

Abstract
Chimeric antigen receptor T cell therapy (CAR-T) has been rolled out as a new treatment for hematological malignancies. For solid tumor treatment, CAR-T has been disappointing so far. Challenges include the quantification of CAR-T trafficking, expansion and retention in tumors, activity at target sites, toxicities, and long-term CAR-T survival. Non-invasive serial in vivo imaging of CAR-T using reporter genes can address several of these challenges. For clinical use, a non-immunogenic reporter that is detectable with exquisite sensitivity by positron emission tomography (PET) using a clinically available non-toxic radiotracer would be beneficial. Here, we employed the human sodium iodide symporter to non-invasively quantify tumor retention of pan-ErbB family targeted CAR-T by PET. We generated and characterized traceable CAR T cells and examined potential negative effects of radionuclide reporter use. We applied our platform to two different triple-negative breast cancer (TNBC) models and unexpectedly observed pronounced differences in CAR-T tumor retention by PET/CT (computed tomography) and confirmed data ex vivo. CAR-T tumor retention inversely correlated with immune checkpoint expression in the TNBC models. Our platform enables highly sensitive non-invasive PET tracking of CAR-T thereby addressing a fundamental unmet need in CAR-T development and offering to provide missing information needed for future clinical CAR-T imaging.
Keywords: cell tracking, chimeric antigen receptor, CAR, immunotherapy, PET imaging, reporter gene, tumour retention, immune checkpoint, triple-negative breast cancer
Graphical Abstract
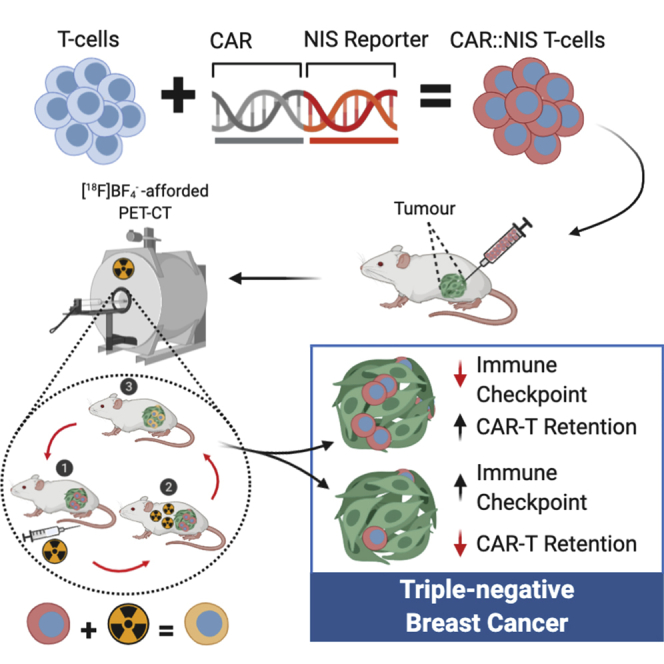
CAR-T immunotherapy remains challenging in solid tumors, not least as quantitation of CAR-T in vivo distribution, activity, and fate remains elusive. Fruhwirth and colleagues developed and validated a platform for non-invasive in vivo CAR-T tracking by highly sensitive PET imaging and demonstrated its application in triple-negative breast cancer models. Beyond supporting preclinical CAR-T development, they provide a clinically compatible CAR-T monitoring tool.
Introduction
Chimeric antigen receptor (CAR) T cell therapy (CAR-T) relies on the genetic modification of a patient’s T cells to express artificial plasma membrane receptors with specificities against the patient’s cancer. This immunotherapy approach has resulted in unprecedented clinical outcomes in certain hematological cancers and is now in routine clinical use.1, 2, 3, 4 However, in solid tumors CAR-T has been disappointing due to barriers in CAR-T tumor infiltration, as well as the impairment of CAR T cell efficacy by metabolic stresses, immune checkpoint molecules, and/or cytokines rendering them apoptotic, exhausted, or anergic.5 Notably, CAR T cell longevity, distribution, and potential re-distribution, as well as their overall survival across tumor sites and other tissues, are critical parameters that need careful evaluation before these therapeutics can reach their full potential in solid tumors. For example, on-target off-site effects have led to severe adverse events6,7 and been associated with life-threatening side-effects and fatalities in clinical trials.8,9 Nevertheless, clinical trials are still largely performed without knowledge about the in vivo distribution and fate of the administered CAR T cells, making it is impossible to adequately assess their in vivo distribution, safety, and fate.
Importantly, non-invasive cell tracking using sensitive whole-body in vivo imaging has great potential to aid CAR-T development and clinical application by elucidating several of the above aspects.10, 11, 12 Multi-modality imaging combining radionuclide technologies such as single-photon computed tomography (SPECT) or positron emission tomography (PET) with anatomical imaging by either computed tomography (CT) or magnetic resonance imaging (MRI) has great potential in this context. Current clinical instrumentation for PET is superior to SPECT in both sensitivity and resolution.13,14 Cells need to be rendered in vivo traceable over days/weeks. Therefore, genetic engineering to ectopically express a reporter gene that enables contrast formation in vivo upon administration of a matching contrast agent holds great promise (e.g., a reporter protein enabling the uptake or the binding of a contrast agent).12,15 Radionuclide reporter genes suitable for immune cell imaging must be non-immunogenic while providing excellent contrast and be endogenously expressed only in a very limited number of tissues and ideally at low levels to achieve good contrast.11 Few proteins fulfil these criteria with a prostate-specific membrane antigen variant (tPSMAN9del)16 and the human sodium iodide symporter (NIS)17 being the most promising, both of which have previously been used before for tracking cytolytic T cells. Both can be matched with clinically used/trialed PET contrast agents, although T cell tracking by PET has not yet been demonstrated for NIS.
Several CAR-T with diverse targets have been trialed for the treatment of solid tumors,18 but none has been approved for clinical use yet. Among these targets are the epidermal growth-factor receptors (ErbB family), which can be pharmacologically targeted (e.g., small molecules, antibodies) but this has frequently resulted in resistance and subsequent therapeutic failure. Consequently, a broadly applicable CAR-based immunotherapy was developed, the T1E28z CAR, in which the propensity of solid tumors to dysregulate the ErbB receptor family network was targeted.19 The T1E28z CAR is already trialed clinically in head and neck cancer (NCT01818323), although applied intratumorally rather than systemically due to potential safety concerns. This immunotherapy could also be applicable for difficult-to-treat triple-negative breast cancers (TNBC)20 or mesothelioma,21 provided in vivo distribution and fate of this immunotherapy are better understood.
Here, we developed a platform enabling highly sensitive non-invasive in vivo PET tracking of CAR-T using NIS. We applied it to the T1E28z CAR to determine CAR-T retention in TNBC xenograft models. Our aims were to provide a tool and demonstrate its application to facilitate preclinical development of new CARs and to support with this clinically compatible tool future clinical CAR-T imaging.
Results
Generation of Reporter Gene-Expressing Pan-ErbB Family-Targeted CAR T Cells
We built a platform for co-expression of a CAR and a radionuclide-fluorescence reporter gene, wherein the radionuclide reporter enables non-invasive in vivo cell tracking and the fluorescence reporter facilitates CAR T cell generation. As a CAR, we used the locally developed multi-specific pan-ErbB T1E28z CAR, which targets most homo- and heterodimers of ErbB family members.19 Additionally, we included the previously reported interleukin-4Rα (IL-4Rα):IL-2/15Rβ chimera (4αβ) intended for selective expansion of transduced CAR T cells in the presence of IL-4.22 This was previously combined with the T1E28z CAR using 2A technology,23 demonstrating that the resultant short N-terminal Pro-Met addition impaired neither CAR trafficking to the plasma membrane nor CAR function. To additionally include the radionuclide-fluorescence fusion reporter human NIS-monomeric TagRFP (NIS-RFP), which was previously characterized,24 we also aimed to utilize a 2A cleavage peptide. Therefore, we first validated the extent to which NIS-RFP would tolerate N-terminal modifications and found that the minimal possible modification, Pro-Met, did not impair NIS function (Figure S1). We then generated lentiviral vectors for the transfer of expression cassettes including 4αβ and the T1E28z CAR with or without the NIS-RFP reporter (T4NT and T4, respectively; Figure 1A). As an additional control, we generated a non-functional CAR lacking the CD3ζ domain and implemented it within the platform at the expense of T1E28z (T4ΔNT; Figure 1A). Lentiviruses were produced from these constructs and used to transduce purified human T cells, which were subsequently cultured in the presence of IL-4 to selectively expand CAR T cells (exploiting 4αβ) as previously reported.22 CD4+/CD8+ ratios depended on human donors (typical example of CAR T cells: Figures 1C and 1D; all batches Figure S2). The expansion characteristics of T4NT and T4ΔNT CAR T cells in the presence of IL-4 was comparable to the proliferation of untransduced T cells in the presence of IL-2, demonstrating no major impact on growth after transduction (Figure S3).
Figure 1.

Generation of Reporter Expressing pan-ErbB Family CAR T Cells
(A) Scheme of the construct platform developed for combined gene transfer of CAR, reporter, and auxiliary genes. Expression is driven from the spleen focus-forming virus promoter (PSFFV). Selective expansion of clones is enabled by 4αβ. The functional CAR is T1E28z while the truncated CAR T1E28Δz is functionally deficient. The radionuclide reporter NIS is fused to the fluorescent protein mTagRFP (TRFP) in these preclinical cassettes; for clinical applications, the fluorescent protein can be omitted. Proteins are separated by self-cleaving peptide sequences (T2A), whereby trimming of the 4αβ to remove T2A remnants was enabled via an additional furin site. (B) Exposure with corresponding viruses resulted in transduction of primary human T cells as assessed by flow cytometry. Selection of positively transduced clones using IL-4 permitted significant enrichment of CAR-T cells without the need of FACS sorting. (C and D) Phenotypes of transduced T4NT CAR T cells after selection demonstrating purity of T cell features (CD3) and the ratio of CD4+ and CD8+ T cells. CD4+:CD8+ dependent on donors (see Figure S2 for examples). Typical data are shown in (B)–(D).
In Vitro Characterization of Pan-ErbB Family-Targeted CAR T Cells
For correct function of both NIS-RFP reporter and CAR, it was important that these proteins were localized at the cellular plasma membrane. Using confocal microscopy, we found for all CARs and the NIS-RFP reporter the expected cellular localization patterns on T cell plasma membranes as indicated by co-localization with the general plasma membrane marker wheat germ agglutinin (WGA; Figure 2A).
Figure 2.

In Vitro Characterization of Traceable pan-ErbB Family CAR T Cells
(A) Confocal microscopy demonstrated correct plasma membrane localization of the CAR and the NIS-RFP fusion reporter through co-localization with the plasma membrane marker WGA (conjugated to the fluorophore AlexaFluor488). Typical micrographs are shown; scale bars is 10 μm, image dimensions are identical for all images. (B) NIS-RFP reporter function in T4NT CAR T cells was validated by uptake of either the PET radiotracer [18F]BF4− or the SPECT radiotracer - [99mTc]TcO4−. Uptake was comparable with the two different NIS radiotracers and was 0.13 ± 0.01 and 0.13 ± 0.03, for [18F]BF4− and [99mTc]TcO4− respectively, compared to larger cancer cells overexpressing NIS-RFP,24 which served as experimental reference. CAR T cells lacking the reporter (T4) did not show any radiotracer uptake. Error bars are SD; n = 3 different CAR T cells batches generated from different donors. (C and D) Validation of CAR function by BCC monolayer killing assays. (C) BCC cell viability was assessed after incubation with indicated CAR T cells or T cells alone, as indicated. A negative control (black) was obtained by not adding immune cells. Addition of the reporter NIS-RFP did not impact CAR function (no difference between red and yellow) while dysfunctional CAR (red-brown) and untransduced T cells (gray) did not mediate tumor cell killing. Error bars are SD; n ≥ 3 different CAR T cell batches each. (D) Cell death was determined in the presence of different amounts of either T4NT or untransduced T cells from the same batches (1:1 or 10:1) at the indicated time points. Error bars are SD; n = 3 independent experiments. Significant differences compared to untransduced T cell experiments at same ratios indicated as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Furthermore, uptake of either the SPECT radiotracer [99mTc]TcO4– or the PET radiotracer [18F]BF4– revealed correct reporter gene function in T4NT CAR T cells (Figure 2B), a pre-requisite for in vivo tracking of these cells. Notably, [99mTc]TcO4– and [18F]BF4– displayed similar relative uptake values compared to a reference cancer cell line expressing high reporter levels (Figure 2B;24). Repeated radiotracer uptake in the same T4NT cells also resulted in comparable cellular radioactivity levels (Figure S4). The latter demonstrated that even with the longer half-life radiotracer ([99mTc]TcO4– [t1/2(99mTc) = 6.01 h versus t1/2(18F) = 110 min]) repeat measurements 24 h apart were feasible without radiotracer from the first uptake affecting uptake measurements of a second uptake experiment. Control CAR T cells lacking the radionuclide reporter (T4) did not take up the radiotracers, thereby demonstrating specificity of the assay (Figure 2B). These results provided a rationale for the interchangeable use of both radiotracers for the in vitro NIS functional characterization.
Next, we determined whether reporter expression affected the function of the pan-ErbB family-targeted CAR. Therefore, we determined the cytotoxicity of T4NT CAR T cells and various control T cells toward different human breast cancer cells (BCCs). We used BCC lines that expressed different combinations of ErbB family proteins including MDA-MB-231, MDA-MB-436 (both triple-negative, i.e., HER2–/ER–/PR–), MCF-7, and HCC1954 cells (for relative ErbB family receptor status, see Figure S5). Cell killing of BCC monolayers was assessed after addition of either T4NT (functional CAR), T4ΔNT (non-functional CAR), T4 (functional CAR but no reporter), or untransduced T cells (no CAR and no reporter). Results confirmed CAR-mediated BCC killing in the presence of functional CARs (Figure 2C) and that the reporter NIS-RFP did not negatively impact on CAR function (Figure 2C; no significant differences between yellow and red bars). While in Figure 2C the quantified parameter was BCC survival, we complemented this data with direct measurements of cell death, which also showed T4NT-mediated BCC killing in a dose-dependent manner (Figure 2D).
In summary, these data demonstrated correct function of both CAR and reporter and that the reporter did not impact on CAR function or T cell proliferation.
Effects of Radiotracer Uptake and Decay on CAR T Cells
Importantly, the uptake of radiotracers has the potential to exert radiation-induced DNA damage and thereby negatively impact on the cells taking them up. We first investigated whether CAR T cell viability was affected by radiotracer uptake. Therefore, T4NT CAR T cells were exposed to either [99mTc]TcO4– or [18F]BF4– and cell viability was quantified five days after radiotracer uptake, at a time point when all radioactivity had decayed, and the cells had received the full dose. We did not find any significant impact on T4NT CAR T cell viability upon uptake of either radiotracer (Figure 3A).
Figure 3.

Quantification of Radiotracer Impact on In Vivo Traceable T4NT CAR T Cells
(A) CAR T cell viability as assessed by Alamar Blue fluorescence. Cells were radiolabeled with the indicated NIS radiotracers; error bars are SD; n = 3 different experiments. (B) T4NT CAR T cell function was determined in cancer cell monolayer killing assays with indicated breast cancer cell lines (over 72 h post T4NT radiolabeling). T4NT CAR T cells were either not radiolabeled (white) or radiolabeled with the indicated NIS radiotracers; untransduced T cells served as negative control. (C) T4NT CAR T cell function as quantified by interferon-γ release (by ELISA 72 h after addition of radiolabeled T4NT to monolayers); T4NT treated as in (B). For both (B) and (C), error bars represent SD from n = 3 different T4NT batches. (D, left) DNA damage in radiolabeled T4NT CAR T cells. T4NT were radiolabeled with [99mTc]TcO4− resulting in the listed amounts of cellular radioactivity (shades of blue). Unlabelled cells served as controls (white). Cells were fixed at indicated time points after radiotracer administration and stained for double strand breaks (detected as γH2AX foci), which were counted per cell. (D, right) Untransduced T cells (striped) were exposed to identical settings and conditions as T4NT in (D, left). (E) Analogous experiment as in (D) but with [18F]BF4−. For (D) and (E), error bars are SD; n = 3 with a total >35 cells each. Significant differences (p < 0.05) between highest dose at 2 h and ($) lower doses at the same time point, (#) same dose at later time points (24 h, 72 h), and (&) all samples with untransduced T cells.
Furthermore, we quantified whether CAR T cells were affected in their BCC killing function by radiolabeling. We subjected the same BCC lines as in Figure 2B to T4NT CAR T cells that had been incubated with either [99mTc]TcO4−, [18F]BF4−, or vehicle, and found no differences in their tumor cell killing capacity (Figure 3B). Active CAR T cells also release interferon-γ into the cell culture medium upon tumor cell killing. Notably, we did not find any significant differences between CAR T cells that were exposed to radiotracers or vehicle (Figure 3C).
Next, we determined the level of radiation-induced DNA damage to CAR T cells. Therefore, we exposed T4NT CAR T cells to the longer half-life radiotracer [99mTc]TcO4− or vehicle, and stained cell aliquots for the double-strand break (DSB) marker γH2AX. At the highest administered radiotracer level, which resulted in 130 mBq [99mTc]TcO4– per cell after uptake, we found a significantly increased number of DSB foci per cell 2 h after radiotracer administration compared to cells that had received no radioactivity (Figure 3D/left). At lower administered radioactivity levels resulting in 51 mBq [99 mTc]TcO4− per cell, a similar trend was observed; when we analyzed the top 15% reporter-expressing T4NT CAR T cells from the population separately (as determined by fluorescence microscopy during the assay), differences were significant to vehicle-treated T4NT cells, indicating dose dependency (Figure S6A). At all lower radioactivity levels, there was no change compared to vehicle-treated cells. Importantly, the observed radiation-induced DSBs were repaired in the cells over time, resulting in no changes between DSB foci per cell after 24 h or 72 h after radiotracer administration (Figure 3D, left). Notably, untransduced T cells from the same batch, which lacked the reporter NIS-RFP, did not take up radioactivity and also did not show any increase in DSB foci, even when exposed to the highest level of radioactivity (Figure 3D, right). Moreover, we performed analogous experiments with the shorter half-life PET radiotracer [18F]BF4– for which we obtained similar results (Figure 3E).
These data demonstrated that the consequences of NIS use, i.e. cellular uptake of both NIS imaging radiotracers ([99mTc]TcO4− and [18F]BF4−) and the dose received by cells due to their complete decay, did not negatively impact on CAR T cell function or viability. In summary, this validated the approach and provided the basis for subsequent in vivo imaging.
Non-invasive Determination of CAR T Cell Retention in TNBC Models
First, we established detectability of traceable T4NT CAR T cells on our PET instrumentation. Therefore, we admixed different amounts of T4NT CAR T cells to untransduced T cells, subjected each cell mixture to an uptake assay with the PET radiotracer [18F]BF4−, and quantified radioactivity signals on the PET scanner. Detection sensitivity was determined to be just above 3,000 cells (Figure 4).
Figure 4.

Detection Sensitivity of T4NT CAR T Cells
(Top) PET images of indicated CAR T cell numbers acquired 15 minutes after radiolabeling with [18F]BF4− (see Materials and Methods). Typical images are shown from one T4NT batch. (Bottom) Cumulative analysis of obtained imaging data from three T4NT batches (error bars are SD). Dashed line indicates background signals, with gray shade indicating background mean ± three times SD. The limit of detection was defined as background mean plus three times SD of the background mean and is marked by the upper dotted line. Cell detection limit is ~3,000 T4NT CAR T cells.
We then established orthotopic human xenograft TNBC models using the well-characterized cell lines MDA-MB-436 and MDA-MB-231.25 To decouple retention measurements from tumor targeting effects, we administered the CAR T cells intratumorally. Traceable pan-ErbB T4NT CAR T cells were imaged in vivo by PET using the radiotracer [18F]BF4− to detect NIS-expressing CAR T cells, followed by ex vivo analyses.
In MDA-MB-436 xenograft tumors, we detected by PET intratumorally administered T4NT CAR T cells in tumors on the day after administration (day 1) and also on subsequent imaging days (days 7 and week 2; Figure 5). Quantitation of in vivo imaging data revealed that observed signals stemming from T4NT CAR T cells over the 2-week observation period were always significantly higher than in control animals (Figure 5C) without major changes of signals within tumors over time. These data were corroborated by ex vivo radioactivity measurements by γ-counting of whole tumors harvested after two weeks, with tumors from animals that had received T4NT CAR T cells showing significantly higher radioactivity levels compared to tumors from control animals (Figure 5D).
Figure 5.

Tracking T4NT CAR T Cell Retention in MDA-MB-436 Xenograft Tumors
(A) Experimental scheme. (B) 5 × 106 T4NT CAR T cells (red frame) or vehicle (blue frame) were intratumorally administered (yellow arrows) on day zero and animals were PET/CT imaged (5 MBq [18F]BF4−; 40 min post administration) on indicated days. Endogenous signals are recorded in the thyroid and salivary glands (Th+SG), the stomach (S) and at much lower levels in some regions of the mammary glands. Tracer excretion happened renally, which is why in some animals, depending on their bladder fill status before imaging, signals from the bladder were recorded (B). T4NT CAR T cells are clearly visible in the tumor in all imaging sessions. Anatomical imagines (CT; grayscale) are overlayed with PET images (hue). Coronal and sagittal slices from a representative animal are shown; all PET images are on the same scale. (C) Cumulative quantitative analysis of tumor radioactivity from in vivo images of all animals. Box shows range from 25th to 75th percentile, whiskers minimum and maximum values; each dot represents one mouse (red, T4NT; blue, control). p values calculated by Mann-Whitney test. (D) Ex vivo analysis of radioactivity in harvested tissues quantified by γ-counting. Cumulative data of indicated organs from all animals are shown with error bars representing SEM. Inset shows zoom-in on tumors. p value calculated by Mann-Whitney test.
Interestingly, in a second TNBC model, MDA-MB-231, we observed a different behavior of the T4NT CAR T cells over time. T4NT CAR T cells were detectable on the day after administration and for the following time point, but signals in tumors were not significantly different from control animals that did not receive CAR T cells after two weeks (Figure 6). Ex vivo radioactivity measurements by γ-counting of whole tumors confirmed in vivo imaging results after two weeks and demonstrated that there were no significant differences between tumors that had received T4NT CAR T cells and control tumors (Figure 6D). We performed additional experiments with dedicated cohorts culled after administration and after two weeks, respectively, and subjected harvested tumors to ex vivo γ-counting measurements and histology. Results confirmed our data from in vivo imaging experiments demonstrating that radioactivity signals from T4NT CAR T cells, as well as their presence in tissues (determined by anti-human CD3 staining), were only detectable in tumors at the first time point (Figures 7A–7C). Overall, there was no effect of CAR T cell administration on tumor sizes in MDA-MB-231 models (Figure S7), which was in line with loss of therapeutic cells over time.
Figure 6.

Tracking T4NT CAR T Cell Retention in MDA-MB-231 Xenograft Tumors
(A) Experimental scheme. (B) 5 × 106 T4NT CAR T cells (red frame) or vehicle (blue frame) were intratumorally administered (yellow arrows) on day zero and animals were PET/CT imaged (5 MBq [18F]BF4−; 40 min post administration) on indicated days. Endogenous signals are recorded in the thyroid and salivary glands (Th+SG), the stomach (S) and at much lower levels in some regions of the mammary glands. Tracer excretion happened renally, which is why in some animals, depending on their bladder fill status before imaging, signals from the bladder were recorded (B). T4NT CAR T cells are clearly visible in the tumor in all imaging sessions. Anatomical imagines (CT; grayscale) are overlayed with PET images (hue). Coronal and sagittal slices from a representative animal are shown; all PET images are on the same scale. (C) Cumulative quantitative analysis of tumor radioactivity from in vivo images of all animals. Box shows range from 25th to 75th percentile, whiskers minimum and maximum values; each dot represents one mouse (red, T4NT; blue, control). p values calculated by Mann-Whitney test. (D) Ex vivo analysis of radioactivity in harvested tissues quantified by γ-counting. Cumulative data of indicated organs from all animals are shown with error bars representing SEM. Inset shows zoom-in on tumors. p value calculated by Mann-Whitney test.
Figure 7.

Ex Vivo Analyses of Additional MDA-MB-231.GFP-CaaX Tumor-Bearing Cohorts
(A) Experimental scheme. (B) Ex vivo analysis of radioactivity in harvested tissues from three different cohorts: animals that received T4NT CAR T cells intratumorally and were sacrificed either on the day after administration (red) or two weeks later (light red), and control animals that received untransduced T cells and were culled two weeks after administration. Shown are cumulative quantified by γ-counting data from all animals. Signal levels after 15 days were indistinguishable between animals that had received CAR T cells and untransduced T cells (p = 0.4425) while differences compared to day 1 were significant (p < 0.05 for both); error bars are SD. (C) Immunofluorescence histology demonstrating T4NT CAR T cell presence at injection sites on day 1 after administration (by anti-human CD3 staining. Tumor cells are visualized by GFP staining. Representative micrographs are shown. Data in (B) and (C) confirm observations from Figure 6. (D) Typical magnified micrograph of a MDA-MB-231.GFP-CaaX tumor on day 1 after T4NT CAR T cell administration. It shows that T4NT CAR T cells did not infiltrate into the adjacent tumor mass and a virtual boundary (white dashed line) was drawn indicating the two zones. Reduced GFP-CaaX signals in the area where T4NT CAR T cells were present indicated loss of tumor cells in this area. All scale bars in (C) and (D) are 100 μm.
Together, these data clearly demonstrate a different behavior of the pan-ErbB CAR T cells in these two TNBC xenograft models. As host immunity is very low in NSG mice,26 this pointed toward differences likely to be intrinsic to the tumor cells.
Intratumoral CAR T Cell Infiltration and Expression of Immune Checkpoints
Through in vivo imaging, we had also observed that T4NT CAR T cells largely remained within MDA-MB-436 tumors, while they were lost in MDA-MB-231 tumors over time. On day 3, we could still detect few T4NT CAR T cells and interestingly the intratumoral signal distribution appeared similar to day 1 (Figure 6B). Histology of sections from early time points after administration further corroborated this by revealing clear margins between tumor areas in which T4NT CAR T cells were injected and adjacent areas, which did not contain cells staining positive for human CD3 (Figure 7D). Tumor cells were easily identifiable in these sections as MDA-MB-231 cells expressing a plasma membrane-targeted GFP were employed (MDA-MB-231.GFP-CaaX). Notably, T4NT CAR T cells appeared to be active in regions where they were present as judged from the loss of tumor cells (loss of GFP fluorescence) in these confined spaces. Moreover, we systemically administered either T4NT CAR T cells or untransduced T cells to animals with MDA-MB-231 tumors (Figure S8). We found that T4NT CAR T cells homed into tumors within 24 h of administration, but they did not uniformly infiltrate the tumor there.
Various immune checkpoints have emerged in regulating anti-cancer T cell activity, thereby providing a rationale for analyzing their potential expression differences between MDA-MB-231 and MDA-MB-436 cells.27 We selected several of them for analysis including the programmed death-ligand 1 (PD-L1), the TIM-3 ligands galectin-9 and CD66a/CAECAM-1, the LAG-3 ligand HLA-DR (a representative of MHC class II), and the T cell immunoreceptor with Ig (ITIM) and immunoreceptor tyrosine-based inhibition motif (TIGIT) domains ligands CD155/PVR and CD112/nectin-2. Furthermore, we investigated potential differential expression of B7-H3, B7-H4, and B7-H5/VISTA. We found significantly higher PD-L1 expression in MDA-MB-231 compared to MDA-MB-436 cells (Figure 8A, top). Moreover, we found that a higher percentage of MDA-MB-231 cells expressed CD112 but a lower percentage of them expressed CD155, both of which are ligands for TIGIT (Figure 8A, middle and bottom). There were also differences in the expression of the B7-class inhibitory molecules B7-H3 (a similar proportion of positive cells but at lower median fluorescence intensities; Figure 8, left and Figure S10) and B7-H4 (higher expression in MDA-MB-231 cells; Figure 8, right). The other analyzed molecules were not expressed in both cell lines (Figures S11 and S12). As the observed PD-L1 differences were the most pronounced, we also validated them independently by immunoblot analysis, which confirmed flow cytometry results (Figure S13). In xenograft tumors established from these two TNBC cell lines, we also found significant differences in PD-L1 staining between the two models with higher staining in MDA-MB-231 tumors compared to MDA-MB-436 tumors (Figure 8C). Notably, in MDA-MB-231 tumors PD-L1 was predominantly found in the plasma membranes of tumor cells suggesting PD-L1 availability for interaction with T cells.
Figure 8.

Analysis of Immune Checkpoint and B7-Class Inhibitory Molecule Expression in MDA-MB-231 and MDA-MB-436 Models
(A) BCC lines were stained with antibodies directed against the indicated cell-surface proteins known to constitute one part of an immune checkpoint axis. Analysis was by flow cytometry using (left) FMO controls to categorize cells into populations positive or negative for the indicated molecule, while (right) obtained mean fluorescence intensities (MFI; median) values were compared between cell lines. (B) Analysis as in (A) but for indicated B7-class inhibitory molecules. (A and B) Notably, additional molecules were analyzed but if no differences were found between MDA-MB-231 and MDA-MB-436 cell lines, then data are shown in the Supplemental Information. For representative corresponding histograms see Supplemental Information. Error bars represent SD; n ≥ 3 different experiments. (C) Immunofluorescence staining for human PD-L1 in tumors established from the indicated cell lines. (Top) Representative micrographs are shown; scale bars are 100 μm. (Bottom) Cumulative quantitative intensity analysis of tumor tissues stained for anti-human PD-L1. Error bars are SD; n = 6 different tumors; all comparisons are significant (p < 0.0001 by ANOVA with Tukey’s multiple comparison test) except for controls (no primary and secondary antibody only; p = 0.6128), which was expected. Data demonstrated that in vitro differences in PD-L1 expression in these cell lines were retained in orthotopic in vivo tumor models.
Together, this data showed an inverse correlation between T4NT CAR T cell retention and the immune checkpoint inhibitor PD-L1 expression in these two TNBC models. While PD-L1 was found to be the most pronounced difference of the analyzed checkpoints, the TIGIT ligands CD112 and CD155 and the B7-class molecules B7-H3 and B7-H4 are likely to also contribute to the differences observed, highlighting the multifactorial challenges facing CAR-T therapy in solid tumors.
Discussion
Despite recent successes of CAR T cell therapy in hematologic malignancies, there are still many challenges for its application to solid tumors. To accelerate CAR T cell therapy development and characterize emerging toxicities, non-invasive whole-body imaging of CAR T cell in vivo distribution and fate is invaluable. Long-term imaging is best achieved using reporter gene methodology, which also reliably reflects cell viability, particularly if employing a reporter like NIS that generates contrast based on its activity. The ideal reporter for cell tracking applications in human settings is (1) detected with exquisite sensitivity using clinical imaging modalities, (2) does not elicit immunogenic reactions, (3) shows very limited endogenous expression and thus high specificity for traceable cells, (4) can be detected with a widely available contrast agent, and (5) does not confer any biological effect.
Our study is the first PET-afforded immune cell tracking study using NIS as a non-immunogenic radionuclide reporter. While NIS has been used as a reporter gene previously,24,28, 29, 30, 31, 32, 33 only recently was it employed for CAR T cell tracking, and then only using SPECT,17 which is more difficult to quantify than PET. We and others have previously shown cancer cell tracking by NIS-PET using the easily accessible short half-life radiotracer [18F]BF4–.30,33,34 We focused on PET detection as clinical PET is currently the most sensitive imaging modality for cell tracking. A first-in-man study has recently reported good contrast (for endogenous NIS expressing tissues such as the thyroid and stomach), generally low uptake in other organs, and safety of [18F]BF4–.35 NIS is a transporter and compared to tracer binding reporters such as SSTR2 or PSMA intrinsically provides signal amplification. Importantly, NIS transport activity depends on the cellular Na+/K+ gradient across the plasma membrane, which is fueled by cellular ATP,36 thereby reflecting cell viability better than a reporter which is detected based solely on its presence. This feature is important for determining the fate of therapeutic cells in vivo.
Our data demonstrate not only that ectopic expression of NIS was feasible and had no detrimental effects on anti-ErbB family CAR T cells (Figure 2) but also that the use of NIS was safe for the CAR T cells. We characterized viability, tumor cell killing capacity, interferon-γ release, and radiation damage for CAR T cells that had been exposed to both the longer half-life NIS-SPECT tracer [99mTc]TcO4− and the shorter half-life NIS-PET tracer [18F]BF4– with complete decay of the radioisotopes in the cells before analysis of CAR and reporter functions (Figure 3). DNA damage experiments were performed at cellular radioactivity concentrations that were comparable (see Materials and Methods) to amounts we had previously found in vivo; values were calculated from quantitative in vivo PET data obtained with [18F]BF4− in small cancer cell deposits.30 Notably, at the highest used concentrations (which were less than an order of magnitude larger than what above calculations had yielded) there was DNA damage detected in CAR T cells 2 h after radiotracer administration. Importantly, our data also demonstrated that radiation-induced damage was repaired within 24 h with no long-lasting effects onto the CAR T cells (Figures 3D and 3E). The obtained data clearly revealed that both PET and SPECT radiotracers were feasible options for longitudinal CAR T cell imaging with negligible damage to administered CAR T cells through radionuclide imaging. Notably, while [99mTc]TcO4– is more available as it is generator-produced, [18F]BF4– has advantages over [99mTc]TcO4− due to its shorter half-life, its decay-dependent chemical decomposition, and because PET is more sensitive than SPECT. Moreover, upcoming total-body PET scanners will provide up to 40-fold increases in sensitivity over current PET instrumentation.14
We applied our CAR T cell tracking approach to quantify CAR T cell retention in vivo over time. While our in vivo traceable CAR T cells had the capacity to home to tumors (e.g., MDA-MB-231 tumors as shown in Figure S8), we chose to administer the CAR T cells intratumorally to exclude variability caused by different kinetics or extent of tumor targeting. Moreover, the intratumoral route is relevant for this CAR, which is administered in this manner clinically (NCT01818323). We performed in vivo tracking of anti-ErbB family CAR T cells in two different orthotopic TNBC models and, unexpectedly, found a stark difference in CAR T cell retention between MDA-MB-231 and MDA-MB-436 TNBC models (Figures 5 and 6). Retention differences inversely correlated with the expression levels of PD-L1 expressed on the surface of these TNBC cells (Figure 8A), but PD-L1 was not the only differentially expressed immune checkpoint molecule. Interestingly, we found differences in the expression of the TIGIT ligands CD112 and CD155 (Figure 8A) and the B7-class molecules B7-H3 and B7-H4 (Figure 8B) but not for B7-H5/VISTA (Figure S11). Both CD112 and CD155 on tumor cells but also antigen presenting cells (e.g., dendritic cells) can provide co-inhibitory signals to tumor-infiltrating T cells through TIGIT, which has been found to be expressed highly on CD8+ T cells in many cancers.37 Notably, CD226 competes with TIGIT for binding of CD112 and CD155, but while CD226 binds with lower affinity to these molecules than TIGIT, this interaction provides co-stimulatory signals.27 A high TIGIT-to-CD226 surface expression ratio on regulatory T cells in the tumor microenvironment has been correlated with a poor prognosis.38,39 TIGIT interactions are complex and not fully understood, but our data showing a higher percentage of MDA-MB-231 cells expressing CD112 and fewer MDA-MB-231 cells expressing CD155 but at a significantly higher level compared to CD155-expressing MDA-MB-436 cells, provide an interesting anchor for further mechanistic studies. The role of the ubiquitously expressed B7-H3 molecule in immune evasion is somewhat controversial27 and while a large proportion of both TNBC cell lines express this molecule, MDA-MB-231 cells express less than MDA-MB-436 cells. In contrast, the cell-surface expression and thus interaction capacity of B7-H4 has been found to be tightly regulated40 and despite its receptor remaining elusive,27 it is interesting that we found its expression to be inversely correlated to T4NT retention, and thus showing a similar correlation as PD-L1 in our TNBC models.
Furthermore, we found that in MDA-MB-231 tumors, CAR T cells killed tumor cells in their direct vicinity at injection sites but did not infiltrate further into the tumor mass adjacent to injection sites (Figure 7). The tumor microenvironment has been extensively characterized and found to be hostile to CAR T cells, on the one hand because the glycolytic metabolism of tumor cells renders it hypoxic, acidic, low in nutrients, and prone to oxidative stress,41 while on the other hand multiple additional molecular and cellular factors suppress the T cell immune response.18 CAR T cells are derived from patients’ T cells; hence, they only differ from host T cells by expression of the CARs but remain otherwise unaltered and consequently responsive to the conditions in the tumor microenvironment. The most pronounced difference in immune checkpoint inhibitor expression between out TNBC models was PD-L1 (Figure 8A). We found high-level PD-L1 expression in MDA-MB-231 cells and much lower levels in MDA-MB-436 cells in vitro and, importantly also ex vivo (Figure 8C). Notably, tumors are also capable of adaptive immune resistance, a reaction of cancer cells resulting in the expression of molecules that actively turn off an otherwise effective antitumor immune response.42 For example, PD-L1 can be upregulated by cancer cells because of exposure to T cell-derived interferon-γ resulting in PD-1 expressing T cells to be turned off, and the cancer cells evading destruction.43 MDA-MB-231 cells have previously been reported to respond to interferon-γ by PD-L1 upregulation.44 We found that T4NT CAR T cells were not retained in MDA-MB-231 tumors (Figure 6), while we also found signs of them being functional initially within these tumors (Figure 7). We also found in vitro that T4NT CAR T cells produced interferon-γ when co-cultured with MDA-MB-231 cells (Figure 3C) and it is therefore not unlikely that this also happened in vivo and contributed to MDA-MB-231 tumors not retaining T4NT CAR T cells and thus not responding to treatment. In contrast, MDA-MB-436 tumors retained the immunotherapy and responded, hence it is unlikely that there was a major impact of interferon-γ-only-mediated adaptive immune resistance in this model.
Preclinically, combining CARs with immune checkpoint inhibitors has shown promising results. For example, CAR T cells engineered to secrete anti-PD-L1 antibodies showed significantly improved activity compared to standard CAR T cells.45 Other studies demonstrated enhanced anti-tumor activities by engineering CAR T cells to secrete anti-PD-1.46,47 CAR T cells engineered to secrete PD-1 or PD-L1 antibodies have been in clinical trials for EGFR, EGFRvIII, mucin-1, and mesothelin expressing cancers.48 Our study was neither dedicated to the development of a new CAR nor to discovery of precise CAR T cell disarming mechanisms, but instead to demonstrate in vivo CAR T cell tracking by non-invasive and highly sensitive PET using a non-immunogenic reporter. The revealed differences in tumor retention in the two TNBC models serve as an application example and expand the utility of in vivo CAR T cell tracking beyond tumor targeting to the quantification of tumor retention. Notably, while we focused on PD-L1 in our ex vivo analyses, our in vitro cell line data suggest that additional other immune checkpoints, in particular TIGIT and hitherto unknown receptor(s) binding to B7-H4, might be involved in the observed CAR T cell retention differences. In summary, we demonstrated the power of the imaging-guided approach for the preclinical development of new or the optimization of existing CAR T cell therapies.
Importantly, our NIS-based PET-afforded CAR T cell tracking approach is directly translatable to the clinic. The platform is versatile and, upon omission of the fluorescent protein RFP, fully compliant with clinical translation. Importantly, clearance of the NIS-PET radiotracer [18F]BF4− from circulation is faster and, crucially, completely reaches lower background levels compared to radioactive iodide tracers for NIS.34 Tracking NIS expressing CAR T cells in organs with high endogenous NIS expression, i.e. thyroid and stomach, is not feasible and signals from these high-expressing organs could potentially also affect low signals in the vicinity of these organs. A potential solution, at least for regions close to the stomach, would be oral administration of a substance absorbing radiation, for example barium sulfate, which has long been in clinical use as a CT contrast agent.49 Here, we also showed that radiotracer-induced damage to CAR T cells during non-invasive imaging is negligible, and therefore repeat-imaging is not a concern for function or survival of the administered traceable therapeutic cells. As [18F]BF4– can be produced in GMP-quality and as it has already been used safely in humans,35 all components for NIS-afforded CAR-T tracking by PET in humans are now available. It will be beneficial by enabling spatiotemporal and non-invasive CAR-T monitoring in patients providing information predictive of therapy success (e.g. extent of CAR-T trafficking to tumors, kinetics of CAR-T retention at lesions, heterogeneity of behavior between lesions, CAR-T survival) and safety-relevant data (i.e. significant on-target off-tumor accumulation). The latter will enable clinicians to control infused cells, whereby CAR-T imaging will act as a biomarker and trigger for cell therapy ablation via suicide gene technology.50 In conclusion, NIS-afforded spatiotemporal CAR T cell tracking by highly sensitive PET imaging is now realistically within reach for human use.
Materials and Methods
Information regarding the generation of constructs, adherent cells, lentivirus production, flow cytometry, confocal microscopy, and immunoblotting is detailed in the Supplemental Information.
Reagents
Reagents were from Merck, New England Biolabs, Sigma-Aldrich, Thermo-Fisher or VWR unless otherwise stated. Tissue culture materials were from Corning, Sarstedt or TPP. [99mTc]TcO4− was generator-eluted (on-site King’s Health Partners’ Radiopharmacy) as sodium salt solution and used within two half-lives. The NIS radiotracer [18F]BF4– was produced on the day of use as previously described.30
Animals
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Charles River UK. All mice were maintained under sterile conditions with food and water available ad libitum. All procedures were performed in accordance with all legal, ethical, and institutional requirements (UK Home Office PPL 70/8879).
Isolation and Culture of Human T Cells
T cells were isolated from human peripheral blood donated by anonymous healthy volunteers via the National Blood Service (NHS Blood and Transplantation) with informed consent and ethical approval from King’s College London Research Ethics Committee (Study Reference HR-16/17-3746). Peripheral blood mononuclear cells (PBMCs) were isolated by low-density centrifugation on Ficoll (Sigma). T cells were cultured in RPMI 1640 containing 5% (v/v) human serum (HS; BioSera), 2 mM L-glutamine and 100 IU/mL penicillin/streptomycin at 37°C in the presence of 5% (v/v) CO2 in a humidified incubator with admixtures as indicated in the text. T cells were activated for 48 h with CD3/CD28-coated Dynabeads (1:1 bead:cell ratio) in growth medium and cultured in 100 IU/ml IL-2 (untransduced T cells; IL-2 from Proleukin-Novartis) or 30 ng/mL IL-4 (transduced T cells, cf. below; IL-4 from Miltenyi Biotec) with growth media replenishment every other day.
Transduction of Human T Cells
Isolated T cells were thawed and cultured in normal growth medium over night before being washed twice with growth medium containing 5 U/mL DNase I. 24 h later, T cells were activated with anti-CD3/CD28 Dyna beads (3 × 106 cells/mL; 1:1 cell:bead ratio) and 24 h later 100 IU/mL IL-2 (Proleukin-Novartis) was added. Tissue culture plates were coated with 50 μg/μL retronectin (TakaraBio) overnight at 4°C. On the next day, 5 × 105 T cells/well and lentiviruses were added to retronectin-coated wells and plates were centrifuged at 350 × g for 1 h at room temperature (RT). Transduced T cells were incubated in growth medium supplemented with IL-4 (30 ng/mL; Miltenyi Biotec) for selective expansion, while untransduced control cells were cultured in growth medium with 100 IU/mL IL-2. Cells were expanded and cultured for up to 21 d before administration to mice.
In Vitro Radiotracer Uptake in NIS-Expressing T Cells
106 indicated CAR T cells were transferred into Eppendorf tubes, washed with ice-cold PBS, and resuspended in 1 mL growth medium. 100 kBq [99mTc]TcO4− or 100 kBq [18F]BF4− were added to each tube and cells were incubated for 30 min at 37°C in a humidified incubator. Subsequently, cells were pelleted, supernatant was collected, and cells were washed twice times with 1 mL PBS before being resuspended in growth medium for γ-counting and further culture if desired. Cells, supernatants and wash solutions were subjected to radioactivity analysis using an automated γ-counter with built-in decay correction (1282 Compugamma, LKB-Wallac). The percentage of radioactivity taken up by the cells was calculated according to Equation 1, wherein Cpm represents decay-corrected radioactivity counts per minute.
| (Equation 1) |
Results of every uptake assay were normalized to a reference cell line, which was run as an additional sample set. The reference cell line was the previously used NIS reporter-expressing rat adenocarcinoma cell line 3E.Δ.NIS.34
T Cell Proliferation
5 × 104 untransduced T cells or CAR T cells (T4NT or T4ΔNT) were plated in four 96-well-plates in 100 μL of cytokine supplemented media and incubated at 37°C, 5% CO2 for up to 4 days (IL-2, 100 IU/mL for T cells or IL-4, 30 ng/mL; 1 plate per day). Cell viability was measured after addition and incubation of 44 μM Alamar Blue at 37°C for 2 h. Individual plates were analyzed for fluorescence at 580 nm (emission; excitation at 530 nm) at each indicated time point.
Determination of T Cell Viability after Radiotracer Exposure
Untransduced T cells or CAR T cells (T4NT or T4ΔNT) were resuspended at 3 × 105 cells per 280 μL fully supplemented growth medium. Either 20 μL PBS (control) or 20 μL containing 60 kBq of one of the indicated NIS radiotracers ([18F]BF4− or [99mTc]TcO4−) were added and cells incubated for 72 h in a tissue culture incubator in humidified air containing 5% (v/v) CO2. Reducing equivalents as a measure of cell viability were then determined using the Alamar Blue fluorescence assay (44 μM, 2 h, 37°C; fluorescence readout at 530/580 nm ex/em).
CAR T Cell Function by Cancer Cell Killing
Two independent assays were performed as indicated. BCC viability assay was performed as follows: BCCs were seeded in a 96-well tissue culture plate at 104 cells/100 μL growth medium/well and incubated overnight. Next, 104 CAR T cells or untransduced T cells were added in 100 μL growth medium per well with or without admixtures of 40 kBq [18F]BF4− or [99mTc]TcO4−, and incubated for 72 h. CAR T Cells or untransduced T cells were gently removed with the supernatant, BCC were twice PBS-washed, and BCC viability was determined with by Alamar Blue (44 μM, 2 h, 37°C; fluorescence readout at 530/580 nm ex/em). Cell death assay was performed as follows: BCCs were seeded in a 96-well tissue culture plate at 104 cells/50 μL growth medium per well and incubated overnight. Untransduced T cells or T4NT CAR-T cells were added at the indicated effector:target ratio. Cell death was measured using the CellTox Green Cytotoxicity Assay (Promega) according to the manufacturer’s instructions. Plates were measured at 24 h and 48 h after starting the co-culture using a GloMax-Multi Microplate Multimode Reader (Promega; fluorescence readout at 490/520 nm ex/em).
CAR T Cell Function by Interferon-γ Release
Experimental design was identical to cancer cell monolayer killing assays but for subsequent determination of interferon-γ instead of cancer cell viability. Culture supernatants were collected and subjected to analysis by interferon-γ ELISA (Invitrogen; minimum detection sensitivity 2 pg/mL) according to the manufacturer’s instructions.
Determination of Cellular DNA Damage
Cellular DNA damage is dependent on the type of radiation, the amount of radioactivity in the cells and the intracellular distribution (cf. distance to the vulnerable molecules). We adopted a function-based approach to radiation effects and aimed to treat cells with radioactivity amounts that were previously reached in non-invasive imaging experiments designed to track cells using NIS as a reporter gene. Small metastases from previous experiments served as the closest available model for inferring radiotracer uptake in the envisaged CAR T cell deposits in this study. We previously found amounts of up to 1.7 kBq/μL of [18F] BF4– in small lung metastases.30 T cell volumes of healthy humans were found to be 166 μm3, (range [126;216];51) equating to ∼6 million T cells fitting into 1 μL. Combining both numbers and omitting any other factors (e.g., presence of other cells, interstitial volumes, activated T cells being larger than naive ones, etc.) this would result in ∼0.28 mBq/T cell. Above factors would lead to less cells in this volume and thereby larger uptake values, which we accommodated by performing experiments such that up to 100-times higher radioactivity-per-cell values were experimentally studied. The SPECT radiotracer [99mTc]TcO4− (25 MBq per mouse) is generally used at 5-times larger radioactivity values during imaging than [18F] BF4− (5 MBq per mouse). Assuming similar in vivo behavior we used a 5-fold higher base for the experiments, i.e., 1.4 mBq/T cell.
Untransduced T cells or T4NT CAR T cells were resuspended at 106 cells/mL in growth medium and incubated in the presence of either 1.5 MBq, 500 kBq, 150 kBq, or 50 kBq of [18F]BF4− or 9 MBq, 3 MBq, 1 MBq, or 300kBq [99mTc]TcO4− for 30 min at 37°C, followed by analysis of cellular radioactivity uptake as described above. Resultant cellular radioactivity amounts are indicated in each corresponding figure legend and were 120-, 50-, 21-, and 7-times ([18F]BF4−) or were 92, 36, 3.5, and 2 times ([99mTc]TcO4−) larger than estimated to be reached during non-invasive imaging (see above). Cells were then resuspended in growth medium in a poly-L-lysine-coated 96-well plate, centrifuged at 650 × g for 10 min, and incubated until fixing with 4% (w/v) paraformaldehyde (ice-cold, 15 min) at indicated time points (times indicate period from start of radioactive uptake until fixation). Fixed samples were kept at 4°C until staining for double-strand DNA breaks using an anti-γH2AX antibody. Subsequently, cells were permeabilized in 0.5% Triton X-100/0.5% IGEPAL CA-640, blocked with 1% donkey serum in 2% BSA for 1 h, incubated with a monoclonal mouse anti-γH2AX antibody (Ser139; clone JBW301 from Merck 05-636; 1:1,600 in 3% BSA), and stained with a donkey anti-mouse-Alexa Fluor 488 secondary antibody (Jackson ImmunoResearch 715-545-150; 0.5 μg/mL in 3% BSA) and Hoechst 33342 (1 μg/mL). Images were acquired on an Operetta CLS system (PerkinElmer) and analyzed by the Harmony v4.1 software using an automated analysis pipeline enabling the identification of γH2AX foci in nuclei of NIS-RFP-expressing CAR T cells or untransduced T cells; >35 nuclei per replicate were analyzed as a minimum for each condition.
Determination of Detection Sensitivity of Traceable CAR T Cells
To determine the detection sensitivity of NIS-RFP expressing transduced T cells in the nanoPET/CT scanner (Mediso), we prepared cell pellets consisting of pre-labeled NIS-RFP-positive (57 ± 3 mBq [99mTc]TcO4−/cell) and NIS-RFP-negative untransduced T cells. The total cell number per pellet was kept constant at 106 cells. This procedure has previously been used to determine the detection limit of reporter gene expressing cells in preclinical SPECT and PET instruments.24,34 Briefly, cell mixtures were prepared in 50 μL PBS+/+ in Eppendorf Tubes, pelleted, and immediately scanned in the nanoSPECT/CT equipment for 30 min. Reconstructed images were analyzed using Vivoquant software, whereby radioactivity was measured in volumes of interest drawn with the help of CT information (tube walls as boundaries). Measurements were performed in triplicate. In line with standard analytical procedures, we defined the limit of detection (LOD) to be three times the standard deviation above background signals (Figure S3).
TNBC Xenograft Models
Young adult (5- to 6-week-old) female NSG mice were used to establish orthotopic mammary tumors with the indicated breast cancer cell lines (106 cells injected into the mammary fat pad). Tumors were monitored using callipers. Five million CAR T cells or vehicle were administered intratumorally in animals bearing tumors of ∼50 mm3 volume (animals were randomly allocated to cohorts).
In Vivo Imaging and Image Analysis
Mice were anesthetized with 2% (v/v) isoflurane/O2 and received 5 MBq [18F]BF4− (i/v in 100 μL sterile PBS) prior to PET/CT imaging (NanoPET/CT; Mediso). After radiotracer administration, animals were placed anaesthetized onto scanner beds and CT was performed (55 kVp tube voltage, 1,200 ms exposure time, 360 projections). 40 min after radiotracer administration, animals were PET imaged. Repeat imaging with the same radiotracer was not affected by tracer amounts from prior imaging sessions due to the short radioactive half-life of 18F (110 min).34 PET/CT data were reconstructed using Tera-Tomo (Mediso) with corrections for attenuation, detector dead time, and radioisotope decay in place as needed. All images were analyzed using VivoQuant software (inviCRO) enabling the delineation of regions of interest (ROIs) for quantification of radioactivity. CT images were used to draw ROIs and provide the volumes required for standard uptake value calculations. The total activity in the whole animal (excluding the tail) at the time of tracer administration was defined as the injected dose (ID).
Ex Vivo Tissue Analyses
For analysis of radioactivity in harvested tissues, tissues were weighed, and radioactivity was quantified using a γ-counter (1282-Compugamma, LKB-Wallac), together with calibration standards. Data were expressed as %ID/g. For histology, tumors were harvested and frozen in Optimal Cutting Temperature Medium (OCT) followed by tissue sectioning (5 μm). Thawed sections were fixed in 4% PFA in PBS for 10 min, permeabilized (0.2% [v/v] Triton X-100/PBS), washed, and blocked (PBS containing 2% [v/v] goat serum, 0.1% fish skin gelatin and 0.2% [v/v] Tween-20) before being incubated with the indicated combinations of the following primary antibodies (2 μg/mL) overnight at 4°C: anti-human CD3 (monoclonal rabbit; [SP7], Abcam) and anti-PD-L1 (monoclonal rabbit; [28-8]; Abcam). After three PBS washes, sections were then stained with the corresponding secondary antibody (1 μg/mL) for 45 min at RT in the dark: goat anti-rabbit-Cy5, (Jackson ImmunoResearch). Nuclei were stained with Hoechst 33342 (1 μg/mL in PBS; 15 min at RT). Sections were washed twice with PBS and deionized water each before being mounted onto microscope slides using Mowiol-488 containing 2.5% DABCO. All solutions were sterile filtered before use. Samples were dried overnight in the dark and imaged on a Nikon Eclipse Ti2 wide-field fluorescence microscope equipped with the following filter sets (ex/em; all BP) for imaging Hoechst 33342 (AT350/50×; T400lp; ET460/50 m), GFP/Alexa 488 (ET470/40×; T495lpxr; ET525/50 m), TagRFP (ET539/21×; T556lpxr; ET576/31 m) and Cy5/Alexa 647 (ET640/30×; T660lpxr; ET690/50 m). Fiji/ImageJ v1.5× software was used for all analyses.
Statistical Analysis
Statistical analysis was performed using Prism v7 software (GraphPad) with details added to figure legends and text.
Author Contributions
The author contributions are listed in alphabetical order. Conceptualization: A.V. and G.O.F. Data Curation: A.V., C.A.-H., C.L., E.K., L.L., and P.J. Formal Analysis: A.V., C.L., E.K., E.S., L.L., and P.J. Funding Acquisition: G.O.F. and R.T.M.d.R. Investigation: A.V., C.A.-H., C.L., E.K., and L.L. Methodology: A.V., C.A.-H., F.M., and G.O.F. Project Administration: A.V. and G.O.F. Supervision: G.O.F. Validation: A.V., C.A.-H., C.L., E.K., L.L., and P.J. Visualization: A.V., C.L., G.O.F., and L.L. Writing – Original Draft: G.O.F. Writing – Review & Editing: A.V., C.A.-H., C.L., E.S., F.M., G.O.F., P.J., and R.T.M.d.R.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors sincerely thank Dr. John Maher (King’s College London) for gifting to them the DNA encoding the pan-ErbB CAR T1E28z and Dr. Erik Sahai (Francis Crick Institute, London) for the viral plasmid encoding GFP-CaaX. A.V., C.L., F.M., and L.L. were supported by a Cancer Research UK grant (Multidisciplinary Project Award C48390/A21153) to G.O.F. and R.T.M.R. E.K. was supported by a Worldwide Cancer Research grant (16-1135) to G.O.F. L.L. was co-supported by a King's Health Partners grant (Research and Development Challenge R150101) to G.O.F. E.S. was supported by a PhD studentship from the Comprehensive Cancer Imaging Centre at King’s College London and UCL, funded by Cancer Research UK and EPSRC. C.A.-H was supported by a PhD studentship from King's Health Partners. The authors received further support from the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London and the Wellcome/EPSRC Centre for Medical Engineering at King’s College London (WT 203148/Z/16/Z). The views expressed are those of the authors and not necessarily those of the NIHR, the National Health Service, or the Department of Health.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.06.028.
Supplemental Information
References
- 1.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts Z.J., Better M., Bot A., Roberts M.R., Ribas A. Axicabtagene ciloleucel, a first-in-class CAR T cell therapy for aggressive NHL. Leuk. Lymphoma. 2017;59:1785–1796. doi: 10.1080/10428194.2017.1387905. [DOI] [PubMed] [Google Scholar]
- 3.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prasad V. Immunotherapy: Tisagenlecleucel - the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat. Rev. Clin. Oncol. 2018;15:11–12. doi: 10.1038/nrclinonc.2017.156. [DOI] [PubMed] [Google Scholar]
- 5.Kosti P., Maher J., Arnold J.N. Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors. Front. Immunol. 2018;9:1104. doi: 10.3389/fimmu.2018.01104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamers C.H., Klaver Y., Gratama J.W., Sleijfer S., Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem. Soc. Trans. 2016;44:951–959. doi: 10.1042/BST20160037. [DOI] [PubMed] [Google Scholar]
- 7.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linette G.P., Stadtmauer E.A., Maus M.V., Rapoport A.P., Levine B.L., Emery L., Litzky L., Bagg A., Carreno B.M., Cimino P.J. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saudemont A., Jespers L., Clay T. Current Status of Gene Engineering Cell Therapeutics. Front. Immunol. 2018;9:153. doi: 10.3389/fimmu.2018.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krekorian M., Fruhwirth G.O., Srinivas M., Figdor C.G., Heskamp S., Witney T.H., Aarntzen E.H.J.G. Imaging of T-cells and their responses during anti-cancer immunotherapy. Theranostics. 2019;9:7924–7947. doi: 10.7150/thno.37924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashmore-Harris C., Iafrate M., Saleem A., Fruhwirth G.O. Non-invasive Reporter Gene Imaging of Cell Therapies, including T Cells and Stem Cells. Mol. Ther. 2020;28:1392–1416. doi: 10.1016/j.ymthe.2020.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iafrate M., Fruhwirth G.O. How non-invasive in vivo cell tracking supports the development and translation of cancer immunotherapies. Front. Physiol. 2020;11:154. doi: 10.3389/fphys.2020.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khalil M.M., Tremoleda J.L., Bayomy T.B., Gsell W. Molecular SPECT Imaging: An Overview. Int. J. Mol. Imaging. 2011;2011:796025. doi: 10.1155/2011/796025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cherry S.R., Jones T., Karp J.S., Qi J., Moses W.W., Badawi R.D. Total-Body PET: Maximizing Sensitivity to Create New Opportunities for Clinical Research and Patient Care. J. Nucl. Med. 2018;59:3–12. doi: 10.2967/jnumed.116.184028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volpe A., Kurtys E., Fruhwirth G.O. Cousins at work: How combining medical with optical imaging enhances in vivo cell tracking. Int. J. Biochem. Cell Biol. 2018;102:40–50. doi: 10.1016/j.biocel.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minn I., Huss D.J., Ahn H.H., Chinn T.M., Park A., Jones J., Brummet M., Rowe S.P., Sysa-Shah P., Du Y. Imaging CAR T cell therapy with PSMA-targeted positron emission tomography. Sci Adv. 2019;5:eaaw5096. doi: 10.1126/sciadv.aaw5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emami-Shahri N., Foster J., Kashani R., Gazinska P., Cook C., Sosabowski J., Maher J., Papa S. Clinically compliant spatial and temporal imaging of chimeric antigen receptor T-cells. Nat. Commun. 2018;9:1081. doi: 10.1038/s41467-018-03524-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez M., Moon E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies D.M., Foster J., Van Der Stegen S.J., Parente-Pereira A.C., Chiapero-Stanke L., Delinassios G.J., Burbridge S.E., Kao V., Liu Z., Bosshard-Carter L. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012;18:565–576. doi: 10.2119/molmed.2011.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou R., Yazdanifar M., Roy L.D., Whilding L.M., Gavrill A., Maher J., Mukherjee P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019;10:1149. doi: 10.3389/fimmu.2019.01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klampatsa A., Achkova D.Y., Davies D.M., Parente-Pereira A.C., Woodman N., Rosekilly J., Osborne G., Thayaparan T., Bille A., Sheaf M. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017;393:52–59. doi: 10.1016/j.canlet.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Wilkie S., Burbridge S.E., Chiapero-Stanke L., Pereira A.C., Cleary S., van der Stegen S.J., Spicer J.F., Davies D.M., Maher J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010;285:25538–25544. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J.H., Lee S.R., Li L.H., Park H.J., Park J.H., Lee K.Y., Kim M.K., Shin B.A., Choi S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE. 2011;6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fruhwirth G.O., Diocou S., Blower P.J., Ng T., Mullen G.E. A whole-body dual-modality radionuclide optical strategy for preclinical imaging of metastasis and heterogeneous treatment response in different microenvironments. J. Nucl. Med. 2014;55:686–694. doi: 10.2967/jnumed.113.127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iorns E., Drews-Elger K., Ward T.M., Dean S., Clarke J., Berry D., El Ashry D., Lippman M. A new mouse model for the study of human breast cancer metastasis. PLoS ONE. 2012;7:e47995. doi: 10.1371/journal.pone.0047995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shultz L.D., Lyons B.L., Burzenski L.M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S.D., King M., Mangada J. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 27.Andrews L.P., Yano H., Vignali D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat. Immunol. 2019;20:1425–1434. doi: 10.1038/s41590-019-0512-0. [DOI] [PubMed] [Google Scholar]
- 28.Ashmore-Harris C., Blackford S.J., Grimsdell B., Kurtys E., Glatz M.C., Rashid T.S., Fruhwirth G.O. Reporter gene-engineering of human induced pluripotent stem cells during differentiation renders in vivo traceable hepatocyte-like cells accessible. Stem Cell Res. (Amst.) 2019;41:101599. doi: 10.1016/j.scr.2019.101599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merron A., Peerlinck I., Martin-Duque P., Burnet J., Quintanilla M., Mather S., Hingorani M., Harrington K., Iggo R., Vassaux G. SPECT/CT imaging of oncolytic adenovirus propagation in tumours in vivo using the Na/I symporter as a reporter gene. Gene Ther. 2007;14:1731–1738. doi: 10.1038/sj.gt.3303043. [DOI] [PubMed] [Google Scholar]
- 30.Volpe A., Man F., Lim L., Khoshnevisan A., Blower J., Blower P.J., Fruhwirth G.O. Radionuclide-fluorescence Reporter Gene Imaging to Track Tumor Progression in Rodent Tumor Models. J. Vis. Exp. 2018;133:e57088. doi: 10.3791/57088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moroz M.A., Zhang H., Lee J., Moroz E., Zurita J., Shenker L., Serganova I., Blasberg R., Ponomarev V. Comparative Analysis of T Cell Imaging with Human Nuclear Reporter Genes. J. Nucl. Med. 2015;56:1055–1060. doi: 10.2967/jnumed.115.159855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terrovitis J., Kwok K.F., Lautamäki R., Engles J.M., Barth A.S., Kizana E., Miake J., Leppo M.K., Fox J., Seidel J. Ectopic expression of the sodium-iodide symporter enables imaging of transplanted cardiac stem cells in vivo by single-photon emission computed tomography or positron emission tomography. J. Am. Coll. Cardiol. 2008;52:1652–1660. doi: 10.1016/j.jacc.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vandergaast R., Khongwichit S., Jiang H., DeGrado T.R., Peng K.W., Smith D.R. Enhanced noninvasive imaging of oncology models using the NIS reporter gene and bioluminescence imaging. Cancer Gene Ther. 2019;27:179–188. doi: 10.1038/s41417-019-0081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diocou S., Volpe A., Jauregui-Osoro M., Boudjemeline M., Chuamsaamarkkee K., Man F., Blower P.J., Ng T., Mullen G.E.D., Fruhwirth G.O. [18F]tetrafluoroborate-PET/CT enables sensitive tumor and metastasis in vivo imaging in a sodium iodide symporter-expressing tumor model. Sci. Rep. 2017;7:946. doi: 10.1038/s41598-017-01044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Doherty J., Jauregui-Osoro M., Brothwood T., Szyszko T., Marsden P.K., O’Doherty M.J., Cook G.J.R., Blower P.J., Lewington V. 18F-Tetrafluoroborate, a PET Probe for Imaging Sodium/Iodide Symporter Expression: Whole-Body Biodistribution, Safety, and Radiation Dosimetry in Thyroid Cancer Patients. J. Nucl. Med. 2017;58:1666–1671. doi: 10.2967/jnumed.117.192252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Portulano C., Paroder-Belenitsky M., Carrasco N. The Na+/I- symporter (NIS): mechanism and medical impact. Endocr. Rev. 2014;35:106–149. doi: 10.1210/er.2012-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whelan S., Ophir E., Kotturi M.F., Levy O., Ganguly S., Leung L., Vaknin I., Kumar S., Dassa L., Hansen K. PVRIG and PVRL2 Are Induced in Cancer and Inhibit CD8+ T-cell Function. Cancer Immunol. Res. 2019;7:257–268. doi: 10.1158/2326-6066.CIR-18-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chauvin J.M., Pagliano O., Fourcade J., Sun Z., Wang H., Sander C., Kirkwood J.M., Chen T.H., Maurer M., Korman A.J., Zarour H.M. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Invest. 2015;125:2046–2058. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fourcade J., Sun Z., Chauvin J.M., Ka M., Davar D., Pagliano O., Wang H., Saada S., Menna C., Amin R. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight. 2018;3:121157. doi: 10.1172/jci.insight.121157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sica G.L., Choi I.H., Zhu G., Tamada K., Wang S.D., Tamura H., Chapoval A.I., Flies D.B., Bajorath J., Chen L. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003;18:849–861. doi: 10.1016/s1074-7613(03)00152-3. [DOI] [PubMed] [Google Scholar]
- 41.Renner K., Singer K., Koehl G.E., Geissler E.K., Peter K., Siska P.J., Kreutz M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017;8:248. doi: 10.3389/fimmu.2017.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ribas A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015;5:915–919. doi: 10.1158/2159-8290.CD-15-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soliman H., Khalil F., Antonia S. PD-L1 expression is increased in a subset of basal type breast cancer cells. PLoS ONE. 2014;9:e88557. doi: 10.1371/journal.pone.0088557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suarez E.R., Chang K., Sun J., Sui J., Freeman G.J., Signoretti S., Zhu Q., Marasco W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7:34341–34355. doi: 10.18632/oncotarget.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rafiq S., Yeku O.O., Jackson H.J., Purdon T.J., van Leeuwen D.G., Drakes D.J., Song M., Miele M.M., Li Z., Wang P. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018;36:847–856. doi: 10.1038/nbt.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie J., Zhou Z., Jiao S., Li X. Construction of an anti-programmed death-ligand 1 chimeric antigen receptor and determination of its antitumor function with transduced cells. Oncol. Lett. 2018;16:157–166. doi: 10.3892/ol.2018.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoon D.H., Osborn M.J., Tolar J., Kim C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018;19:340. doi: 10.3390/ijms19020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suksanpaisan L., Pham L., McIvor S., Russell S.J., Peng K.W. Oral contrast enhances the resolution of in-life NIS reporter gene imaging. Cancer Gene Ther. 2013;20:638–641. doi: 10.1038/cgt.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones B.S., Lamb L.S., Goldman F., Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014;5:254. doi: 10.3389/fphar.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuse R., Schuster S., Schübbe H., Dix S., Hausmann K. Blood lymphocyte volumes and diameters in patients with chronic lymphocytic leukemia and normal controls. Blut. 1985;50:243–248. doi: 10.1007/BF00320301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.