

Abstract
The GM2 gangliosidoses, Tay-Sachs disease (TSD) and Sandhoff disease (SD), are fatal lysosomal storage disorders caused by mutations in the HEXA and HEXB genes, respectively. These mutations cause dysfunction of the lysosomal enzyme β-N-acetylhexosaminidase A (HexA) and accumulation of GM2 ganglioside (GM2) with ensuing neurodegeneration, and death by 5 years of age. Until recently, the most successful therapy was achieved by intracranial co-delivery of monocistronic adeno-associated viral (AAV) vectors encoding Hex alpha and beta-subunits in animal models of SD. The blood-brain barrier crossing properties of AAV9 enables systemic gene therapy; however, the requirement of co-delivery of two monocistronic AAV vectors to overexpress the heterodimeric HexA protein has prevented the use of this approach. To address this need, we developed multiple AAV constructs encoding simultaneously HEXA and HEXB using AAV9 and AAV-PHP.B and tested their therapeutic efficacy in 4- to 6-week-old SD mice after systemic administration. Survival and biochemical outcomes revealed superiority of the AAV vector design using a bidirectional CBA promoter with equivalent dose-dependent outcomes for both capsids. AAV-treated mice performed normally in tests of motor function, CNS GM2 ganglioside levels were significantly reduced, and survival increased by >4-fold with some animals surviving past 2 years of age.
Keywords: AAV9, Tay-Sachs disease, Sandhoff disease, gene therapy, intravenous delivery, GM2 gangliosidosis
Graphical Abstract
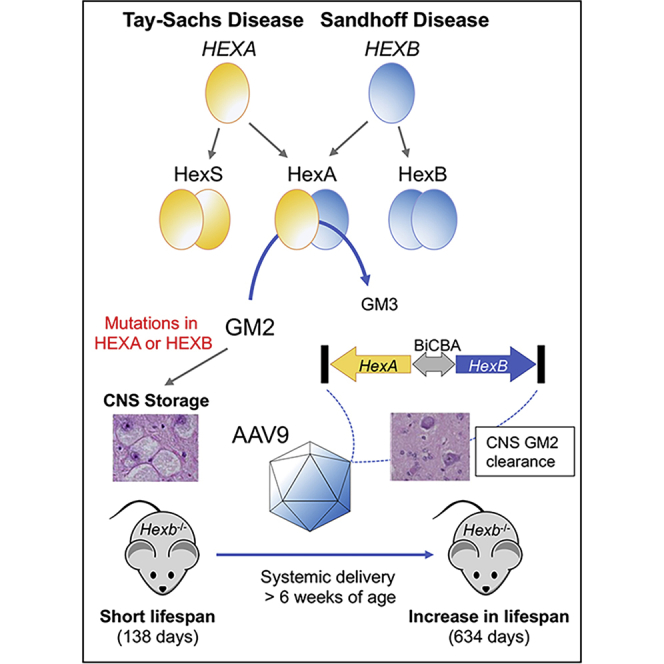
Tay-Sachs and Sandhoff diseases are caused by mutations in the HEXA and HEXB genes, respectively, which impact the activity of the heterodimeric lysosomal enzyme β-hexosaminidase A, and result in the accumulation of GM2 ganglioside in CNS. HexA is the only isozyme in human that hydrolyzes GM2 ganglioside, and co-expression of both proteins is necessary to produce high enzyme levels for cross-correction. An AAV9 vector expressing both proteins from a central promoter was administered systemically in Sandhoff mice and shown to reduce GM2 ganglioside levels, improve behavioral performance, and extend median survival to 634 days.
Introduction
Tay-Sachs disease (TSD) and Sandhoff disease (SD) are autosomal recessive diseases caused by mutations in the HEXA and HEXB genes, respectively, that reduce the activity of the heterodimeric enzyme β-N-acetylhexosaminidase A (HexA). Disease presentation in TSD and SD is classically categorized in three forms based on the age of onset: infantile patients manifest symptoms at ∼6 months of age, late-infantile/juvenile at 2–6 years, and late-onset usually in the late teens. The severity of symptoms and rate of disease progression are correlated with the residual activity of mutant HexA. Whereas the disease progresses over several decades in late-onset patients with 2%–4% of normal HexA activity, infantile patients usually have less than 0.1%. The diagnoses of infantile patients usually occur within the first year of life, because they fail to meet developmental milestones. Symptoms include seizures, inability to sit, difficulty swallowing, and eventual loss of the few acquired developmental milestones. Quality of life is severely compromised for children suffering with this disease, with palliative measures only extending life to a maximum of 5 years. Prior to gastric tubes becoming an available option to feed GM2 children, the lifespan was 2–2.5 years of age due to the loss of swallowing ability.1 However, this technological solution to prevent dying from starvation or aspiration pneumonia has had no impact in neurological function. Currently there is no treatment for GM2 gangliosidoses.
The HexA enzyme is a heterodimer of α and β subunits encoded by the HEXA and HEXB genes, respectively. Two additional homodimeric isozymes, HexB (ββ) and HexS (αα) are also formed and have different substrate specificities. HexA is the only isozyme that hydrolyzes GM2 ganglioside in humans, but both HexA and HexB hydrolyze a broad variety of substrates with β linked terminal N-acetylgalactosamine residues in glycosaminoglycans, glycolipids, and glycoproteins. The biochemical hallmark of GM2 gangliosidoses (TSD and SD) is the progressive storage of GM2 ganglioside in neurons in the central nervous system causing lysosomal dysfunction and ultimately neuronal death by molecular mechanisms that remain largely unknown. Similar to other neurodegenerative diseases, disease progression in GM2 gangliosidoses is accompanied by microglial and astroglial activation.2, 3, 4
Experimental treatments for GM2 gangliosidoses have explored a number of options including enzyme replacement therapy,5 substrate reduction therapy (SRT),6,7 neural stem cell transplantation,8,9 anti-inflammatory drugs combined with SRT,10 bone marrow transplantation combined with SRT,11 and caloric restriction,12 or ketogenic diet combined with SRT.13 Some of these approaches extended lifespan of SD mice significantly by a maximum of ∼2-fold, but the few that have progressed to clinical trials, such as SRT (miglustat), showed no effect in patients.14
Adeno-associated virus (AAV) gene therapy has emerged as the most promising approach for developing an effective treatment for Tay-Sachs and Sandhoff diseases to date, despite challenges associated with the need to deliver both subunits for successful production of HexA. Intracranial co-administration of two AAV1 vectors encoding the human alpha or beta subunits restored enzyme activity in the CNS, corrected neuropathology and neuroinflammation, and, importantly, extended lifespan dramatically.15, 16, 17 These results translated to SD cats, where intracranial injection of monocistronic AAVrh8 vectors encoding feline subunits resulted in improved neurochemistry, reduced neuroinflammation, and extended lifespan dramatically.4,18,19 Similarly, Tay-Sachs sheep survived longer than untreated animals with normalization of storage and neuroinflammation in the brain.20
A less invasive approach to gene therapy is appealing, but expressing both HEXA and HEXB in a single AAV is difficult due to the limited capacity for transgenes and regulatory elements (∼4.7 kb). Several studies have shown efficacy of neonatal systemic delivery of AAV9-HEXB vectors in SD mice,21,22 but despite unequivocal disease amelioration in SD mice, their relevance for translation to a gene therapy for patients is less clear. This is because mice have an alternative metabolic pathway that catabolizes GM2 ganglioside via GA2, which can be hydrolyzed to lactosylceramide by the HexB isozyme.23 This pathway is not functional in humans. Therefore, delivery of HEXB encoding the β-subunit alone, which produces the HexB homodimer exclusively, works only in mice.
Here we developed and characterized the therapeutic efficacy of two new bicistronic AAV vectors, AAV-Bic and AAV-P2I, encoding simultaneously the α and β subunits of mouse HexA. The AAV-Bic vector, which carries a bidirectional mini CBA promoter (BiCBA) to drive expression of the two proteins in opposite directions from the center of the vector genome, proved to be exceptionally efficient when injected systemically in 4-week-old SD mice. To our knowledge, this is the first report of treatment of SD mice by systemic administration of an AAV9 vector encoding both Hex subunits, which extended survival by >4-fold with some treated mice reaching 2 years of age (the maximum allowable lifespan).
Results
Co-expression of the hexosaminidase α and β subunits is thought to be a fundamental requirement for the treatment of Tay-Sachs and Sandhoff diseases and depends largely on overexpression of HexA in a relatively small percentage of transduced cells with the current generation of AAV technology.
Vector Design Iterations
Given the ∼4.7 kb transgene capacity of AAV, incorporating both Hexa and Hexb cDNAs in a single vector has been challenging. We tested numerous designs using a single promoter to drive expression of both proteins with viral24 and synthetic25 internal ribosome entry sites (IRES), as well as various 2A peptides.26,27 Although many AAV vector designs were effective in overexpressing HexA and HexB isozymes in transfected HEK293T cells, none were effective in vivo (data not shown). The latest design iterations incorporate a bidirectional promoter (BiCBA) to drive transgene expression in opposite directions (AAV-Bic) toward the inverted terminal repeats (ITR), or a small promoter (P2) and intron cassette (Figure S1) cloned next to each ITR in inverted positions to drive gene expression toward the center of the vector genome (AAV-P2I). The therapeutic efficacy of AAV-PHP.B and AAV9 vectors of both designs were tested in 4- to 6-week-old Sandhoff disease (SD) mice (Hexb−/−) treated by intravenous administration (Figure 1B). AAV-PHP.B vectors were administered at 3 × 1011 and 1 × 1012 vg/mouse (1.5–5 × 1013 vg/kg) and AAV9 vectors at 1 × 1012 and 4 × 1012 vg/mouse (0.5-2 × 1014 vg/kg). Controls included untreated SD mice and phenotypically normal littermates with heterozygote (HZ; hexb+/−) and wild-type (WT; hexb+/+) genotypes.
Figure 1.

Systemic Treatment with Bicistronic AAV Vectors Extends the Survival of SD Mice
(A) Design of AAV-Bic and AAV-P2I vectors. (B) Vector dosing and median survival for all groups. Kaplan-Meier survival curves for SD mice treated systemically with (C) AAV-PHP.B-Bic, (D) AAV-PHP.B-P2I, (E) AAV9-Bic, and (F) AAV9-P2I vector. Only one control group of untreated SD (UT) and normal (WT+HZ) mice was included in the experiment and their survival curves shown in all graphs (C–F) for comparison purposes. Log-rank (Mantel-Cox) test showed that survival of all treatment groups was significantly (p < 0.0001) increased compared to untreated SD controls.
Treatment with Bicistronic AAV Vectors Improves Survival
All AAV vectors improved survival significantly compared to untreated SD mice (Figure 1B). AAV-Bic vectors (Figures 1E and 1F) proved to be more efficient than AAV-P2I vectors (Figures 1C and 1D), a finding that was particularly evident for AAV9 vectors. Treatment with AAV9-Bic vector increased median survival from 138 days for untreated SD mice to more than 600 days of age (Figure 1E), and at least two mice survived past 2 years of age and were euthanized at the experimental endpoint. In comparison, treatment with 1 × 1012 vg of AAV9-P2I improved median survival modestly to 154 days of age (p < 0.0001), while treatment with 4 × 1012 vg increased median survival to 313 days of age (Figure 1F). The impact of AAV-PHP.B on survival was comparable to AAV9 for AAV-Bic (Figures 1C and 1E), but increased survival significantly compared to AAV9 for AAV-P2I (p < 0.0001; Figures 1D and 1F).
Stabilization of Motor Function
The motor performance of SD mice is comparable to that of normal littermates until 90–100 days of age but declines rapidly thereafter.28 The motor performance, strength, and coordination of SD mice treated with AAV-Bic and AAV-P2I vectors, untreated SD mice, and age matched normal controls (WT and HZ) was assessed in the rotarod and inverted screen tests (Figure 2). Similar to previous reports, the performance of untreated SD mice in both tests was stable until 90 days of age but declined rapidly in the two subsequent test ages of 105 and 120 days. The humane endpoint of all untreated SD mice occurred before 150 days of age (maximum survival was 142 days). The motor performance of SD mice treated with AAV-Bic and AAV-P2I vectors (AAV9 and AAV-PHP.B capsids) remained comparable to that of WT and HZ mice until at least 150 days of age (Figure 2).
Figure 2.

Motor Performance, Strength, and Coordination of Treated SD Mice Remain Normal Until at Least 150 Days of Age
The performance of AAV-treated SD mice and controls was assessed starting at 60 days of age until 150 days of age in the (A) accelerating rotarod test (4–40 rpm) and (B) inverted screen test. The performance of untreated (UT) SD mice declined rapidly after 90 days of age with the last testing point at 120 days of age. Data are represented as mean ± SD
Increased Hexosaminidase Activity in CNS and Liver
One subset of mice (n = 6) in the high dose cohorts (4 × 1012 vg for AAV9 vectors and 1 × 1012 vg for AAV-PHP.B vectors) of AAV treatment groups was euthanized at 150 days of age to assess biochemical (enzyme activity and GM2 ganglioside levels) and histological outcome measures (below). Hexosaminidase (Hex) activity was measured in cerebrum, cerebellum, brain stem, spinal cord, and liver using two artificial substrates, MUG to determine total Hex activity (HexA, HexB, and HexS), and MUGS to determine HexA activity (Figure 3). As anticipated based on the differential CNS gene transfer efficiency between AAV-PHP.B and AAV9,29 hexosaminidase activities in the brain were higher in SD mice treated with AAV-PHP.B vectors. Total Hex and HexA activities in CNS structures were significantly higher in AAV-PHP.B-Bic- and AAV-PHP.B-P2I-treated cohorts compared to untreated SD controls, but nonetheless enzyme activity was still lower than in normal age matched controls (Figure 3A). The exception was in cerebellum of AAV-PHP.B-Bic-treated SD mice where HexA activity reached ∼3-fold above normal levels. Interestingly, the PHP.B-P2I HexA activities were not significantly different from untreated SD mice (Figure 3A).
Figure 3.

Hexosaminidase Activity Is Restored in the CNS and Liver of Treated SD Mice
Mice (n = 6) from the high-dose cohorts were used (4 × 1012 vg for AAV9 vectors and 1 × 1012 vg for AAV-PHP.B vectors). Hexosaminidase activity (fold normal) in (A) CNS and (B) liver using artificial substrates 4-MUG and 4-MUGS hydrolyzed by all isozymes (total hexosaminidase) and HexA, respectively. (C) Isozyme analysis in liver of AAV treated SD mice and controls show peaks of 4-MUG activity in fractions 3, 15, and 22 corresponding to HexB (ββ), HexA (αβ), and HexS (αα), respectively. Treatment groups were compared to untreated SD mice using one-way ANOVA with Dunnett’s multiple comparison test for each tissue: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
In liver, total Hex and HexA activities in the AAV9-Bic vector treatment group were 16.9 ± 5.7-fold and 47.9 ± 19.3-fold over normal, respectively (p < 0.001; Figure 3B). Total Hex and HexA activities in the liver of AAV-PHP.B-Bic-treated SD mice were significantly lower than in the AAV9-Bic group (MUG, p < 0.01; MUGS, p < 0.022; Figure 3B). Liver hexosaminidase activity in non-AAV9-Bic treatment groups was below normal, which suggests a difference in functionality between BiCBA and P2I promoters in liver. In addition to the heterodimeric HexA isozyme (αβ; the isozyme that hydrolyzes GM2 in humans), the Hex subunits can also form HexS (αα) or HexB (ββ) homodimers. To determine the amount of HexA (αβ) formation, we analyzed the isozyme profile in liver samples (Figure 3C). Treatment with AAV9-Bic and AAV9-P2I vectors showed a normal isozyme profile (presence of HexB, HexA, and HexS peaks)20 (Figure 3C). We found no evidence of hepatic tumors in AAV-treated mice.
Reduction in GM2-Ganglioside Content in the CNS
GM2 ganglioside is present at very low, often undetectable levels in normal postnatal CNS, but accumulates to exceptionally high levels in SD mice,12 and in this study ranged from 28 to 51 ng GM2/mg protein (black bars, Figure 4). The GM2 ganglioside content in the cerebrum, cerebellum, brain stem, and spinal cord of AAV-treated SD mice was 5- to 10-fold lower than untreated SD mice (p < 0.0001 for all structures and treatment groups; Figure 4). Only trace levels were measured in the CNS of SD mice treated with AAV-PHP.B-Bic or AAV-PHP.P2I vectors (<0.5 ng GM2/mg protein), and the effect of AAV9-P2I vector on storage was less profound. In AAV9-Bic treated group, 4 of 6 animals had GM2 ganglioside levels in the cerebrum comparable to that in AAV-PHP.B groups, and the other two mice had levels below 4 ng GM2/mg protein. In the other CNS structures of SD mice treated with AAV9-Bic vector, the GM2 ganglioside levels were below 0.5 ng GM2/mg protein.
Figure 4.

Reduction in GM2 Ganglioside Content in the CNS of Treated SD Mice
GM2 ganglioside was quantified in cerebrum, cerebellum, brainstem, and spinal cord of SD mice in the high dose cohorts at 150 days of age or humane endpoint (<150 days of age) for untreated SD mice using LC-MS/MS mass spectrometry. Treatment groups were compared to untreated SD mice using two-way ANOVA with Dunnett’s multiple comparison test for each tissue: ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
Treatment with Bicistronic AAV Vectors Attenuates Microglial Activation
The onset of symptoms in SD mice coincides with neuroinflammation characterized by increased levels of several cytokines (tumor necrosis factor alpha [TNF-α], transforming growth factor β1 [TGF-β1], and interleukin-1β [IL-1β]), as well as microglial activation and reactive astrogliosis.3 Immunofluorescence staining of brain sections for CD68 revealed a marked increase in activated microglia throughout the brain and spinal cord of untreated SD mice at the humane endpoint as previously described2 but an absence in normal mice at 150 days of age (Figure 5). Microglial activation was reduced in the CNS of SD mice treated with AAV9-Bic vector compared to untreated knockout (KO) controls (Figure 5A), and reached statistical significance in the brain stem and spinal cord (p < 0.05; Figure 5B), as well as a trend in the thalamus (p = 0.07; Figure 5B). The apparent differences in signal intensity in cortex and cerebellum were not statistically different between groups.
Figure 5.

Reduction of Microglial Activation in Brain and Spinal Cord of SD Mice Treated with AAV9-Bic Vector
(A) Microglial activation was assessed by immunofluorescence staining for CD68 (red) and DAPI to identify nuclei (blue) in brain and spinal cord sections from untreated SD mice (untreated KO), SD mice treated with AAV9-Bic vector (high dose), as well as normal controls (WT). (B) Quantification of CD68 staining intensity showed statistically significant reductions in AAV9-Bic treated mice compared (white bars) to untreated SD mice (black bars) in brainstem and spinal cord (p < 0.05), as well as a trend toward lower levels in thalamus (p = 0.07). No significant differences between groups, including normal controls (gray bars) were apparent in cortex and cerebellum). Abbreviations are as follows: CT, cortex; TH, thalamus; BS, brainstem; CB, cerebellum; SC, spinal cord. Scale bars, 150 μm. Two tailed t tests were used to determine statistical significance (∗p < 0.05).
Effect of Age on Therapeutic Efficacy and Dose Response
Previous AAV gene therapy studies based on intracranial delivery of two monocistronic AAV vectors have shown that therapeutic efficacy declines with treatment age, and by 12 weeks of age there is no survival benefit.17 To assess the impact of age on the efficacy of treatment with AAV9-Bic vector administered systemically, we treated SD mice at 8, 10, and 12 weeks of age with 1 × 1012 vg and followed until the humane endpoint (Figure 6A). The median survival for SD mice treated at 10 and 12 weeks was 165 days (p < 0.0001) and 158 days (p < 0.005), respectively, which is significantly longer than 121 days for untreated SD mice (maximum survival for this cohort of untreated SD mice was 136 days). The majority of SD mice treated at 8 weeks of age remain alive at >375 days of age.
Figure 6.

Systemic Treatment with AAV9-Bic Vector: Maximum Effective Treatment Age and Minimum Effective Dose
(A) SD mice were treated with 1 × 1012 vg AAV9-Bic at 8 (n = 10), 10 (n = 9), and 12 weeks (n = 8) of age and survival followed until the humane endpoint. (B) Dose escalation study in 4-week-old SD mice treated at doses of 1 × 1011 (n = 11), 3 × 1011 (n = 8), and 1 × 1012 vg (n = 4). Kaplan-Meier survival curves are shown for treatment groups compared to a common group of untreated (UT) SD mice (n = 6). Log-rank (Mantel-Cox) test was used to compare survival of treatment groups to that of untreated SD mice: ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
4-week-old SD mice were treated with 1 × 1011, 3 × 1011, or 1 × 1012 vg of AAV9-Bic vector to determine the minimum dose that shows therapeutic benefit (Figure 6B). The median survival of SD mice treated with 1 × 1011 vg was 128 days, which is not significantly different than 121 days for untreated animals. In contrast, the majority of SD mice treated with 3 × 1011 vg remain alive (5 of 8) at 290–350 days of age. The SD mice treated with 1 × 1012 vg, which were included in this experiment as a control since this dose was shown to be efficacious in the initial therapeutic study (Figure 1), remain alive at 350 days of age.
Discussion
In this study, treatment of adult SD mice by systemic administration of AAV9-Bic vector extended survival to ∼2 years of age in some animals, for the first time matching the therapeutic efficacy achieved by intra-striatal and deep cerebellar nuclei administration of AAV gene therapy.15, 16, 17 The exceptional therapeutic results after parenchymal brain delivery showed that most transduced cells were co-infected with both AAV vectors, achieving restoration of the normal isozyme expression profile in SD mice, SD cats, and TSD sheep.16,20,30,31 This was reproduced in this study after intravascular delivery and is considered to be an important feature of an efficacious treatment strategy for TSD and SD because HexA heterodimerizes in the endoplasmic reticulum and excess monomers are rapidly degraded.32,33
Despite the success of intraparenchymal delivery of two monocistronic AAV vectors, a two-vector strategy is not compatible with vascular or cerebral spinal fluid (CSF) delivery. Rapid dilution in blood or CSF reduces AAV concentrations to a point where co-transduction of target cells by two viruses is unlikely. A single AAV vector encoding both HexA α- and β-subunits obviates this limitation, but the approximately 1.6 kb size of the HEXA and HEXB cDNAs imposes significant design constraints. In the course of developing a single AAV vector encoding both subunits, we tested various designs expressing both proteins from a single promoter using viral24 or artificial internal ribosomal entry sites,25 and 2A self-cleaving peptides,26,27 as well as furin cleavage sites fusing the two proteins,34 but all failed to produce the normal isozyme expression profile in transfected HEK293 cells or mice (data not shown). Unlike our previous attempts, the AAV-Bic vector reported here shows a clear indication for translation through restoration of the normal hexosaminidase isozyme profile and a marked increase in survival, with some treated SD mice reaching more than 2 years of age.
The AAV-Bic vector is clearly more efficient than AAV-P2I, with differential efficacy likely related to their functionality in specific regions or cell types (e.g., CNS, or hepatocytes). The P2I promoter-intron combination was generated using a small promoter and a truncated version of the chimeric intron (chicken beta-actin intron fused to part of rabbit beta-globin intron 2) present in the original CAG/CBA promoter.35 The small promoter described in US patent 20020164783A1, which we named here P2, incorporates promoter sequences from the rat housekeeping Atp1a1 gene,36 which encodes the Na,K-ATPase α1 subunit, fused to the cytomegalovirus immediate early gene (CMV) promoter TATA box and transcription initiation sequences (Figure S1). This small promoter was shown to have ∼51-fold higher activity than the WT AAV2 ITR alone (US patent 20020164783A1), which has been shown to have promoter activity in vivo.37 As the Atp1a1 gene is broadly expressed in most cells in the mouse brain (Allen Brain Atlas, http://mouse.brain-map.org/gene/show/11714 and http://www.brainrnaseq.org), its small promoter appeared ideal to drive gene expression in a bicistronic vector where transgene space is limited. The low (or absent) enzyme activity in the cerebellum and spinal cord may be due to the P2 promoter only carrying a small portion of the rat Atp1a1 promoter, which may not contain all transcriptional elements necessary to direct broad gene expression in CNS. Alternatively, it is possible the antisense inward orientation (in relation to the ITRs) of the two transgene cassettes in the AAV-P2I vector may lead to interference between them resulting in decreased activity. Further studies will be necessary to fully characterize the regional functionality of the P2 promoter in the CNS in an AAV vector with a single transgene cassette. Regardless of molecular mechanisms, the clear efficacy of the AAV-Bic vector makes it a candidate for translation into clinical trials.
The CNS gene transfer efficiency and tropism of AAV9 delivered systemically in mice changes after the neonatal period where neuronal transduction is observed, to a predominantly astrocytic profile soon after birth.38,39 The AAV9 neuronal tropism is unique to neonatal mice and has not been reproduced in other species, even when treatment is initiated early.40,41 To avoid overrepresenting transduction and therapeutic efficacy, we conducted most studies here in 4-week-old SD mice. Moreover, we demonstrate the treatment to have a beneficial impact on survival when initiated in 8-week-old SD mice. Previous studies of systemic AAV9 delivery in neonatal SD mice yielded excellent therapeutic results,21,22 but these data should be interpreted carefully because of the age of treatment and the alternative biochemical pathway for GM2 ganglioside degradation that is absent in humans.23
An interesting approach bypassing the need to encode two different proteins in a single AAV vector was the design of a new hybrid μ-subunit (HEXM) by grafting amino acids from the β-subunit into the C terminus of the α-subunit, enabling stable homodimers that interact with the GM2 activator (GM2A) protein.42 AAV delivery of HEXM in neonatal SD mice showed promising results with increased survival and considerable reduction in GM2 ganglioside content in Sandhoff and Tay-Sachs mice.42,43 Ultimately, efficacy of HEXM was limited by an immune response, which will likely be overcome by an appropriate adjunct immunosuppression protocol.44 Another bicistronic AAV vector, carrying human HEXB and HEXA subunits fused by a 2A peptide, showed improved mean survival of SD mice to 170 days of age after neonatal systemic administration.45 The limited impact on survival was unexpected, because the subunits appeared to be properly processed and cells expressed mature, functional HexA and HexB isozymes. Another interesting alternative for SD and TSD is intravascular administration of two monocistronic vectors encoding human HEXA and HEXB using a new brain endothelium targeted AAV capsid and high-expressing CBA promoter. This strategy extended median survival to 189 days in SD mice after neonatal delivery.46 Further improvements in AAV capsid technology (e.g., AAV-PHP.B29 and AAV-PHP.eB47) may eventually make a systemic treatment with a two-vector system viable, but clinical translation will still require manufacturing of two clinical grade vectors and thus higher costs.
An intravascular approach is intrinsically less invasive than intracranial injection and the dose response in this study indicates that a dose of 3 × 1011 vg in 4-week-old SD mice (∼20 g), or 1.5 × 1013 vg/kg, is sufficient to drive a significant survival benefit. In ongoing human clinical trials, AAV doses below 1 × 1014 vg/kg appear to be well tolerated when T cell responses are controlled by co-administration of prednisolone for 40–90 days48, 49, 50 or by a combination of rituximab and sirolimus.51 Treatment with AAV9-Bic at 4 × 1012 vg in 4-week-old SD mice was also efficacious and well tolerated, indicating that systemic AAV9-Bic delivery has a broad therapeutic range of 1.5 × 1013 to 2 × 1014 vg/kg. The dose range established here is below or comparable to the 1.1 × 1014 vg/kg recommended for ZOLGENSMA (package insert, https://www.fda.gov/media/126109/download), the FDA approved AAV9 vector for treatment of spinal muscular atrophy (SMA) by systemic administration. As this is a product on the market for an indication with a considerable number of patients worldwide, one surmises that production of clinical grade AAV9-Bic vector for the relatively small numbers of Tay-Sachs and Sandhoff disease patients is well within the capability of the current AAV production platforms.
Additionally, for a fast progressing disease such as infantile-onset Tay-Sachs disease, the timing of intervention and time to therapeutic effect will be critical in determining the effectiveness of a systemic therapy. Post-symptomatic intravenous administration of these vectors was slightly less efficacious than previously reported results using the intracranial monocistronic vector approach,17 and differences in the therapeutic window could be related to promoter strength, HexA expression levels, and/or the ability of parenchymal injections to distribute rapidly via axonal transport and perivascular fluid flow.16 Additionally, all SD mice were treated with 1 × 1012 vg regardless of age, which corresponds to an effective lowering of dose (vg/kg body weight) in older animals.
In conclusion, intravascular delivery of the AAV9-Bic vector is on par with intracranial administration of two monocistronic vectors, which embodies the best preclinical results in the treatment of TSD and SD to date. These results make the AAV9-Bic vector an excellent candidate for clinical translation in the treatment of Tay-Sachs and Sandhoff diseases. Additionally, the minimally invasive intravascular delivery route can be easily implemented in most clinical centers, and the modest doses make it compatible with current AAV production technologies.
Materials and Methods
Vectors and Dosage
Two different plasmids were employed in this study, AAV-Bic and AAV-P2I. Expression in the AAV-Bic vector is driven by the BiCBA promoter, which is composed of a central CMV enhancer flanked by two chicken beta-actin promoters in opposite directions, with no introns. The mouse HexA subunit is flanked by an SV40 poly(A) on one side of the promoter, while the mouse HexB subunit is flanked by an RBG poly(A) on the other side of the promoter. The AAV-P2 plasmid uses a small synthetic promoter plus an intron sequence on either side of the Hex subunits, with a central BGH poly(A) flanking the alpha subunit and SV40 poly(A) flanking the beta subunit. Each plasmid was packaged with AAV9 and PHP.B capsids to generate a total of 4 vectors in the study. Each vector group contained animals treated with a low dose and animals treated with a high dose. For AAV9 vectors, low dose was 1 × 1012 vg and high dose was 4 × 1012 vg. For PHP.B vectors, low dose was 3 × 1011 vg and high dose was 1 × 1012 vg. Virus was administered by tail vein in a total volume of 300 μL.
Virus Production and Purification
AAV vectors were prepared by transient transfection of HEK293T cells and purification by iodixanol gradient ultracentrifugation as described.52
Animals
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC). Hexb−/− mice bred at the University of Massachusetts animal facility were used in this study. Intravenous administration of AAV was performed via the tail vein. Behavioral testing was performed at 60, 90, 105, 120, and 149 (±2) days of age for animals receiving a high dose treatment. Each vector group contained 22 animals, with 14 receiving a high dose treatment and 8 animals receiving a low dose treatment. Six PBS-injected GM2 mice and 12 PBS-injected WT mice were used as controls. Half of the high dose animals (n = 44) and half of the WT controls were sacrificed at 150 days of age, while the remaining high dose animals, all low dose animals, and remaining control animals were left for survival until humane endpoint, which was defined as hind-limb paralysis or the inability to right themselves within 15 s when turned on their back.
Behavioral Testing
Inverted screen testing was performed by placing animals on a square wire mesh screen and slowly inverting until the screen reached a 60° angle. Latency to fall (2 min maximum) was assessed during the first trial. In the subsequent three trials, latency to fall and hindlimb movements were recorded over the course of 2 min. Latency to fall and number of hindlimb movements in trials 2–4 were averaged and reported. Rotarod performance measured latency to fall on an accelerating rotarod 4 to 40 rpm over 5 min. The amount of time spent on the rotarod was recorded for three trials, averaged, and reported. If it were the animal’s first time ever performing rotarod, they were subjected to a training trial during which the rotarod accelerated at a rate of 2 to 20 rpm over 5 min. Mice were given minimum 15 min of rest between trials on inverted screen and rotarod.
Necropsy
Animals received an overdose of a ketamine/xylazine solution and then were perfused transcardially with ice-cold PBS. The right side of the brain was embedded in Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetek USA, Torrance, CA) in a plastic mold and rapidly frozen by immersion in a dry ice-methylbutane bath. The left brain hemisphere was separated into cerebrum, cerebellum, and brain stem, and frozen on dry ice. The spinal cord was separated into cervical, thoracic, and lumbar regions and each region sub-divided in half for histological (fresh frozen in OCT) and biochemical (frozen on dry ice) analyses. Liver, kidney, heart, lung, and skeletal muscle were also frozen on dry ice.
Hexosaminidase Activity
Tissues were lysed in 0.1% Triton X-100 in 0.01 M phosphate citrate buffer pH 4.4 using a TissueLyser II (QIAGEN, Germantown, MD) with 5 mm stainless steel beads at 20 Hz for 30 s with three pulses. Lysates underwent three freeze-thaws alternating between a dry ice-ethanol bath and 37°C water bath. Lysates were centrifuged at 20,000 × g for 15 min at 4°C, and the supernatant was transferred to a new microcentrifuge tube and stored at −80°C. Hexosaminidase activity was measured using 4-methylumbelliferyl N-acetyl-β-D-glucosaminide (MUG, Sigma, St. Louis, MO) and 4-methylumbelliferyl 6-Sulfo-2-acetamido-2-deoxy-β-D-glucopyranoside Potassium Salt (MUGS, Toronto Research Chemicals, Toronto, Canada) and normalized to total protein content measured by QuickStart Bradford Protein Assay (Bio-Rad, Hercules, CA) with serial dilutions of bovine serum albumin as protein standard.
GM2 Ganglioside Content
GM2 ganglioside content in the CNS was measured by liquid chromatography-tandem mass spectrometry (LC-MS/ MS) using the University of Massachusetts Medical School Proteomics and Mass Spectrometry Facility. Briefly, 0.04 or 0.1 mg/μL of tissue homogenate was diluted in 0.01 M phosphate citrate buffer with 0.1% Triton X-100 (VWR) pH 4.4 to 25 or 50 μL, respectively. Each sample was spiked with 200 ng of d3-labeled GM2 ganglioside formulated in methanol (Matreya, State College, PA). Standard calibration curves were made with GM2-ganglioside formulated in methanol (Matreya) over the range of 10–800 ng in phosphate citrate buffer with 0.1% Triton X-100 and also spiked with 200 ng of d3-GM2 ganglioside and also put through the extraction process. Total lipids were extracted twice by vortexing and sonicating samples for 30 s with 1 mL isopronal:ethyl acetate:water (6:3:1), combining both supernatants after centrifugation. Samples were dried and then resuspended in 100 μL of 1 part 0.1% (v/v) formic acid (mobile phase A) to 4 parts methanol:2-propanol:0.1% formic acid (47.5:47.5: 4.9, mobile phase B). For each sample, 6 μL was injected in duplicate onto a 2.1 × 50 mm Kinetex (Phenomenex, Torrance, CA) C18 (1.7 μ,100 Å) column using an Acquity HPLC (Waters, Milford, MA) coupled to a Quattro Premier XE (Waters) mass spectrometer operating in the negative ion electrospray mode. Gangliosides were eluted at 270 μL/min using the following gradient: 0–1 min (80% B); 1–5 min (80%–100% B); 5–7 min (100% B); 7.1–12 min (80% B). Multiple reaction monitoring (MRM) transitions for GM2 (fatty acids: 18:0, d3-18:0, 20:0) were monitored by following corresponding (M-H)- precursor ions to the common sialic acid anion fragment at m/z 290 (cone voltage, 90 V; collision energy, 70 eV; collision gas pressure, 2.2 μbar). The area of all the individual GM2 lipid species (18:0, 20:0) were combined and normalized to the d3-18:0 GM2 lipid species internal standard. Briefly, 11.06% of the area of 18:0 GM2 lipid was subtracted from the area of d3-18:0 GM2, to correct for d3-18:0 GM2/18:0 GM2 overlap. The ratio of the sum of the area of 18:0 GM2 and 20:0 GM2 to the corrected d3-18:0 GM2 was calculated and ng of GM2 lipid was determined from the standard curve. Total GM2 ganglioside concentrations were then normalized to total protein content in tissue homogenate, measured by QuickStart Bradford Protein Assay (Bio-Rad) with serial dilutions of bovine serum albumin as protein standard.
Immunostaining and Image Acquisition
Brain and spinal cord, embedded in OCT, were cut into 20 μm sections. Immunofluorescent staining was performed for glial fibrillary acidic protein (GFAP) (ab7260, Abcam, Cambridge, MA) and CD68 (MCA1957, Bio-Rad). Sections were thawed at room-temperature (30 min), formalin fixed (30 min), and permeabilized (10 min) with 0.5% Triton X-100 in 1× PBS. Sections were incubated for 1 h at room temperature in blocking solution (GFAP: 5% FBS + 5% donkey serum, CD68: 5% FBS + 5% goat serum in PBS), followed by overnight primary incubation (GFAP 1:500 and CD68 1:1,000 in PBS with 3% respective serum + 1% FBS +0.05% Triton X-100). Sections were washed with PBS and incubated with 1:1,000 dilution of Alexa 555-conjugated secondary antibodies in PBS for 1 h at room temperature (GFAP: Alexa 555-conjugated donkey anti-rabbit; CD68: Alexa 555-conjugated goat anti-rat, Invitrogen, Carlsbad, CA). Sections were washed with PBS and mounted using Vectashield mounting medium containing 2-(4-amidinophenyl)-1H -indole-6-carboxamidine (DAPI) (H-1200, Vector Laboratories, Burlingame, CA). Fluorescent images were acquired using a Leica DM5500 B upright microscope (Leica Microsystems, Buffalo Grove, IL). Three to four images (10× magnification, 8 bit, gray scale) per CNS region in each animal (at least n = 3 animals/group) were used to calculate signal intensities using ImageJ Software (NIH). The background signal was determined from images of parallel tissue sections stained with secondary antibody only. Background signal was subtracted from the intensity of each image, and the mean intensity calculated for each structure in each group. Statistical analysis was performed with Graphpad Prism 8.0 (GraphPad Software, San Diego, CA) using two tailed t tests to determine the significance of differences between AAV9-Bic treatment and untreated SD groups.
DEAE Cellulose Anion Exchange Chromatography
Relative levels of each isoenzyme (HexA, HexB, and HexS) in liver were measured by diethylaminoethyl cellulose chromatography, as described.20 Fractions were eluted from the column with increasing concentrations of sodium chloride (0–400 mmol/L), and total Hex activity in each fraction was determined with the fluorogenic substrate MUG. To resolve the HexA and HexS peaks better, we added additional steps of 104, 108, 112, and 116 mM of sodium chloride between the standard 100 and 120 mM elutions, for a total of 30 fractions.
Authors Contributions
H.G.L., C.J.W., D.G., C.M.I., E.H., T.T., P.R., A.R.B., L.E.E., and M.H. executed the experiments and analyzed data. D.R.M., H.G.-E., and M.S.-E. contributed to experimental design, oversaw experiments, and analyzed data. H.G.L., H.G.-E., and M.S.-E drafted the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The work was supported by NINDS/NIH grant R01NS093941 (D.R.M. and M.S.-E.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.06.021.
Supplemental Information
References
- 1.Bley A.E., Giannikopoulos O.A., Hayden D., Kubilus K., Tifft C.J., Eichler F.S. Natural history of infantile G(M2) gangliosidosis. Pediatrics. 2011;128:e1233–e1241. doi: 10.1542/peds.2011-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wada R., Tifft C.J., Proia R.L. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeyakumar M., Thomas R., Elliot-Smith E., Smith D.A., van der Spoel A.C., d’Azzo A., Perry V.H., Butters T.D., Dwek R.A., Platt F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003;126:974–987. doi: 10.1093/brain/awg089. [DOI] [PubMed] [Google Scholar]
- 4.Bradbury A.M., Gray-Edwards H.L., Shirley J.L., McCurdy V.J., Colaco A.N., Randle A.N., Christopherson P.W., Bird A.C., Johnson A.K., Wilson D.U. Biomarkers for disease progression and AAV therapeutic efficacy in feline Sandhoff disease. Exp. Neurol. 2015;263:102–112. doi: 10.1016/j.expneurol.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Specht B.U., Geiger B., Arnon R., Passwell J., Keren G., Goldman B., Padeh B. Enzyme replacement in Tay-Sachs disease. Neurology. 1979;29:848–854. doi: 10.1212/wnl.29.6.848. [DOI] [PubMed] [Google Scholar]
- 6.Platt F.M., Neises G.R., Reinkensmeier G., Townsend M.J., Perry V.H., Proia R.L., Winchester B., Dwek R.A., Butters T.D. Prevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycin. Science. 1997;276:428–431. doi: 10.1126/science.276.5311.428. [DOI] [PubMed] [Google Scholar]
- 7.Baek R.C., Kasperzyk J.L., Platt F.M., Seyfried T.N. N-butyldeoxygalactonojirimycin reduces brain ganglioside and GM2 content in neonatal Sandhoff disease mice. Neurochem. Int. 2008;52:1125–1133. doi: 10.1016/j.neuint.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Lee J.P., Jeyakumar M., Gonzalez R., Takahashi H., Lee P.J., Baek R.C., Clark D., Rose H., Fu G., Clarke J. Stem cells act through multiple mechanisms to benefit mice with neurodegenerative metabolic disease. Nat. Med. 2007;13:439–447. doi: 10.1038/nm1548. [DOI] [PubMed] [Google Scholar]
- 9.Arthur J.R., Lee J.P., Snyder E.Y., Seyfried T.N. Therapeutic effects of stem cells and substrate reduction in juvenile Sandhoff mice. Neurochem. Res. 2012;37:1335–1343. doi: 10.1007/s11064-012-0718-0. [DOI] [PubMed] [Google Scholar]
- 10.Jeyakumar M., Smith D.A., Williams I.M., Borja M.C., Neville D.C., Butters T.D., Dwek R.A., Platt F.M. NSAIDs increase survival in the Sandhoff disease mouse: synergy with N-butyldeoxynojirimycin. Ann. Neurol. 2004;56:642–649. doi: 10.1002/ana.20242. [DOI] [PubMed] [Google Scholar]
- 11.Jeyakumar M., Norflus F., Tifft C.J., Cortina-Borja M., Butters T.D., Proia R.L., Perry V.H., Dwek R.A., Platt F.M. Enhanced survival in Sandhoff disease mice receiving a combination of substrate deprivation therapy and bone marrow transplantation. Blood. 2001;97:327–329. doi: 10.1182/blood.v97.1.327. [DOI] [PubMed] [Google Scholar]
- 12.Denny C.A., Kasperzyk J.L., Gorham K.N., Bronson R.T., Seyfried T.N. Influence of caloric restriction on motor behavior, longevity, and brain lipid composition in Sandhoff disease mice. J. Neurosci. Res. 2006;83:1028–1038. doi: 10.1002/jnr.20798. [DOI] [PubMed] [Google Scholar]
- 13.Denny C.A., Heinecke K.A., Kim Y.P., Baek R.C., Loh K.S., Butters T.D., Bronson R.T., Platt F.M., Seyfried T.N. Restricted ketogenic diet enhances the therapeutic action of N-butyldeoxynojirimycin towards brain GM2 accumulation in adult Sandhoff disease mice. J. Neurochem. 2010;113:1525–1535. doi: 10.1111/j.1471-4159.2010.06733.x. [DOI] [PubMed] [Google Scholar]
- 14.Shapiro B.E., Pastores G.M., Gianutsos J., Luzy C., Kolodny E.H. Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet. Med. 2009;11:425–433. doi: 10.1097/GIM.0b013e3181a1b5c5. [DOI] [PubMed] [Google Scholar]
- 15.Cachón-González M.B., Wang S.Z., Lynch A., Ziegler R., Cheng S.H., Cox T.M. Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proc. Natl. Acad. Sci. USA. 2006;103:10373–10378. doi: 10.1073/pnas.0603765103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cachon-Gonzalez M.B., Wang S.Z., McNair R., Bradley J., Lunn D., Ziegler R., Cheng S.H., Cox T.M. Gene transfer corrects acute GM2 gangliosidosis–potential therapeutic contribution of perivascular enzyme flow. Mol. Ther. 2012;20:1489–1500. doi: 10.1038/mt.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cachón-González M.B., Wang S.Z., Ziegler R., Cheng S.H., Cox T.M. Reversibility of neuropathology in Tay-Sachs-related diseases. Hum. Mol. Genet. 2014;23:730–748. doi: 10.1093/hmg/ddt459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCurdy V.J., Rockwell H.E., Arthur J.R., Bradbury A.M., Johnson A.K., Randle A.N., Brunson B.L., Hwang M., Gray-Edwards H.L., Morrison N.E. Widespread correction of central nervous system disease after intracranial gene therapy in a feline model of Sandhoff disease. Gene Ther. 2015;22:181–189. doi: 10.1038/gt.2014.108. [DOI] [PubMed] [Google Scholar]
- 19.Rockwell H.E., McCurdy V.J., Eaton S.C., Wilson D.U., Johnson A.K., Randle A.N., Bradbury A.M., Gray-Edwards H.L., Baker H.J., Hudson J.A. AAV-mediated gene delivery in a feline model of Sandhoff disease corrects lysosomal storage in the central nervous system. ASN Neuro. 2015;7:7. doi: 10.1177/1759091415569908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray-Edwards H., Randle A.N., Maitland S., Benatti H., Hubbard S., Canning P., Vogel M.B., Brunson B.L., Hwang M., Ellis L.E. AAV gene therapy in a sheep model of Tay-Sachs disease. Hum. Gene Ther. 2017;29:312–326. doi: 10.1089/hum.2017.163. [DOI] [PubMed] [Google Scholar]
- 21.Walia J.S., Altaleb N., Bello A., Kruck C., LaFave M.C., Varshney G.K., Burgess S.M., Chowdhury B., Hemming R. Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol. Ther. 2015;23:414–422. doi: 10.1038/mt.2014.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niemir N., Rouvière L., Besse A., Vanier M.T., Dmytrus J., Marais T., Astord S., Puech J.P., Panasyuk G., Cooper J.D. Intravenous administration of scAAV9-Hexb normalizes lifespan and prevents pathology in Sandhoff disease mice. Hum. Mol. Genet. 2018;27:954–968. doi: 10.1093/hmg/ddy012. [DOI] [PubMed] [Google Scholar]
- 23.Sango K., Yamanaka S., Hoffmann A., Okuda Y., Grinberg A., Westphal H., McDonald M.P., Crawley J.N., Sandhoff K., Suzuki K., Proia R.L. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 1995;11:170–176. doi: 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- 24.Jang S.K., Kräusslich H.G., Nicklin M.J., Duke G.M., Palmenberg A.C., Wimmer E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 1988;62:2636–2643. doi: 10.1128/jvi.62.8.2636-2643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chappell S.A., Dresios J., Edelman G.M., Mauro V.P. Ribosomal shunting mediated by a translational enhancer element that base pairs to 18S rRNA. Proc. Natl. Acad. Sci. USA. 2006;103:9488–9493. doi: 10.1073/pnas.0603597103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Felipe P., Martín V., Cortés M.L., Ryan M., Izquierdo M. Use of the 2A sequence from foot-and-mouth disease virus in the generation of retroviral vectors for gene therapy. Gene Ther. 1999;6:198–208. doi: 10.1038/sj.gt.3300811. [DOI] [PubMed] [Google Scholar]
- 27.Furler S., Paterna J.C., Weibel M., Büeler H. Recombinant AAV vectors containing the foot and mouth disease virus 2A sequence confer efficient bicistronic gene expression in cultured cells and rat substantia nigra neurons. Gene Ther. 2001;8:864–873. doi: 10.1038/sj.gt.3301469. [DOI] [PubMed] [Google Scholar]
- 28.Jeyakumar M., Butters T.D., Cortina-Borja M., Hunnam V., Proia R.L., Perry V.H., Dwek R.A., Platt F.M. Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc. Natl. Acad. Sci. USA. 1999;96:6388–6393. doi: 10.1073/pnas.96.11.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golebiowski D., van der Bom I.M.J., Kwon C.S., Miller A.D., Petrosky K., Bradbury A.M., Maitland S., Kühn A.L., Bishop N., Curran E. Direct Intracranial Injection of AAVrh8 Encoding Monkey β-N-Acetylhexosaminidase Causes Neurotoxicity in the Primate Brain. Hum. Gene Ther. 2017;28:510–522. doi: 10.1089/hum.2016.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradbury A.M., Cochran J.N., McCurdy V.J., Johnson A.K., Brunson B.L., Gray-Edwards H., Leroy S.G., Hwang M., Randle A.N., Jackson L.S. Therapeutic response in feline sandhoff disease despite immunity to intracranial gene therapy. Mol. Ther. 2013;21:1306–1315. doi: 10.1038/mt.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahuran D.J. Beta-hexosaminidase: biosynthesis and processing of the normal enzyme, and identification of mutations causing Jewish Tay-Sachs disease. Clin. Biochem. 1995;28:101–106. doi: 10.1016/0009-9120(95)00003-r. [DOI] [PubMed] [Google Scholar]
- 33.Mahuran D.J. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim. Biophys. Acta. 1999;1455:105–138. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 34.Foti S.B., Samulski R.J., McCown T.J. Delivering multiple gene products in the brain from a single adeno-associated virus vector. Gene Ther. 2009;16:1314–1319. doi: 10.1038/gt.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyazaki J., Takaki S., Araki K., Tashiro F., Tominaga A., Takatsu K., Yamamura K. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene. 1989;79:269–277. doi: 10.1016/0378-1119(89)90209-6. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki-Yagawa Y., Kawakami K., Nagano K. Housekeeping Na,K-ATPase alpha 1 subunit gene promoter is composed of multiple cis elements to which common and cell type-specific factors bind. Mol. Cell. Biol. 1992;12:4046–4055. doi: 10.1128/mcb.12.9.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flotte T.R., Afione S.A., Conrad C., McGrath S.A., Solow R., Oka H., Zeitlin P.L., Guggino W.B., Carter B.J. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc. Natl. Acad. Sci. USA. 1993;90:10613–10617. doi: 10.1073/pnas.90.22.10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., Kaspar B.K. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Bevan A.K., Duque S., Foust K.D., Morales P.R., Braun L., Schmelzer L., Chan C.M., McCrate M., Chicoine L.G., Coley B.D. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 2011;19:1971–1980. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duque S., Joussemet B., Riviere C., Marais T., Dubreil L., Douar A.M., Fyfe J., Moullier P., Colle M.-A., Barkats M. Intravenous Administration of Self-complementary AAV9 Enables Transgene Delivery to Adult Motor Neurons. Mol. Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tropak M.B., Yonekawa S., Karumuthil-Melethil S., Thompson P., Wakarchuk W., Gray S.J., Walia J.S., Mark B.L., Mahuran D. Construction of a hybrid β-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol. Ther. Methods Clin. Dev. 2016;3:15057. doi: 10.1038/mtm.2015.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karumuthil-Melethil S., Nagabhushan Kalburgi S., Thompson P., Tropak M., Kaytor M.D., Keimel J.G., Mark B.L., Mahuran D., Walia J.S., Gray S.J. Novel Vector Design and Hexosaminidase Variant Enabling Self-Complementary Adeno-Associated Virus for the Treatment of Tay-Sachs Disease. Hum. Gene Ther. 2016;27:509–521. doi: 10.1089/hum.2016.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kot S., Chen Z., Vyas M., Mitchell M., Deschenes N.M., Osmon K.J.L. Induction of Immune Tolerance towards a Human Protein- HexM Expressed through AAV Gene Transfer in Sandhoff Mice. Mol. Ther. 2018;26:105–106. [Google Scholar]
- 45.Woodley E., Osmon K.J.L., Thompson P., Richmond C., Chen Z., Gray S.J., Walia J.S. Efficacy of a Bicistronic Vector for Correction of Sandhoff Disease in a Mouse Model. Mol. Ther. Methods Clin. Dev. 2018;12:47–57. doi: 10.1016/j.omtm.2018.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dogbevia G., Grasshoff H., Othman A., Penno A., Schwaninger M. Brain endothelial specific gene therapy improves experimental Sandhoff disease. J. Cereb. Blood Flow Metab. 2019;40:1338–1350. doi: 10.1177/0271678X19865917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 51.Corti M., Liberati C., Smith B.K., Lawson L.A., Tuna I.S., Conlon T.J., Coleman K.E., Islam S., Herzog R.W., Fuller D.D. Safety of Intradiaphragmatic Delivery of Adeno-Associated Virus-Mediated Alpha-Glucosidase (rAAV1-CMV-hGAA) Gene Therapy in Children Affected by Pompe Disease. Hum. Gene Ther. Clin. Dev. 2017;28:208–218. doi: 10.1089/humc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Batista A.R., King O.D., Reardon C.P., Davis C., Shankaracharya, Philip V., Gray-Edwards H., Aronin N., Lutz C., Landers J., Sena-Esteves M. Ly6a Differential Expression in Blood-Brain Barrier Is Responsible for Strain Specific Central Nervous System Transduction Profile of AAV-PHP.B. Hum. Gene Ther. 2020;31:90–102. doi: 10.1089/hum.2019.186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.