccotti, G. (2003). Constant pressure-constant temperature molecular dynamics: A correct constrained NPT ensemble using the molecular virial. Molecular Physics, 101(6), 765–778. 10.1080/0026897021000044025 [DOI] [Google Scholar]
- Kazuma, K., Noda, N., & Suzuki, M. (2003). Flavonoid composition related to petal color in different lines of Clitoria ternatea. Phytochemistry, 64(6), 1133–1139. 10.1016/S0031-9422(03)00504-1 [DOI] [PubMed] [Google Scholar]
- Keyhan, S. O., Fallahi, H. R., & Cheshmi, B. (2020). Dysosmia and dysgeusia due to the 2019 Novel Coronavirus; a hypothesis that needs further investigation. Maxillofacial Plastic and Reconstructive Surgery, 42(1), 9. 10.1186/s40902-020-00254-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J., Abel, R., Zhu, K., Cao, Y., Zhao, S., & Friesner, R. A. (2011). The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins: Structure, Function, and Bioinformatics, 79(10), 2794–2812. 10.1002/prot.23106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J., Jiang, Y., & Tu, P. F. (2005). Tricornoses A-L, oligosaccharide multi-esters from the roots of Polygala tricornis. Journal of Natural Products, 68(5), 739–744. 10.1021/np050023x [DOI] [PubMed] [Google Scholar]
- Lin, L. T., Hsu, W. C., & Lin, C. C. (2014). Antiviral natural products and herbal medicines. Journal of Traditional and Complementary Medicine, 4(1), 24–35. 10.4103/2225-4110.124335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, H., Tang, Q., Shang, Y., Liang, S., Yang, M., Robinson, N., & Liu, J. P. (2020). Can Chinese medicine be used for prevention of Corona Virus Disease 2019 (COVID-19)? A review of historical classics, research evidence and current prevention programs. Chinese Journal of Integrative Medicine, 26(4), 243–250. 10.1007/s11655-020-3192-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani, J. S., Johnson, J. B., Steel, J. C., Broszczak, D. A., Neilsen, P. M., Walsh, K. B., & Naiker, M. (2020). Natural product-derived phytochemicals as potential agents against coronaviruses: A review. Virus Research. Elsevier B.V. 10.1016/j.virusres.2020.197989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark, P., & Nilsson, L. (2001). Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. The Journal of Physical Chemistry A, 105(43), 9954–9960. 10.1021/jp003020w [DOI] [Google Scholar]
- Masetti, M., Falchi, F., Gioia, D., Recanatini, M., Ciurli, S., & Musiani, F. (2020). Targeting the protein tunnels of the urease accessory complex: A theoretical investigation. Molecules, 25(12), 2911. 10.3390/molecules25122911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuoka, T., Hidaka, H., & Eida, T. (1987). Effect of fructo‐oligosaccharides on intestinal microflora. Food / Nahrung, 31(5–6), 427–436. 10.1002/food.19870310528 [DOI] [PubMed] [Google Scholar]
- Miyase, T., & Ueno, A. (1993). Sucrose derivatives from the roots of Polygala tenuifolia. Japanese Journal of Pharmacognosy, 47(3), 267–278. [Google Scholar]
- Miyase, T., Noguchi, H., & Chen, X. M. (1999). Sucrose esters and xanthone C-glycosides from the roots of Polygala sibirica. Journal of Natural Products, 62(7), 993–996. 10.1021/np990084t [DOI] [PubMed] [Google Scholar]
- Orhan, F., Çeker, S., Anar, M., Agar, G., Arasoglu, T., & Gulluce, M. (2016). Protective effects of three luteolin derivatives on aflatoxin B1-induced genotoxicity on human blood cells. Medicinal Chemistry Research, 25(11), 2567–2577. 10.1007/s00044-016-1681-0 [DOI] [Google Scholar]
- Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y., & Jung, S. H. (2016). An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. Journal of Medicinal Chemistry, 59(14), 6595–6628. 10.1021/acs.jmedchem.5b01461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- QikProp Descriptors and Properties PISA . (2015). 2–4.
- Qiu, Z. K., Liu, C. H., Gao, Z. W., He, J. L., Liu, X., Wei, Q. L., & Chen, J. S. (2016). The inulin-type oligosaccharides extract from Morinda officinalis, a traditional Chinese herb, ameliorated behavioral deficits in an animal model of post-traumatic stress disorder. Metabolic Brain Disease, 31(5), 1143–1149. 10.1007/s11011-016-9853-7 [DOI] [PubMed] [Google Scholar]
- Rocchetti, G., Chiodelli, G., Giuberti, G., Masoero, F., Trevisan, M., & Lucini, L. (2017). Evaluation of phenolic profile and antioxidant capacity in gluten-free flours. Food Chemistry, 228, 367–373. 10.1016/j.foodchem.2017.01.142 [DOI] [PubMed] [Google Scholar]
- Rothan, H. A., & Byrareddy, S. N. (2020). The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. Journal of Autoimmunity, 109, 102433. 10.1016/j.jaut.2020.102433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabater-Molina, M., Larqué, E., Torrella, F., & Zamora, S. (2009). Dietary fructooligosaccharides and potential benefits on health. Journal of Physiology and Biochemistry, 65(3), 315–328. 10.1007/BF03180584 [DOI] [PubMed] [Google Scholar]
- Schrodinger. (2019). Schrödinger Release 2019-1: LigPrep. Schrödinger, LLC. [Google Scholar]
- Schrödinger Release . (2019. a). Desmond molecular dynamics system. Maestro-Desmond interoperability tools. D. E. Shaw Research. [Google Scholar]
- Schrödinger Release . (2019. b). Schrödinger Release 2019-1: LigPrep. Schrödinger, LLC. [Google Scholar]
- Schrödinger Release . (2019. c). Schrödinger Release 2019-1: Maestro. Schrödinger, LLC. [Google Scholar]
- Schrödinger Release . (2019. d). Schrödinger Release 2019-1: QikProp. Schrödinger, LLC. [Google Scholar]
- Schrödinger Release . (2019. e). Schrödinger Release 2019-1: Schrödinger Suite 2019-1 Protein Preparation Wizard; Epik. Schrödinger, LLC. [Google Scholar]
- Sterling, T., & Irwin, J. J. (2015). ZINC 15 - Ligand discovery for everyone. Journal of Chemical Information and Modeling, 55(11), 2324–2337. 10.1021/acs.jcim.5b00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunseri, J., & Koes, D. R. (2016). Pharmit: Interactive exploration of chemical space. Nucleic Acids Research, 44(W1), W442–W448. 10.1093/nar/gkw287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, M. S., & Walker, J. B. (1970). Streptomycin biosynthesis and metabolism. The Journal of Biological Chemistry, 245(24), 6683–6689. [PubMed] [Google Scholar]
- Walls, A. C., Park, Y.-J., Tortorici, M. A., Wall, A., McGuire, A. T., & Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell, 181(2), 281–292. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., Hu, Y., Tao, Z.-W., Tian, J.-H., Pei, Y.-Y., Yuan, M.-L., Zhang, Y.-L., Dai, F.-H., Liu, Y., Wang, Q.-M., Zheng, J.-J., Xu, L., Holmes, E. C., & Zhang, Y.-Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X., Liang, L., Zou, Y., Zhao, T., Zhao, J., Li, F., & Yang, L. (2011). Aqueous two-phase extraction, identification and antioxidant activity of anthocyanins from mulberry (Morus atropurpurea Roxb.). Food Chemistry, 129(2), 443–453. 10.1016/j.foodchem.2011.04.097 [DOI] [PubMed] [Google Scholar]
- Xia, S., Liu, M., Wang, C., Xu, W., Lan, Q., Feng, S., Qi, F., Bao, L., Du, L., Liu, S., Qin, C., Sun, F., Shi, Z., Zhu, Y., Jiang, S., & Lu, L. (2020). Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Research, 30(4), 343–355. 10.1038/s41422-020-0305-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, S., Zhu, Y., Liu, M., Lan, Q., Xu, W., Wu, Y., & Lu, L. (2020). Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cellular and Molecular Immunology. Springer Nature. 10.1038/s41423-020-s0374-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., Islam, M. S., Wang, J., Li, Y., & Chen, X. (2020). Traditional Chinese medicine in the treatment of patients infected with 2019-new coronavirus (SARS-CoV-2): A review and perspective. International Journal of Biological Sciences, 16(10), 1708–1717. 10.7150/ijbs.45538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J., Hua, Xin, H., Liang, Xu, Y., Ming, Shen, Y., He, Y. Q., Hsien-Yeh., & Du, J. (2018). Morinda officinalis How. – A comprehensive review of traditional uses, phytochemistry and pharmacology. Journal of Ethnopharmacology, 213, 230–255. 10.1016/j.jep.2017.10.028 [DOI] [PubMed] [Google Scholar]
- Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., & Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. Q., Yuan, L., Yang, M., Luo, Z. P., & Zhao, Y. M. (2002). The effect of Morinda officinalis How, a Chinese traditional medicinal plant, on the DRL 72-s schedule in rats and the forced swimming test in mice. Pharmacology Biochemistry and Behavior, 72(1–2), 39–43. 10.1016/S0091-3057(01)00730-4 [DOI] [PubMed] [Google Scholar]
- Zhu, X., Zhang, H., & Lo, R. (2004). Phenolic compounds from the leaf extract of artichoke (Cynara scolymus L.) and their antimicrobial activities. Journal of Agricultural and Food Chemistry, 52(24), 7272–7278. 10.1021/jf0490192 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.