

Abstract
Patients with relapsed or refractory acute myeloid leukemia (AML) have a dismal prognosis and limited treatment options. Chimeric antigen receptor (CAR) T cells have achieved unprecedented clinical responses in patients with B cell leukemias and lymphomas and could prove highly efficacious in AML. However, a significant number of patients with AML may not receive treatment with an autologous product due to manufacturing failures associated with low lymphocyte counts or rapid disease progression while the therapeutic is being produced. We report the preclinical evaluation of an off-the-shelf CAR T cell therapy targeting Fms-related tyrosine kinase 3 (FLT3) for the treatment of AML. Single-chain variable fragments (scFvs) targeting various epitopes in the extracellular region of FLT3 were inserted into CAR constructs and tested for their ability to redirect T cell specificity and effector function to FLT3+ AML cells. A lead CAR, exhibiting minimal tonic signaling and robust activity in vitro and in vivo, was selected and then modified to incorporate a rituximab-responsive off-switch in cis. We found that allogeneic FLT3 CAR T cells, generated from healthy-donor T cells, eliminate primary AML blasts but are also active against mouse and human hematopoietic stem and progenitor cells, indicating risk of myelotoxicity. By employing a surrogate CAR with affinity to murine FLT3, we show that rituximab-mediated depletion of FLT3 CAR T cells after AML eradication enables bone marrow recovery without compromising leukemia remission. These results support clinical investigation of allogeneic FLT3 CAR T cells in AML and other FLT3+ hematologic malignancies.
Keywords: allogeneic CAR T therapy, FLT3, acute myeloid leukemia, chimeric antigen receptor
Graphical Abstract
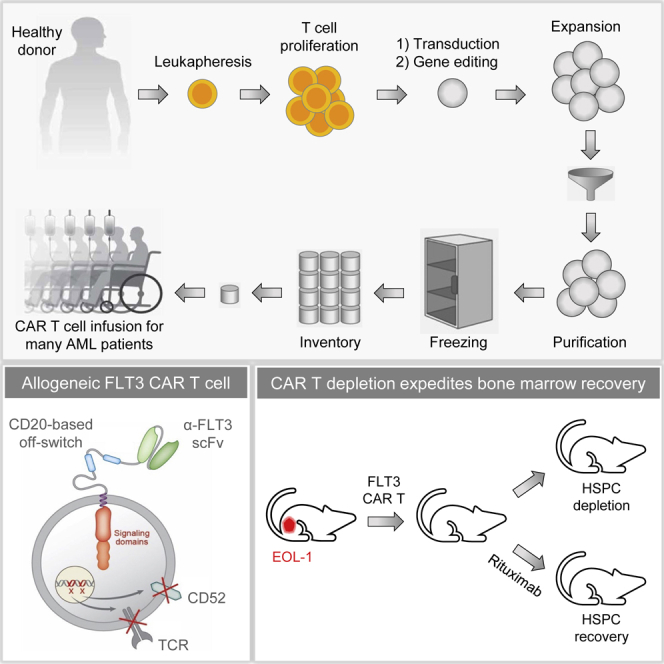
Patients with relapsed or refractory AML have poor prognosis and limited treatment options. Sommer et al. demonstrate potent antileukemic activity of allogeneic FLT3 CAR T cells generated from healthy donor T cells and investigate the possibility to selectively eliminate the engineered cells after AML eradication to mitigate potential hematopoietic toxicity.
Introduction
Acute myeloid leukemia (AML) is the most common acute leukemia in adults and accounts for roughly 20% of pediatric leukemias. Although most patients achieve remission after one or two rounds of chemotherapy, relapses occur frequently and are associated with poor prognosis, resulting in an estimated 5-year survival rate of 28%.1 Consolidation with hematopoietic stem cell (HSC) transplantation (HSCT) remains the only curative therapy, particularly in the high-risk group. Outcomes for patients undergoing HSCT while in morphologic remission, however, vary widely, with minimal residual disease (MRD) levels before transplant being a strong prognostic indicator of the risk of relapse.2 Novel and more potent therapies capable of achieving MRD-negative remissions could substantially improve patient outcomes3 and are urgently needed.
Chimeric antigen receptor (CAR) T cell therapies have achieved impressive clinical responses in patients with leukemia, lymphoma, and multiple myeloma4, 5, 6 and could provide benefit in AML. Support for this notion comes from preclinical studies evaluating CAR T cells directed toward AML-associated antigens, such as CD33, CD123, CLL-1, and Fms-related tyrosine kinase 3 (FLT3).7, 8, 9, 10, 11, 12, 13 Translating these promising findings into clinically feasible autologous therapies is associated with some challenges. Peripheral blood T cells in patients with AML often display poor abundance or abnormalities in activation and function14,15 that could lead to poor cellular fitness16 or production failures. Given the rapid progression rate of AML upon relapse, a 2–4 week wait necessary for manufacturing and release of an autologous product may be extremely problematic. Taking this into account, allogeneic CAR T cell therapies, which utilize T cells derived from healthy donors for large-scale manufacturing of off-the-shelf products and are intended to be readily available to patients,17,18 may be particularly attractive for the treatment of AML.
Identification of a truly AML-specific cell surface antigen has thus far remained elusive. Most antigens are also expressed in normal myeloid progenitors and/or mature hematopoietic cells,13,19 posing the risk of prolonged myeloid cell aplasia,8,11,20 cytokine release syndrome (CRS), and potentially other toxicities that may limit therapeutic efficacy. Among commonly pursued targets, FLT3 appears promising for the development of immunotherapies in AML.21 Expression of FLT3 is reported to be restricted to the hematopoietic lineage, including a subset of hematopoietic stem and progenitor cells (HSPCs) and plasmacytoid dendritic cells (pDCs).22,23 FLT3 has also been detected in Purkinje cells in the cerebellum, although it was reported to be intracellular.24 FLT3 is found expressed at high density on the surface of AML blasts as well as in leukemic cells of B cell acute lymphoblastic leukemia (B-ALL) patients.25,26 FLT3 is the most commonly mutated gene in AML patients27,28 and is implicated in the pathogenesis and progression of the disease. Mutations involving an internal tandem duplication (ITD) in the juxtamembrane domain of FLT3 are particularly prevalent and result in ligand-independent overactive signaling that confers a survival and proliferation advantage to blast cells.29 Adoptive cell therapies with FLT3-directed CAR T cells, which rely on recognition of the extracellular region of the receptor to effect antitumor activity, are therefore relevant for patients with either wild-type or mutated FLT3 since the extracellular portion of the molecule typically remains intact.30 These observations highlight the potential of FLT3 as a clinically actionable target for T cell immunotherapies and have prompted studies investigating CARs that incorporate single-chain variable fragments (scFvs) derived from murine anti-human FLT3 antibodies.10,12 While preclinical evidence for the antileukemic efficacy of FLT3 CAR T cells exists, the associated risks of on-target myelotoxicity and neurotoxicity are not completely understood.10,12,13
We report the preclinical evaluation of an allogeneic FLT3 CAR T cell therapy for the treatment of AML. Fully human scFvs binding to different domains of the FLT3 protein were isolated from a phage-display library and inserted into CAR constructs that were tested through in vitro assays and in an orthotopic mouse model of AML. A lead CAR was selected based on exhibiting minimal tonic signaling and potent antitumor activity in vitro and in vivo and then engineered to contain an off-switch responsive to rituximab. Allogeneic FLT3 CAR T cells were generated by transducing healthy donor-derived T cells with the lead construct followed by transcription activator-like effector nuclease (TALEN)-mediated inactivation of the T cell receptor α constant (TRAC) and CD52 genes. The gene-edited cells were resistant to treatment with a lymphodepleting anti-CD52 antibody, allowing the use of anti-CD52 to deplete the patient’s immune cells to delay rejection of the graft, potentially resulting in enhanced persistence and antitumor efficacy. We found that FLT3 CAR T cells were highly active against patient-derived AML blasts but also showed on-target activity against a subset of human HSPCs in vitro and mouse HSPCs in vivo. Activation of the off-switch with rituximab provided a means to deplete circulating CAR T cells after AML clearance, allowing for subsequent bone marrow recovery from remaining hematopoietic progenitors without compromising leukemia remission. The high antileukemic activity of allogeneic FLT3 CAR T cells combined with a safety mechanism to mitigate potential off-tumor effects may facilitate clinical translation in this patient population with a high unmet medical need.
Results
Functional Screening of Anti-FLT3 scFvs and Selection of a Lead CAR with Optimal Activity In Vitro and In Vivo
The extracellular segment of the FLT3 receptor consists of five immunoglobulin (Ig)-like domains that are potentially accessible to targeting with a CAR.31 A synthetic human scFv phage library was screened to identify clones demonstrating binding to recombinant human FLT3 as previously described.21 Epitope mapping experiments using surface plasmon resonance assays indicated that all five Ig domains of FLT3 were targeted by at least one of the nine selected binders with affinities in the nanomolar range (Table S1). To identify an optimal anti-FLT3 CAR for clinical development, codon-optimized sequences of selected scFvs were inserted into lentiviral CAR constructs in orientation variable heavy chain (VH)-linker-variable light chain (VL) followed by a CD8α hinge/transmembrane domain fused with 4-1BB and CD3ζ intracellular signaling elements. The CD8α hinge was selected based on preliminary studies indicating more consistent degranulation and cytotoxic activity compared with shorter Fcγ receptor-based hinges (data not shown). Co-expression of blue fluorescent protein (BFP) from the same vector enabled easy detection of transduced cells by flow cytometry. A close correlation between BFP expression and CAR expression was observed by staining transduced Jurkat cells with recombinant soluble FLT3 (sFLT3) protein, indicating proper folding and cell surface expression of all CARs (Figure S1A).
Primary T cells isolated from three healthy donors were activated and effectively transduced with all constructs (Figure 1A). Ligand-independent CAR signaling has been associated with accelerated T cell differentiation and reduced antitumor efficacy.32,33 To investigate scFv-dependent tonic signaling, the expression of the activation marker 4-1BB (CD137) and the differentiation markers CD45RO and CD62L was measured 6 and 9 days after transduction, respectively. Overall, candidate FLT3 CARs exhibited limited tonic signaling, with less than 15% of CAR+ cells showing upregulation of 4-1BB (Figure 1B). T cells expressing P1A5, P12B6, and P14G2 showed the highest frequencies of 4-1BB+ cells (Figure 1B) and increased differentiation in both the CD4+ and CD8+ subsets (Figure 1C). No FLT3 expression was detected on activated T cells by flow cytometry (Figure S1B), indicating that any activation was not due to FLT3 target recognition. P12B6 and P14G2 CAR T cells exhibited some killing of a FLT3-negative control cell line (MOLP-8) in long-term co-culture assays (Figure S2), suggesting that the increased activation could in part be attributed to non-specific activity.
Figure 1.

Screening of a Library of FLT3-Targeted scFvs and Selection of a High-Performing FLT3 CAR
(A) Activated primary human T cells were effectively transduced with lentiviral vectors encoding a range of FLT3 CARs. Bar graph shows the frequencies of transduced cells determined by flow cytometry analysis 3 days after transduction (mean ± SEM; n = 3 donors). (B) Target-independent “tonic” signaling was evaluated by measuring the frequency of transduced cells positive for the activation marker 4-1BB 6 days after transduction (mean ± SEM; n = 3 donors). (C) Higher tonic signaling observed with some scFvs (P1A5, P12B6, P14G2) was associated with a decrease in the TSCM cell subset indicative of increased CAR T cell differentiation. CAR T cells were analyzed by flow cytometry 9 days after transduction and phenotypes were determined according to CD62L and CD45RO expression within the CAR+ cell population: stem cell memory (CD45RO−/CD62L+), central memory (CD45RO+/CD62L+), effector memory (CD45RO+/CD62L−), and effector (CD45RO−/CD62L−) cells (mean ± SEM; n = 3 donors). (D) CAR T cells exhibited expansion upon repeated exposure to target. The proliferative capacity of CAR T cells was determined by counting viable CAR+ cells 20 days after co-culture with EOL-1 target cells. Fold-expansion is expressed as mean ± SEM relative to day 0; n = 2 donors. (E) Assessment of antitumor activity in an orthotopic model of AML led to effective differentiation of candidate scFvs and selection of the lead FLT3 CAR. NSG mice received a single dose of 2.5 × 106 or 5 × 106 FLT3 CAR T cells 10 days after injection of luciferase-labeled EOL-1 cells and tumor burden was assessed by bioluminescence imaging (BLI) over time (n = 10 mice/group). Values are expressed as mean ± SEM (∗∗∗∗p < 0.0001 by one-way ANOVA with Dunnett’s post hoc test for multiple comparisons versus non-transduced control T cells). Tumor growth kinetics for the P4C7 (2.5 × 106) and P9B5 (2.5 × 106) groups was comparable to control T cells, and their curves are not shown for simplicity.
To attempt to distinguish CARs conferring superior target-specific T cell expansion and effector function, CAR T cells were co-cultured with AML target cell lines expressing varying levels of wild-type and/or mutant FLT3 (Figure S2A), with fresh target cells being subsequently added every 2–3 days to test long-term CAR T cell activity. In these repeated antigen stimulation assays, all candidate CAR T cells displayed proliferation (Figure 1D; Figure S2B) and maintained robust cytotoxic activity (Figure S2C). Four CARs (P4C7, P9B5, P3A1, P3E10) were then selected for in vivo evaluation based on low tonic signaling, a less differentiated T cell phenotype, appropriate target-specific T cell expansion, and cytotoxicity in vitro and which moreover bound to a range of Ig domains of FLT3 (domains 1–4; Table S1) and therefore likely recognized diverse epitopes. The two CARs with specificity to Ig domain 5 of FLT3 (P12B6 and P14G2) were excluded from further analysis, as they had shown higher tonic signaling and non-specific killing in vitro. Mice orthotopically engrafted with luciferase-expressing EOL-1 cells received a single dose of FLT3 CAR T cells, and changes in tumor burden were monitored by bioluminescent imaging (BLI). Whereas T cells expressing the P4C7 or the P9B5 CAR showed no apparent efficacy in this model at the doses tested (Figure 1E), potent antitumor activity was seen for P3A1 and P3E10 at a single dose of 5 × 106 CAR T cells, with the latter also inducing rapid and sustained tumor eradication at a dose of 2.5 × 106 CAR T cells. These in vivo differences in activity were confirmed using CAR T cells derived from a second donor (data not shown). Based on these findings, the P3E10 CAR directed to Ig domain 4 of FLT3 was selected as the lead CAR for further development.
FLT3 CAR T Cells Containing an Intra-CAR Off-Switch Maintain Their Function and Can Be Selectively Depleted with Rituximab
Adoptive cell therapies have the potential to elicit on-target and off-target toxicities to normal tissues, in addition to cytokine release syndrome caused by rapid immune cell activation induced by CAR T cell activity.34 In the case of allogeneic CAR T cell therapies, there is an added risk of graft-versus-host disease (GvHD), although to date only a few cases of mild GvHD have been encountered in clinical trials using allogeneic CD19 CAR T cells.35 In order to have the flexibility to decrease or abrogate FLT3 CAR T cell activity, an inducible system that relies on the administration of a clinically approved antibody was evaluated. Incorporation of short linear mimotopes for the anti-CD20 antibody rituximab in the extracellular portion of the CAR has been shown to render CAR T cells sensitive to complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC), providing a means to selectively ablate CAR T cells on-demand.36 The lead CAR, P3E10, was therefore engineered to contain two rituximab (R) mimotopes (R2 off-switch) between the hinge region and the scFv (Figure 2A). T cells transduced with this construct (hereafter referred to as FLT3 CAR-R2) were efficiently co-stained with rituximab and sFLT3 (Figure 2A), demonstrating proper folding of the scFv and the off-switch. Inclusion of the off-switch did not lead to noticeable changes in transduction efficiency, CD4/CD8 ratio, or CAR T cell phenotype (Figures S3A–S3C). FLT3 CAR-R2 T cells maintained a high proportion of stem cell-like memory T (TSCM) and central memory T (TCM) cells following the expansion phase (Figure S3C) and exhibited specific cytotoxic effector function that was equivalent to cells transduced with the same CAR lacking the off-switch (Figure S3D). In vivo, FLT3 CAR-R2 T cells displayed high antitumor efficacy at a single dose of 2.5 × 106 cells (Figure S3E), although the kinetics of AML clearance relative to FLT3 CAR T cells lacking the off-switch was slightly different. In line with our previous observations, FLT3 CAR-R2 T cells exhibited superior efficacy compared to T cells expressing the P3A1 CAR engineered to incorporate the R2 switch (P3A1-R2; data not shown), suggesting that scFv-related differences in in vivo activity were preserved in this format.
Figure 2.

Effective Depletion of FLT3 CAR T Cells Containing an Intra-CAR Off-Switch Inducible by Rituximab
(A) Left: schematic of the FLT3 CAR-R2 construct containing two rituximab mimotopes (R2) in the extracellular domain. Right: FLT3 CAR-R2 T cells can be effectively detected with rituximab. Flow cytometry analysis of transduced T cells using rituximab and biotinylated soluble FLT3 (sFLT3) demonstrates a distinct double-positive cell population. (B) FLT3 CAR-R2 T cells are depleted via complement-dependent cytotoxicity (CDC) in the presence of rituximab. CAR T cells were cultured in the presence of complement and/or rituximab for 3 h, and residual viable cells were enumerated by flow cytometry analysis of sFLT3-labeled cells mixed with counting beads. Results are the mean ± SEM of triplicate wells for a single experiment, representative of three separate experiments. Differences in viability between treatment groups were compared using the Student’s t test (∗∗∗∗p < 0.0001). (C) FLT3 CAR-R2 T cells are depleted in vivo following rituximab administration. NSG mice were engrafted with luciferase-labeled EOL-1 cells and treated with 2.5 × 106 FLT3 CAR-R2 T cells at day 9, followed by intraperitoneal administration of rituximab or control antibody after tumor clearance (day 21), as determined by BLI analysis. Mice administered rituximab remained tumor-free until the end of the study (day 37). Representative images of mice taken at different time points are shown (n = 4 mice/group). (D) Enumeration of FLT3 CAR-R2 T cells was performed by flow cytometry analysis of peripheral blood from the treated mice at days 3 and 16 after initiation of rituximab treatment (mean ± SEM; ∗∗p < 0.01, ∗∗∗p < 0.001 by unpaired t test).
To test the functionality of the off-switch, FLT3 CAR-R2 T cells were cultured with complement and rituximab for 3 h and their viability was measured by flow cytometry. A significant decrease in CAR T cell viability relative to cells that received complement alone was observed (Figure 2B), demonstrating that rituximab can mediate depletion of FLT3 CAR-R2 T cells via CDC. To validate these results in vivo, mice engrafted with luciferase-expressing EOL-1 cells were treated with FLT3 CAR-R2 T cells and then administered rituximab for 5 consecutive days after AML clearance (Figure 2C). To increase the number of circulating CAR T cells that could be detected by flow cytometry analysis before and after rituximab administration, mice were given intermittent injections of interleukin (IL)-15, which has been shown to increase the persistence of adoptively transferred human CAR T cells in immunocompromised mice.37 Compared with the control group, a marked depletion of FLT3 CAR-R2 T cells was seen in the peripheral blood of mice that received rituximab, as measured at days 3 and 16 after initiation of rituximab treatment (Figure 2D). Notably, CAR T cell depletion did not seem to compromise leukemia remission in this model, indicating effective eradication of EOL-1 cells at the time of rituximab administration. EOL-1 cells stained negative for CD20 (data not shown), ruling out any contribution of rituximab to killing of residual EOL-1 cells and prevention of AML relapse.
Allogeneic FLT3 CAR-R2 T Cells Generated via Targeted Disruption of the TRAC and CD52 Genes Display Robust Antileukemic Activity
Previous studies have demonstrated the feasibility of using TALEN gene-editing technology to produce allogeneic CAR T cells for multiple targets.37,38 CAR T cells lacking expression of the TCRαβ receptor have a decreased risk of inducing GvHD. Disrupting expression of CD52 is intended to increase the window of allogeneic CAR T cell persistence before rejection by the host immune system in patients preconditioned with an anti-CD52 antibody, which can efficiently deplete host lymphocytes while leaving the infused CAR T cells untouched.18,38 TALEN designed to specifically inactivate the TRAC and CD52 genes were used to generate allogeneic FLT3 CAR-R2 T cells. Efficient knockout (KO) of TRAC and CD52 was verified by flow cytometry analysis of the expression of CD3ε and CD52 in TALEN-treated cells before and after magnetic isolation of the TCRαβ− cell population (Figure 3A). As expected, absence of CD52 expression rendered allogeneic FLT3 CAR-R2 T cells resistant to an anti-CD52 antibody, whereas non-gene-edited (TCR/CD52 wild-type [WT]) CAR T cells were rapidly eliminated under the same conditions (Figure 3B). To assess any detrimental effects of TALEN treatment on CAR T cells, allogeneic FLT3 CAR-R2 T cells were tested for their effector function immediately after thawing and 7 days after repeated exposure to antigen. Allogeneic FLT3 CAR-R2 T cells showed cytotoxicity against the AML cell lines EOL-1, MOLM-13, and MV4-11, but negligible cytolytic activity against the FLT3-negative cell line MOLP-8 (Figure S4). There were no differences in killing capacity between allogeneic (TCR/CD52 KO) and non-gene-edited (TCR/CD52 WT) CAR T cells (Figure S4). Furthermore, allogeneic and non-gene-edited FLT3 CAR-R2 T cells displayed similar antitumor activity in vivo (Figure 3C). Next, the antitumor efficacy and proliferative capacity of allogeneic FLT3 CAR-R2 T cells were further investigated in two distinct AML xenograft models. Treatment of mice with low disease burden resulted in complete and durable responses (Figure 3D). In mice with higher disease burden, allogeneic FLT3 CAR-R2 T cells showed dose-dependent antitumor activity, although responses were transient (Figures 3E and 3F). The deeper responses seen in the EOL-1 model relative to MV4-11 were consistent with the higher sensitivity of EOL-1 cells to FLT3 CAR-R2 T cells in vitro (Figure S4). Flow cytometry analysis of peripheral blood revealed dose-dependent peak CAR T cell levels on day 7 and 3 after CAR T cell infusion for EOL-1 and MV4-11, respectively, which corresponded to maximal antileukemic activity (Figures 3E and 3F).
Figure 3.

Allogeneic FLT3 CAR-R2 T Cells Are Resistant to Anti-CD52 Antibody Treatment and Exhibit Dose-Dependent Antitumor Activity and Expansion in Orthotopic Models of Human AML
(A) Generation of allogeneic FLT3 CAR-R2 T cells by targeted inactivation of the TRAC and CD52 genes using TALEN. Flow cytometry analysis shows expression of CD3ε (as a surrogate for the TCRαβ complex) and CD52 in control (TCR/CD52 WT, left plot) and gene-edited FLT3 CAR-R2 T cells before (middle plot) and after (right plot) TCRαβ− cell isolation. (B) Allogeneic FLT3 CAR-R2 T cells are resistant to anti-CD52 antibody-mediated CDC whereas non-gene-edited CAR T cells are efficiently depleted. CAR T cells were incubated for 3 h with complement and anti-CD52 antibody, and cell viability was measured using flow cytometry. Results are expressed as mean ± SEM (n = 3 donors, ∗∗∗∗p < 0.0001 by unpaired t test). (C) Gene-editing does not affect the antitumor activity of FLT3 CAR-R2 T cells. NSG mice were engrafted with luciferase-labeled EOL-1 cells and treated with a single dose of 2.5 × 106 TCR/CD52 KO or TCR/CD52 WT FLT3 CAR-R2 T cells 6 days later. Tumor burden was assessed by bioluminescence imaging (BLI) over time (n = 10 mice/group). Values are expressed as mean ± SEM. (D) Allogeneic FLT3 CAR-R2 T cells induce durable responses in mice with lower tumor burden. NSG mice were engrafted with luciferase-labeled EOL-1 cells and treated with a single dose of 5 × 106 or 1 × 107 TCR/CD52 KO FLT3 CAR-R2 T cells 4 days later. Tumor burden was monitored by BLI over time (n = 10 mice/group). Values are expressed as mean ± SEM. (E and F) Allogeneic FLT3 CAR-R2 T cells exhibit dose-dependent antitumor activity and expansion in two orthotopic models of human AML. NSG mice were inoculated with 5 × 104 luciferase-labeled EOL-1 cells (E) or 5 × 106 luciferase-labeled MV4-11 cells (F) and then treated with the indicated numbers of CAR T cells or control non-transduced T cells 6 days (EOL-1) or 7 days (MV4-11) later. CAR T cells in blood were enumerated using flow cytometry twice a week. Results are expressed as mean ± SEM and are representative of two independent experiments using CAR T cells from two separate healthy donors (n = 10 mice/group; ∗∗p < 0.01, ∗∗∗p < 0.001 by one-way ANOVA with Dunnett’s post hoc test for multiple comparisons versus non-transduced control T cells).
The effector function of allogeneic FLT3 CAR-R2 T cells was further evaluated in assays with primary AML blasts. Flow cytometry analysis of patient-derived peripheral blood mononuclear cells (PBMCs) confirmed cell surface expression of FLT3 in a high percentage of AML cells (60%–90%) in all samples tested (representative plots shown in Figure 4A). No apparent differences in FLT3 surface expression between FLT3 wild-type and FLT3 mutant samples were found (Figures 4A and 4B). Co-culture of allogeneic FLT3 CAR-R2 T cells with AML PBMCs at a 1:1 effector-to-target (E:T) ratio resulted in upregulation of the activation marker CD25 and degranulation in both CD4+ and CD8+ CAR T cell populations (Figure 4C; Figure S5). Moreover, FLT3 CAR-R2 T cells exhibited enhanced tumor necrosis factor (TNF)-α, interleukin (IL)-2, and interferon (IFN)-γ production relative to control non-transduced T cells (Figure 4C; Figure S5). Whereas minimal AML cell killing was observed with control T cells, FLT3 CAR-R2 T cells eliminated more than 80% of AML blasts in 48 h (Figures 4D and 4E).
Figure 4.

Allogeneic FLT3 CAR-R2 T Cells Eliminate Primary AML Blasts In Vitro
(A) Cell surface expression of FLT3 was detected in primary AML samples irrespective of the mutation status of the gene. AML blasts were gated using side scatter (SSC)low and CD45dim characteristics, and residual T cells were used to set negative and positive gates for FLT3. Two representative samples are shown. (B) Percentages of FLT3+ blasts were consistently high in both FLT3WT and FLT3MUT AML samples (mean ± SEM; n = 4/group). (C) Allogeneic FLT3 CAR-R2 T cells displayed markers of effector function in response to exposure to primary AML cells. Representative histograms show increased expression of markers of target-dependent activation (CD25), degranulation (CD107a), and cytokine secretion (TNF-α, IL-2, IFN-γ) in FLT3 CAR-R2 T cells compared to non-transduced control T cells after co-culture with primary AML cells. (D) FLT3 CAR-R2 T cells were cytotoxic to primary AML cells. Effector and target cells were co-cultured at a 1:1 ratio for 48 h, and residual AML cells were identified as SSClowCD45dim and enumerated using flow cytometry. (E) Scatterplot shows the percentage of lysed AML cells for three separate primary AML patient samples (mean ± SEM).
FLT3 CAR T Cell Treatment Results in Hematopoietic Toxicity
FLT3 has a well-characterized role in human hematopoiesis and is thought to be expressed in a subset of early progenitors and HSCs,22 indicating the possibility of on-target off-tumor hematopoietic toxicity. Conflicting results regarding the potential impact of FLT3 CAR T cells on hematopoiesis have nonetheless been reported in preclinical studies.10,12 The prevalence of FLT3 expression was therefore investigated in freshly isolated CD34+ bone marrow mononuclear cells obtained from healthy donors using flow cytometry (Figures S6A and S6B). On average, FLT3 expression was detected in more than 50% of HSCs and nearly half of the multipotent progenitor (MPP) cell population, with less than 20% of common lymphoid progenitors (CLPs) on average staining positive for FLT3 (Figure 5A; Figure S6B). In addition, FLT3 was present in a high proportion of granulocyte-monocyte progenitors (GMPs) and roughly 50% of a cell subset corresponding to common-myeloid progenitors (CMPs) and megakaryocyte-erythrocyte progenitors (MEPs) (Figure 5A). Analysis of the expression of CD123 in additional donors allowed differentiation of the CMP and MEP populations (Figures S6A and S6B). Consistent with this expression pattern, co-culture of CD34+ bone marrow cells with FLT3 CAR-R2 T cells led to a significant reduction in HSCs, MPPs, and GMPs, whereas the CLP population was minimally affected (Figure 5B). Allogeneic T cells expressing a CAR with specificity to CD33, a marker of AML that is broadly expressed on HSPCs,9 were generated following the standard protocol and tested side by side as a comparator. Both FLT3 CAR-R2 and CD33 CAR T cells displayed similar toxicity in this assay, except for the CMP + MEP subset, which was found to be significantly depleted with CD33 CAR T cells but only moderately reduced in the presence of FLT3 CAR-R2 T cells (Figure 5B).
Figure 5.

FLT3 CAR T Cells Exhibit On-Target Off-Tumor Activity against Hematopoietic Stem and Progenitor Cells In Vitro and In Vivo
(A) Distribution of FLT3 expression in adult hematopoietic stem and progenitor cells (HSPCs). CD34+ bone marrow mononuclear cells were isolated from six healthy donors and analyzed for the expression of FLT3 by flow cytometry. The percentage of FLT3+ cells was measured within the indicated HSPC subpopulations. HSC, hematopoietic stem cell; MPP, multipotent progenitor; CLP, common lymphoid progenitor; GMP, granulocyte-monocyte progenitor; CMP, common myeloid progenitor; MEP, megakaryocyte-erythroid progenitor. (B) FLT3 CAR-R2 T cells showed differential toxicity to progenitor subsets in vitro. CD34+ bone marrow mononuclear cells were co-cultured with FLT3 CAR-R2 T cells at 1:1 (E:T) for 24 h, and surviving HSPCs were identified by flow cytometry analysis and enumerated using counting beads. T cells expressing a CAR directed against CD33 and non-transduced T cells were used as positive and negative control, respectively. Results are shown as mean ± SEM of five separate bone marrow donors (∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by unpaired t test). (C) Study design for assessing hematopoietic toxicity in mice infused with cross-reactive FLT3 CAR T cells. NSG mice were injected with luciferase-labeled EOL-1 cells and randomized based on tumor burden. Animals received a single dose of 3 × 106 T cells expressing either an anti-mouse/human-FLT3-reactive CAR (P7H3) or an anti-human-FLT3 CAR (P3E10) 7 days after EOL-1 cell injection and tumor burden was monitored by BLI. Where indicated, 500 ng of IL-15 was administered intraperitoneally. (D) The antitumor efficacy of mouse cross-reactive FLT3 CAR T cells in vivo was dependent on frequent administration of IL-15, whereas T cells expressing the P3E10 CAR did not require IL-15 to boost efficacy in the same model. BLI values are expressed as mean ± SEM (∗∗p < 0.01, ∗∗∗p < 0.001 by one-way ANOVA with Dunnett’s post hoc test for multiple comparisons versus non-transduced control T cells; n = 10 mice/group). (E) Mouse cross-reactive FLT3 CAR T cells exhibited on-target off-tumor effects in the bone marrow only in the presence of IL-15. Percentages of bone marrow HSPCs were measured by flow cytometry analysis at days 13 and 26 after CAR T cell infusion. HSPCs were defined as the Lin−/Sca-1+/cKit+ (LSK) cell population. Results are expressed as percentage of HSPCs from total live bone marrow cells (mean ± SEM; n = 5 mice/group; ∗∗p < 0.01, ∗∗∗∗p < 0.0001 by one-way ANOVA with Dunnett’s post hoc test for multiple comparisons). (F) Decreased cellularity was observed in hematoxylin and eosin-stained sections of femur taken from IL-15-supplemented mice that received cross-reactive FLT3 CAR T cells. No apparent microscopic lesions were seen in mice treated with human FLT3-specific CAR T cells (day 33). Immunohistochemical analysis of human CD3+ T cells in brain and cerebellum tissue sections from the same mice revealed minimal T cell infiltration in both groups and no apparent tissue damage.
To investigate on-target off-tumor effects of FLT3 CAR T cells in vivo, a surrogate FLT3 CAR containing an scFv with affinity to murine FLT3 (clone P7H3; Table S1) was utilized. Preliminary studies had shown suboptimal in vivo efficacy of P7H3 CAR T cells relative to P3E10, and therefore supplementation of mice with exogenous human IL-15 was used to augment CAR T cell persistence and antitumor activity, as described recently.37 Tumor-bearing mice received a single dose of either control non-cross-reactive FLT3 CAR T cells (anti-human FLT3 CAR) or cross-reactive FLT3 CAR T cells (anti-mouse/human FLT3 CAR) followed by periodic injections of either IL-15 or vehicle (Figure 5C). Hematopoietic toxicity was assessed by measuring the lineage−Sca-1+cKit+ (LSK) population (Figure S6C), representative of HSPCs, in bone marrow samples taken at days 13 and 26 after CAR T cell infusion. Whereas both CAR T cells achieved AML eradication in the presence of IL-15 (Figure 5D), only mice treated with cross-reactive FLT3 CAR T cells showed signs of hematopoietic toxicity, evidenced by a significant reduction of HSPCs relative to the control group (Figure 5E). The partial antitumor response observed with cross-reactive FLT3 CAR T cells in the absence of cytokine support was not accompanied by a reduction of HSPCs, indicating a correlation between the magnitude of on-tumor activity and the off-tumor effects. The lack of apparent toxicity seen in mice receiving control non-cross-reactive CAR T cells (with or without IL-15) suggests that the off-tumor effects of anti-murine FLT3 CAR T cells can be attributed to direct killing of normal hematopoietic progenitors expressing the target antigen rather than to a secondary “bystander” effect associated with killing of EOL-1 cells in the bone marrow. No evidence of target-dependent CAR T cell infiltration or activity in other tissues that express FLT3, including brain and cerebellum, was found (Figure 5F). Collectively, these findings indicate that off-tumor effects of cross-reactive FLT3 CAR T cells in mice are restricted to the hematopoietic lineage and correlate with antitumor activity.
Rituximab-Mediated Depletion of FLT3 CAR-R2 T Cells after AML Eradication Facilitates Bone Marrow Recovery in Mice
The presence of FLT3 in a subset of human HSCs and myeloid progenitors and the susceptibility of human and mouse HSPCs to FLT3 CAR T cells observed in the in vitro assays and our in vivo model indicate risk of hematopoietic toxicity associated with uncontrolled FLT3 CAR T cell expansion and reactivity. Therefore, the feasibility of rituximab-mediated depletion of FLT3 CAR-R2 T cells to facilitate bone marrow repopulation from residual HSPCs in the absence of leukemia remission was investigated (Figure 6A). To this end, the cross-reactive FLT3 CAR was engineered to contain the R2 off-switch inducible by rituximab. As expected, tumor-bearing mice receiving anti-mouse/human FLT3 CAR-R2 T cells and periodic injections of IL-15 showed complete tumor elimination (Figure 6B) and a concomitant reduction in bone marrow HSPCs (Figure 6C). Mice were then administered rituximab or a control antibody for 4 consecutive days. Partial depletion of CAR T cells in the rituximab group was confirmed by flow cytometry analysis of peripheral blood (Figure 6D), indicating that rituximab-mediated elimination was not as efficient for this surrogate CAR as it was for the lead candidate. Despite the lower efficiency of CAR T cell clearance, four out of five mice that had received rituximab showed hematopoietic progenitor cell repopulation to levels seen in naive, untreated mice (Figure 6E). In this orthotopic model of AML with exogenous cytokine support, CAR T cell depletion did not result in AML recurrence at later time points (Figure 6B), indicative of effective elimination of EOL-1 cells prior to rituximab administration. These findings were confirmed with a different anti-murine FLT3 CAR (data not shown). These results demonstrate that depletion of FLT3 CAR-R2 T cells after leukemia eradication in mice can expedite bone marrow recovery from residual HSPCs.
Figure 6.

Rituximab Administration after AML Clearance Results in Depletion of Mouse Cross-Reactive FLT3 CAR-R2 T Cells and Expedited Bone Marrow Recovery
(A) Study design. NSG mice were injected with luciferase-labeled EOL-1 cells and randomized based on tumor burden. Animals received a single dose of 3 × 106 T cells expressing either an anti-mouse/human-FLT3 CAR modified to contain the off-switch or the non-cross-reactive FLT3 CAR-R2 6 days after EOL-1 cell injection, and tumor burden was assessed by BLI. Mice received periodic injections of IL-15 and, where indicated, rituximab was administered once daily. (B) Complete clearance of AML was seen in all groups. BLI values are expressed as mean ± SEM. (C) The percentage of HSPCs was significantly reduced in mice treated with cross-reactive FLT3 CAR-R2 T cells compared to mice treated with control CAR-R2 T cells, as measured by flow cytometry analysis before rituximab administration (day 17). Results are expressed as percentage of HSPCs from total live bone marrow cells (mean ± SEM; n = 3-4 mice/group; ∗∗∗p < 0.001 by unpaired t test). (D) Rituximab administration led to a reduction in circulating CAR-R2 T cells, as measured by flow cytometry analysis of peripheral blood cells. Results are expressed as mean ± SEM (n = 10 mice/group; ∗p < 0.05 by unpaired t test). (E) Recovery of HSPC frequencies to levels seen in naive mice was observed in mice treated with cross-reactive FLT3 CAR-R2 T cells that received rituximab. HSPC frequencies in the group that did not receive rituximab remained low. Results are expressed as percentage of HSPCs from total live bone marrow cells (mean ± SEM; ∗p < 0.05 by unpaired t test; n = 5 mice/group for all groups except naive).
Discussion
Despite extensive research during the last decades, no standard regimen for treating relapsed or refractory AML has emerged,39 and the prognosis of these patients remains extremely poor. Allogeneic (allo-)HSCT is the only potentially curative treatment; however, many patients with persistent disease are considered transplantation ineligible or have an adverse outcome after HSCT compared with patients who achieve MRD-negative status.3 In these settings, investigational therapies such as CAR T cells, which have shown encouraging therapeutic effects in hematologic malignancies, may prove effective in preventing or delaying AML relapse or as a bridging therapy between relapse and transplantation.40
In this study, we developed an allogeneic CAR T cell therapy for the treatment of AML by engineering healthy donor T cells to express a high-performing fully human FLT3 CAR and to eliminate endogenous TCR expression, thereby minimizing the risk of alloreactivity. Allogeneic FLT3 CAR-R2 T cells exhibited target-dependent expansion and potent anti-AML activity in vitro and in vivo. Furthermore, incorporating a rituximab-inducible off-switch in the FLT3 CAR had no detrimental effects on potency and provided a mechanism to deplete CAR T cells after leukemia eradication in mice, limiting hematopoietic toxicity and facilitating bone marrow recovery from residual HSPCs.
Previous studies had relied on murine anti-FLT3 antibodies to derive scFvs for CAR design.10,12 scFv-associated differences in CAR expression and activity, either spontaneous or triggered upon binding of the target antigen, can ultimately affect antitumor efficacy. Likewise, the binding properties of the scFv and the location of its cognate epitope relative to the plasma membrane of the target cell could have profound effects on CAR T cell effector function.41 Therefore, we took a comprehensive approach by screening a library of novel fully human binders targeting all five domains of the extracellular region of FLT3 in the context of a second-generation CAR. Within the limitations of our scFv library and the assays performed, we did not find strong correlations between scFv affinity or cognate epitope location and T cell potency. However, our strategy helped to identify and exclude scFvs with potential target-independent activity and led to the selection of a CAR conveying limited tonic signaling and that was highly effective in vitro and in vivo. Systematic examination of affinity variants of the lead scFv generated through mutagenesis of complementary-determining regions and fine-tuning of the CAR modules42 could be explored if required to further improve CAR activity.
Whereas scFvs displaying affinity to both human and murine FLT3 showed favorable characteristics in vitro, their performance was significantly diminished when tested in orthotopic models of AML. It is possible that on-target off-tumor activity of CAR T cells expressing scFvs that cross-react with the murine ortholog of FLT3 could compromise their antitumor efficacy in vivo. These observations underline the need to consider potential binding of candidate scFvs to mouse tissues, either on-target or off-target, when interpreting the outcome of in vivo efficacy studies. In addition, our findings indicate that it may be possible to overcome the reduced antitumor efficacy secondary to off-tumor activity of CAR T cells by enhancing their persistence and/or function via cytokine supplementation. Such a strategy could be used to improve clinical responses when the off-tumor effects are confined to non-vital tissues, such as B cells, or in the context of CAR T cell-induced myeloablative regimens prior to allo-HSCT.43
In agreement with a previous report,37 we found no noticeable differences in antitumor efficacy between TCR/CD52 KO CAR T cells and non-gene-edited CAR T cells. Maintenance of T cell fitness in gene-edited CAR T cells is paramount, as the intended genetic modifications are required for successful implementation of an allogeneic therapy. Inactivation of the TRAC locus has been shown to abolish alloreactivity in preclinical studies.37,38 Accordingly, clinical trials with allogeneic CAR T cells have reported only mild cases of GvHD that occurred at low incidence.35 Abrogation of CD52 expression provides an opportunity to utilize a CD52-depleting antibody as part of the preparative regimen prior to CAR T cell infusion.38 We found that whereas CD52-expressing T cells were rapidly depleted by an anti-CD52 antibody in vitro, allogeneic FLT3 CAR-R2 T cells were resistant to high concentrations of the same antibody and would likely be spared in this setting, potentially resulting in increased persistence in patients.
Primary and secondary resistance to CAR T cell therapies has been reported in hematologic malignancies and solid tumors.44, 45, 46, 47 In addition to patient-specific and disease-related factors, it is likely that their incidence in AML will be influenced by the relevance of the CAR T targets in leukemic cell proliferation and survival. Although several AML-associated antigens have been exploited for the development of immunotherapies, we reasoned that targeting of FLT3, a tyrosine kinase receptor with prognostic value and an established role in disease progression, may limit the ability of malignant cells to escape therapy by target downregulation. In addition, the low expression of FLT3 in mature myeloid cells21 may reduce the risk of extensive cytokine release reported for other common AML antigens in preclinical studies,20 potentially enhancing the therapeutic profile of FLT3 CAR T cells. We confirmed high cell-surface expression of FLT3 in primary AML cells regardless of the presence or absence of mutations in the intracellular domain of the receptor. In vitro, allogeneic FLT3 CAR-R2 T cells demonstrated polyfunctionality and eliminated patient-derived leukemic cells. Although FLT3 is uniformly expressed in AML, it is currently unknown if and how changes in target density, spontaneous or induced by its ligand, may influence clinical responses to FLT3 CAR T cell therapies. Targeting of a second AML antigen via co-administration of a CAR T cell product with a different specificity or by engineering bicistronic vectors or bispecific (tandem) CARs may overcome potential antigen loss or downregulation.48 An allogeneic, off-the-shelf platform could expedite clinical evaluation of dual-targeting approaches in AML, should they be required to improve patient outcomes.
Prior studies addressing the potential risk of myelosuppression associated with uncontrolled activity of FLT3 CAR T cells had reported conflicting results, possibly due to the use of different experimental models and/or sources of human CD34+ cells.10,12 We investigated FLT3 expression in human HSPCs and found that it was present at higher levels on HSC, MPP, and GMP subsets, which also showed higher sensitivity to FLT3 CAR T cells in vitro. Additional in vivo studies using a surrogate CAR recognizing the mouse FLT3 protein confirmed depletion of murine HSPCs by CAR T cells and revealed an association between antitumor activity and off-tumor effects. Besides the anticipated depletion of bone marrow progenitors, no other signs of toxicity or tissue damage were observed. In particular, even though FLT3 expression has been detected in mouse cerebellum,49 no apparent neurological symptoms or increased infiltrations of CAR T cells in brain tissue relative to the control group were seen. The recent finding that FLT3 protein expression in Purkinje cells in human cerebellum is cytoplasmic24 and the lack of apparent neurotoxicity observed in our studies and in non-human primates administered a FLT3/CD3 bispecific antibody21 suggest that the off-tumor effects of FLT3 CAR T cells may be confined to the hematopoietic tissue.
Although the expression level of FLT3 has been reported to be higher in AML versus normal HSPCs,25 we did not find that HSPCs were less susceptible to FLT3 CAR T cells in in vitro assays. Thus, it is likely that FLT3 CAR T cell expansion and activity in patients responding to the therapy will be accompanied by some degree of hematopoietic toxicity. One way to mitigate off-tumor toxicities is the utilization of suicide switches, and we found that cells expressing the lead FLT3 CAR modified to contain a rituximab-inducible off-switch were sensitive to rituximab treatment in vitro and in vivo. Furthermore, depletion of cross-reactive FLT3 CAR-R2 T cells after AML eradication enabled bone marrow recovery from remaining HSPCs in mice without compromising the antitumor activity. Given the differences in FLT3 expression between mice and humans,50,51 it is unknown whether this approach could be equally successful in a clinical setting. Protracted FLT3 CAR T cell activity in patients could lead to stem cell depletion, increasing the likelihood of myeloid cell aplasia. Termination of CAR T cell activity via rituximab administration could enable bone marrow reconstitution from residual HSCs, albeit with a concomitant increase in the risk of relapse, or allow rescue allo-HSCT, as suggested previously.12,43
In summary, our findings suggest that allogeneic FLT3 CAR T cells represent a novel therapeutic approach for the treatment of AML. The convenience of an off-the-shelf CAR T cell product may result in improved clinical translatability and faster time to treatment, ultimately increasing patient access to these therapies. The presence of an off-switch in the lead CAR offers the possibility to regulate CAR T cell activity to allow AML eradication and then hematological recovery, either as a route to allow patients to qualify for transplant or as a standalone treatment.
Materials and Methods
Construction of Lentiviral Vectors Encoding FLT3 CARs
scFvs were obtained by panning a synthetic human scFv phage-display library against a recombinant protein consisting of the ectodomain of the human FLT3 protein (residues 27–541) fused to a C-terminal histidine tag.21 Anti-FLT3 binders were isolated and reformatted as human IgG antibodies. Nine antibodies that bound to various epitopes of FLT3 with high affinities, as determined by surface plasmon resonance, were chosen for further evaluation (Table S1; Supplemental Materials and Methods). Codon-optimized sequences of selected scFvs were inserted into CAR constructs in orientation VH-linker-VL followed by a CD8α hinge/transmembrane domain fused with 4-1BB and CD3ζ intracellular signaling elements. A sequence encoding BFP was included upstream of the CAR for co-expression via a porcine teschovirus-1 2A (P2A) co-translation peptide to enable detection of transduced cells. In all lentiviral vectors, CAR expression was under regulatory control of the EF1α promoter and the WPRE element.
CAR T Cell Production
T cells were isolated from human PBMCs obtained by density gradient centrifugation (Ficoll Paque, GE Healthcare, Pittsburgh, PA, USA) using a Pan T cell isolation kit (Miltenyi Biotec, Auburn, CA, USA) and cryopreserved. T cells were activated immediately after recovery from cryopreservation in X-VIVO 15 medium (Lonza, Basel, Switzerland) supplemented with 10% fetal calf serum (FCS) (GE Healthcare, Pittsburgh, PA, USA) and 20 ng/mL IL-2 (Miltenyi Biotec, Auburn, CA, USA) using T cell TransAct reagent (Miltenyi Biotec, Auburn, CA, USA), as recommended by the manufacturer. T cells were transduced with 50% (v/v) lentiviral supernatant 2 days after activation and then cultured in X-VIVO 15 medium supplemented with 5% human AB serum (Gemini Bio-Products, West Sacramento, CA, USA). IL-2 was supplemented every 2–3 days. Six days after activation, T cells were transfected with 25 μg/mL per TRAC and CD52 TALEN monomer mRNA (TriLink Biotechnologies, San Diego, CA, USA) using AgilePulse MAX electroporators (BTX, Holliston, MA, USA). Fourteen days after activation, depletion of TCRαβ-positive cells was performed using a TCRαβ cell isolation kit (Miltenyi Biotec, Auburn, CA, USA) and T cells were cryopreserved in 90% FCS/10% DMSO using rate-controlled freezing chambers and stored in a liquid nitrogen vapor phase. All functional assays were performed with cells after recovery from cryopreservation.
Flow Cytometry Analysis
Mouse blood samples were treated twice with red blood cell lysis buffer (BioLegend, San Diego, CA) and washed in phosphate-buffered saline (PBS). Mouse bone marrow mononuclear cells were flushed out from mouse femurs with PBS and filtered through a 70-μm nylon mesh (Thermo Fisher Scientific, Waltham, MA, USA). Cells were stained using standard flow cytometry protocols in brilliant stain buffer (BD Biosciences, San Jose, CA, USA) supplemented with human TruStain Fc blocking reagent (BioLegend, San Diego, CA, USA). A list of the antibodies used is provided in the Supplemental Materials and Methods. Dead cells were detected using Sytox dead cell stains (Thermo Fisher Scientific, Waltham, MA, USA). Where indicated, samples were supplemented with counting beads (Thermo Fisher Scientific, Waltham, MA, USA) prior to analysis using LSRFortessa II (BD Biosciences, San Jose, CA, USA) or CytoFLEX (Beckman Coulter, Brea, CA, USA) flow cytometers.
Short-Term Cytotoxicity Assay
3 × 104 luciferase-expressing target cells were co-cultured with different numbers of effector cells in RPMI 1640 medium supplemented with 10% FCS at defined E:T ratios for 24 h. Target cell survival was assessed by measuring residual luciferase activity using Bright-Glo reagent (Promega, Madison, WI, USA) following the manufacturer’s protocol.
Long-Term Cytotoxicity and Proliferation Assay
To quantify CAR T cell proliferation upon repeated exposure to target, 1 × 106 luciferase-expressing target cells and 2.5 × 105 CAR T cells were co-cultured in RPMI 1640 medium supplemented with 10% FCS in 24-well G-rex plates (Wilson Wolf, Saint Paul, MN, USA). Every 2–3 days, target cell viability was measured by luminescence and 1 × 106 fresh target cells were added to the culture. On day 20, CAR T cells were stained using sFLT3 and quantified by flow cytometry and viable cell count. In some experiments, CAR T cells were collected on day 7 and tested in the short-term cytotoxicity assay.
CDC Assay
CAR T cells were diluted in RPMI 1640 medium supplemented with 10% FCS and incubated for 3 h in the presence or absence of 12.5% baby rabbit complement (Cedarlane, Burlington, ON, Canada) and an anti-CD52 antibody (100 μg/mL) or the anti-CD20 antibody rituximab (100 μg/mL). Following incubation, CAR T cells were stained with sFLT3 and cell viability was analyzed using flow cytometry.
Primary AML Cell Cytotoxicity Assay
De-identified primary human AML samples were obtained from the Fred Hutchinson Cancer Research Center and the University of Washington Leukemia Repository under an institutionally approved protocol. Cytotoxicity assays were conducted by coculturing allogeneic FLT3 CAR T cells and AML cells for 48 h. Residual AML blasts, identified as side scatter (SSC)lowCD45dim, were quantitated by flow cytometry analysis at the end of the culture period. The effector function of CAR T cells was also assessed by flow cytometry analysis of activation (CD25) and degranulation (CD107a) markers and the cytokines IL-2, TNF-α, and IFN-γ. A Cytofix/Cytoperm fixation/permeabilization kit (BD Biosciences, San Jose, CA, USA) was used for intracellular staining of cytokines, as described by the manufacturer.
HSPC Cytotoxicity Assay
Expression of FLT3 on HSPC subsets was determined by flow cytometry analysis of freshly isolated CD34+ bone marrow cells (HemaCare, Northridge, CA, USA) stained for expression of lineage-specific markers and FLT3. For the cytotoxicity assays, 1 × 105 CAR T cells or control cells were co-cultured with the same number of CD34+ HSPCs for 24 h and residual HSPCs were identified and enumerated by flow cytometry analysis using counting beads and normalized to the control group (non-transduced T cells).
Mouse Studies
All animal studies were performed in accordance with regulations and established guidelines and were reviewed and approved by an Institutional Animal Care and Use Committee. 8- to 12-week-old nonobese diabetic (NOD).Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Luciferase-expressing EOL-1 cells (5 × 104) or MV4-11 cells (5 × 106) were intravenously injected into mice, and tumor burden was monitored using an in vivo imaging system (IVIS) Spectrum instrument (PerkinElmer, Boston, MA, USA) twice weekly. Mice were randomized based on total body bioluminescence 4 days (EOL-1, lower disease burden), 6–10 days (EOL-1, higher disease burden), or 7 days (MV4-11) after tumor cell injection, and CAR T cells were infused immediately after thawing. Total T cell numbers were kept constant across all groups by normalizing with non-transduced T cells, unless stated otherwise. Where indicated, animals received intraperitoneal injections of 10 mg/kg rituximab or isotype control antibody (IgG1). In some studies, mice received intraperitoneal injections of human IL-15 (PeproTech, Rocky Hill, NJ, USA; 0.5 μg per mice) and IL-15Rα (R&D Systems, Minneapolis, MN, USA; 3 μg per mice) twice weekly to boost CAR T cell persistence. Mouse blood was collected into EDTA-coated tubes using submandibular bleeds and 50 μL was used for flow cytometry. Mice were euthanized when they exhibited disease model-specific endpoints such as hindleg paralysis, ruffled fur, or 20% of body weight loss. In some experiments, sternums and brains were harvested from euthanized mice and fixed with formalin. The fixed tissues were sectioned and stained with H&E or an anti-human CD3 antibody for CAR T cell identification. Histopathology and CAR T cell infiltration were evaluated by qualified pathologists.
Statistical Analysis
Statistical analysis was performed using the Prism software package (GraphPad, San Diego, CA, USA), and statistical tests used are indicated in the figure legends. p < 0.05 was considered significant.
Author Contributions
C.S., H.-Y.C., D.N., D.D., and J. Sutton, performed experiments. Y.A.Y. created antibodies and protein reagents. C.S., H.-Y.C., D.D., Y.A.Y., M.H., J.V., J. Smith, I.D., J.C.-R, and B.J.S. participated in study design and interpreted the data. C.S. wrote the manuscript with support from H.-Y.C. and B.J.S.
Conflicts of Interest
All authors are current or former employees of Allogene Therapeutics, Inc., Pfizer Inc., Cellectis SA, or Cellectis, Inc.
Acknowledgments
We would like to acknowledge the Fred Hutchinson Cancer Research Center and the University of Washington Leukemia Repository for providing the AML samples utilized in our studies. We thank the protein expression and purification group, the flow cytometry group, and the vivarium staff at Allogene Therapeutics, Inc. and Pfizer Inc., for their support. The FLT3 target is exclusively licensed from Cellectis, and the TALEN gene-editing technology is pioneered and owned by Cellectis.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.06.022.
Contributor Information
Cesar Sommer, Email: cesar.sommer@allogene.com.
Barbra J. Sasu, Email: barbra.sasu@allogene.com.
Supplemental Information
References
- 1.National Institutes of Health Cancer stat facts: leukemia—acute myeloid leukemia (AML) https://seer.cancer.gov/statfacts/html/amyl.html
- 2.Campana D., Leung W. Clinical significance of minimal residual disease in patients with acute leukaemia undergoing haematopoietic stem cell transplantation. Br. J. Haematol. 2013;162:147–161. doi: 10.1111/bjh.12358. [DOI] [PubMed] [Google Scholar]
- 3.Araki D., Wood B.L., Othus M., Radich J.P., Halpern A.B., Zhou Y., Mielcarek M., Estey E.H., Appelbaum F.R., Walter R.B. Allogeneic hematopoietic cell transplantation for acute myeloid leukemia: time to move toward a minimal residual disease-based definition of complete remission? J. Clin. Oncol. 2016;34:329–336. doi: 10.1200/JCO.2015.63.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raje N., Berdeja J., Lin Y., Siegel D., Jagannath S., Madduri D., Liedtke M., Rosenblatt J., Maus M.V., Turka A. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019;380:1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mardiros A., Dos Santos C., McDonald T., Brown C.E., Wang X., Budde L.E., Hoffman L., Aguilar B., Chang W.C., Bretzlaff W. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill S., Tasian S.K., Ruella M., Shestova O., Li Y., Porter D.L., Carroll M., Danet-Desnoyers G., Scholler J., Grupp S.A. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenderian S.S., Ruella M., Shestova O., Klichinsky M., Aikawa V., Morrissette J.J., Scholler J., Song D., Porter D.L., Carroll M. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29:1637–1647. doi: 10.1038/leu.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L., Mao H., Zhang J., Chu J., Devine S., Caligiuri M.A., Yu J. Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia. 2017;31:1830–1834. doi: 10.1038/leu.2017.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tashiro H., Sauer T., Shum T., Parikh K., Mamonkin M., Omer B., Rouce R.H., Lulla P., Rooney C.M., Gottschalk S., Brenner M.K. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to C-type lectin-like molecule 1. Mol. Ther. 2017;25:2202–2213. doi: 10.1016/j.ymthe.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jetani H., Garcia-Cadenas I., Nerreter T., Thomas S., Rydzek J., Meijide J.B., Bonig H., Herr W., Sierra J., Einsele H., Hudecek M. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. 2018;32:1168–1179. doi: 10.1038/s41375-018-0009-0. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y., Xu Y., Li S., Liu J., Xing Y., Xing H., Tian Z., Tang K., Rao Q., Wang M., Wang J. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J. Hematol. Oncol. 2018;11:60. doi: 10.1186/s13045-018-0603-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Dieu R., Taussig D.C., Ramsay A.G., Mitter R., Miraki-Moud F., Fatah R., Lee A.M., Lister T.A., Gribben J.G. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood. 2009;114:3909–3916. doi: 10.1182/blood-2009-02-206946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knaus H.A., Berglund S., Hackl H., Blackford A.L., Zeidner J.F., Montiel-Esparza R., Mukhopadhyay R., Vanura K., Blazar B.R., Karp J.E. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight. 2018;3:120974. doi: 10.1172/jci.insight.120974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Bruggen J.A.C., Martens A.W.J., Fraietta J.A., Hofland T., Tonino S.H., Eldering E., Levin M.D., Siska P.J., Endstra S., Rathmell J.C. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8+ T cells and impede CAR T-cell efficacy. Blood. 2019;134:44–58. doi: 10.1182/blood.2018885863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham C., Jozwik A., Pepper A., Benjamin R. Allogeneic CAR-T cells: more than ease of access? Cells. 2018;7:E155. doi: 10.3390/cells7100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Depil S., Duchateau P., Grupp S.A., Mufti G., Poirot L. “Off-the-shelf” allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 2020;19:185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 19.Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir A.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leong S.R., Sukumaran S., Hristopoulos M., Totpal K., Stainton S., Lu E., Wong A., Tam L., Newman R., Vuillemenot B.R. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood. 2017;129:609–618. doi: 10.1182/blood-2016-08-735365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeung Y.A., Krishnamoorthy V., Dettling D., Sommer C., Poulsen K., Ni I., Pham A., Chen W., Liao-Chan S., Lindquist K. An optimized full-length FLT3/CD3 bispecific antibody demonstrates potent anti-leukemia activity and reversible hematological toxicity. Mol. Ther. 2020;28:889–900. doi: 10.1016/j.ymthe.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kikushige Y., Yoshimoto G., Miyamoto T., Iino T., Mori Y., Iwasaki H., Niiro H., Takenaka K., Nagafuji K., Harada M. Human Flt3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J. Immunol. 2008;180:7358–7367. doi: 10.4049/jimmunol.180.11.7358. [DOI] [PubMed] [Google Scholar]
- 23.Tsapogas P., Mooney C.J., Brown G., Rolink A. The cytokine Flt3-ligand in normal and malignant hematopoiesis. Int. J. Mol. Sci. 2017;18:E1115. doi: 10.3390/ijms18061115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Çakmak-Görür N., Radke J., Rhein S., Schumann E., Willimsky G., Heppner F.L., Blankenstein T., Pezzutto A. Intracellular expression of FLT3 in Purkinje cells: implications for adoptive T-cell therapies. Leukemia. 2019;33:1039–1043. doi: 10.1038/s41375-018-0330-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milne P., Wilhelm-Benartzi C., Grunwald M.R., Bigley V., Dillon R., Freeman S.D., Gallagher K., Publicover A., Pagan S., Marr H. Serum Flt3 ligand is a biomarker of progenitor cell mass and prognosis in acute myeloid leukemia. Blood Adv. 2019;3:3052–3061. doi: 10.1182/bloodadvances.2019000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmied B.J., Lutz M.S., Riegg F., Zekri L., Heitmann J.S., Bühring H.J., Jung G., Salih H.R. Induction of NK cell reactivity against B-cell acute lymphoblastic leukemia by an Fc-optimized FLT3 antibody. Cancers (Basel) 2019;11:E1966. doi: 10.3390/cancers11121966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A., Hoadley K., Triche T.J., Jr., Laird P.W., Baty J.D., Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papaemmanuil E., Gerstung M., Bullinger L., Gaidzik V.I., Paschka P., Roberts N.D., Potter N.E., Heuser M., Thol F., Bolli N. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown P., Levis M., Shurtleff S., Campana D., Downing J., Small D. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood. 2005;105:812–820. doi: 10.1182/blood-2004-06-2498. [DOI] [PubMed] [Google Scholar]
- 30.Staudt D., Murray H.C., McLachlan T., Alvaro F., Enjeti A.K., Verrills N.M., Dun M.D. Targeting oncogenic signaling in mutant FLT3 acute myeloid leukemia: the path to least resistance. Int. J. Mol. Sci. 2018;19:3198. doi: 10.3390/ijms19103198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verstraete K., Vandriessche G., Januar M., Elegheert J., Shkumatov A.V., Desfosses A., Van Craenenbroeck K., Svergun D.I., Gutsche I., Vergauwen B., Savvides S.N. Structural insights into the extracellular assembly of the hematopoietic Flt3 signaling complex. Blood. 2011;118:60–68. doi: 10.1182/blood-2011-01-329532. [DOI] [PubMed] [Google Scholar]
- 32.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomes-Silva D., Mukherjee M., Srinivasan M., Krenciute G., Dakhova O., Zheng Y., Cabral J.M.S., Rooney C.M., Orange J.S., Brenner M.K., Mamonkin M. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Rep. 2017;21:17–26. doi: 10.1016/j.celrep.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonifant C.L., Jackson H.J., Brentjens R.J., Curran K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain N., Graham C., Konopleva M., Yallop D., Jozwik A., Patten P., Ellard R., Stewart O., Cuthill K., Potter V. 2018. UCART19, an allogeneic anti-CD19 CAR T-cell product, in high risk adult patients with CD19+ relapsed/refractory B-cell acute lymphoblastic leukemia: preliminary results of phase I CALM study.https://library.ehaweb.org/eha/2018/stockholm/214674/reuben.benjamin.ucart19.an.allogeneic.anti-cd19.car.t-cell.product.in.high.html?f=media=3∗c_id=214674∗listing=3∗browseby=8 [Google Scholar]
- 36.Valton J., Guyot V., Boldajipour B., Sommer C., Pertel T., Juillerat A., Duclert A., Sasu B.J., Duchateau P., Poirot L. A versatile safeguard for chimeric antigen receptor T-cell immunotherapies. Sci. Rep. 2018;8:8972. doi: 10.1038/s41598-018-27264-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sommer C., Boldajipour B., Kuo T.C., Bentley T., Sutton J., Chen A., Geng T., Dong H., Galetto R., Valton J. Preclinical evaluation of allogeneic CAR T cells targeting BCMA for the treatment of multiple myeloma. Mol. Ther. 2019;27:1126–1138. doi: 10.1016/j.ymthe.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poirot L., Philip B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 39.Döhner H., Estey E., Grimwade D., Amadori S., Appelbaum F.R., Büchner T., Dombret H., Ebert B.L., Fenaux P., Larson R.A. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forman S.J., Rowe J.M. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121:1077–1082. doi: 10.1182/blood-2012-08-234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.James S.E., Greenberg P.D., Jensen M.C., Lin Y., Wang J., Till B.G., Raubitschek A.A., Forman S.J., Press O.W. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol. 2008;180:7028–7038. doi: 10.4049/jimmunol.180.10.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feucht J., Sun J., Eyquem J., Ho Y.J., Zhao Z., Leibold J., Dobrin A., Cabriolu A., Hamieh M., Sadelain M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019;25:82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tasian S.K., Kenderian S.S., Shen F., Ruella M., Shestova O., Kozlowski M., Li Y., Schrank-Hacker A., Morrissette J.J.D., Carroll M. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood. 2017;129:2395–2407. doi: 10.1182/blood-2016-08-736041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown C.E., Badie B., Barish M.E., Weng L., Ostberg J.R., Chang W.C., Naranjo A., Starr R., Wagner J., Wright C. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015;21:4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orlando E.J., Han X., Tribouley C., Wood P.A., Leary R.J., Riester M., Levine J.E., Qayed M., Grupp S.A., Boyer M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018;24:1504–1506. doi: 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- 47.Shah N.N., Fry T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrov J.C., Wada M., Pinz K.G., Yan L.E., Chen K.H., Shuai X., Liu H., Chen X., Leung L.H., Salman H. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia. 2018;32:1317–1326. doi: 10.1038/s41375-018-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.deLapeyrière O., Naquet P., Planche J., Marchetto S., Rottapel R., Gambarelli D., Rosnet O., Birnbaum D. Expression of Flt3 tyrosine kinase receptor gene in mouse hematopoietic and nervous tissues. Differentiation. 1995;58:351–359. doi: 10.1046/j.1432-0436.1995.5850351.x. [DOI] [PubMed] [Google Scholar]
- 50.Pietras E.M., Reynaud D., Kang Y.A., Carlin D., Calero-Nieto F.J., Leavitt A.D., Stuart J.M., Göttgens B., Passegué E. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell. 2015;17:35–46. doi: 10.1016/j.stem.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mooney C.J., Cunningham A., Tsapogas P., Toellner K.M., Brown G. Selective expression of Flt3 within the mouse hematopoietic stem cell compartment. Int. J. Mol. Sci. 2017;18:1037. doi: 10.3390/ijms18051037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.