

ABSTRACT
Introduction
The ongoing life-threatening pandemic of coronavirus disease 2019 (COVID-19) has extensively affected the world. During this global health crisis, it is fundamentally crucial to find strategies to combat SARS-CoV-2. Despite several efforts in this direction and continuing clinical trials, no vaccine has been approved for it yet.
Methods
To find a preventive measure, we have computationally designed a multi-epitopic subunit vaccine using immuno-informatic approaches.
Results
The structural proteins of SARS-CoV-2 involved in its survival and pathogenicity were used to predict antigenic epitopes. The antigenic epitopes were capable of eliciting a strong humoral as well as cell-mediated immune response, our predictions suggest. The final vaccine was constructed by joining the all epitopes with specific linkers and to enhance their stability and immunogenicity. The physicochemical property of the vaccine was assessed. The vaccine 3D structure prediction and validation were done and docked with the human TLR-3 receptor. Furthermore, molecular dynamics simulations of the vaccine-TLR-3 receptor complex are employed to assess its dynamic motions and binding stability in-silico.
Conclusion
Based on this study, we strongly suggest synthesizing this vaccine, which further can be tested in-vitro and in-vivo to check its potency in a cure for COVID-19.
KEYWORDS: SARS-CoV-2, vaccine, epitopes, spike glycoprotein, nucleocapsid, envelope
1. Introduction
Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2) is the causative agent for the associated disease named as COVID-19, which had spread throughout the world and declared as a pandemic [1,2]. SARS-CoV-2 has been demonstrated to have a strong affinity for human respiratory receptors (hACE2), which makes it more vulnerable to humans globally [3]. SARS-coronavirus infections are commonly found to be associated with multiple disorders, such as respiratory, enteric, hepatic, and neurological complications [4]. In general, coronaviruses (CoV) belong to a family Coronaviridae, which consists of enveloped positive-sense, single-stranded RNA viruses [5,6]. Among the four genera of CoVs, SARS-CoV, MERS-CoV, and SARS-CoV-2 belong to Betacoronaviruses [7]. Inside host cells, SARS-CoV-2 genome is translated into two large polyproteins: replicase polyprotein 1a (pp1a) and 1ab (pp1ab) [8]. The larger pp1ab comprises 16 non-structural proteins (Nsp1–Nsp16), which are responsible for constructing a viral replication-transcription complex [9]. Alongside the viral RNA transcribes subgenomic RNAs, which encode four structural proteins; spike (S), envelope (E), membrane (M), and nucleocapsid (N) and several accessory proteins [10]. While accessory proteins are engaged in host immune suppression, including downregulation of the interferon pathway, structural proteins shape the virion, facilitating genome encapsulation, viral particle assembly, and release [11,12]. Recently, one health concept is used and hypothesized that the prior exposure of humans with pet animals lowers the adverse effect of SARS-CoV-2. The N and S protein epitopes in comparison with taxonomically related coronaviruses’ epitopes share high sequence homology, which has high implications for vaccine designing against SARS-CoV-2 [13,14]. Additionally, there are repurposing of drugs are also important to cobat this disease [15,16].
Since the beginning of the 21st century, two outbreaks of SARS-CoV and MERS-CoV have led the researchers into designing vaccines against SARS coronaviruses. The third outbreak, wherein SARS-CoV-2 infects promiscuously to humans, has given the need for speedy research into developing a broad-range vaccine against these coronaviruses. Evidence shows that humoral and cell-mediated immune response plays a protective role against coronavirus infections, specifically against S and N proteins [17–19]. Although the effective antibody response is short-lived, the T cell responses are found to provide long-term protection [20,21]. Similarly, TC cell response against the structural proteins S and N are long-lasting [22]. Previously, immunoinformatics approaches have been used to construct the vaccines against the Zika virus Nipah virus, and Human papillomavirus, where in-vivo studies of the Human papillomavirus showed promising results [23–25]. Hence, in this study, we have used immunoinformatic approaches to predict highly antigenic epitopes from SARS-CoV-2 structural proteins that would evoke a strong immune response in humans. For this purpose, we have used the structural proteins: Spike, Envelope, and nucleocapsid to predict B-cell, cytotoxic T lymphocyte (CTL) and helper T lymphocyte (HTL) epitopes for construction of vaccine. Thus, the multi-epitopic subunit vaccine would be capable of eliciting humoral as well as cell-mediated immune responses. We have also performed the docking and molecular dynamic simulations (MDS) between the vaccine and human Toll-like Receptor-3 (TLR-3) to study their binding stability. The outcome of the present study will result in a novel and immunogenic vaccine, which may be further accessed against SARS-CoV-2.
2. Material and methods
2.1. Protein sequence retrieval
Translated sequences of three structural proteins of SARS-CoV-2: Spike glycoprotein [Protein ID: QHD43416.1], Envelope [Protein ID: QHD43418.1], and Nucleocapsid protein [Protein ID: QHD43423.2] were obtained from the GenBank database.
2.2. B lymphocytes epitopes prediction
The IEDB tools (http://tools.iedb.org/bcell/) were used to identify the B cell epitope and for verifying antigenicity, BepiPred linear epitope analysis was performed. According to the BepiPred linear epitope prediction method, residues with scores above the threshold (default value 0.35) are predicted to be part of an epitope having 49% sensitivity and 75% specificity [26].
2.3. Helper T – cells epitopes prediction
The IEDB server (http://tools.iedb.org/mhcii/) was used for the prediction of MHC II antigenic binders in translated sequences. Top 10 HTL epitopes were sorted based on their percentile rank and IC50 value. Epitopes with the lowest percentile rank were considered an excellent binding affinity, whereas, peptides with IC50 values in lower (<50 nM), middle (<500 nM), and higher (<5000 nM) range corresponds to the highest, intermediate, and lowest binding affinity, for the T-cell, respectively [27].
2.4. Cytotoxic T- cells epitopes prediction
MHC class I binding/CTL epitopes for supertype A3 were predicted for all three proteins by using an online server NetCTL 1.2 (http://www.cbs.dtu.dk/services/NetCTL/) [28]. Default threshold of 0.75 was chosen for vaccine construction in this study (>1.25 corresponds to 54% sensitivity and 99.30% specificity; >1.00 shows 70% sensitivity and 98.5% specificity; >0.90 shows 74% sensitivity and 98% specificity). MHC class I binding and proteasomal cleavage prediction was performed using artificial neural networks. Accordingly, TAP transport efficiency was predicted using a weight matrix.
2.5. IFN‐γ inducing epitopes prediction
MHC class II binder epitopes/HTL, with the ability to induce cell‐mediated immunity, were identified using the IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/). The prediction was performed by a motif and support vector machine (SVM) hybrid approach [29]. All the positive epitopes able to induce interferon‐γ (IFN‐γ) were selected for vaccine construct.
2.6. Multi-epitopic vaccine construction
Selected high-scoring CTLs, high-affinity HTLs, and B-cell epitopes were used to generate the vaccine sequence. The different epitopes were linked together using linkers: CTL linker (AAY), HTL linker (GPGPG), and B epitope linker (KK). To improve the immunogenicity of the vaccine, β-defensin 1 (Uniprot Id: P60022) as an adjuvant was added through an EAAAK linker at the N-terminal of the construct.
2.7. Prediction of the antigenicity and allergenicity of the vaccine candidate
2.7.1. Antigenicity prediction
The servers ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) and VaxiJen v2.0 (http://www.ddg‐pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) were used for the antigenicity evaluation of the final vaccine construct [30,31]. The VaxiJen 2.0 server predicts the antigenicity of the multi-epitope vaccine peptide based on the physicochemical properties of the input protein. Whereas, ANTIGENpro server predicts the antigenicity of the multi-epitopic vaccine based on the protein microarray data analysis of the target organism.
2.7.2. Allergen prediction
The server AllerTOP v2.0 (http://www.ddg-pharmfac.net/AllerTOP) and AllergenFP were used to predict the allergenicity of multi-epitopic vaccine [32,33]. AllerTOP v2.0 classifies the vaccine protein sequence by machine learning methods for the classification of allergens. AllergenFP is an alignment-free online server that uses SVM module to predict the allergenic and non-allergenic nature of proteins.
2.8. Vaccine features
2.8.1. Physiochemical properties of the constructed vaccine
Vaccine construct was accessed for its physicochemical properties using the ProtParam server (http://web.expasy.org/protparam/) that predicts the theoretical pI, instability index, half‐life, stability profiling, aliphatic index, and Grand Average of Hydropathy of the sequence [34].
2.8.2. Secondary structure prediction
Protein secondary structure was determined using PSIPRED, a web-based freely accessible online server (http://bioinf.cs.ucl.ac.uk/psipred/) [35] which also gives information of contact analysis, fold recognition, structure modeling, function prediction, protein intrinsic disorder prediction and domain prediction of a query sequence [36].
2.8.3. Disorder profile generation
The disorder profile of the vaccine construct was generated using PONDR pool of four predictors (http://original.disprot.org/metapredictor.php) [37–39] and IUPRED 2A predictor [40] as described in our previous reports [41–43].
2.8.4. Tertiary structure prediction and validation
The tertiary structure of the vaccine construct was generated using the I-TASSER web server [44]. It provides an energy minimized model through iterative template-based fragment assembly simulations. The structure was further optimized upon adding hydrogen and missing side-chain atoms followed by minimization in Schrodinger’s protein preparation wizard using OPLS 2005 forcefield [45]. This produced a high-quality model for protein-protein docking and molecular simulations. RAMPAGE [Mordred.bioc.cam.ac.uk/~rapper/rampage.php] and PROCHECK [46] web servers were used for evaluation of the vaccine model based on the distribution of amino acids in the Ramachandran plot.
2.8.5. Molecular docking of the final vaccine with immune receptors
PIPER program embedded in the BioLuminate module of Schrodinger for protein-protein docking was implemented for docking of vaccine model and TLR-3 (PDB ID: 20AZ) and TLR-5 (PDB ID: 3J0A) receptors [47,48]. It is an efficient tool that reduces false positive poses and performs a global search with Fast-Fourier Transform (FFT) approach. Using 1000 conformations of input structures, the top 50 clusters were selected with cluster radius 9 Å, which then centralized, and the outcomes of the docking based on cluster size were evaluated. With the largest cluster size, the docked complex out of 10 complexes was selected for molecular dynamics simulation.
2.8.6. Molecular dynamics simulation of vaccine-immune receptors
The ~86kDa vaccine model in complex with TLR-3 receptor was evaluated for its binding stability in an aqueous environment for 20ns. With a total of 6,66,607 molecules of TIP3P water model, 60 neutralizing Cl− ions, and 0.15 M concentration of salt, the complex was incorporated in a cubic box. Recently updated forcefield CHARMM36 was implemented to generate the topologies of the protein-protein complex. The generated system was minimized for 50,000 steps of the steepest descent algorithm. Further, the equilibration process under NPT and NVT conditions for 1ns was done with Parrinello-Rahman and V-rescale methods for coupling of pressure and temperature, respectively. Lastly, production MD run for all three systems was performed in periodic boundary conditions for 20ns. LINCS algorithm was used for calculating bond parameters. Particle Mesh Ewald (PME) for long-range electrostatics with fourier spacing of 0.16 was used for production MD. An analysis of MD trajectory, Root Mean Square Deviation (RMSD), and Root Mean Square Fluctuation (RMSF) was calculated with gmx rms, and rmsf commands.
2.9. In-silico cloning of final vaccine
For the production of final vaccine in large quantity, Java Codon Adaptation Tool (http://www.jcat.de/) was used for codon optimization and attached with expression vector. The optimized codon sequence includes Codon Adaption Index (value 1.0) and %GC content (30–70%), which denotes the high level of protein expression and DNA stability. Finally, restriction cloning was performed using tool SnapGene v3.3.4. The restriction sites NdeI and XhoI were added to the N and C terminals of the optimized complementary DNA sequences followed by their insertion within the pET‐21a(+) vector to ensure the polyprotein synthesis within the E coli expression system.
3. Results
3.1. Prediction of B cell and T cell epitope
HTL epitopes for all structural proteins were predicted using IEDB server for human MHC-II alleles. Among these, top 10 epitopes based on their least percentile rank depicting their high affinity were selected. The final selection of epitopes was made on the basis of overlapping sequences, which were further assessed for their IFN inducing potential. Epitopes with positive IFN inducing potential were finally selected for vaccine construction (Table 1). For each structural protein, CTL epitopes for supertype A3 were predicted using an online server NetCTL 1.2. A total of 40, 5, and 4 epitopes were predicted for S, N, and E proteins, respectively (Table 2). Linear B-cell epitopes in all three structural proteins were predicted using another online server BCPREDS. Epitopes selected on the basis of prediction scores were further screened for their non-allergic and antigenicity potential. Two epitopes each for S and N with high antigenicity scores were selected for final vaccine construction are tabulated in Table 3. However, no B cell epitopes were found for the E protein.
Table 1.
Predicted HTL specific epitopes obtained from IEDB.
| S. No. | SARS-CoV-2 protein | Position | Allele | HTL epitopes | Prediction result for the IFN |
|---|---|---|---|---|---|
| 1. | Spike Glycoprotein | 113–131 | HLA-DRB1 | KTQSLLIVNNATNVVIKVC | Negative |
| 691–707 | HLA-DRB1 | SIIAYTMSLGAENSVAY | Positive | ||
| 115–130 | HLA-DRB3 | QSLLIVNNATNVVIKV | Negative | ||
| 2. | Nucleocapsid | 301–320 | HLA-DRB1 | WPQIAQFAPSASAFFGMSRI | Positive |
| 310–326 | HLA-DRB1 | SASAFFGMSRIGMEVTP | Positive | ||
| 3. | Envelope | 34–50 | HLA-DRB1 | LTALRLCAYCCNIVNVS | Positive |
| 45–60 | HLA-DRB1 | NIVNVSLVKPSFYVYS | Negative | ||
| 55–71 | HLA-DRB1 | SFYVYSRVKNLNSSRVP | Negative |
Table 2.
Predicted CTL epitopes for A3 super type obtained from NetCTL 1.2 server.
| S. No. | SARS-CoV-2 protein | Position | Allele supertype | CTL epitopes | Score |
|---|---|---|---|---|---|
| 1. | Spike Glycoprotein | 30 | A3 | NSFTRGVYY | 0.9914 |
| 41 | A3 | KVFRSSVLH | 1.3419 | ||
| 69 | A3 | HVSGTNGTK | 1.1575 | ||
| 89 | A3 | GVYFASTEK | 1.4615 | ||
| 142 | A3 | GVYYHKNNK | 1.3335 | ||
| 162 | A3 | SANNCTFEY | 0.8377 | ||
| 187 | A3 | KNLREFVFK | 0.7504 | ||
| 270 | A3 | LQPRTFLLK | 1.0511 | ||
| 296 | A3 | LSETKCTLK | 0.8213 | ||
| 302 | A3 | TLKSFTVEK | 1.3483 | ||
| 311 | A3 | GIYQTSNFR | 1.1119 | ||
| 349 | A3 | SVYAWNRKR | 1.1423 | ||
| 357 | A3 | RISNCVADY | 1.1734 | ||
| 361 | A3 | CVADYSVLY | 1.1344 | ||
| 370 | A3 | NSASFSTFK | 1.3454 | ||
| 372 | A3 | ASFSTFKCY | 0.8899 | ||
| 378 | A3 | KCYGVSPTK | 1.2722 | ||
| 409 | A3 | QIAPGQTGK | 1.0483 | ||
| 454 | A3 | RLFRKSNLK | 1.7563 | ||
| 458 | A3 | KSNLKPFER | 0.9792 | ||
| 529 | A3 | KSTNLVKNK | 0.9809 | ||
| 550 | A3 | GVLTESNKK | 0.9827 | ||
| 559 | A3 | FLPFQQFGR | 0.8186 | ||
| 604 | A3 | TSNQVAVLY | 1.1959 | ||
| 725 | A3 | EILPVSMTK | 1.2271 | ||
| 733 | A3 | KTSVDCTMY | 0.8544 | ||
| 757 | A3 | GSFCTQLNR | 1.0783 | ||
| 787 | A3 | QIYKTPPIK | 1.4526 | ||
| 817 | A3 | FIEDLLFNK | 0.8027 | ||
| 827 | A3 | TLADAGFIK | 1.2451 | ||
| 925 | A3 | NQFNSAIGK | 1.0796 | ||
| 939 | A3 | SSTASALGK | 1.0678 | ||
| 956 | A3 | AQALNTLVK | 1.0252 | ||
| 975 | A3 | SVLNDILSR | 0.9214 | ||
| 1020 | A3 | ASANLAATK | 1.2193 | ||
| 1039 | A3 | RVDFCGKGY | 0.7565 | ||
| 1065 | A3 | VTYVPAQEK | 1.3960 | ||
| 1099 | A3 | GTHWFVTQR | 1.0342 | ||
| 1237 | A3 | MTSCCSCLK | 1.3360 | ||
| 1264 | A3 | VLKGVKLHY | 0.9774 | ||
| 2. | Nucleocapsid | 229 | A3 | QLESKMSGK | 1.1019 |
| 249 | A3 | KSAAEASKK | 1.4421 | ||
| 311 | A3 | ASAFFGMSR | 1.0610 | ||
| 361 | A3 | KTFPPTEPK | 1.4314 | ||
| 379 | A3 | TQALPQRQK | 0.9357 | ||
| 3. | Envelope | 30 | A3 | TLAILTALR | 1.1790 |
| 45 | A3 | NIVNVSLVK | 0.9921 | ||
| 55 | A3 | SFYVYSRVK | 1.0275 | ||
| 61 | A3 | RVKNLNSSR | 1.0581 |
Table 3.
Predicted B-cell specific epitopes obtained from server BCPREDS.
| S. No. | SARS-CoV-2 protein | B-cell epitopes | Antigenic score |
|---|---|---|---|
| 1. | Spike Glycoprotein | VRQIAPGQTGKIAD | 0.68 |
| LTPGDSSSGWTAG | 0.90 | ||
| 2. | Nucleocapsid | AFGRRGPEQTQGNFG | 0.56 |
| TGPEAGLPYGANK | 0.81 |
3.2. Multi-epitope vaccine candidate
The designed construct comprising of 48 CTL epitopes, 4 HTL epitopes, and 4 B-cell epitopes were then joined using linkers. Additionally, in order to improve the immunogenicity of multi-epitope vaccine, the construct was tagged with an adjuvant (β-defensin-TLR-3 agonist) at N terminal region, which enhances the natural immune response.
3.3. Prediction of antigenicity and allergenicity of constructed vaccine
The vaccine must be antigenic and non-allergic in nature and also induce humoral as well as cell-mediated immune responses against the targeted virus. Our vaccine was observed to be antigenic with a respective probability score of 0.566 and 0.845 predicted by VaxiJen v2.0 and ANTIGENPro servers. The construct was also detected as a non-allergen, checked using the AllerTOP v2.0 server.
3.4. Physiochemical characterization of designed vaccine
The vaccine construct is composed of 799 amino acids and has a molecular weight of 86.35 kDa. It is basic in nature (theoretical pI 9.70) and has a half-life of 30 hrs in mammalian reticulocytes (in vitro). The instability index is estimated to be 32.21, which denotes the stability of the protein. The aliphatic index (68.42) showed that the protein is thermostable, whereas grand average of hydropathicity is observed to be negative (−0.084), indicating the hydrophilic nature of vaccine.
3.5. Prediction of secondary structure and disordered residues
The secondary structure was predicted using PESIPRED 4.0 server. As shown in Figure 1, the vaccine contains several α-helices together with many the β-strands. Six long helices from residues 3–19, 65–102, 119–142, 433–448, 475–541, and 597–614 with a long β-strands of 14 amino acids from residues 401–414 can be observed.
Figure 1.

PSIPRED graphical result of secondary structure prediction of vaccine is represented by different colors: α-helix (pink), β-sheet (yellow), and coil (gray).
3.6. Evaluation of tertiary structure of vaccine construct and docking with TLR-3 and TLR-5 receptors
The 799-residue long vaccine construct was modeled using I-TASSER. The lowest energy predicted model was selected for further refinement and docking studies. However, the validation on Ramachandran plot by PROCHECK server suggested that 88% residues of the constructed vaccine model lies in the favored and allowed regions while the rest 12% are a part of generously allowed and disallowed regions (Figure 2(b)). Similarly, RAMPAGE server also mapped nearly 90.7% residues in favored and allowed regions, while 9.3% residues were present in coordinates portraying the outlier region (Figure 2(a)). As predicted by secondary structure and disorder-based structure analysis, the tertiary structure of the construct has a high number of unstructured regions. Based on disorder prediction data, it is largely disordered at its terminals.
Figure 2.
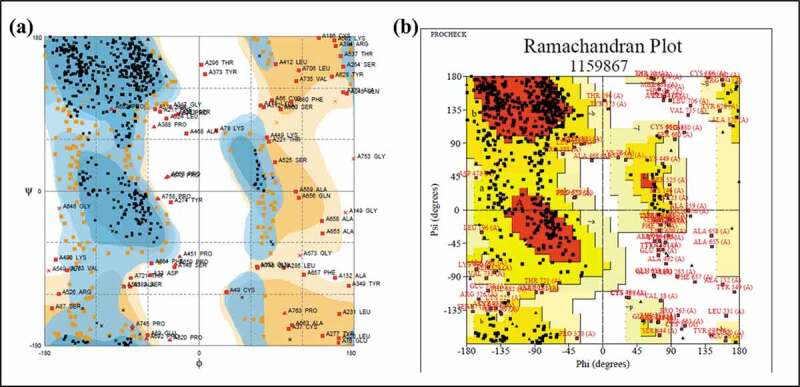
Ramachandran plot evaluation by RAMPAGE (a) and PROCHECK (b) servers.
From the PIPER program, with highest cluster size 28, out of 10 complexes ranging till 15, the complex 1, also with greatest minimal local energy, was chosen for further analysis. The vaccine model interacts with the TLR-3 receptor (PDB: 20AZ) through its N-terminal and the middle regions (Figure 3). The resultant interacting residues of vaccine-TLR-3 complex are shown in Table 4. At the interface of docked complex, Asn617 of vaccine is observed to form a H-bond with Asp153 residue of the receptor. Additionally, a few more aromatic H-bonds between Val621, Leu620 residues of vaccine, and His60 of TLR-3 are identified. The complex if further stabilized by a pi-cation interaction between Lys595 of vaccine and Tyr283 of TLR-3 (Table 4). Figure 4 depicts a close view of vaccine-TLR-3 interface. Several amino acids (red) of the receptor (green) are found engaged with vaccine residues (blue).
Figure 3.
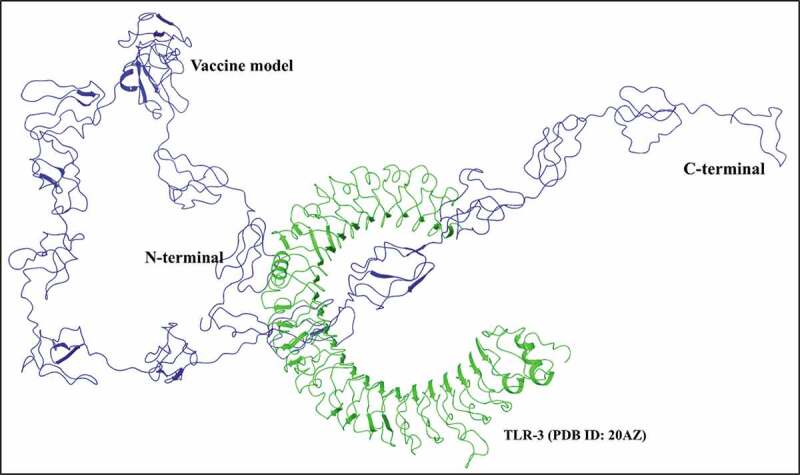
Molecular docking of vaccine construct with human TLR-3 receptor: The 799 residues long vaccine construct (blue) is found interacting with TLR-3 receptor (PDB: 20AZ) (green) through its residues at N-terminal and middle regions.
Table 4.
A list of Interacting residues of docked vaccine-TLR-3 complex.
| Vaccine model | TLR-3 receptor | Distance | Buried SASA | Vaccine model | TLR-3 receptor | Distance | Buried SASA |
|---|---|---|---|---|---|---|---|
| B:13:Leu | A:287:AsnA:288:Val | 2.1 A3.5 A | 60.70% | B:572:Thr | A:278:Met | 3.9 A | 76.10% |
| B:38:Val | A:247:Asn | 3.0 A | 8.20% | B:574:Arg | A:227:PheA:229:AsnA:254:SerA:203:Glu | 1.5 A1.8 A2.7 A3.9 A | 89.30% |
| B:39:Ser | A:247:Asn | 3.2 A | 24.80% | B:582:Cys | A:39:His | 3.9 A | 28.10% |
| B:40:Ser | A:273:TrpA:247:AsnA:272:Lys | 2.7 A3.0 A3.7 A | 73.80% | B:594:Val | A:283:Tyr | 3.4 A | 59.30% |
| B:41:Gly | A:272:Lys | 3.5 A | 0.10% | B:595:Lys | A:230:AsnA:229:AsnA:256:SerA:257:Asn | 2.0 A3.3 A3.5 A3.9 A | 82.70% |
| B:42:Gly | A:272:Lys | 1.5 A | 56.70% | B:596:Leu | A:206:SerA:230:AsnA:180:Asn | 2.5 A3.2 A3.4 A | 59.50% |
| B:43:Gln | A:272:Lys | 2.9 A | 15.90% | B:606:Leu | A:60:His | 2.9 A | 71.30% |
| B:44:Cys | A:273:Trp | 3.5 A | 57.60% | B:608:Ala | A:108:His | 3.0 A | 21.90% |
| B:45:Leu | A:296:TrpA:269:Leu | 3.1 A3.4 A | 52.50% | B:612:Ala | A:156:His | 3.0 A | 69.00% |
| B:46:Tyr | A:269:Leu | 3.5 A | 1.50% | B:613:Tyr | A:180:AsnA:156:His | 3.0 A3.5 A | 77.30% |
| B:47:Ser | A:269:Leu | 3.3 A | 59.50% | B:614:Asn | A:156:HisA:180:AsnA:179:SerA:203:Glu | 2.7 A3.0 A3.4 A4.0 A | 100.00% |
| B:48:Ala | A:273:Trp | 2.6 A | 97.90% | B:615:Ile | A:203:GluA:201:LysA:227:Phe | 1.4 A2.6 A3.8 A | 94.80% |
| B:49:Cys | A:243:Leu | 3.5 A | 31.90% | B:617:Asn | A:177:LeuA:153:Asp | 3.3 A3.5 A | 53.90% |
| B:50:Pro | A:243:LeuA:246:Ala | 1.8 A3.4 A | 94.20% | B:619:Ser | A:131:MetA:107:GlnA:156:His | 3.3 A3.5 A3.9 A | 99.00% |
| B:51:Ile | A:243:LeuA:244:Glu | 3.6 A3.9 A | 41.10% | B:620:Leu | A:84:PheA:108:HisA:107:Gln | 1.1 A3.1 A3.8 A | 87.80% |
| B:52:Phe | A:244:GluA:218:HisA:243:LeuA:215:Gly | 2.7 A2.8 A3.6 A3.8 A | 98.30% | B:621:Val | A:84:PheA:60:HisA:107:Gln | 2.2 A2.3 A3.4 A | 55.90% |
| B:55:Ile | A:215:Gly | 3.9 A | 49.50% | B:622:Lys | A:60:His | 3.6 A | 0.00% |
| B:59:Cys | A:273:Trp | 3.7 A | 59.00% | B:623:Ala | A:39:HisA:60:His | 1.6 A1.6 A | 99.60% |
| B:524:Leu | A:325:Arg | 3.5 A | 32.50% | B:624:Ala | A:39:His | 1.7 A | 59.90% |
| B:550:Tyr | A:358:GluA:382:Lys | 1.7 A2.1 A | 64.20% | B:625:Tyr | A:41:LysA:39:His | 1.9 A3.2 A | 41.90% |
| B:563:Ala | A:380:Asn | 3.6 A | 66.40% | B:626:Ser | A:41:Lys | 2.9 A | 5.90% |
| B:564:Ala | A:356:Cys | 3.9 A | 85.50% | B:627:Phe | A:41:Lys | 3.6 A | 0.00% |
| B:566:Gly | A:325:ArgA:358:Glu | 2.9 A4.0 A | 90.00% | B:654:Ile | A:41:LysA:62:Gln | 0.9 A3.0 A | 68.40% |
| B:567:Thr | A:325:Arg | 4.0 A | 0.00% | B:655:Ala | A:40:LeuA:41:Lys | 2.5 A2.5 A | 92.90% |
| B:568:His | A:302:TyrA:301:Glu | 2.2 A2.3 A | 50.50% | B:664:Phe | A:27:Lys | 1.4 A | 12.60% |
| B:569:Trp | A:302:Tyr | 2.7 A | 22.90% | B:665:Phe | A:27:LysA:26:ThrA:28:Cys | 1.3 A2.6 A2.7 A | 77.60% |
| B:570:Phe | A:302:Tyr | 3.2 A | 27.10% | B:667:Met | A:26:Thr | 3.1 A | 67.00% |
Figure 4.
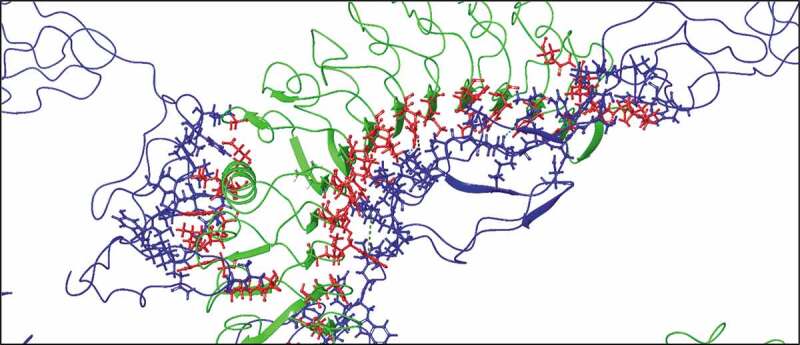
Detailed molecular interaction between epitopes vaccine and TLR-3 receptor. The residues at the interface are represented as lines and sticks. H-bonds (wine color), pi-cation (green), and non-covalent interactions (aromatic H-bond in cyan) between two molecules are displayed using dashed arrows.
Additionally, we have also performed docking with another toll-like receptor (TLR-5) which is known to recognize the flagellin of bacteria. Similar to TLR-3, the vaccine model has shown significant interactions with TLR-5 through its N-terminal as well as residues of middle region where the N-terminal folds in the constructed model, as shown in Figure 5. Residues such as Tyr5, Glu17 from N-terminal, and Thr511, Lys514 from middle regions of vaccine construct interacts with residues Asp607, Ala674, Cys585, and Glu584 of TLR-5, respectively, through aromatic hydrogen bonds, salt bridge, and hydrogen bonds.
Figure 5.
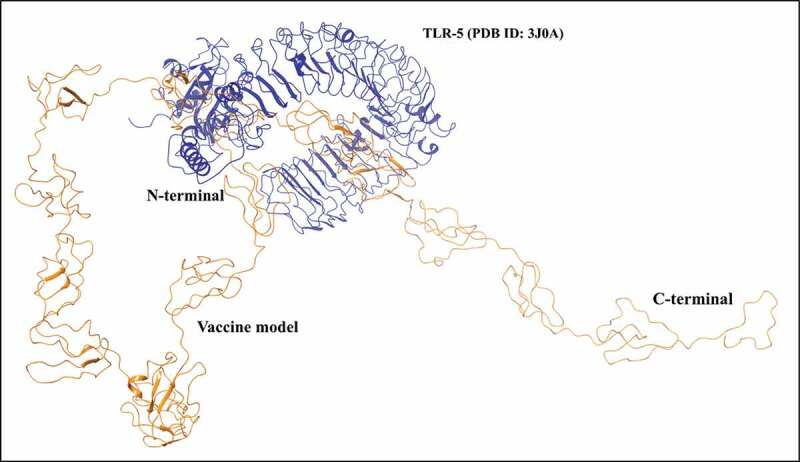
Vaccine-TLR-5 interaction identified through docking. As observed in the interacting complex, the vaccine construct interacts through its N-terminal region with TLR-5.
3.7. Molecular dynamics simulations
To analyze the stability and dynamic motions of docked complex, we performed a 20ns long MD simulation using Gromacs v5.1. The simulation system comprised ~7 lacs atoms in a cubic box with proteins, solvent, and ions. Based on the RMSD data, the complex was found to have an upward trend ranging from ~1 to 2.5 nm till 20ns with small fractions of stability in between (Figure 6(a)). However, according to RMSF values depicted in the graph of Figure 6(b), the fluctuation in residues of TLR-3 receptor was very less as compared to the docked structure of the vaccine, which had fluctuations in terminal regions. The interacting part of vaccine (i.e. N-terminal and middle regions) with TLR had minimal fluctuations in comparison with the other residues. Overall, the average fluctuation in the residues of the vaccine model was near ~1.2 nm.
Figure 6.
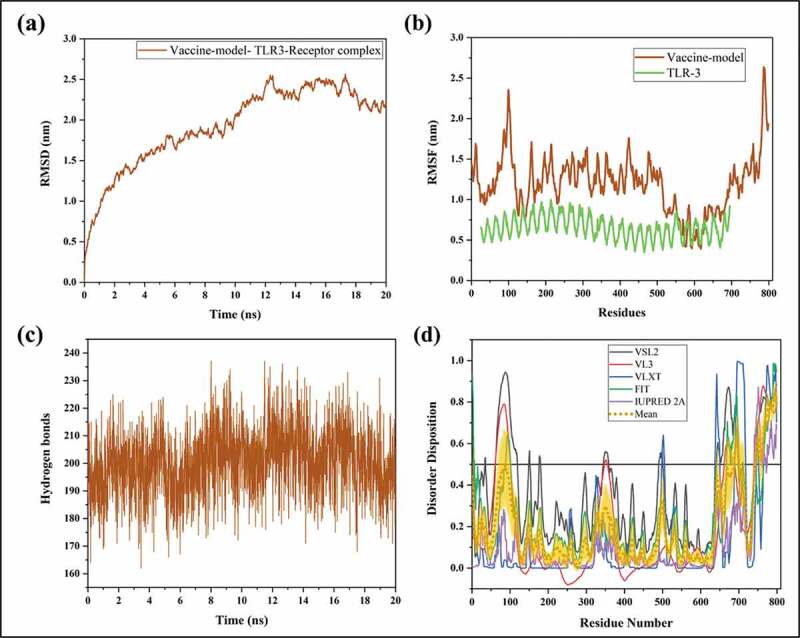
Illustration of docked vaccine model and TLR-3 complex through molecular dynamics simulations till 20ns. (a) RMSD, (b) RMSF, and (c) Hydrogen bonds. (d) Represents the predisposition of intrinsic disorder in residues of vaccine construct as predicted by PONDR® pool of predictors: PONDR-FIT, PONDR-VSL2B, PONDR-VL3, PONDR-VLXT, and IUPRED 2A. The mean of all predictors is shown in short dotted lines in yellow, and the shadowed area over the mean plot represents its standard error.
Moreover, the regions interacting with TLR-3 receptor are not disordered as per our analysis in Figure 1(b) and had stability throughout the simulation. As calculated in visual molecular dynamics, the average H-bonds were found to be 200 in vaccine-TLR-3 complex during MD simulations (Figure 6(c)). Additionally, disordered regions based on the sequence of vaccine were also predicted. The graph in Figure 6(d) represents the moderate amount of disorder (~20% calculated as the mean of all predictors) within residues 50–100 and 650–799 near N- and C-terminal regions. The regions interacting with TLR-3 receptor are not disordered as per our analysis in Figure 6(d) and had stability throughout the simulation.
3.8. In-silico cloning of final vaccine
The CAI improved value for codon optimized sequence was 1.0, while GC content obtained for our vaccine construct was 49.56%. Overall, the vaccine sequence lies in a suitable category for its expression in E coli. Finally, the vaccine construct was cloned with restriction sites NdeI and XhoI in the E coli pET‐21a(+) vectors. Finally, a cloned construct was generated, having a sequence length of 7767 base pairs (Figure 7).
Figure 7.
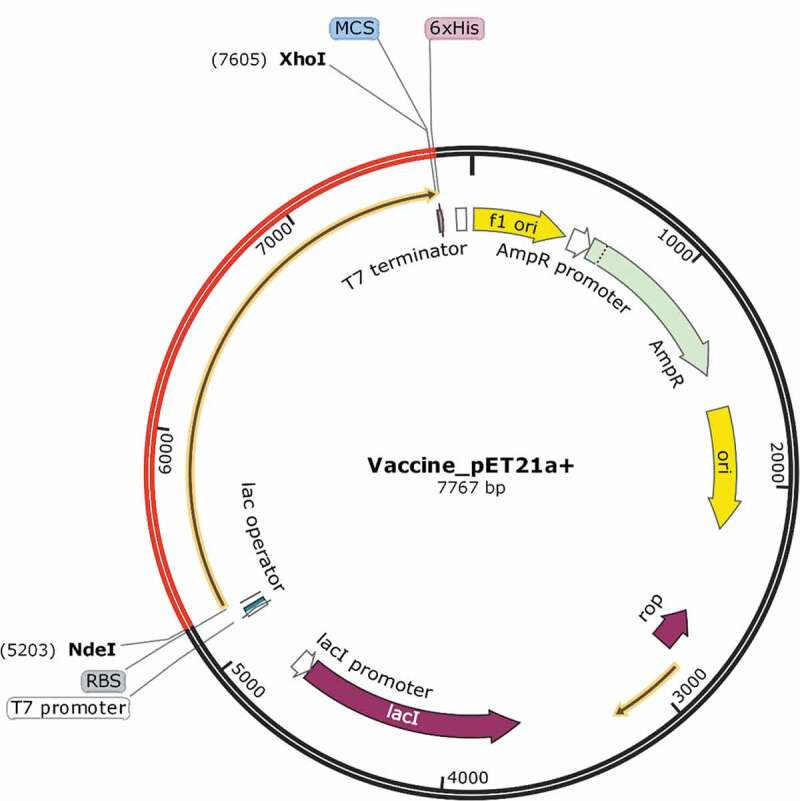
In-silico cloning to express final vaccine construct representing pET‐21a(+) vector. The optimized codon of final vaccine construct is in Red.
4. Discussion
SARS-CoV-2 is highly pathogenic, which has caused more than 4 lakh deaths globally until the writing of this manuscript. Particularly among all, Coronaviridae structural proteins S, M, E, and N are the main structural proteins and can be employed for protein/peptide-based vaccines. Different human infecting coronaviruses like OC43, HKU1, and MERS-CoV use a diverse set of lipids and receptors to enter into target cells [49,50]. In SARS-CoV, the spike glycoprotein interacts with ACE-2 receptor and facilitates the fusion of viral membrane with the lungs membrane [11,51,52]. E is an integral membrane protein with N-terminal region, a transmembrane domain (TMD), and a flexible hydrophilic C-terminal tail containing a PZD-binding motif in the last four amino acids [53,54]. Envelope protein forms pentamer to lead viroporin like structure that exhibits a membrane-destabilizing ion-channel activity on the host membrane [55]. CoV envelope, which plays a vital role during virion assembly, consists mainly of M protein and only a small fraction of E protein [56]. Whereas N protein shows interaction with M protein in lipid membrane of infected cells forming a double-layered vesicular structure in a secreted form that further interacts with host proteins [57,58].
Multiple vaccines are in clinical trial, but till date, no vaccine is approved against the SARS-CoV-2. In a recent in-silico report, the vaccine model using spike glycoprotein has been constructed but limited to the docking with TLR 5 receptor [59,60]. It was reported that Human coronaviruses OC43 recognize the MHC class I supertype A3 [61]. Based on these reports, we have chosen the A3 super type for epitope prediction, which will help the vaccine construct to act significantly. According to information obtained from the World Health Organization (WHO), one vaccine is in Phase 2, and two vaccines (DNA and RNA based) are in Phase 12 clinical trial. Moreover, 67 vaccines are in pre-clinical trials [62]. Previously, studies showed that the whole virus vaccine and virus-like particle vaccine against SARS-CoV are able to cure infection but leads to the pulmonary immunopathologic type lung disease [63]. Therefore, it is needed that the vaccine is selected on the basis of multi factors, i.e., antigenicity, immunogenicity, toxicity, allergenicity. In the present study, we have designed the vaccine construct by using immune-informatics approaches. Three structural proteins (spike glycoprotein, nucleocapsid, and envelope) were selected to construct a multi-epitope vaccine, which is capable of eliciting the humoral and cell-mediated immune response. Furthermore, the vaccine is antigenic, non-allergen, nontoxic, immunogenic in nature, and capable of IFN-γ production. The vaccine epitopes were joined with the help of specific linkers to enhance stability and immunogenicity. The vaccine has a molecular weight of 86.35 kDa, basic in nature, and half-life of 30 hrs in mammalian reticulocytes (in-vitro). Moreover, vaccine was found to be highly stable and hydrophilic in nature. Finally, the build vaccine construct was found to be moderately disordered, containing few disordered residues at N-terminal and relatively a vast region at C-terminus. As previously known, the vaccine candidates often have an ample amount of protein intrinsic disorder as they feature in the immune system [64]. Further, we also checked the interaction of vaccine construct through a modeled structure docked with two Toll-like Receptors (TLR-3 and TLR-5) structure. The adjuvant β-defensin 1 showed interaction with TLR-3 receptor and few residues from the same region also showed interaction with TLR-5. Further, the stability of the vaccine-TLR-3 complex was monitored by implementing molecular dynamics simulations till 20ns. Several residues at the N-terminal and middle region of vaccine construct were found to form stable interactions. Finally, the cloned sequence vector was generated in-silico for the expression of the vaccine.
5. Conclusion and expert opinion
Newly emerged SARS-CoV-2 is declared as pandemic and scientists all over the world running a race against the time to find vaccine/drugs against COVID-19. Despite that, many obstacles like allergenicity, immunogenicity, toxicity, etc. limit the progress of vaccine development. In recent times, advancement in immunoinformatics approaches led researchers to speed up vaccine development. In this context, by precise prediction and selection of multi-epitope, we strongly suggest to experimentaly validate this multi-subunit vaccine, in-vitro and in-vivo.
Acknowledgments
The authors would like to thank IIT Mandi for research facilities. RG is thankful to DBT, Govt. of India (BT/11/IYBA/2018/06) to RG. AK is supported from DBT, Govt. of India grant (BT/11/IYBA/2018/06). PK and SK were supported by MHRD-India for their funding. KUS and TB are grateful to the ICMR SRF and DST INSPIRE PhD fellowships, respectively.
Funding Statement
This paper was not funded.
Author contribution
AK, PK, KUS, SK, TB: acquisition and interpretation of data, writing, and editing of the manuscript.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Cascella M, Rajnik M, Cuomo A, et al. Features, evaluation and treatment Coronavirus (COVID-19). StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020. cited 2020 March22. Available from: http://www.ncbi.nlm.nih.gov/books/NBK554776/ [Google Scholar]
- 2.Giri R, Bhardwaj T, Shegane M, et al. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell Mol Life Sci [Internet]. 2020; Available from: https://doi.org/10.1007/s00018-020-03603–x. [DOI] [PMC free article] [PubMed]
- 3.Xu X-W, Wu -X-X, Jiang X-G, et al. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. BMJ [Internet]. 2020. cited 2020 March22;368. Available from: https://www.bmj.com/content/368/bmj.m606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakraborty C, Sharma AR, Sharma G, et al. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur Rev Med Pharmacol Sci. 2020;24:4016–4026. [DOI] [PubMed] [Google Scholar]
- 5.Chan JFW, Li KSM, To KKW, et al. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J Infect. 2012;65:477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zumla A, Chan JFW, Azhar EI, et al. Coronaviruses — drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15:327–347. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This important review on coronavirus details about drug discovery.
- 7.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses . The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Wit E, van Doremalen N, Falzarano D, et al. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fehr AR, Perlman S.. Coronaviruses: an overview of their replication and pathogenesis. In: Maier HJ, Bickerton E, Britton P, editors. Coronaviruses methods protoc [Internet]. New York, NY: Springer; 2015. p. 1–23. DOI: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92:418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3:237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tilocca B, Soggiu A, Sanguinetti M, et al. Comparative computational analysis of SARS-CoV-2 nucleocapsid protein epitopes in taxonomically related coronaviruses. Microbes Infect [Internet]. 2020. cited 2020 June1;22:188–194. Available from: http://www.sciencedirect.com/science/article/pii/S1286457920300538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tilocca B, Soggiu A, Musella V, et al. Molecular basis of COVID-19 relationships in different species: a one health perspective. Microbes Infect. 2020;22:218–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bzówka M, Mitusińska K, Raczyńska A, et al. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. Int J Mol Sci. 2020;21:3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saha A, Sharma AR, Bhattacharya M, et al. Probable molecular mechanism of remdesivir for the treatment of COVID-19: need to know more. Arch Med Res [Internet]. 2020. cited 2020 June1. Available from: http://www.sciencedirect.com/science/article/pii/S0188440920306998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 Coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z, Kong W, Huang Y, et al. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428:561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This important paper detail about vaccine development.
- 19.Lin Y, Shen X, Yang RF, et al. Identification of an epitope of SARS-coronavirus nucleocapsid protein. Cell Res. 2003;13:141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng H, Yang L, Wang L, et al. Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology. 2006;351:466–475. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This important paper detail about T cell response against coronavirus nucleocapsid protein.
- 21.Tang F, Quan Y, Xin Z-T, et al. Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: a six-year follow-up study. J Immunol. 2011;186:7264–7268. [DOI] [PubMed] [Google Scholar]
- 22.Channappanavar R, Fett C, Zhao J, et al. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J Virol. 2014;88:11034–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar Pandey R, Ojha R, Mishra A, et al. Designing B- and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J Cell Biochem. 2018;119:7631–7642. [DOI] [PubMed] [Google Scholar]
- 24.Ojha R, Pareek A, Pandey RK, et al. Strategic development of a next-generation multi-epitope vaccine to prevent nipah virus zoonotic infection. ACS Omega. 2019;4:13069–13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Namvar A, Bolhassani A, Javadi G, et al. In silico/In vivo analysis of high-risk papillomavirus L1 and L2 conserved sequences for development of cross-subtype prophylactic vaccine. Sci Rep. 2019;9:15225. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This important paper detail about insilico vaccine development followed by in vivo studies.
- 26.Larsen JEP, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larsen MV, Lundegaard C, Lamberth K, et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magnan CN, Zeller M, Kayala MA, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinforma Oxf Engl. 2010;26:2936–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimitrov I, Flower DR, Doytchinova I. AllerTOP - a server for in silico prediction of allergens. BMC Bioinformatics. 2013;14:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimitrov I, Naneva L, Doytchinova I, et al. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinforma Oxf Engl. 2013;30:846–851. [DOI] [PubMed] [Google Scholar]
- 34.Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Proteomics Protoc. Handb: Humana Press; 2005. pp. 571–607. DOI: 10.1385/1-59259-890-0:571. [DOI] [Google Scholar]
- 35.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. [DOI] [PubMed] [Google Scholar]
- 36.Buchan DWA, Jones DT. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019;47:W402–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obradovic Z, Peng K, Vucetic S, et al. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61(Suppl 7):176–182. . [DOI] [PubMed] [Google Scholar]
- 38.Obradovic Z, Peng K, Vucetic S, et al. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003;53(Suppl 6):566–572. . [DOI] [PubMed] [Google Scholar]
- 39.Xue B, Dunbrack RL, Williams RW, et al. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mészáros B, Erdos G, Dosztányi Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;46:W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gadhave K, Gehi BR, Kumar P, et al. The dark side of Alzheimer’s disease: unstructured biology of proteins from the amyloid cascade signaling pathway. Cell Mol Life Sci CMLS. 2020. DOI: 10.1007/s00018-019-03414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar A, Kumar P, Kumari S, et al. Folding and structural polymorphism of p53 C-terminal domain: one peptide with many conformations. Arch Biochem Biophys. 2020;684:108342. [DOI] [PubMed] [Google Scholar]
- 43.Kumar D, Singh A, Kumar P, et al. Understanding the penetrance of intrinsic protein disorder in rotavirus proteome. Int J Biol Macromol. 2020;144:892–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Yan R, Roy A, et al. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madhavi Sastry G, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. [DOI] [PubMed] [Google Scholar]
- 46.Laskowski RA, MacArthur MW, Moss DS, et al. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 47.Chuang G-Y, Kozakov D, Brenke R, et al. DARS (Decoys As the Reference State) potentials for protein-protein docking. Biophys J. 2008;95:4217–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kozakov D, Brenke R, Comeau SR, et al. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65:392–406. [DOI] [PubMed] [Google Scholar]
- 49.Hulswit RJG, Lang Y, Bakkers MJG, et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc Natl Acad Sci USA. 2019;116:2681–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Hulswit RJG, Widjaja I, et al. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc Natl Acad Sci USA. 2017;114:E8508–E8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belouzard S, Millet JK, Licitra BN, et al. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4:1011–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Haan CAM, Te Lintelo E, Li Z, et al. Cooperative involvement of the S1 and S2 subunits of the murine coronavirus spike protein in receptor binding and extended host range. J Virol. 2006;80:10909–10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liao Y, Yuan Q, Torres J, et al. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology. 2006;349:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teoh K-T, Siu Y-L, Chan W-L, et al. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol Biol Cell. 2010;21:3838–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This imporatant paper detail about envelope protein and their role in pathogenesis.
- 55.Wilson L, McKinlay C, Gage P, et al. SARS coronavirus E protein forms cation-selective ion channels. Virology. 2004;330:322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vennema H, Godeke GJ, Rossen JW, et al. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. Embo J. 1996;15:2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He R, Dobie F, Ballantine M, et al. Analysis of multimerization of the SARS coronavirus nucleocapsid protein. Biochem Biophys Res Commun. 2004;316:476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuo L, Hurst-Hess KR, Koetzner CA, et al. Analyses of coronavirus assembly interactions with interspecies membrane and nucleocapsid protein chimeras. J Virol. 2016;90:4357–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhattacharya M, Sharma AR, Patra P, et al. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): immunoinformatics approach. J Med Virol. 2020;92:618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chakraborty C, Sharma AR, Bhattacharya M, et al. Consider TLR5 for new therapeutic development against COVID-19. J Med Virol [Internet]. cited 2020 June1. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jmv.25997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Collins AR. Human coronavirus OC43 interacts with major histocompatibility complex class I molecules at the cell surface to establish infection. Immunol Invest. 1994;23:313–321. [DOI] [PubMed] [Google Scholar]
- 62.WHO . WHO | Coronavirus disease (COVID-2019) R&D [Internet]. World Health Organization; [cited 2020. April 22]. Available from: http://www.who.int/blueprint/priority-diseases/key-action/novel-coronavirus/en/
- 63.Tseng C-T, Sbrana E, Iwata-Yoshikawa N, et al. Immunization with SARS Coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE [Internet]. 2012. cited 2020 April22;7. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3335060/ [DOI] [PMC free article] [PubMed] [Google Scholar]; • This important paper detail about the infection after vaccination.
- 64.Guy AJ, Irani V, MacRaild CA, et al. Insights into the immunological properties of intrinsically disordered malaria proteins using proteome scale predictions. PloS One. 2015;10:e0141729. [DOI] [PMC free article] [PubMed] [Google Scholar]