

Abstract
A new strain of coronavirus (CoV) has been identified as SARS-CoV-2, which is responsible for the recent COVID-19 pandemic. Currently, there is no approved vaccine or drug available to combat the pandemic. COVID-19 main protease (Mpro) is a key CoV enzyme, which plays an important role in triggering viral replication and transcription, turns it into an attractive target. Therefore, we aim to screen natural products library to find out potential COVID-19 Mpro inhibitors. Plant-based natural compounds from Sigma-Aldrich plant profiler chemical library have been screened through virtual molecular docking and molecular dynamics simulation to identify potential inhibitors of COVID Mpro. Our virtual molecular docking results have shown that there are twenty-eight natural compounds with a greater binding affinity toward the COVID-19 Mpro inhibition site as compared to the co-crystal native ligand Inhibitor N3 (-7.9 kcal/mol). Also, molecular dynamics simulation results have confirmed that Peonidin 3-O-glucoside, Kaempferol 3-O-β–rutinoside, 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-β-D-xylopyranosyl-β-D-glucopyranosyl)oxy]-2H-1-benzopyran-2-one, Quercetin-3-D-xyloside, and Quercetin 3-O-α-L-arabinopyranoside (selected based on the docking score) possess a significant amount of dynamic properties such as stability, flexibility and binding energy. Our In silco results suggests that all the above mention natural compounds have the potential to be developed as a COVID-19 Mpro inhibitor. But before that, it must go through under the proper preclinical and clinical trials for further scientific validation.
Communicated by Ramaswamy H. Sarma
Keywords: Coronavirus (SARS-CoV-2), COVID-19 main protease (Mpro), natural compounds, in silico molecular docking (Autodock vina), molecular dynamics (MD) simulation (Gromacs)
Introduction
Last year, in December 2019 rapidly spreading viral Pneumonia cases were found in the city of Wuhan (China) (Wu et al., 2020; Zhou et al., 2020). Later on, a new strain of coronavirus was identified responsible for the outbreak, named SARS-CoV-2 (Gorbalenya et al., 2020), because the RNA genome of this new virus is 82% identical to the SARS coronavirus (SARS-CoV) and both the viruses belong to clade b of the genus Betacoronavirus (Wu et al., 2020; Zhou et al., 2020). On February 11, 2020, the World Health Organization (WHO) officially named the disease COVID-19 (coronavirus disease 2019). As human to human transmission of this virus headed to exponential growth globally. The World Health Organization (WHO) declared the outbreak a pandemic on 11th March 2020. According to the current situation report (WHO) on Aug 14th, 2020 there are 20,730,456 cumulative confirmed cases globally with a 3.62% death rate (World Health Organisation (WHO), 2020).
Currently, there is no available therapy to treat COVID-19. Therefore, drugs are needed which can inhibit the SARS-CoV-2. One of the best drug targets to combat the coronaviruses is the main protease (Mpro) (Anand et al., 2003) (Figure 1). As Mpro plays a pivotal role in the translation of polyproteins from viral RNA. The functional polypeptides are release from two overlapping polyproteins, pp1a and pp1ab through an expanded proteolytic process, majorly by the Mpro. It operates at no less than 11 cleavage sites on the large polyprotein 1ab (replicase 1ab, ∼790 kDa); the recognition sequence at most sites is Leu–Gln ↓(Ser, Ala, Gly) (↓ marks the cleavage site) (Zhang et al., 2020). Therefore, blocking the activity of this enzyme would inhibit viral replication and transcription. Also, no proteases with a similar cleavage specificity are known to be found in human, therefore inhibitors are more likely to be non-toxic (Zhang et al., 2020). Regarding that, target-based screening of bioactive compounds could be an option to identify potential Mpro inhibitor for SARS-CoV-2. For this purpose, computation (In silico) methods like virtual molecular docking and molecular dynamic simulation could be utilized efficiently. Later, screened potential compounds can be validated through In vitro/In vivo experiments. Thus, we can speed up the process of drug discovery and development. As plant-based natural compounds have a large range of structural diversity, we have tried to screen (In silico) natural compounds from Sigma-Aldrich plant profiler chemical library against the target (COVID-19 Mpro), and found a natural compound Rutin as a hit from garlic (Allium sativum) with a basic structure of anthocyanin. Later on, we have tried to find structurally similar natural compounds to Rutin and also screened (In silico) them against the target.
Figure 1.

Schematic diagram of SARS-CoV-2 life-cycle.
Materials and methods
Target preparation
The 3 D crystal structure of COVID-19 main protease (Mpro) has been obtained from RCBS Protein Data Bank (PDB) (https://www.rcsb.org/) using PDB ID: 6LU7 (Jin et al., 2020). The crystal structure has two chains: A and C. Chain A of the macromolecule has been selected as the target receptor. The native co-crystal ligand present in the crystal structure is n-[(5-methylisoxazol-3-yl)carbonyl]alanyl-l-valyl-n∼1∼-((1r,2z)-4-(benzyloxy)-4-oxo-1-{[(3r)-2-oxopyrrolidin-3-yl] methyl}but-2-enyl)-l-leucinamide (Inhibitor N3). Then, the selected target receptor has been prepared (add hydrogen and charge: AMBER ff14SB) for docking using Dock Prep tool of UCSF Chimera software.
Inhibition site selection
COVID-19 main protease (Mpro) 3 D crystal structure (PDB ID: 6LU7) is a complex structure with an inhibitor N3. In Discovery Studio Visualizer 2017 R2, it has been observed that this protease crystal structure has five cavities site and the Inhibitor N3 is bound to the protease at site 1 (Figure S9). Therefore, we have selected site 1 as an inhibition site (X = −13.670, Y = 14.677, Z = 73.814).
Ligand selection
Initially, we have screened various natural compounds present in Sigma-Aldrich plant profiler chemical library (https://www.sigmaaldrich.com/life-science/nutrition-research/learning-center/plant-profiler.html). Performing virtual molecular docking, we found that Rutin from garlic (Allium sativum) possesses a higher binding affinity than native co-crystal ligand inhibitor N3 toward COVID-19 main protease. After that, structurally similar compounds of rutin has been searched using SwissSimilarity (http://www.swisssimilarity.ch/) web tool (Zoete et al., 2016). From here, 40 compounds have been successfully docked to COVID-19 (Mpro) inhibition site (Table 1).
Table 1.
Virtual molecular docking score.
| Sl. No. | Rank | Ligand | PubChem CID | Binding Energy (kcal/mol) |
|---|---|---|---|---|
| 1. | 1. | Peonidin 3-O-glucoside | 443654 | −9.4 |
| 2. | 2. | Kaempferol 3-O-β -rutinoside | 25201364 | −9.3 |
| 3. | 3. | Rutin | 5280805 | −9.2 |
| 4. | 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-β-D-xylopyranosyl- β-D-glucopyranosyl)oxy]- 2H-1-benzopyran-2-one | 329824875 | ||
| 5. | 4. | Quercetin-3-D-xyloside | 5320863 | −9.1 |
| 6. | 5. | Quercetin 3-O-α-L-arabinopyranoside | 329766687 | −9.0 |
| 7. | 6. | Kaempferol 3-rutinoside 4′-glucoside | 329824889 | −8.9 |
| 8. | Apigenin 7-O-neohesperidoside | 24891380 | ||
| 9. | 7. | Quercetin 3-O-(6″-O-malonyl)-β-D-glucoside | 329751366 | −8.8 |
| 10. | Idaein | 44256700 | ||
| 11. | Callistephin | 44256621 | ||
| 12. | 8. | Malvin | 441765 | −8.7 |
| 13. | 9. | Luteolin 7-rutinoside | 44258082 | −8.6 |
| 14. | 10. | Cyanin | 441688 | −8.5 |
| 15. | Apiin | 5280746 | ||
| 16. | Mearnsitrin | 6918652 | ||
| 17. | Hispidulin 7-glucuronide | 44258434 | ||
| 18. | 5-Glucopyranosyloxy-3′,4′,7-trihydroxyneoflavone | 14134093 | ||
| 19. | 11. | Kaempferol 7-O-neohesperidoside | 5483905 | −8.4 |
| 20 | 12. | Rhamnetin 3-sophoroside | 71341751 | −8.3 |
| 21. | 5-Hydroxy-3-(4-methoxyphenyl)-4-oxo-4H-chromen-7-yl 6-O- (6-deoxyhexopyranosyl)hexopyranoside | 97952641 | ||
| 22. | Kaempferol 3-O-D-galactoside | 5488283 | ||
| 23. | Baicalin | 64982 | ||
| 24. | 13. | Myricetin 3-O-β-D-Galactopyranoside | 329824902 | −8.2 |
| 25. | Wogonoside | 3084961 | ||
| 26. | 14. | 2"-O-alpha-L-Rhamnopyranosyl-isovitexin | 329824876 | −8.2 |
| 27. | 15. | Hesperidin methylchalcone | 6436550 | −8.0 |
| 28. | Undulatoside A | 5321494 | ||
| 29. | 16. | Inhihitor N3 (Native co-crystal Ligand) | – | -7.9 |
| 30. | 17. | Isoprunetin 7-O-glucoside | 189922 | −7.7 |
| 31. | 18. | Ononin | 442813 | −7.6 |
| 32. | Daidzin | 107971 | ||
| 33. | 19. | Hispidulin 4′-glucoside | 44258466 | −7.5 |
| 34. | 4-Methylumbelliferyl β-D-cellobioside | 126287 | ||
| 35. | 20. | Sissotrin | 5280781 | −7.4 |
| 36. | 3-O-Methylquercetin | 5280681 | ||
| 37. | 21. | 3,7,2′,4′,5′-pentamethoxyflavone | 45933946 | −7.2 |
| 38. | 22. | Rhamnazin | 5320945 | −7.1 |
| 39. | 4′,5,7-Trihydroxy-3,6-dimethoxyflavone | 5352032 | ||
| 40. | 23. | Isokaempferide | 5280862 | −7.0 |
| 41. | 3-hydroxy-7,3′,4′,5′-tetramethoxyflavone | 21721930 |
Ligand preparation
All the ligands have been obtained from PubChem chemical library in SDF format. Later on, it have been converted into mol2 file format for docking purposes using Discovery Studio Visualizer 2017 R2. Before docking, all the ligands (mol2 format) have been prepared (add hydrogen and charge: Gasteiger) using Dock Prep tool of UCSF Chimera software.
Virtual molecular docking
Virtual molecular docking and analysis have been performed according to the previously described method (Majumder et al., 2019). All the small molecules have been docked at COVID-19 Mpro inhibition site by Autodock vina (Trott & Olson, 2009) using UCSF Chimera gui.
Molecular dynamics simulation
Explicit solvent molecular dynamics (MD) simulations have been executed for the further investigation of virtual molecular docking results. GROMACS v.2019.4 package has been used to run 100 ns MD simulation of protease-ligand complexes according to the previously described method (Das et al., 2020; Panda et al., 2020). GROMOS96 43a1 in single point charge (SPC) water models (Berendsen et al., 1995) and PRODRG server (van Aalten et al., 1996) have been used to generate force field and parameter files for protease and ligands. The receptor-ligand complexes have been to be placed in a periodic cubic solvated box (x = 9.631, y = 9.631, z = 9.631) with at least 10 Å distance from the edge of the box. Periodic boundary conditions have been employed using the particle mesh Ewald (PME) method for long-range electrostatics interactions. All the receptor-ligand complex systems have been neutralized by adding 4 NA+ counter ions. Prior to the running of real dynamics, energy minimization and equilibration of all systems has been executed through following three steps: (i) Energy minimization of all systems containing ions, solvent, receptor, and ligand has been executed through using the steepest descent minimization algorithm with 5000 steps to achieve stable system with maximum force < 1000 kJ mol−1 nm−1. (ii) Position restrains have been applied to receptor and ligand of the each systems for 100 ns throughout heating (300 K) utilizing NVT (No. of atoms, Volume, Temperature) ensemble with leap-frog integrator, a time step of 2 fs and LINCS holonomic constraints. (iii) NPT (No. of atoms, Pressure, Temperature) ensemble has been applied at a constant pressure (1 bar) and temperature (300 K) for 100 ps using a time step of 2 fs for NPT equilibration phase. After the energy minimization and equilibration of all systems, MD production run has been executed without any restrain for 100 ns with a time step of 2 fs, and after every 10 ps coordinates of the structure have been saved. After the completion of 100 ns MD simulation, the trajectories have been used for various dynamics analysis such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), number of hydrogen bonds, Gibbs free landscape and secondary structural analysis by different inbuilt scripts of GROMACS. Also, the MM-PBSA binding free energy of receptor-ligand complexes have been calculated by utilizing the Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) method which utilized the MD simulation trajectories and is one of the best-used methods in this kind of analysis (Kumari et al., 2014).
Results
Virtual molecular docking
Initially, native co-crystal ligand: Inhibitor N3 of the COVID-19 main protease (Mpro) complex (crystal structure) has been redocked to legitimize the virtual molecular docking protocol for its accuracy. The root mean square deviation (RMSD) value between the co-crystal and redocked native ligand: Inhibitor N3 pose is 1.094 Å. The RMSD value within 2 Å is considered as a successful docking protocol (Rao et al., 2007). It has been observed that the interaction of co-crystal and redocked native ligand: Inhibitor N3 with COVID-19 Mpro inhibition site residues are mostly identical (Figure 2). After that, forty natural compounds have been screened as a potential inhibitor (Table 1). Redocking of native co-crystal ligand: Inhibitor N3 shows that it has a high amount of binding affinity (-7.9 kcal/mol) toward inhibition site. Therefore, we have analysed the interaction of those small molecules which have lesser binding energy that means higher binding affinity toward inhibition site as compared to native co-crystal ligand: Inhibitor N3. We have found twenty-eight small molecules that possess higher binding affinity as compared to native co-crystal ligand: Inhibitor N3 (Figure S1-S8). Also, these twenty-eight small molecules and native co-crystal ligand: Inhibitor N3 interactions with inhibition site amino residues of COVID-19 (Mpro) have been depicted in Table 2.
Figure 2.

Superimpose interaction of co-crystal (Green) and redocked (Red) native ligand: Inhibitor N3 at inhibition site of COVID-19 main protease.
Table 2.
Inhibition site amino residues interaction.*
| Rank | Compounds (Formula) |
Structure | Source | Binding Energy (kcal/mol) | H-Bond interaction with receptor amino acid residues | Hydrophobic interaction with receptor amino acid residues |
|---|---|---|---|---|---|---|
| 1. | Peonidin 3-O-glucoside (C22H23O11+) |
 |
Fruits, berries, red grapes and red wines (Landrault et al., 2001) | −9.4 | Leu141, Asn142, Ser144, His163, Glu166, | His41, Met49, Cys145, Met165, Arg187, Arg188, Gln189 |
| 2. | Kaempferol 3-O-β –rutinoside (C33H40O20) |
 |
Selliguea feei rhizomes (Baek et al., 1994), Solanum Campanoforme leaves (“kaempferol-3-rutinoside (CHEBI:69657),” n.d.) | −9.3 | Thr25, Ser46, Tyr54, Glu166, | Leu27, His41, Met49, Cys145, Met165, Asp187 |
| 3. | Rutin (C27H30O16) |
 |
Buckwheat, most fruits and vegetables (Patel & Patel, 2019) | −9.2 | Asn142, Thr190 | His41, Met49, Glu166, Asp187, Arg188, Gln189 |
| 4-(3,4-Dihydroxyphenyl)-7-methoxy-5- [(6-O-β-D-xylopyranosyl-β-D-glucopyranosyl)oxy]- 2H-1-benzopyran-2-one (C27H30O15) |
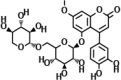 |
Hintonia standleyana, Hintonia latiflora leaves (Cristians et al., 2009) | Thr26, Gly143, Ser144, Cys145, His163 | Thr25, His41, Met49, Met165, Glu166, Asp187, Gln189 | ||
| 4. | Quercetin-3-D-xyloside (C20H18O11) |
 |
Prunus salicina , Prunus domestica peels (Jang et al., 2018) | −9.1 | His41, Leu141, Asn142, Ser144, His163, Glu166, Thr190 | Met165, Pro168, Asp187, Arg188, Gln189 |
| 5. | Quercetin 3-O-α-L-arabinopyranoside (C20H18O11) |
 |
Ruprechtia polystachya leaves (Bruzual De Abreu et al., 2011) | −9.0 | Tyr53, His163, Glu166, Asp187 | His41, Met49, Phe140, Leu141, Arg188, Gln189 |
| 6. | Kaempferol 3-rutinoside 4′-glucoside (C33H40O20) |
 |
Prunus armeniaca (Jang et al., 2018) | −8.9 | Thr24, Thr26, Tyr54, Phe140, Leu141, Ser144, Cys145, His163, Glu166 | His41, Met49, Asn142, Gly143, Met165, Gln189 |
| Apigenin 7-O-neohesperidoside (C27H30O14) |
 |
Bitter orange, Bergamot, Grapefruit, Lemon, Lupines (Refaat et al., 2015) | Leu141, Gly143, Ser 144, His163 | Asn142, Cys145, Met165, Glu166, Pro168, Gln189 | ||
| 7. | Quercetin 3-O-(6″-O-malonyl)-β-D-glucoside (C24H22O15) |
 |
Mulberry leaves (Ju et al., 2018) | −8.8 | Leu141, Ser144, Glu166 | His41, Met49, Asn142, Cys145, Met165, Asp187, Arg188, Gln189 |
| Idaein (C21H21O11+ ) |
 |
Chinese hawthorn fruits (Liu et al., 2011) | Thr26, His163 | His41, Met49, Leu141, Cys145, Met165, Asp187, Arg188, Gln189 | ||
| Callistephin (C21H21O10+) |
 |
Pomegranate (Hernández et al., 1999), Straw berries (Mullen et al., 2008) | Thr26, His163, Glu166 | His41, Met49, Leu141, Cys145, Met165, Asp187, Arg188, Gln189 | ||
| 8. | Malvin (C29H35O17+) |
 |
Grapes and wine (Jackson, 2017) | −8.7 | Leu141, Ser144, His163, Thr190 | Leu27, His41, Met49, Gly143, Cys145, Met165, Glu166, Gln189 |
| 9. | Luteolin 7-rutinoside (C27H30O15) |
 |
Cyclopia subternata (Yoon et al., 2015) | −8.6 | Thr26, Ser144 | His41, Met49, Asn142, Cys145, Met165, Glu166, Asp187, Asp188, Gln189 |
| 10. | Cyanin (C27H31O16+) |
 |
Berries, Grapes, Apples, Purple Cabbage, Corn (Kamenickova et al., 2013) | −8.5 | Thr26, Ser144, His163, Arg188 | His41, Met49, Met165, Gln189 |
| Apiin (C26H28O14) |
 |
Winter-hardy plants parsley and celery (Meyer et al., 2006), (Gupta & Seshadri, 1952) | Ser144, Cys145, His163, Thr190 | Thr25, Met49, Gly143, Glu166, Pro168, Gln189 | ||
| Mearnsitrin (C22H22O12) |
 |
Myrcia uniflora (Ferreira et al., 2006) | Gly143, His163 | His41, Met49, Met165, Glu166, Gln189 | ||
| Hispidulin 7-glucuronide (C22H20O12) |
 |
Salvia plebia (Weng & Wang, 2000) | Thr24, Thr25, Thr26, Ser46, Gly143, Ser144, Cys145 | Leu27, Met49, Glu166 | ||
| 5-Glucopyranosyloxy-3′,4′,7-trihydroxyneoflavone (C21H20O11) |
 |
Hintonia latiflora stem bark (Mata et al., 1992) | Asn142, Ser144, Cys145, His163 | His41, Met49, Gly143, Met165, Gln189 | ||
| 11. | Kaempferol 7-O-neohesperidoside (C27H30O15) |
 |
Litchi chinensis fruit (X. Xu et al., 2011) | −8.4 | Thr26, Ser46, Gly143, Glu166, Thr190 | Thr25, Leu27, Thr45, Met49, Cys145, Met165, Gln189 |
| 12. | Rhamnetin 3-sophoroside (C28H32O17) |
 |
Nasturtium officinale (water cress) (“Human Metabolome Database: Showing metabocard for Rhamnetin 3-sophoroside (HMDB0038303),” n.d.) | −8.3 | Ser144, Cys145, His163, Met165, Asp187 | His41, Tyr54, Glu166, Arg188 |
| 5-Hydroxy-3-(4-methoxyphenyl)- 4-oxo-4H-chromen-7-yl 6-O-(6-deoxyhexopyranosyl)hexopyranoside (C28H32O14) |
 |
Mirabilis jalapa (Eneji Sadiq, 2016) | His41, Arg188, Thr190, Gln192 | Thr24, Thr45, Met49, Asn142, Cys145, Met165, Glu166, Gln189 | ||
| Kaempferol 3-O-D-galactoside (C21H20O11) |
 |
Camptotheca acuminate (Li et al., 2005), Euphorbia condylocarpa (Roshchin, 1977), Consolida oliveriana (Díaz et al., 2008) | Thr26, Ser46, Gly143, Glu166, Thr190 | Thr25, Leu27, Thr45, Met49, Cys145, Met165, Gln189 | ||
| Baicalin (C21H18O11) |
 |
Scutellaria baicalensis roots (Habtemariam, 2019) | Glu166, Arg188, Thr190 | Thr25, Leu27, His41, Gly143, Cys145, Met165, Gln189 | ||
| 13. | Myricetin 3-O-β-D-Galactopyranoside (C21H20O13) |
 |
Nelumbo nucifera Gaertn (Xu et al., 2011) |
−8.2 | Leu141, Ser144, Met165, Thr190, Gln192 | Glu166, Glu189 |
| Wogonoside (C22H20O11) |
 |
Scutellaria baicalensis (Grzegorczyk-Karolak et al., 2016) | His41 | Met49, Asn142, Gly143, Cys145, Met165, Pro168, Arg188, Gln189 | ||
| 14. | 2"-O-alpha-L-Rhamnopyranosyl-isovitexin (C27H30O14) |
 |
Mimosa xanthocentra (Camargo et al., 2012) | −8.2 | Leu141, Gly143, Ser144 | Met49, Phe140, Asn142, Cys145, His163, Met165, Pro168, Gln189, Thr190, Ala191, |
| 15. | Hesperidin methylchalcone (C29H36O15) |
 |
lemons and sweet oranges (Zanwar et al., 2013) | −8.0 | Thr24, Thr25, Thr26, Cys44, Asn142 | Met49, Cys145, Met165, Gln189 |
| Undulatoside A (C16H18O9) |
 |
Dryopteris fragrans (Peng et al., 2016) | Phe140, Leu141, Gly143, Ser144, Cys145, His163, Glu166 | His41, Met165, Gln189 | ||
| 16. | Inhihitor N3 (Native co-crystal Ligand) |  |
Synthetic construct (Jin et al., 2020) | -7.9 | Phe140, Gly143, Cys145, His163, His164, Glu166, Gln189, Thr190 | Thr25, Thr26, His41, Leu141, Met165, Pro168 |
*Identical amino acid residues interaction of COVID-19 main protease with native co-crystal ligand: Inhibitor N3 and other ligands at inhibition site are highlighted in bold front.
Molecular dynamic simulation
For the further investigation of virtual molecular docking results, top six docked ligand (binding energy ≤ −9.0 kcal/mol) complex (COVID-19 MPRO - ligand) and crystal protease complex with the Inhibitor N3 (COVID-19 MPRO, PDB ID: 6LU7), have been run for 100 ns molecular dynamics simulation. From that MD simulation trajectory, we have analysed root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), number of hydrogen bond, Gibbs free energy landscape and secondary structural to check receptor-ligand conformational properties such as stability, and flexibility. Besides that, the binding energy of receptor-ligand complexes throughout the whole MD simulation has also calculated using MMPBSA methods.
RMSD analysis
Root mean square deviation (RMSD) value is an indicator of the stability of the receptor-ligand complex. Here, the RMSD values of all six docked COVID-19 Mpro–ligand complexes (ligands docked at inhibition site) have been compared with COVID-19 Mpro–Inhibitor N3 complex (crystal structure) (Figure 3a). It is visible that RMSD of COVID-19 Mpro complexes docked with Peonidin 3-O-glucoside (0.303 nm), Quercetin-3-D-xyloside (0.363 nm), 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-ß-D-xylopyranosyl-ß-D-glucopyranosyl)oxy]-2H-1-benzopyran-2-one (0.434 nm), Quercetin 3-O-α-L-arabinopyranoside (0.436 nm), and Kaempferol 3-O-ß –rutinoside (0.586 nm) respectively are significantly stable as compared to the native co-crystal ligand Inhibitor N3 (0.617 nm). It indicates that all of these bioactive molecules have formed stable complexes with COVID-19 Mpro. We have found that RMSD value of Rutin (0.822 nm) is higher when it binds with COVID-19 Mpro as compared to the native co-crystal ligand Inhibitor N3. It suggests that COVID-19 Mpro-Rutin complex is slightly less stable.
Figure 3.

(a) RMSD analysis for the COVID-19 MPRO -ligand complexes. (b) RMSF analysis for the COVID-19 MPRO -ligand complexes. (c) Radius of gyration (Rg) analysis for the COVID-19 MPRO -ligand complexes. (d) Estimation of the hydrogen bond number during the 50 ns MD simulations of COVID-19 Mpro-ligand complexes.
RMSF analysis
Root mean square fluctuation (RMSF) is an indicator of amino acid residual mobility. The high RMSF values of amino acid residues indicate a large degree of mobility and instability in the receptor. On the other hand, the low RMSF values of amino acid residues define stable and rigid receptor. The RMSF values of all docked complexes have been calculated and depicted in Figure 3b. The overall average RMSF values of COVID-19 Mpro amino acid residues has been calculated for 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-ß-D-xylopyranosyl-ß-D-glucopyranosyl)oxy]-2H-1-benzopyran-2-one, Kaempferol 3-O-ß-rutinoside, Quercetin-3-D-xyloside, Peonidin 3-O-glucoside, Quercetin 3-O-α-L-arabinopyranoside, Rutin, Kaempferol 3-O-ß –rutinoside, and native ligand Inhibitor N3 are 0.118, 0.134, 0.141, 0.149, 0.151, 0.157, and 0.190 nm respectively. Here, it can be observed that overall average RMSF values of receptor amino acid residues are lower when it binds with the bioactive natural compounds respectively as compared to the native ligand Inhibitor N3. It suggests that COVID-19 Mpro has formed stable and rigid complexes with bioactive natural compounds.
Radius of gyration (Rg) analysis
Rg is the indicator of compactness, stability, and folding of structure. We have calculated the Rg based on the intrinsic dynamics of protease-ligand complexes (Figure 3c). All the complexes have a steady average Rg of ∼2.10 nm over the 100 ns MD simulation. Such results indicate that majority of ligands have formed compact and stable complexes with COVID-19 MPRO as compared to the native co-crystal ligand Inhibitor N3.
Hydrogen bond (H-bond) analysis
The number of Hydrogen bonds (H-bond), which are the main stabilizing interaction factor between two molecules, have been calculated to investigate the nature of H-bond at the inhibition site of COVID-19 Mpro. The H-bonds have been recorded throughout the 100 ns of the receptor-ligand MD simulations (Figure 3d). Consequently, it has been observed that native co-crystal ligand Inhibitor N3 has formed an average of 1.06 H-bonds, while other docked ligands such as Kaempferol 3-O-b-rutinoside, Quercetin-3-D-xyloside, Peonidin 3-O-glucoside, 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-ß-D-xylopyranosyl-ß-D-glucopyranosyl)oxy]-2H-1-benzopyran-2-one, Quercetin 3-O- α-L-arabinopyranoside, and Rutin have produced an average of 7.18, 6.16, 5.67, 4.97, 4.87, and 4.17 H-bonds respectively at COVID Mpro inhibition site. Such results indicate that docked ligands have formed a higher number of stable H-bonds as compared to the native co-crystal ligand: Inhibitor N3 at inhibition site of COVID-19 Mpro during the whole MD simulation.
Free energy landscape
Gibbs free energy landscape (FEL) plot for COVID-19 MPRO- ligand complexes have been generated by calculating the first two principal components (PC1 and PC2) as reaction coordinates, using GROMACS inbuilt scripts (g_covar, g_anaeig, and g_sham). The primary function of g_sham is to plot Gibbs free energy landscapes by Bolzmann inverting multi-dimensional histograms using PC1 and PC2 as input. The FEL can depict the global minima energy conformation of a structure (receptor-ligand complex). If the receptor-lignad interaction is very weak or unstable, it can achieve multiple minimum energy clusters; whereas a strong and stable interaction can bring almost a single conformation cluster in the potential energy distribution. All the contour 2 D and 3 D plots of receptor-ligand complexes during 100 ns MD simulation have been depicted in Figure 4. Each receptor-ligand complex has shown a distinct pattern for FEL. Dark violet/blue spots reflect the energy minima and energetically favoured structural conformation and red/yellow spots indicate the unfavourable structural confirmation. The shallow and narrow energy basin indicates the low stability of structural conformation. It can be observed from the Figure 4 that the COVID-19 main protease (Mpro) complexes with Peonidin 3-O-glucoside, Rutin, 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-ß-D-xylopyranosyl-ß-D-glucopyranosyl)oxy]-2H-1-benzopyran-2-one, and Quercetin-3-D-xyloside respectively, have shown a noticeable single large global energy minima basin related to its conformational state. It suggests a strong and stable conformation of the receptor-ligand complex. On other hand, the COVID-19 main protease (Mpro) complexes with native ligand Inhibitor N3, Kaempferol 3-O-β –rutinoside, and Quercetin 3-O-α-L-arabinopyranoside respectively, have shown split of the basin into two relative energy minima which indicates the conformational insability of receptor-ligand complex. It is important to mention that though natural compounds like Kaempferol 3-O-β –rutinoside, and Quercetin 3-O-α-L-arabinopyranoside have formed energetically less stable structural conformation with receptor Mpro, as FEL shows two energy minima region (Figure 4c and 4g) but two regions are not entirely separated by energy barrier region like crystal structure of COVID-19 Mpro-Inhibitor N3 complex (Figure 4a). It suggests these two natural compounds have formed more strong, stable, and energetically favourable structural conformation when they bind with COVID-19 Mpro as compared to native ligand Inhibitor N3.
Figure 4.

The Gibbs free energy landscape plot of COVID-19 MPRO complexes with (a) Inhibitor N3, (b) Peonidin 3-O-glucoside, (c) Kaempferol 3-O-β-rutinoside, (d) Rutin, (e) 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-ß-D-xylopyranosyl-ß-D-glucopyranosyl)oxy]-2H-1-benzopyran-2-one, (f) Quercetin-3-D-xyloside, (g) Quercetin 3-O-α-L-arabinopyranoside.
MM/PBSA binding free energy calculations
To calculate a more accurate binding free energy between COVID-19 main protease (COVID-19 Mpro) and selected ligands at receptor inhibition site, the MM/PBSA based method has been used. Here, the binding free energy defines the total of non-bonded interaction energies (van der Wall, electrostatic, polar solvation, and SASA energy) between receptor and ligand throughout the whole MD simulation (100 ns). Lower binding free energy indicates a better binding between receptor and ligand. We have estimated binding free energy of docked ligands from 100 ns MD simulation trajectories and it suggests that Kaempferol 3-O-β–rutinoside (-343.212 +/- 33.998 kJ/mol), Peonidin 3-O-glucoside (-274.623 +/- 27.613 kJ/mol), Quercetin-3-D-xyloside (-268.184 +/- 31.135 kJ/mol), 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-β-D-xylopyran (-259.970 +/- 46.377 kJ/mol), and Quercetin 3-O-α-L-arabinopyranoside (-229.655 +/- 27.4 kJ/mol) have a higher binding affinity towards receptor (COVID-19 Mpro) inhibition site as compared to co-crystal ligand inhibitor N3 (-224.101 +/- 28.124 kJ/mol) (Figure 5a and Table 3). We have found that Rutin (-164.299 +/- 38.698 kJ/mol) has a lower binding affinity towards COVID-19 Mpro inhibition site as compared to co-crystal ligand inhibitor N3. It may be due to the higher RMSD value of Rutin at the receptor binding/inhibition site as compared to other ligands.
Figure 5.

(a) Gibbs free energy calculation (MMPBSA) of receptor-ligand complexes. (b) The quantification of the individual amino acid residues of receptor to the total binding energies toward ligands.
Table 3.
Van der Waals, electrostatic, polar solvation, SASA and binding energy for the docked compounds into Mpro inhibition site.
| Compounds | Van der Waal energy (kJ/mol) | Electrostattic energy (kJ/mol) | Polar solvation energy (kJ/mol) | SASA energy (kJ/mol) | Binding energy (kJ/mol) |
|---|---|---|---|---|---|
| Kaempferol 3-O-β -rutinoside | −304.878 +/− 23.757 | −220.815 +/− 49.635 | 205.931 +/− 32.348 | −23.451 +/− 1.898 | −343.212 +/− 33.998 |
| Peonidin 3-O-glucoside | −231.392 +/− 22.967 | −202.404 +/− 48.521 | 178.379 +/− 26.368 | −19.206 +/− 1.292 | −274.623 +/− 27.613 |
| Quercetin-3-D-xyloside | −217.810 +/− 21.114 | −199.746 +/− 43.194 | 167.472 +/− 17.962 | −18.099 +/− 1.164 | −268.184 +/− 31.135 |
| 4-(3,4-Dihydroxyphenyl)-7- methoxy-5-[(6-O-β-D-xylopyran | −228.732 +/− 34.957 | −216.638 +/− 68.161 | 205.946 +/− 37.868 | −20.545 +/− 2.841 | −259.970 +/− 46.377 |
| Quercetin 3-O-α-L-arabinopyranoside | −218.131 +/− 16.581 | −160.942 +/− 42.139 | 166.954 +/− 23.152 | −17.536 +/− 1.080 | −229.655 +/− 27.400 |
| Inhibitor N3 | −271.825 +/− 27.798 | −41.168 +/− 20.409 | 110.185 +/− 21.426 | −21.293 +/− 2.040 | −224.101 +/− 28.124 |
| Rutin | −178.043 +/− 26.061 | −136.565 +/− 55.091 | 168.270 +/− 40.575 | −17.961 +/− 2.093 | −164.299 +/− 38.698 |
Figure 5.

The snapshot of COVID-19 Mpro complexes with (a) Native co-crystal ligand: Inhibitor N3, (b) Peonidin 3-O-glucoside, (c) Kaempferol 3-O-β -rutinoside, (d) Rutin, (e) 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-ß-D-xylopyranosyl-ß-D-glucopyranosyl)oxy]-2H-1-benzopyran-2-one Robinin, (f) Quercetin -3-D-xyloside, (g) Quercetin 3-O-α-L-arabinopyranoside, (h) over the 10 ns interval of the 50 ns MD simulation trajectory.
Later on, we have analyzed the binding contribution energy of each amino acid residue of the receptor (COVID-19 Mpro) in the binding of ligands at the inhibition site (Figure 5b). There are several amino acid residues (THR-24, HIS-41, MET-49, TYR-54, CYS-145, GLU-166, ASP-187, and GLN-189), which have shown favorable contribution energy when they bind with natural ligands as compared to native ligand Inhibitor N3. We have found that amino acid residue GLU-166 has involved in highest contribution energy during the receptor-ligand binding. It has been reported that amino acid residue GLU-166 involves in the construction of a biologically active main protease (Mpro) dimer form (Anand et al., 2003). It suggests bioactive natural ligands like Kaempferol 3-O-β –rutinoside, Peonidin 3-O-glucoside, Quercetin-3-D-xyloside, etc. could effectively bind to COVID-19 Mpro amino acid residue GLU-166 and inhibit the dimerization process of COVID-19 Mpro. It could lead to higher efficacy in the inhibition of the COVID-19 Mpro.
Secondary structural analysis during simulation
An extensive study has been done to understand the structural evolution of the COVID-19 Mpro-ligand during the 100 ns simulation. Moreover, how the ligand is sticking to the inhibition site of COVID-19 Mpro is a crucial point to understand the stability of the ligands. Five snapshots of the COVID-19 Mpro-ligand complexes have been taken at every 25 ns interval (25 ns, 50 ns, 75 ns, and 100 ns). It is observed that the secondary structure elements (helix and beta-sheets) remained conserved throughout the simulation process (Figure 5), which highlights the stability and reliability of COVID-19 Mpro after binding to the ligands. The N-terminal and the C-terminal coils are observed to be fluctuating a bit, but structurally, no significant changes have been seen. It has been also observed that the ligands are constantly attached to the inhibition site without any structural modification, which implies that the ligands are highly stable.
The Secondary structural analysis with gmx_do_dssp has shown no significant level of overall conformational change in ligand-bound COVID-19 Mpro complexes as compared to the crystal structure of COVID-19 MPRO in complex with an inhibitor N3 (Figure 6 and Table 4).
Figure 6.

The secondary structure content of COVID-19 Mpro complexes with (a) Inhibitor N3, (b) Peonidin 3-O-glucoside, (c) Kaempferol 3-O-ß –rutinoside, (d) Rutin, (e) 4-(3,4-Dihydroxyphenyl) – 7 – methoxy – 5 - [(6-O-ß-D-xylopyranosyl-ß-D - glucopyranosyl) oxy] -2H-1-benzopyran-2-one Quercetin-3-D-xyloside, (f) Quercetin-3-D-xyloside, (g), Quercetin 3-O-a-L-arabinopyranoside.
Table 4.
The overall percentage of secondary structure elements in COVID-19 Mpro-ligand complexes.
| Compound | Structure (A-Helix + B-Sheet + B-Bridge + Turn)% | Coil% | B-Sheet% | B-Bride% | Bend% | Tunr% | A-Helix% | 5-Helix% | 3-Helix% |
|---|---|---|---|---|---|---|---|---|---|
| Inhibitor N3 | 54 | 30 | 24 | 1 | 14 | 7 | 22 | 1 | 0 |
| Peonidin 3-O-glucoside | 59 | 26 | 26 | 2 | 12 | 9 | 23 | 0 | 2 |
| Kaempferol 3-O-β -rutinoside | 59 | 28 | 26 | 1 | 12 | 9 | 23 | 0 | 2 |
| Rutin | 60 | 27 | 25 | 1 | 12 | 9 | 24 | 0 | 2 |
| 4-(3,4-Dihydroxyphenyl)- 7-methoxy-5-[(6-O-β-D-xylopyran | 59 | 28 | 25 | 1 | 11 | 9 | 24 | 0 | 2 |
| Quercetin-3-D-xyloside | 60 | 27 | 26 | 2 | 12 | 8 | 25 | 0 | 1 |
| Quercetin 3-O-α-L-arabinopyranoside | 56 | 28 | 25 | 2 | 13 | 8 | 22 | 1 | 2 |
Discussion
Our In silico studies reveals that natural compounds like Rutin and its structurally similar compounds with a basic structure of anthocyanin (Peonidin 3-O-glucoside, Kaempferol 3-O-b-rutinoside, Quercetin- 3-D-xyloside, Quercetin 3-O-α-L-arabinopyranoside, etc.) may inhibit the COVID-19 main protease (Mpro), which is essential to block the replication and growth of the novel coronavirus (SARS-CoV-2). Our virtual molecular docking score suggests that the top twenty-eight compounds (Table 1) have a higher amount of binding affinity toward inhibition site of COVID-19 Mpro as compared to the native co-crystal ligand: Inhibitor N3. Furthermore, molecular dynamic simulation studies of the top six compounds which have a very high amount of binding affinity (binding energy ≤ −9 kcal/mol from docking score) toward inhibition site reveal that compounds like Kaempferol 3-O-b-rutinoside, Peonidin 3-O-glucoside, Quercetin-3-D-xyloside, 4-(3,4-Dihydroxyphenyl)-7-methoxy-5-[(6-O-ß-D-xylopyranosyl-ß-D-glucopyranosyl)oxy]-2H-1-benzopyran-2-one, and Quercetin 3-O-a-L-arabinopyranoside have a highly favorable conformational stability, flexibility, and binding energy when they have docked into the inhibition site of COVID-19 Mpro compared to the crystal structure of COVID-19 MPRO in complex with an Inhibitor N3.
Overall, our In silico results indicate that the above-mentioned compounds may have the potential to be developed as an anti-COVID-19 main protease drug to combat the novel coronavirus but before that, it must go through under the proper preclinical and clinical trials for further scientific validation.
Supplementary Material
Acknowledgements
We would like to acknowledge the Indian Institute of Technology Kharagpur, India, Indian Council of Medical Research, Council of Scientific and Industrial Research, and J C Bose Fellowship (SERB, India) for providing structural and financial support.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R., & Hilgenfeld, R. (2003). Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science (New York, N.Y.).), 300(5626), 1763–1767. 10.1126/science.1085658 [DOI] [PubMed] [Google Scholar]
- Baek, N. I., Kennelly, E. J., Kardono, L. B. S., Tsauri, S., Padmawinata, K., Doel Soejarto, D., & Douglas Kinghorn, A. (1994). Flavonoids and a proanthocyanidin from rhizomes of Selliguea feei. Phytochemistry, 36(2), 513–518. 10.1016/S0031-9422(00)97105-X [DOI] [Google Scholar]
- Berendsen, H. J. C., van der Spoel, D., & van Drunen, R. (1995). GROMACS: A message-passing parallel molecular dynamics implementation. Computer Physics Communications, 91(1–3), 43–56. 10.1016/0010-4655(95)00042-E [DOI] [Google Scholar]
- Camargo, L. M. D. M., Férézou, J. P., Tinoco, L. W., Kaiser, C. R., & Costa, S. S. (2012). Flavonoids from Mimosa xanthocentra (Leguminosae: Mimosoideae) and molecular modeling studies for isovitexin-2″-O-α-l-rhamnopyranoside rotamers. Phytochemistry Letters, 5(3), 427–431. 10.1016/j.phytol.2012.03.015 [DOI] [Google Scholar]
- Cristians, S., Guerrero-Analco, J. A., Pérez-Vásquez, A., Palacios-Espinosa, F., Ciangherotti, C., Bye, R., & Mata, R., (2009). Hypoglycemic activity of extracts and compounds from the leaves of hintonia standleyana and H. latiflora: Potential alternatives to the use of the stem bark of these species. Journal of Natural Products, 72(3), 408–413. 10.1021/np800642d [DOI] [PubMed] [Google Scholar]
- Das, P., Majumder, R., Mandal, M., & Basak, P. (2020). In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (M pro) from flavonoid based phytochemical constituents of Calendula officinalis. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1796799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzual De Abreu, M. B., Temraz, A., Malafronte, N., Gonzalez-Mujica, F., Duque, S., & Braca, A. (2011). Phenolic derivatives from ruprechtia polystachya and their inhibitory activities on the glucose-6-phosphatase system. Chemistry & Biodiversity, 8(11), 2126–2134. 10.1002/cbdv.201000334 [DOI] [PubMed] [Google Scholar]
- Díaz, J. G., Carmona, A. J., Fernando Torres, A. T., Quintana, J., Estévez, F., & Herz, W. (2008). Cytotoxic activities of flavonoid glycoside acetates from Consolida oliveriana. Planta Medica, 74(2), 171–174. 10.1055/s-2008-1034278 [DOI] [PubMed] [Google Scholar]
- Ferreira, A. C. F., Neto, J. C., Da Silva, A. C. M., Kuster, R. M., & Carvalho, D. P. (2006). Inhibition of thyroid peroxidase by Myrcia uniflora flavonoids. Chemical Research in Toxicology, 19(3), 351–355. 10.1021/tx0501684 [DOI] [PubMed] [Google Scholar]
- Gorbalenya, A. E., Baker, S. C., Baric, R. S., de Groot, R. J., Drosten, C., Gulyaeva, A. A., … Ziebuhr, J. (2020). The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology, 5, 536–544. 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzegorczyk-Karolak, I., Kuźma, Ł., & Wysokińska, H. (2016). In vitro cultures of Scutellaria alpina as a source of pharmacologically active metabolites. Acta Physiologiae Plantarum, 38(1), 1–9. 10.1007/s11738-015-2024-3 [DOI] [Google Scholar]
- Gupta, S. R., & Seshadri, T. R. (1952). A study of apiin from the parsley seeds and plant. Proceedings of the Indian Academy of Sciences - Section A, 35(5), 242–248. 10.1007/BF03172503 [DOI] [Google Scholar]
- Habtemariam, S. (2019). Chapter 26 - Antidiabetic herbal medicines rebranded as dietary supplements. In S. Habtemariam (Ed.), Medicinal foods as potential therapies for type-2 diabetes and associated diseases (pp.1049-1134). Academic Press. ISBN 9780081029220, 10.1016/b978-0-08-102922-0.00026-2 [DOI] [Google Scholar]
- Hernández, F., Melgarejo, P., Tomás-Barberán, F. A., & Artés, F. (1999). Evolution of juice anthocyanins during ripening of new selected pomegranate (Punica granatum) clones. European Food Research and Technology , 210(1), 39–42. 10.1007/s002170050529 [DOI] [Google Scholar]
- Human Metabolome Database: Showing metabocard for Rhamnetin 3-sophoroside (HMDB0038303). (n.d.). Retrieved April 11, 2020, from https://hmdb.ca/metabolites/HMDB0038303
- Jackson, R. S. (2017). Chapter 2 - Visual perceptions. In R. S. Jackson (Ed.), Wine tasting(pp.19-40).Academic press. ISBN 9780128018132, 10.1016/b978-0-12-801813-2.00002-1 [DOI] [Google Scholar]
- Jang, G. H., Kim, H. W., Lee, M. K., Jeong, S. Y., Bak, A. R., Lee, D. J., & Kim, J. B. (2018). Characterization and quantification of flavonoid glycosides in the Prunus genus by UPLC-DAD-QTOF/MS. Saudi Journal of Biological Sciences, 25(8), 1622–1631. 10.1016/j.sjbs.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020). Structure of M pro from COVID-19 virus and discovery of its inhibitors. Nature, 582(7811), 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Ju, W. T., Kwon, O. C., Kim, H. B., Sung, G. B., Kim, H. W., & Kim, Y. S. (2018). Qualitative and quantitative analysis of flavonoids from 12 species of Korean mulberry leaves. Journal of Food Science and Technology, 55(5), 1789–1796. 10.1007/s13197-018-3093-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- kaempferol-3-rutinoside (CHEBI:69657 ). (n.d.). Retrieved April 11, 2020, from https://www.ebi.ac.uk/chebi/searchId.do;15942DE7427998FC235B9F100186349A?printerFriendlyView=true&locale=null&chebiId=69657&viewTermLineage=null&structureView=&
- Kamenickova, A., Anzenbacherova, E., Pavek, P., Soshilov, A. A., Denison, M. S., Zapletalova, M., Anzenbacher, P., & Dvorak, Z. (2013). Effects of anthocyanins on the AhR-CYP1A1 signaling pathway in human hepatocytes and human cancer cell lines. Toxicology Letters, 221(1), 1–8. 10.1016/j.toxlet.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari, R., Kumar, R., & Lynn, A. (2014). g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling, 54(7), 1951–1962. 10.1021/ci500020m [DOI] [PubMed] [Google Scholar]
- Landrault, N., Poucheret, P., Ravel, P., Gasc, F., Cros, G., & Teissedre, P. L. (2001). Antioxidant capacities and phenolics levels of French wines from different varieties and vintages. Journal of Agricultural and Food Chemistry, 49(7), 3341–3348. 10.1021/jf010128f [DOI] [PubMed] [Google Scholar]
- Li, S., Zhang, Z., Cain, A., Wang, B., Long, M., & Taylor, J. (2005). Antifungal activity of camptothecin, trifolin, and hyperoside isolated from Camptotheca acuminata. Journal of Agricultural and Food Chemistry, 53(1), 32–37. 10.1021/jf0484780 [DOI] [PubMed] [Google Scholar]
- Liu, P., Kallio, H., Lü, D., Zhou, C., & Yang, B. (2011). Quantitative analysis of phenolic compounds in Chinese hawthorn (Crataegus spp.) fruits by high performance liquid chromatography-electrospray ionisation mass spectrometry. Food Chemistry, 127(3), 1370–1377. 10.1016/j.foodchem.2011.01.103 [DOI] [PubMed] [Google Scholar]
- Majumder, R., Parida, P., Paul, S., & Basak, P. (2019). In vitro and in silico study of Aloe vera leaf extract against human breast cancer. Natural Product Research, 34(16), 2363–2366. 10.1080/14786419.2018.1534848 [DOI] [PubMed] [Google Scholar]
- Mata, R., Del Rayo Camacho, M., Mendoza, S., & Del Carmen Cruz, M. (1992). A phenylstyrene from Hintonia latiflora. Phytochemistry, 31(9), 3199–3201. 10.1016/0031-9422(92)83474-D [DOI] [Google Scholar]
- Meyer, H., Bolarinwa, A., Wolfram, G., & Linseisen, J. (2006). Bioavailability of Apigenin from Apiin-Rich Parsley in Humans. Annals of Nutrition & Metabolism, 50(3), 167–172. 10.1159/000090736 [DOI] [PubMed] [Google Scholar]
- Mullen, W., Edwards, C. A., Serafini, M., & Crozier, A. (2008). Bioavailability of pelargonidin-3-O-glucoside and its metabolites in humans following the ingestion of strawberries with and without cream. Journal of Agricultural and Food Chemistry, 56(3), 713–719. 10.1021/jf072000p [DOI] [PubMed] [Google Scholar]
- Panda, P. K., Arul, M. N., Patel, P., Verma, S. K., Luo, W., Rubahn, H.-G., Mishra, Y. K., Suar, M., & Ahuja, R. (2020). Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Science Advances, 6(28), eabb8097. 10.1126/sciadv.abb8097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, K., & Patel, D. K. (2019). Chapter 26 - The beneficial role of rutin, a naturally occurring flavonoid in health promotion and disease prevention: A systematic review and update. In R. R. Watson & V. R. Preedy (Eds.), Bioactive food as dietary interventions for arthritis and related inflammatory diseases(pp.457-479).Academic Press. ISBN 9780128138205, 10.1016/b978-0-12-813820-5.00026-x [DOI] [Google Scholar]
- Peng, B., Bai, R.-F., Li, P., Han, X.-Y., Wang, H., Zhu, C.-C., Zeng, Z.-P., & Chai, X.-Y. (2016). Two new glycosides from Dryopteris fragrans with anti-inflammatory activities. Journal of Asian Natural Products Research, 18(1), 59–64. 10.1080/10286020.2015.1121853 [DOI] [PubMed] [Google Scholar]
- Rao, S. N., Head, M. S., Kulkarni, A., & Lalonde, J. M. (2007). Validation studies of the site-directed docking program libdock. Journal of Chemical Information and Modeling,47(6), 2159-2171. 10.1021/ci6004299 [DOI] [PubMed]
- Refaat, J., Yehia, S. Y., Ramadan, M. A., & Kamel, M. S. (2015). Rhoifolin: A review of sources and biological activities. International Journal of Pharmacognosy, 2(3),102–09. 10.13040/IJPSR.0975-8232.IJP.2(3).102-09 [DOI] [Google Scholar]
- Roshchin, Y. V. (1977). Trifolin from Euphorbia condylocarpa. Chemistry of Natural Compounds, 13(4), 481–482. 10.1007/BF00565849 [DOI] [Google Scholar]
- Sadiqud, M. U. E. (2016). Isolation and spectroscopic identification of some constituents of bioactive fractions of aerial parts of mirabilis Jalapa. IOSR Journal of Applied Chemistry (IOSR-JAC), 9, 12–17. 10.9790/5736-0904021217 [DOI] [Google Scholar]
- Trott, O., & Olson, A. J. (2009). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Aalten, D. M., Bywater, R., Findlay, J. B., Hendlich, M., Hooft, R. W., & Vriend, G. (1996). PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. Journal of Computer-Aided Molecular Design, 10(3), 255–262. 10.1007/BF00355047 [DOI] [PubMed] [Google Scholar]
- Weng, X. C., & Wang, W. (2000). Antioxidant activity of compounds isolated from Salvia plebeia. Food Chemistry, 71(4), 489–493. 10.1016/S0308-8146(00)00191-6 [DOI] [Google Scholar]
- World Health Organisation (WHO). (2020). Coronavirus disease 2019 (COVID-19) situation report – 77. Retrieved April 7, from https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200406-sitrep-77-covid-19.pdf?sfvrsn=21d1e632_2
- Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., Hu, Y., Tao, Z.-W., Tian, J.-H., Pei, Y.-Y., Yuan, M.-L., Zhang, Y.-L., Dai, F.-H., Liu, Y., Wang, Q.-M., Zheng, J.-J., Xu, L., Holmes, E. C., & Zhang, Y.-Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., Sun, Y., Jing, F., Duan, W., Du, J., & Wang, X. (2011). Separation and purification of flavones from Nelumbo nucifera Gaertn. by silica gel chromatography and high-speed counter-current chromatography. Se pu = Chinese Journal of Chromatography, 29(12), 1244–1248. http://www.ncbi.nlm.nih.gov/pubmed/22500455 [PubMed] [Google Scholar]
- Xu, X., Xie, H., Hao, J., Jiang, Y., & Wei, X. (2011). Flavonoid glycosides from the seeds of litchi chinensis. Journal of Agricultural and Food Chemistry, 59(4), 1205–1209. 10.1021/jf104387y [DOI] [PubMed] [Google Scholar]
- Yoon, E.-K., Ku, S.-K., Lee, W., Kwak, S., Kang, H., Jung, B., & Bae, J.-S. (2015). Antitcoagulant and antiplatelet activities of scolymoside. BMB Reports, 48(10), 577–582. 10.5483/bmbrep.2015.48.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanwar, A. A., Badole, S. L., Shende, P. S., Hegde, M. V., & Bodhankar, S. L. (2013). Chapter 76 - Cardiovascular effects of hesperidin: A flavanone glycosidePolyphenols in human health and disease (Vol 2, pp. 989-992). Academic Press. ISBN 9780123984562, 10.1016/B978-0-12-398456-2.00076-1 [DOI] [Google Scholar]
- Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., Becker, S., Rox, K., & Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (New York, N.Y ), 368,409-412. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P., Yang, X.-L., Wang, X.-G., Hu, B., Zhang, L., Zhang, W., Si, H.-R., Zhu, Y., Li, B., Huang, C.-L., Chen, H.-D., Chen, J., Luo, Y., Guo, H., Jiang, R.-D., Liu, M.-Q., Chen, Y., Shen, X.-R., Wang, X., … Shi, Z.-L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete, V., Daina, A., Bovigny, C., & Michielin, O. (2016). SwissSimilarity: A web tool for low to ultra high throughput ligand-based virtual screening. Journal of Chemical Information and Modeling, 56(8), 1399–1404. 10.1021/acs.jcim.6b00174 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.