

Abstract
The main protease (Mpro) of SARS-associated coronavirus (SARS-CoV) had caused a high rate of mortality in 2003. Current events (2019–2020) substantiate important challenges for society due to coronaviruses. Consequently, advancing models for the antiviral activity of therapeutic agents is a necessary component of the fast development of treatment for the virus. An analogy between anti-SARS agents suggested in 2017 and anti-coronavirus COVID-19 agents are quite probable. Quantitative structure-activity relationships for SARS-CoV are developed and proposed in this study. The statistical quality of these models is quite good. Mechanistic interpretation of developed models is based on the statistical and probability quality of molecular alerts extracted from SMILES. The novel, designed structures of molecules able to possess anti-SARS activities are suggested. For the final assessment of the designed molecules inhibitory potential, developed from the obtained QSAR model, molecular docking studies were applied. Results obtained from molecular docking studies were in a good correlation with the results obtained from QSAR modeling.
Keywords: Coronavirus, Severe Acute Respiratory Syndrome, anti-SARS agents, Monte Carlo method, index of ideality of correlation, molecular docking
Graphical Abstract

Communicated by Ramaswamy H. Sarma
Introduction
The genus coronavirus appertains to the plus-strand RNA virus and currently contains about 25 species that are classified into three groups under their genetic and serological relationships (Siddell et al., 2005; Yang et al., 2005). Coronaviruses (CoVs) infect humans and different species of animals, causing severe diseases (Yang et al., 2005). For example, human coronavirus (HCoV) strains 229E (HCoV-229E), NL63 (HCoV-NL63), OC43 (HCoV-OC43), and HKU1 (HCoV-HKU1) cause a significant portion of respiratory tract infections in humans, including bronchiolitis and pneumonia. They have also been involved in otitis, asthma, diarrhea, and neurological disease (Remuzzi & Remuzzi, 2020; Siddell et al., 2005; Yang et al., 2005).
Acute respiratory syndrome is an infectious disease caused by different viruses. Symptoms can be mild to severe. Severe acute respiratory syndrome (SARS) is a highly infective respiratory disease caused by SARS coronavirus (SARS-CoV). The SARS-CoV-2 is a very dangerous virus, according to most experts, and proved to be the etiological agent of a global outbreak of a life-threatening form of pneumonia called severe acute respiratory syndrome (SARS) (Abuhammad et al., 2017; Amin et al., 2020; Jain et al., 2020; Remuzzi & Remuzzi, 2020; Worldometer's COVID-19 data, 2020). The coronavirus COVID-19 also is causing severe respiratory pathology in humans and its spread alarms health organizations around the world (Amin et al., 2020; Remuzzi & Remuzzi, 2020; Worldometer's COVID-19 data, 2020).
An attractive therapeutic target for CoVs is the main protease (Mpro) or 3-chymotrypsin-like cysteine protease (3CLpro), as this enzyme plays a key role in polyprotein processing and is active in a dimeric form. Further, Mpro is highly conserved among various CoVs, and a mutation in Mpro is often lethal to the virus. Thus, drugs targeting the Mpro enzyme significantly reduce the risk of mutation-mediated drug resistance and display broad-spectrum antiviral activity (Goyal & Goyal, 2020).
One can expect some analogies between different coronaviruses. There are available experimental data for inhibitory activity towards SARS-CoV-2 of a set of organic compounds (Abuhammad et al., 2017).
Quantitative structure – activity relationships (QSARs) are a tool to solve problems related to medicinal chemistry when the analysis of the chemical structure becomes important (Abuhammad et al., 2017; Amin et al., 2020; Bhargava et al., 2017, 2019; Halder, 2018; Jain et al., 2020; Makhouri & Ghasemi, 2018; Wang et al., 2017). The approach can be based on the Monte Carlo technique (Abuhammad et al., 2017; Amin et al., 2020; Bhargava et al., 2017; Duhan et al., 2019; Jain et al., 2020; Kumar et al., 2020; Kumar & Kumar, 2020).
Disulfide bonds play key roles for bioactive proteins in the aspect of preferable correct folding (Wang et al., 2017). It is not a simple task to develop a method sensitive to assigned molecular features. CoMFA and CoMSIA were used to build up 3D-QSAR for novel inhibitors of SARS-CoV-2 main protease (Wang et al., 2017). The direct interface between 3D models and the chemical technique of synthesis of considered unsymmetrical aromatic disulfides is an attractive advantage of the above research work (Wang et al., 2017).
In this research in silico method - MD was used to test the inhibition effect of designed molecules, because, the calculated binding energies can correlate with inhibitory potentials (Halperin et al., 2002; Kitchen et al., 2004).
We suppose that these models can provide a bridge to develop antiviral agents for the COVID-19 when the corresponding experimental data will become available.
Method
Data
The data on unsymmetrical aromatic disulfide compounds and their SARS-CoV Mpro inhibitory activities (IC50, µM) are available in the literature (Wang et al., 2017). This data set includes forty compounds. In our study the compounds were randomly splitting into the active training set (25%), passive training set (25%), calibration set (25%), and validation set (25%). The distribution of data into those four sets is a necessary component of building up QSAR models using the CORAL software (Toropova & Toropov, 2017). The details and tasks related to each of the above sets are described in the literature (Toropova & Toropov, 2019).
Optimal descriptor
The optimal descriptor is calculated with molecular features extracted from SMILES (Weininger, 1988):
| (1) |
The Sk is the SMILES-atom, i.e. single symbol (e.g. C, N, O, etc.), or a group of symbols which cannot be examined apart (e.g. Cl, Br, %11, etc.); SSk is a pair of SMILES-atoms; SSSk is a trine of SMILES-atoms. It is to be noted the SMILES-atom can be the traditional atom (e.g. N, S, Br, etc.), but also it can represent covalent bonds (i.e. =, #, @), as well as other molecular phenomena (e.g. the number of rings, 1, 2, %11, etc.). The NS is the number of SMILES-atoms for given SMILES.
Having the correlation weight for all molecular features involved in the modelling process, one can obtain the one-variable model:
| (2) |
Monte Carlo optimization
The scheme of the improved Monte Carlo optimization is described in the number of papers (Kumar & Chauhan, 2017; Manisha et al., 2019; Toropova et al., 2020). The essence of this version of the optimization procedure is in the application of the Index of ideality of correlation (IIC). Models for the inhibitory activity built up here apply the algorithm previously described (Kumar & Chauhan, 2017; Manisha et al., 2019; Toropova et al., 2020). Three models have been developed based on the three splits of data as described above.
Molecular docking
Different host receptors for cellular entry, the structural proteins (antigens), and the high mutation and recombination rates of CoVs are a significant problem in the development of wide-spectrum anti-CoV drugs. The accession number in the Protien Data Bank (PDB, http://www.rcsb.org/pdb/) for each structure of SARS Mpro is individual. The crystal structure of SARS-CoV Mpro in complex with inhibitor (PDB code 2AMD) was obtained from the PDB databank and used for molecular docking studies. All molecules that acted as ligands for docking studies were drawn using Marvin sketch (Marvin 6.1.0, 2013, ChemAxon), and MMFF94 force field implemented in Marvin sketch was used to obtain their optimal 3D geometry. In this research, Molegro Virtual Docker (MVD) was used as the main software for molecular docking studies. MVD can be successfully used in MD studies for obtaining an appropriate geometrical orientation of the ligand inside the active site of the studied enzyme. Also, MVD can identify hydrogen bonds and hydrophobic interactions between flexible studied compounds and rigid amino acids from the enzyme active site. Further, MVD can be used to calculate “scoring” functions related to relevant binding energies and these functions can be applied for the estimation of the studied compounds inhibitory effect (Thomsen & Christensen, 2006; Zivkovic et al., 2020). In this study, the used “scoring” functions were: Hbond, VdW, Steric, Pose energy, MolDock, and Rerank Score and their detailed description is given in the literature (Zivkovic et al., 2020). A published approach was used for molecular docking protocol validation (Amin et al., 2018). Maestro software version 11.6.012. was used for presenting 2D representations of interactions between the studied molecules and amino acids from the studied enzyme active site.
Results and discussion
The models for SARS-CoV Mpro inhibitory activity obtained in the three employed splits are the following:
| (3) |
| (4) |
| (5) |
Table 1 contains the statistical characteristics of these models. Table 1 confirms two important features of the approach: (i) the statistical quality for the validation set is high, the correlation coefficient for each split is larger than 0.90; and (ii) dispersion for all considered criteria is small.
Table 1.
The statistical characteristics of the developed model for SARS-CoV Mpro inhibitory activities for three random splits of data.
| Split | Set | R2 | Q2 | CCC | IIC* | RMSE | MAE |
|---|---|---|---|---|---|---|---|
| 1 | Active training set | 0.6464 | 0.4450 | 0.7852 | 0.956 | 0.795 | |
| Passive training set | 0.7586 | 0.5802 | 0.8205 | 0.886 | 0.610 | ||
| Calibration set | 0.9115 | 0.8512 | 0.9026 | 0.9539 | 0.470 | 0.371 | |
| Validation set | 0.9566 | 0.9320 | 0.9216 | 0.542 | 0.438 | ||
| 2 | Active training set | 0.7439 | 0.5840 | 0.8531 | 0.893 | 0.762 | |
| Passive training set | 0.7654 | 0.6003 | 0.7366 | 1.07 | 0.714 | ||
| Calibration set | 0.9174 | 0.8799 | 0.9358 | 0.9577 | 0.413 | 0.336 | |
| Validation set | 0.9442 | 0.9154 | 0.9479 | 0.454 | 0.309 | ||
| 3 | Active training set | 0.6983 | 0.4234 | 0.8223 | 0.824 | 0.650 | |
| Passive training set | 0.8445 | 0.7742 | 0.5553 | 1.54 | 1.33 | ||
| Calibration set | 0.9124 | 0.8829 | 0.9081 | 0.9548 | 0.345 | 0.267 | |
| Validation set | 0.9175 | 0.8916 | 0.9249 | 0.489 | 0.403 |
IIC = index of ideality of correlation (Toropova & Toropov, 2019); RMSE = root mean squared error; MAE = mean absolute error.
The statistical quality of the model based on 3D representation of the molecular structure suggested in the literature (Wang et al., 2017) is defined as R2=0.916, Q2=0.681. Three compounds were removed as influential outliers (Wang et al., 2017). Thus, the comparison of the above model with the predictive potential of models suggested here confirms that the described approach gives models with quite good predictive potential. Several runs of the Monte Carlo optimization with a different distribution of data into the training and validation sets allow obtaining the statistical and mechanistic interpretation of the model (Table 2). It should be noted that promoters of increase for IC50[µM] have stable prevalence, whereas promoters of decrease are relatively rare ones.
Table 2.
Promoters of increase and decrease of the inhibitory activity of SARS‐CoV Mpro (IC50, μM).
| No. | SMILES attribute | CWs Probe 1 | CWs Probe 2 | CWs Probe 3 | N1 | N2 | N3 |
|---|---|---|---|---|---|---|---|
| Promoters of IC50 increase | |||||||
| 1 | 1……….. | 2.63153 | 1.15486 | 3.78669 | 10 | 10 | 10 |
| 2 | 2……….. | 2.55878 | 1.21207 | 1.64933 | 10 | 10 | 10 |
| 3 | S……….. | 0.48795 | 0.06021 | 2.20339 | 10 | 10 | 10 |
| 4 | S…S……. | 2.02730 | 0.56998 | 2.44765 | 10 | 10 | 10 |
| 5 | c……….. | 0.05582 | 0.34232 | 0.34661 | 10 | 10 | 10 |
| 6 | c…1……. | 0.07471 | 0.40638 | 1.18716 | 10 | 10 | 10 |
| 7 | c…S…S… | 2.96490 | 1.14621 | 2.40403 | 10 | 10 | 10 |
| 8 | c…c……. | 0.04212 | 0.58132 | 0.22205 | 10 | 10 | 10 |
| 9 | n……….. | 0.30264 | 0.00072 | 0.28500 | 10 | 10 | 10 |
| 10 | n…c……. | 1.35271 | 0.05507 | 0.12055 | 10 | 10 | 10 |
| 11 | C…(……. | 0.43671 | 0.17893 | 0.17255 | 9 | 8 | 6 |
| 12 | C……….. | 1.10523 | 0.50393 | 0.47038 | 9 | 8 | 7 |
| 13 | O…(……. | 0.16441 | 0.57525 | 0.88699 | 9 | 7 | 7 |
| 14 | O…=……. | 0.13313 | 0.43790 | 0.28864 | 9 | 7 | 6 |
| 15 | n…c…(… | 0.10720 | 0.32142 | 0.33964 | 8 | 6 | 4 |
| Promoters of IC50 decrease | |||||||
| 1 | N……….. | −0.02294 | −0.17040 | −0.48815 | 6 | 3 | 4 |
| 2 | 3…n…(… | −0.15624 | −0.05464 | −0.16157 | 3 | 2 | 0 |
| 3 | n…3…n… | −0.25960 | −0.10767 | −0.08532 | 3 | 2 | 0 |
| 4 | n…(…O… | −2.54052 | −0.12802 | −2.47903 | 2 | 0 | 0 |
| 5 | 1…n…(… | −2.13926 | −1.32809 | −2.11675 | 1 | 3 | 1 |
| 6 | s…(…1… | −0.05921 | −0.10068 | −0.01523 | 1 | 1 | 2 |
N1, N2, and N3 are frequencies of molecular features in the active training set, passive training set, and calibration set, respectively; CWs are the correlation weight.
The described approach indicates that the molecular features related to nitrogen atoms hint on how to select promising molecular structures (Table 3). In other words, the analysis of various structures based on the suggested CORAL model is transparent and convenient for practical applying.
Table 3.
Examples of proposed modifications for structure #38 together with variations of model values of SARS-CoV Mpro inhibitory activity.
| Structures and SMILES | Model IC50[µM] | Comment |
|---|---|---|
Basis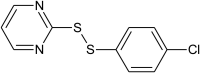 Clc2ccc(SSc1ncccn1)cc2 Experiment IC50[µM] = 0.684 |
0.921 | |
Improvement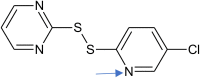 Clc2cnc(SSc1ncccn1)cc2 |
0.850 | Fragment [N] is added |
Improvement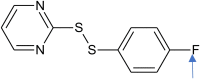 Fc2ccc(SSc1ncccn1)cc2 |
0.909 | Fragment [F] is added |
Improvement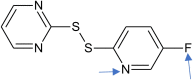 Fc2cnc(SSc1ncccn1)cc2 |
0.838 | Fragments [N] and [F] are added |
Figure 1 represents experimental and calculated with Equations (3)–(5) the inhibitory activity of SARS‐CoV Mpro. One can see (Figure 1), the split (distribution of available data into the training set, and the validation set) have a significant influence on the prediction for a given structure. However, as noted above, in all the described experiments, the statistical quality of models is quite good.
Figure 1.

The experimental and calculated SARS-CoV Mpro inhibitory activities for three random splits.
For the assessment of designed molecules, inhibitory potential MD studies were used. Binding energies of the studies compound with the active site of SARS‐CoV Mpro are presented in Table 4 and the best-calculated poses for all studied compounds inside the SARS‐CoV Mpro were shown in Figure 2.
Table 4.
Score values (kcal/mol) for all computer-aided designed compounds.
| Molecule | Pose Energy | HBond | Steric | VdW | MolDock Score | Rerank Score |
|---|---|---|---|---|---|---|
| A | −95.3978 | 0 | −108.245 | −31.6369 | −93.9852 | −81.9477 |
| A1 | −96.2774 | −2.5 | −109.457 | −21.6198 | −94.3305 | −78.3661 |
| A2 | −96.357 | 0 | −108.868 | −30.6175 | −94.9384 | −82.1304 |
| A3 | −97.1479 | −2.5 | −107.498 | −33.5078 | −93.8624 | −85.0888 |
Figure 2.

Binding energies of the studied compound with the active site of SARS-CoV Mpro.
“Scoring” functions can be associated with the inhibitory potential of the studied compound and according to obtained results for MolDock score function molecule with the highest potential is A2, while results for ReRank score function indicate that molecule with the highest potential is A3. According to results for both above stated “scoring” functions molecule with the lowest inhibitory potential is A. The highest energy interactions related to Van der Waals interactions were determined for molecule A3, while molecule A1 had the highest energy-related to steric interactions. The highest pose energy was calculated for molecule A3. All obtained results for “scoring” functions are in good agreement with results obtained from QSAR modeling. MD studies were applied to identify all interactions between the designed molecules and amino acids from SARS‐CoV Mpro, including hydrogen bonds, hydrophobic, and hydrophilic interactions, and they are presented in Figure 3. Most significant components of the interaction are histidine, valine, and glutamine (Figure 3).
Figure 3.

MD studies are applied to identify all interactions between the designed molecules and amino acids from SARS-CoV Mpro, including hydrogen bonds, hydrophobic, and hydrophilic interactions.
Supplementary materials section contains experimental and calculated SARS-CoV Mpro inhibitory activities for three random splits.
Conclusions
The described approach provides a quite good model for the inhibitory activity of SARS‐CoV Mpro by 50% (IC50, μM). The model is accompanied by the mechanistic interpretation that can help to compare the potentials of different molecular structures as possible antiviral agents. This facilitates the exploration of efficient drug candidates. The CORAL software is freely available on the Internet (www.insilico.eu/coral) and provides a capable tool for QSAR studies. Molecular docking studies were applied to calculate the energy and determine interactions between the designed compounds and amino acids inside the SARS‐CoV Mpro. In presented research calculated “scoring” functions for designed molecules were used as estimators for their inhibitory potential and obtained results were in good correlation with the results obtained from QSAR modeling.
Author contributions
The authors contributed equally to this work.
Supplementary Material
Glossary
Abbreviations
- CoV
Coronaviruses
- COVID-19
(CO: Corona, VI: Virus, D: Disease, 19: 2019)
- CoMFA
Comparative molecular field analysis
- CoMSIA
comparative molecular similarity indices analysis
- DCW
optimal descriptor of correlation weights
- HCoV
human coronavirus
- MD
molecular docking
- Mpro
main protease
- MVD
Molegro Virtual Docker
- PDB
Protein Data Bank
- QSAR
quantitative structure activity relationships
- RNA
ribonucleic acid
- SMILES
simplified molecular input-line entry system
- SARS
Severe acute respiratory syndrome
Funding Statement
A.A.T and A.P.T. are grateful for the contribution of the project LIFE-VERMEER contract (LIFE16 ENV/IT/000167) for the support. A.M.V. would like to thank the Ministry of Education and Science, the Republic of Serbia, under Project Number 172044. J.L. and DL would like to thank the NSF-CREST program for the support (grant HRD #154774).
Disclosure statement
The authors confirm they have no conflict of interest.
References
- Abuhammad, A., Al-Aqtash, R. A., Anson, B. J., Mesecar, A. D., & Taha, M. O. (2017). Computational modeling of the bat HKU4 coronavirus 3CLpro inhibitors as a tool for the development of antivirals against the emerging Middle East Respiratory Syndrome (MERS) coronavirus. Journal of Molecular Recognition, 30 (11), e2644. 10.1002/jmr.2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, S. A., Bhargava, S., Adhikari, N., Gayen, S., & Jha, T. (2018). Exploring pyrazolo[3,4-d]pyrimidine phosphodiesterase 1 (PDE1) inhibitors: A predictive approach combining comparative validated multiple molecular modelling techniques. Journal of Biomolecular Structure & Dynamics, 36 (3), 590–608. 10.1080/07391102.2017.1288659 [DOI] [PubMed] [Google Scholar]
- Amin, S. A., Ghosh, K., Gayen, S., & Jha, T. (2020). Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1780946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava, S., Adhikari, N., Amin, S. A., Das, K., Gayen, S., & Jha, T. (2017). Hydroxyethylamine derivatives as HIV-1 protease inhibitors: A predictive QSAR modelling study based on Monte Carlo optimization. SAR and QSAR in Environmental Research, 28 (12), 973–990. 10.1080/1062936X.2017.1388281 [DOI] [PubMed] [Google Scholar]
- Bhargava, S., Patel, T., Gaikwad, R., Patil, U. K., & Gayen, S. (2019). Identification of structural requirements and prediction of inhibitory activity of natural flavonoids against Zika virus through molecular docking and Monte Carlo based QSAR simulation. Natural Product Research, 33 (6), 851–857. 10.1080/14786419.2017.1413574 [DOI] [PubMed] [Google Scholar]
- Duhan, M., Singh, R., Devi, M., Sindhu, J., Bhatia, R., Kumar, A., & Kumar, P. (2019). Synthesis, molecular docking and QSAR study of thiazole clubbed pyrazole hybrid as α-amylase inhibitor. Journal of Biomolecular Structure and Dynamics, 1–17. 10.1080/07391102.2019.1704885 [DOI] [PubMed] [Google Scholar]
- Goyal, B., & Goyal, D. (2020). Targeting the dimerization of the main protease of coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Combinatorial Science, 22 (6), 297–305. 10.1021/acscombsci.0c00058 [DOI] [PubMed] [Google Scholar]
- Halder, A. K. (2018). Finding the structural requirements of diverse HIV-1 protease inhibitors using multiple QSAR modelling for lead identification. SAR and QSAR in Environmental Research, 29 (11), 911–933. 10.1080/1062936X.2018.1529702 [DOI] [PubMed] [Google Scholar]
- Halperin, I., Ma, B., Wolfson, H., & Nussinov, R. (2002). Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins, 47(4), 409–443. 10.1002/prot.10115 [DOI] [PubMed] [Google Scholar]
- Jain, S., Amin, S. A., Adhikari, N., Jha, T., & Gayen, S. (2020). Good and bad molecular fingerprints for human rhinovirus 3C protease inhibition: Identification, validation, and application in designing of new inhibitors through Monte Carlo-based QSAR study. Journal of Biomolecular Structure & Dynamics, 38 (1), 66–77. 10.1080/07391102.2019.1566093 [DOI] [PubMed] [Google Scholar]
- Kitchen, D. B., Decornez, H., Furr, J. R., & Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews. Drug Discovery, 3(11), 935–949. 10.1038/nrd1549 [DOI] [PubMed] [Google Scholar]
- Kumar, A., & Chauhan, S. (2017). Monte Carlo method based QSAR modelling of natural lipase inhibitors using hybrid optimal descriptors. SAR and QSAR in Environmental Research, 28 (3), 179–197. 10.1080/1062936X.2017.1293729 [DOI] [PubMed] [Google Scholar]
- Kumar, P., & Kumar, A. (2020). Nucleobase sequence based building up of reliable QSAR models with the index of ideality correlation using Monte Carlo method. Journal of Biomolecular Structure & Dynamics, 38 (11), 3296–3306. 10.1080/07391102.2019.1656109 [DOI] [PubMed] [Google Scholar]
- Kumar, A., Sindhu, J., & Kumar, P. (2020). In-silico identification of fingerprint of pyrazolyl sulfonamide responsible for inhibition of N-myristoyltransferase using Monte Carlo method with index of ideality of correlation. Journal of Biomolecular Structure and Dynamics, Published online: 24 Jun 2020, 1–12. 10.1080/07391102.2020.1784286 [DOI] [PubMed] [Google Scholar]
- Makhouri, F. R., & Ghasemi, J. B. (2018). In silico studies in drug research against neurodegenerative diseases. Current Neuropharmacology, 16 (6), 664–725. 10.2174/1570159X15666170823095628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manisha, Chauhan, S., Kumar, P., & Kumar, A. (2019). Development of prediction model for fructose- 1,6- bisphosphatase inhibitors using the Monte Carlo method. SAR and QSAR in Environmental Research, 30 (3), 145–159. 10.1080/1062936X.2019.1568299 [DOI] [PubMed] [Google Scholar]
- Remuzzi, A., & Remuzzi, G. (2020). COVID-19 and Italy: What next? The Lancet, 395 (10231), 1225–1228. 10.1016/S0140-6736(20)30627-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddell, S. G., Ziebuhr, J., & Snijder, E. J. (2005). Coronaviruses, toroviruses, and arteriviruses. In Mahy B. W. J. & ter Meulen V. (Eds.). Topley and Wilson's microbiology and microbial infections (10th ed., pp. 823–856). Hodder Arnold. [Google Scholar]
- Thomsen, R., & Christensen, M. H. (2006). MolDock: A new technique for high-accuracy molecular docking. Journal of Medicinal Chemistry, 49(11), 3315–3321. 10.1021/jm051197e [DOI] [PubMed] [Google Scholar]
- Toropova, A. P., & Toropov, A. A. (2017). Hybrid optimal descriptors as a tool to predict skin sensitization in accordance to OECD principles. Toxicology Letters, 275, 57–66. 10.1016/j.toxlet.2017.03.023 [DOI] [PubMed] [Google Scholar]
- Toropova, A. P., & Toropov, A. A. (2019). Does the index of ideality of correlation detect the better model correctly? Molecular Informatics, 38 (8–9), 1800157. 10.1002/minf.201800157 [DOI] [PubMed] [Google Scholar]
- Toropova, A. P., Toropov, A. A., Leszczynska, D., & Leszczynski, J. (2020). The index of ideality of correlation: Models of the flash points of ternary mixtures. New Journal of Chemistry, 44(12), 4858–4868. 10.1039/D0NJ00121J [DOI] [Google Scholar]
- Wang, L., Bao, B.-B., Song, G.-Q., Chen, C., Zhang, X.-M., Lu, W., Wang, Z., Cai, Y., Li, S., Fu, S., Song, F.-H., Yang, H., & Wang, J.-G. (2017). Discovery of unsymmetrical aromatic disulfides as novel inhibitors of SARS-CoV main protease: Chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. European Journal of Medicinal Chemistry, 137, 450–461. 10.1016/j.ejmech.2017.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weininger, D. (1988). SMILES, a chemical language and information system: 1: Introduction to methodology and encoding rules. Journal of Chemical Information and Modeling, 28 (1), 31–36. 10.1021/ci00057a005 [DOI] [Google Scholar]
- Worldometer's COVID-19 data . 2020. Retrieved July, 2020, from https://www.worldometers.info/coronavirus/?fbclid=IwAR1e00MffPgwU_mGPU11ECyg-5iDzewENoQCpRkcGD2LKcV_n1wJzbC6zDE#page-top.
- Yang, H., Xie, W., Xue, X., Yang, K., Ma, J., Liang, W., Zhao, Q., Zhou, Z., Pei, D., Ziebuhr, J., Hilgenfeld, R., Yuen, K. Y., Wong, L., Gao, G., Chen, S., Chen, Z., Ma, D., Bartlam, M., & Rao, Z. (2005). Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biology, 3 (10), e324. 10.1371/journal.pbio.0030324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivkovic, M., Zlatanovic, M., Zlatanovic, N., Golubović, M., & Veselinović, A. M. (2020). The application of combination of Monte Carlo optimization method based QSAR modeling and molecular docking in drug design and development. Mini-Reviews in Medicinal Chemistry, 20(14), 1389–1402. 10.2174/1389557520666200212111428 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.