

Abstract
Adoptive T cell therapy (ACT) in combination with lymphodepleting chemotherapy is an effective strategy to induce the eradication of tumors, providing long-term regression in cancer patients. Despite that lymphodepleting regimens condition the host for optimal engraftment and expansion of adoptively transferred T cells, lymphodepletion concomitantly promotes immunosuppression during the course of endogenous immune recovery. In this study, we have identified that lymphodepleting chemotherapy initiates the mobilization of hematopoietic progenitor cells that differentiate to immunosuppressive myeloid cells, leading to a dramatic increase of peripheral myeloid-derived suppressor cells (MDSCs). In melanoma and lung cancer patients, MDSCs rapidly expanded in the periphery within 1 week after completion of a lymphodepleting regimen and infusion of autologous tumor-infiltrating lymphocytes (TILs). This expansion was associated with disease progression, poor survival, and reduced TIL persistence in melanoma patients. We demonstrated that the interleukin 6 (IL-6)-driven differentiation of mobilized hematopoietic progenitor cells promoted the survival and immunosuppressive capacity of post-lymphodepletion MDSCs. Furthermore, the genetic abrogation or therapeutic inhibition of IL-6 in mouse models enhanced host survival and reduced tumor growth in mice that received ACT. Thus, the expansion of MDSCs in response to lymphodepleting chemotherapy may contribute to ACT failure, and targeting myeloid-mediated immunosuppression may support anti-tumor immune responses.
Keywords: adoptive cell therapy, cancer immunotherapy, lymphodepletion, cyclophosphamide, fludarabine, myeloid derived suppressor cells
Graphical Abstract
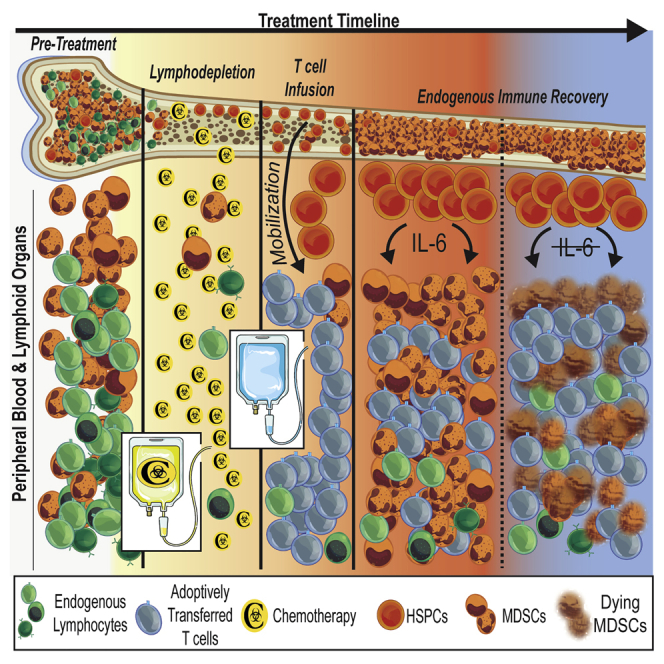
Preparative lymphodepletion transiently reduces the frequency of endogenous leukocytes prior to the infusion of tumor-specific T cells, but the ensuing impact of immune recovery is unclear. In this study, Innamarato et al. report that lymphodepleting chemotherapy promotes immunosuppressive myelopoiesis that restricts the anti-tumor efficacy of adoptive T cell therapy.
Introduction
The success of adoptive T cell therapy (ACT) using tumor-infiltrating lymphocytes (TILs), chimeric antigen receptor (CAR)-T cells, or T cells modified to express a transgenic T cell receptor (TCR) is strongly dependent on pre-conditioning patients with lymphodepleting chemotherapy prior to infusion.1,2 Lymphodepleting regimens eliminate endogenous lymphocytes that compete for homeostatic cytokines, including interleukin 2 (IL-2), IL-7, and IL-15, which are required for the persistence and function of infused T cells.3,4 Patients who undergo cyclophosphamide-fludarabine lymphodepleting regimens concurrent with ACT exhibit increased overall survival and enhanced persistence of infused T cells.2,5,6 While lymphodepleting regimens are required to maximize clinical benefit, most melanoma patients fail to exhibit complete and durable responses to ACT with TILs.7,8 Hence, an understanding of immunosuppressive mechanisms and other modalities of therapeutic failure need to be established in ACT settings to maximize clinical responses.
Our group and others have reported that lymphodepletion by total body irradiation or non-myeloablative chemotherapy initiates the rapid expansion of highly immunosuppressive myeloid-derived suppressor cells (MDSCs).9,10 MDSCs are a heterogeneous population of immature myeloid cells, including polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs), that expand under altered physiologic states associated with infectious diseases and malignancy, but also function in the settings of pregnancy, transplantation, obesity, and aging.11 Despite the phenotypic resemblance to normal physiologic neutrophils and monocytes, PMN-MDSCs and M-MDSCs potently suppress T cells, promote metastatic progression, and stimulate angiogenesis, which are ultimately associated with disease progression and poor survival in cancer patients.12, 13, 14, 15 Nevertheless, the mechanisms dictating the function and accumulation of MDSCs during immune recovery after lymphodepleting chemotherapy are not well known. Moreover, the consequences of MDSC expansion in human cancer patients receiving ACT have been largely unexplored.
In addition to its use for lymphodepleting regimens in patients receiving ACT and its use as a tumor-cytotoxic agent, cyclophosphamide has long been used to mobilize hematopoietic stem and progenitor cells (HSPCs) for autologous hematopoietic stem cell transplantation (HSCT).16 Subsequently, an increased frequency of granulocytes occurs shortly after mobilization treatment regimens.17,18 This process has been described as “reactive” or “emergency” granulopoiesis and myelopoiesis, which is characterized by the rapid egress of bone marrow (BM)-resident neutrophils and monocytes to the periphery coinciding with the mobilization of HSPCs.19 However, emergency myelopoiesis is widely described in non-malignant settings in response to pathologic microbial stimuli, and the immunosuppressive capacity of myeloid cells that accumulate during this immunologic state has not been evaluated.20, 21, 22, 23 In ACT settings, combinatorial approaches with HSCT have been successfully used to treat patients with hematologic malignancies.24,25 In contrast, the benefit of combining ACT with HSCT in solid tumors settings, such as melanoma, remains unclear.26 Consequently, the specific mechanisms regulating the differentiation of immune cells from HSPCs and their ensuing impact on anti-tumor immune functions during the course of ACT for the treatment of cancer remain unknown.
In this study, we aimed to determine how lymphodepletion directly regulates the accumulation of MDSCs. We sought to identify strategies to overcome this detrimental immunosuppressive effect for the purpose of promoting anti-tumor responses evoked by adoptively transferred T cells. Herein, we describe that lymphodepleting chemotherapy leads to the expansion of hematopoietic progenitor cells that exhibit a myeloid-differentiation bias, leading to the rapid accumulation of MDSCs. Furthermore, we show that IL-6 acts as a critical differentiation factor during lymphodepletion recovery, which improves cell survival and imparts resistance to Fas-induced cell death.
Results
The Accumulation of Myeloid Cells Reduces the Therapeutic Efficacy of ACT with TILs
We examined myeloid cell recovery in melanoma patients after treatment with a lymphodepleting regimen and infusion of autologous TILs. As expected, patient peripheral blood mononuclear cell (PBMC) counts reached their nadir (day 0) upon completion of a cyclophosphamide-fludarabine (Cy/Flu) lymphodepleting chemotherapy regimen, which rebounded after infusion with autologous TILs (Figure 1A). Despite the rebound of total PBMC counts, the proportionality of myeloid cells significantly increased in comparison to their respective pre-treatment frequencies (Figures 1B–1D; Figure S1). We found that nearly all patients exhibited increases in CD11b+ myeloid cells and subsets, CD11b+histocompatibility leukocyte antigen (HLA)-DR−/lowCD14− cells and CD11b+HLA-DR−/lowCD14+ cells (M-MDSCs), 1 week after TIL infusion (Figures 1B–1D). Similar trends were observed when we examined whole cell numbers in PBMCs (Figure S2). We confirmed that these myeloid cell populations suppressed T cell proliferation (Figure 1E). Additionally, M-MDSCs isolated from week 1 post-TIL infusion PBMCs suppressed autologous TIL proliferation in response to αCD3/αCD28 stimulation and autologous tumor stimulation (Figures 1F and 1G). We confirmed that PMN-MDSCs (CD11b+CD15+LOX-1+ cells) were elevated in two melanoma patients at week 1 post-TIL infusion (Figures 1H and 1I; Figure S2).15 PMN-MDSCs and M-MDSCs purified from week 1 PBMCs suppressed autologous TIL production of interferon γ (IFN-γ) (Figure 1I). Furthermore, we confirmed that MDSCs collected at pre-treatment or at week 2 post-TIL infusion potently suppressed donor T cell and TIL proliferation (Figure S3). We confirmed these findings by detecting increases of PMN-MDSCs after lymphodepletion and TIL infusion in two non-small-cell lung cancer (NSCLC) patients (Figure S4).
Figure 1.

Immunosuppressive Myeloid Cells Expand during ACT with TILs in Human Cancer Patients
(A) Reduction and recovery of the number of PBMCs in response to lymphodepletion and TIL infusion in melanoma patients (n = 10). (B–D) The frequency of (B) CD11b+ myeloid cells, and subsets (C) CD11b+HLA-DR-/lowCD14- cells, and (D) CD11b+HLA-DR-/lowCD14+ cells were determined by flow cytometry prior to and after treatment with lymphodepleting chemotherapy and TIL infusion in melanoma patients (n = 21). (E) Suppression of donor T cell proliferation in co-cultures with indicated cell subsets isolated from patient PBMCs. (F and G) Suppression of TIL proliferation in co-cultures with autologous M-MDSCs with CD3/CD28 stimulation (F) or cultures with autologous tumor cells (G). (H) Frequency of PMN-MDSCs determined at day −14 and day 7 post-TILs. (I) Suppression of autologous TIL IFN-γ production by the patient’s CD14+ cells and PMN-MDSCs collected from week 1 post-TIL PBMCs. In (B)–(D), p values were determined by a paired two-tailed t test. In (E)–(I), p values were determined by one-way ANOVA or a two-tailed t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. In (E)–(I), individual data points represent technical replicates. Error bars represent standard error of mean (SEM).
Because MDSCs were significantly elevated immediately after TIL infusion, we hypothesized that the abundance of MDSCs would be associated with clinical responses. Retrospective analysis revealed that higher frequencies of CD11b+ myeloid cells (greater than the median of 41%) detected at week 1 post-TIL infusion were associated with worse progression-free survival (PFS) and overall survival (OS) (Figure 2A; Figure S5). We identified similar survival trends when we examined survival associations with myeloid cell subsets (CD11b+HLA-DR−/lowCD14− cells and M-MDSCs) (Figures 2B and 2C). Additionally, an increased ratio of the number of CD11b+ cells relative to CD8+ T cells detected at week 1 post-TIL infusion was associated with poor survival (Figure 2D; Figures S5D and S5E). We identified that the number of TILs infused in each melanoma patient did not correlate with the frequency of CD11b+ cells, suggesting that the magnitude of myeloid cell accumulation after lymphodepletion is independent of TIL infusion (Figure 2E). Likewise, the frequency of CD11b+ cells in patients prior to receiving lymphodepletion and ACT with TILs did not have an impact on PFS, suggesting that the pre-infusion myeloid cell abundance did not predispose patients to worse outcomes (Figure 2F). Because therapeutic responses to treatment with ACT with TILs are associated with the persistence of infused T cells,27,28 we used TCRβ sequencing to identify persistent TIL clones after infusion. Strikingly, the frequency of infused TILs at week 6 was inversely correlated with the frequency of CD11b+ cells at week 1 post-TIL infusion (Figure 2G). Next, we examined the persistence of the top 50 TIL clones from the time of infusion to 6 weeks post-infusion (Figures 2H and 2I). Overall, the proportion of the top 50 TIL clones among the total T cell pool varied greatly among patients (15.6%–96%). Moreover, the sum frequency of the top 50 clones was reduced in nearly all patients by week 6, which was likely due to a dilution caused by the reconstitution of T cell clones that were not present within the infusion product (i.e., endogenous T cell clones) (Figure S6). Intriguingly, the persistence of the top 50 TIL clones was negatively associated with the frequency of CD11b+ cells at week 1 post-TIL infusion. Specifically, patients who had a frequency of myeloid cells above the median (>41% CD11b+) at week 1 post-TIL infusion (CD11bhigh) exhibited a greater reduction in the frequency of the top 50 TIL clones compared to patients who had <41% CD11b+ myeloid cells at week 1 post-TIL infusion (CD11blow) (Figure 2I). We next divided the patients from Figure 2G into two groups to interrogate survival analysis based on both the frequency of TILs at week 6 post-infusion and the matched frequency of CD11b+ cells at week 1 post-infusion. The median sum of TIL frequency was 0.66 among patients (>0.66, TILhigh; <0.66, TILlow). We determined that the combined metrics encompassing both TIL frequency and CD11b+ cell frequency revealed that PFS and OS was poor in patients who exhibited low TIL persistence and a high frequency of CD11b+ cells (TILlowCD11bhigh) in comparison to patients who exhibited high TIL persistence and a low frequency of CD11b+ cells (TILhighCD11blow) (Figures 2J and 2K). Collectively, these data demonstrate that the accumulation of immunosuppressive myeloid cells during treatment limits the efficacy of ACT with TILs, potentially by reducing the persistence of adoptively transferred tumor-specific T cells.
Figure 2.

Myeloid Cell Expansion after Lymphodepletion Is Associated with Poor Patient Outcomes
(A–C) Kaplan-Meier curves of PFS in melanoma patients who received ACT with TILs in relation to the frequency of (A) CD11b+ cells, (B) CD11b+HLA-DR-/lowCD14-, and (C) M-MDSCs in week 1 post-TIL PBMCs. (D) Kaplan-Meier curve representing PFS in association with the number of CD11b+ cells relative to CD8+ T cells in week 1 post-TIL PBMCs. (E) Correlation of the number of TILs infused with the frequency of myeloid cells in week 1 post-TIL patient PBMCs (n = 25). (F) Kaplan-Meier curve of PFS in patients with respect to their pre-treatment myeloid cell abundance. (G) Correlation between sum TIL frequency determined by TCRβ sequencing and myeloid cell frequency in PBMCs (n = 23). (H) Sum frequency of the top 50 TIL clones within each patient’s TIL infusion product and week 6 post-TIL infusion. (I) Fold reduction of TIL frequency from the time of infusion to week 6 post-TIL. (J and K) Kaplan-Meier curves of (J) PFS and (K) OS in patients determined by week 6 TIL frequency in relationship to week 1 CD11b frequency calculated from (H). Statistical data for linear regressions or analysis of Kaplan-Meier plots are shown. In (I), the p value was determined by a two-tailed t test with Welch’s correction. Error bars represent standard deviation (SD).
MDSCs Rapidly Expand after Treatment with Lymphodepleting Chemotherapy in Mice
The use of Cy/Flu-based regimens are widely applied clinically to induce lymphodepletion for ACT.7,29 To examine endogenous immune cell reconstitution, we treated tumor-bearing mice with lymphodepleting doses of Cy/Flu and examined the frequency of immune cells in the spleen and BM at multiple time points. At early time points, the cellularity of spleens and BM remained well below baseline, while at 7 days post-lymphodepletion, total cell numbers were similar to non-treated (NT) mice (Figures 3A–3C). However, the frequency of myeloid cells was dramatically increased in the spleens and BM of lymphodepleted (LD) mice (Figures 3D–3F). As expected, the number of T cells remained significantly depleted at day 7 post-lymphodepletion, while CD11b+Ly6ChighLy6G− M-MDSCs and CD11b+Ly6C+Ly6G+ PMN-MDSCs were present at 4- to 5-fold higher levels than in untreated mice (Figures 3F–3I). A similar trend was exhibited in the BM (Figures 3J and 3K). Furthermore, sorted Gr-1+ cells from the spleens of NT and LD mice suppressed T cell proliferation and IFN-γ production (Figures 3L and 3M). We validated these results in mice bearing Panc02 tumors and confirmed that T cells were effectively depleted, while MDSCs expanded upon lymphodepleting chemotherapy treatment (Figures 3N and 3O) Taken together, these data show that lymphodepleting chemotherapy induces the accumulation of immunosuppressive myeloid cells.
Figure 3.

Lymphodepleting Chemotherapy Induces the Expansion of MDSCs in Mice
(A and B) Whole-cell numbers from spleens (A) and bone marrow extracted from the femurs and tibias (B) from NT or LD B16 tumor-bearing mice. (C) Fold change of whole-cell numbers compared to control NT mice from four independent experiments. (D and E) Representative frequency of CD11b+ cells of total live cells in the spleen (D) and BM (E). (F) Representative gating strategy of MDSC subsets in mice. (G–I) Whole-cell numbers of indicated immune cell populations in the spleens at respective time points. (G and H) Representative experiments showing the depletion of T cells (G) and expansion of MDSC subsets (H) (n = 3–4 mice per group). (I) Fold change of whole-cell numbers compared to control NT mice from four independent experiments. (J) Representative experiment showing MDSC expansion in BM after lymphodepletion. (K) Fold change of whole-cell numbers of MDSCs within BM compared to control NT mice from four independent experiments. (L and M) Suppression of pmel T cell proliferation determined via 3H incorporation (L) and IFN-γ production (M) after co-culture with MDSCs from NT or LD mice. (N) The frequencies of CD3+ T cells and CD11b+ myeloid cells were determined in NT mice and LD mice bearing Panc02 tumors 7 days after lymphodepletion. (O) OT-I T cell proliferation determined via 3H incorporation in cultures with MDSCs from NT mice and LD mice bearing Panc02 tumors. In (L)–(O), data are representative of two to three independent experiments with biological replicates shown. In (I) and (K), each data point is the average fold change from an independent experiment (n = 3–5 mice per group per experiment). p values were determined by a two-tailed t test. Error bars for (C), (I), and (K) represent SD. Errors bars represent SEM for remaining figures.
MDSCs Differentiate from Mobilized Hematopoietic Progenitor Cells in Mice and Humans
We hypothesized that the elevated frequency of MDSCs post-lymphodepletion may arise from an increased number of circulating myeloid progenitor cells that mobilize from the BM because cyclophosphamide can mobilize HSPCs.30 We found that lineage (Lin)−c-kit+Sca-1+ and Lin−c-kit+Sca-1- HSPCs were dramatically increased in the spleens 7 days after lymphodepletion but were depleted 1 day after treatment compared to NT mice (Figures 4A–4C; Figure S7). In contrast, BM HSPCs remained depleted after treatment, suggesting that the progenitor cells egressed from the BM and remained in the periphery (Figure 4C). Likewise, Lin−c-kit+Sca-1−CD16/32−/lowIL-7R− common myeloid progenitors (CMPs) and Lin−c-kit+Sca-1−CD16/32highIL-7R− granulocyte-macrophage progenitors (GMPs) were increased in the spleen at day 7 post-lymphodepletion when mice also exhibited peak MDSC expansion (Figure 4D). To examine the ability of mobilized progenitors to differentiate to MDSCs, we adoptively transferred HSPCs collected from LD congenic CD45.1+ tumor-bearing mice to CD45.2+ recipient tumor-bearing mice that were left untreated or given lymphodepleting Cy/Flu before HSPC transfer (Figures 4E–4K). As expected, LD recipient mice exhibited increases in endogenous CD45.1−CD11b+ cells and CD45.1− HSPCs and reductions in CD45.1− lymphocytes in comparison to NT recipient mice (Figure 4F). The total frequency of donor CD45.1+ cells was similar between NT and LD recipient mice 7 days after transfer (Figure 4G). However, most donor CD45.1+ HSPCs differentiated to CD11b+ cells in LD recipient mice (Figures 4H and 4I). Inversely, lymphocytes (CD3+CD19+NK1.1+CD11b−Ly6C−Ly6G−c-kit−) predominated the proportion of CD45.1+ donor-derived cells in NT recipient mice (Figure 4I). Furthermore, the ratio of CD11b+ cells relative to lymphocytes that differentiated from donor CD45.1+ HSPCs was significantly elevated in LD recipient mice, indicating that the mobilized progenitors preferentially differentiate to myeloid cells in a LD environment (Figure 4J). Indeed, M-MDSCs and PMN-MDSCs derived from donor mice were more prevalent in LD recipient mice compared to NT recipient mice (Figure 4K).
Figure 4.

Lymphodepleting Chemotherapy Mobilizes Hematopoietic Progenitors That Differentiate to MDSCs
(A) Representative dot plots of Lin−c-kit+ cells in spleens in untreated mice or day 7 post-lymphodepletion-treated mice. (B–D) The numbers of indicated HSPC populations were determined in NT mice and LD mice within spleens (B and D) or BM (C) at day 1 or day 7 after lymphodepletion. (E) Experimental design for (F)–(K); n = 10 mice per group. (F) Frequency of CD45.1− endogenous leukocytes in recipient mice after adoptive transfer. (G) Frequency of total donor CD45.1+ cells in the spleens of recipient mice. (H) Representative dot plots showing the percentage of myeloid cells and lymphocytes among donor-derived cells. Arrow indicates directionality of subgating with the frequency of parent gates indicated. (I) Frequency of donor-derived cells in recipient mice after adoptive transfer. (J) Ratio of the number myeloid cells in relationship to lymphocytes that differentiated from donor CD45.1+ cells. (K) Proportion of donor cells that differentiated to MDSCs in recipient mice. (L) Whole-cell number of Lin−CD34+ cells in the blood of melanoma patients who received ACT with TILs. (M) Frequency of Lin−CD34+ of total live cells in one patient at pre-treatment and week 1 after TIL infusion. (N) Allogeneic myeloid suppressor cells were differentiated from Lin−CD34+ cells from (M) and co-cultured with TILs cultured with autologous tumor cells. Suppressor cells were conditioned with or without TCM. IFN-γ production was determined by ELISA after 48 h of culture. In (A)–(D), data are reflective of four individual experiments (n = 4–5 mice per group). In (E)–(K), data representative of two individual experiments are shown. Biological replicates are shown. p values were determined by a two-tailed Student’s t test. Error bars represent SEM. ND, not detected
In human samples, we confirmed that HSPCs (Lin−CD34+ cells) were increased in melanoma patients at week 1 post-TIL infusion compared to pre-infusion levels (Figure 4L; Figure S8). In addition to an increased abundance of Lin−CD34+ cells at week 1 post-infusion, we determined that the phenotype of HSPCs changed after TIL infusion. Nearly, all Lin−CD34+ cells were also CD38+, co-expressing CD45RA and/or CD90, and the abundance of CD34+CD38+CD90+ triple-positive cells increased in patients at the week 1 post-TIL blood draw compared to the pre-treatment PBMCs (Figure S8). We confirmed that immunosuppressive myeloid cells could be generated from mobilized CD34+ cells collected from a melanoma patient who received ACT with TILs (Figure 4M; Figure S9). In this melanoma patient, the frequency of CD34+ cells in PBMCs dramatically increased from 0.11% at pre-infusion to 10.8% at week 1 post-TIL infusion (Figure 4M). We differentiated the CD34+ cells using tumor-conditioned media (TCM) in addition to CC110 (SCF, TPO, Flt3L) in combination with granulocyte colony-stimulating factor (G-CSF) and successfully generated cells resembling CD33+CD15+ PMN-MDSCs displaying elevated expression of CD11b, LOX-1, CD14, and PD-L1 in response to treatment with TCM (Figure S9). To determine the suppressive capacity of these cells, the tumor cells used to produce the TCM were co-cultured with autologous TILs and the in vitro-generated suppressor cells at varying ratios. As expected, TILs produced IFN-γ when cultured with the tumor cells alone. However, the CD15+ suppressor cells generated in the presence of TCM exhibited an enhanced capacity to inhibit IFN-γ production in TIL/tumor co-cultures in comparison to the suppressor cells generated without TCM (Figure 4N). Taken together, these results confirm our findings in murine models demonstrating that lymphodepletion-mobilized HSPCs give rise to immunosuppressive myeloid cells.
IL-6 Promotes MDSC Activity after Lymphodepletion Treatment
To identify factors that contribute to MDSC accumulation after lymphodepletion, we performed RNA sequencing on PMN-MDSCs and M-MDSCs from NT and LD mice (Figure S10). Ingenuity Pathway Analysis (IPA) revealed that multiple functions were enriched due to changes induced by lymphodepletion, including cellular movement, hematological system development and function, cell cycle, cell death, and survival (Figure S10). We then enriched our dataset using IPA upstream regulator analyses and focused on cytokines as regulators (Figures 5A and 5B). Because IL-6 enhances immunosuppressive functions and differentiation in MDSCs,31,32 we investigated the role of IL-6 on MDSCs during lymphodepletion recovery. Indeed, the transcriptional alterations induced by lymphodepletion were predicted to be regulated by multiple cytokines, including IL-6, in both PMN-MDSCs and M-MDSCs collected from LD mice compared to NT mice (Figures 5A and 5B). Likewise, IL-6 was more abundant in the plasma of melanoma patients receiving ACT with TILs at week 1 post-TIL infusion (Figure 5C). To determine the role of IL-6 in ACT models, we lymphodeleted wild-type (WT) and IL-6 knockout (IL-6KO) mice bearing B16 tumors and measured the frequency of MDSCs. We observed significant increases of MDSCs in LD mice, but no difference was observed between WT and IL-6KO mice (Figure 5D). However, the ability of MDSCs from IL-6KO LD mice to suppress T cell proliferation was dampened (Figure 5E). We confirmed these results in mice with a conditional knockout of the IL-6 receptor (IL-6R) in myeloid cells (IL-6RM-KO) (Figure S11). Since MDSCs from LD knockout mice were less immunosuppressive, we hypothesized that ACT would be more efficacious in IL-6KO recipient mice. Indeed, IL-6KO recipient mice exhibited reduced tumor growth and improved survival after ACT in comparison to WT recipient mice (Figures 5F and 5G). The improved efficacy was associated with an increased frequency of circulating donor CD90.1+ pmel T cells (Figure 5H). Similarly, the treatment of ACT in combination with IL-6 receptor blocking antibodies (αIL6R) significantly reduced tumor growth in comparison to mice that received ACT alone (Figures 5I and 5J). To confirm MDSC-mediated suppression of ACT efficacy, MDSCs were purified from donor WT and IL-6KO LD mice and co-transferred with pmel T cells (Figures 5K–5M). Despite an initial reduction in tumor growth, recipient mice that received a co-transfer of pmel T cells with WT MDSCs exhibited a significant acceleration of tumor growth compared to mice that received T cells alone, whereas mice co-transferred with IL-6KO MDSCs and T cells exhibited a similar control of tumor growth compared to mice that received T cells alone (Figure 5L). Moreover, the percentage change in tumor growth from the time of infusion was greatest in mice that received pmel T cells and WT MDSCs, while tumor growth was stabilized or reduced in most recipient mice that received T cells alone or T cells co-transferred with IL-6KO MDSCs (Figure 5M). Collectively, our results demonstrate that IL-6 regulates the suppressive capacity of MDSCs that accumulate after lymphodepletion and that the efficacy of ACT is enhanced upon blockade of IL-6 signaling.
Figure 5.

IL-6 Promotes the Immunosuppressive Capacity of MDSCs Induced by Lymphodepletion
(A and B) IPA upstream regulator analysis enriched in (A) PMN-MDSCs and (B) M-MDSCs from LD mice over PMN-MDSCs and M-MDSCs taken from untreated mice. (C) IL-6 concentration in plasma samples obtained from melanoma patients receiving ACT with TILs (n = 12). (D) MDSC frequency in the spleens of WT and IL-6KO mice. (E) Pmel T cell proliferation determined by 3H-thymidine incorporation in cultures with MDSCs were purified from NT or LD WT and IL-6KO mice. (F and G) Tumor growth (F) and survival (G) after lymphodepletion with and without ACT with pmel T cells in mice with B16 tumors among WT or IL-6KO recipient mice (n = 8–10 mice per group). (H) Donor pmel T cell frequency in blood of mice from (F) and (G). (I) Tumor growth in B16 tumor-bearing LD mice treated with ACT with or without αIL6R blocking antibodies (n = 8–10 mice per group). (J) Waterfall plot showing percentage change in tumor growth in mice from (I) at the termination of the experiment. (K) Experimental design for (L)–(M). Recipient mice received 2.5 × 106 pmel T cells alone or in combination with 2.5 × 106 MDSCs from LD WT or IL-6KO donor mice. (L) Tumor growth for individual mice is shown. (M) Waterfall plot showing percentage change in tumor growth in mice from (L) at the termination of the experiment. In (D) and (E), p values were determined by a paired two-tailed Student’s t test. Data are representative of two independent experiments. Tumor growth curve p values were determined by two-way ANOVA. Data are representative of two independent experiments. Data points represent biological replicates. Error bars represent SEM.
Lymphodepleting Chemotherapy Induces the Production of IL-6 in BM
We established that IL-6 regulates the suppressive capacity of MDSCs in LD mice (Figure 5). To evaluate IL-6 signaling during recovery after lymphodepletion, we measured phosphorylated STAT3 (pSTAT3) in myeloid cells (Figures 6A and 6B). At peak expansion (day 7 post-lymphodepletion), the abundance of pSTAT3+ cells was reduced in myeloid cells from LD mice. In contrast, the percentage of pSTAT3+ cells were elevated in myeloid cells at day 1 post-lymphodepletion (Figure 6B). In response to ex vivo IL-6 stimulation, pSTAT3 was elevated at day 1 and day 3 post-lymphodepletion compared to cells from NT mice, suggesting that IL-6 may be acting on myeloid cells soon after chemotherapy treatment rather than at peak expansion (Figure 6B). We defined that CMPs, GMPs, M-MDSCs, and PMN-MDSCs expressed IL-6R (Figure 6C; Figure S12A). However, the expression of the IL-6R was elevated in CMPs and GMPs in the BM of LD mice at day 1 post-lymphodepletion, while MDSCs in the spleens or BM did not exhibit any change in IL-6R expression compared to NT mice (Figure 6D; Figures S12B and S12C). We then identified that BM CD11b+ cells were the primary producers of IL-6 in NT or LD mice with B16 tumors, and the proportion of IL-6+ cell subsets was similar between both groups (Figures 6E and 6F). However, all BM-derived cells produced more IL-6 within 24 h after lymphodepletion both with and without the presence of ex vivo lipopolysaccharide (LPS) stimulation (Figures 6G and 6H). However, the expression values for lymphocytes and CD45−Lin− cells were lower compared to CD11b+ and HSPCs cells. Nevertheless, both CD11b+ cells and Lin−c-kit+ cells had an enhanced production of IL-6 after lymphodepletion (Figure 6H). Taken together, our data show that lymphodepleting chemotherapy induces the expression of IL-6 in BM-derived cells, which may drive the function of MDSCs.
Figure 6.

IL-6 Expression Is Induced by Lymphodepleting Chemotherapy
(A and B) pSTAT3 expression was determined by flow cytometry among splenic myeloid cells collected from NT or LD mice treated with or without ex vivo IL-6 stimulation. (A) Representative histogram with mean fluorescence intensity (MFI) values indicated adjacent to each histogram. (B) Summarized data from a representative experiment. (C) Representative histogram showing IL-6R expression among BM CMPs and GMPs from NT or LD mice at day 1 after lymphodepletion. (D) Percentage of IL-6R+ CMPs and GMPs at day 1 and day 7 after lymphodepletion. (E) Experimental design for (F)–(H). Bone marrow was collected from NT or LD B16-bearing mice and cultured for 24 h. (F) Stacked bar chart of intracellular IL-6 expression by CD11b+ myeloid cells, Lin−c-kit+ HSPCs, lymphocytes (CD3+/CD19+/NK1.1+), and CD45−Lin− cells as determined by flow cytometry. (G) Representative histograms showing IL-6 expression among individual BM cell subsets with or without ex vivo LPS stimulation. MFI is indicated on the right of each histogram. (H) Fold change in IL-6 expression among individual BM cell subsets. In (A)–(D), data are representative of two to three independent experiments. In (H), normalized data are a compilation of three independent experiments with biological replicates shown. Data were normalized to NT or NT+LPS. p values were determined by a two-tailed Student’s t test. In (B) and (D), errors bars represent SEM. In (H), error bars represent SD.
IL-6 Regulates the Survival and Resistance to Fas-Induced Apoptosis in Lymphodepletion-Induced MDSCs
To elucidate the effect of IL-6 signaling on the suppressive capacity of MDSCs in LD mice, we performed RNA sequencing on MDSC subsets sorted from WT and IL-6KO NT and LD mice (Figure S13). In both MDSC subsets, there were few transcriptional differences between MDSCs from WT NT mice in comparison to MDSCs from IL-6KO NT mice. In M-MDSCs, 600 gene transcripts calculated to be statistically significant were upregulated or downregulated among M-MDSCs from LD mice in comparison to M-MDSCs from WT NT mice. However, there was little difference in M-MDSCs when we compared cells from WT LD mice to IL-6KO LD mice (Figure S13A). In contrast, 146 gene transcripts calculated to be statistically significant were upregulated or downregulated among PMN-MDSCs from WT LD mice in comparison to PMN-MDSCs from IL-6KO LD mice, suggesting that IL-6 may have a broader impact in the development of PMN-MDSCs (Figure S13B). We next performed gene set enrichment analysis on PMN-MDSCs and identified that pathways and functions associated with apoptosis were significantly enriched in PMN-MDSCs from WT LD mice in comparison to PMN-MDSCs from IL-6KO LD mice (Figures S13G and S13H). Indeed, LD mice exhibited an increased frequency of viable MDSCs compared to mice with no treatment (Figure 7A). Moreover, MDSCs from LD mice were sensitive to apoptosis mediated by T cells, which was blocked by the addition of a Fas ligand (FasL) blocking antibody (αFasL) (Figure 7B). Because MDSCs can undergo Fas-induced apoptosis,33 we next compared the survival of MDSCs from WT and IL-6KO mice. First, we observed that Fas expression was reduced in MDSCs from WT LD mice compared to WT NT mice. Conversely, IL-6KO MDSCs from LD mice exhibited an increase in Fas expression compared to WT MDSCs from LD mice (Figures 7C and 7D). Next, we cultured MDSCs from WT and IL-6KO mice with Fas agonistic antibodies (αFas). As expected, MDSCs from WT LD mice exhibited an increased percentage of live cells compared to MDSCs from NT mice after culture, even in the presence of αFas. In contrast, the percentage of live cells from IL-6KO LD mice was reduced compared to MDSCs from WT LD mice both with and without treatment with αFas (Figure 7E). In addition, similar results were observed in LD mice treated in vivo with a JAK/STAT3 inhibitor, JSI-124, compared to mice treated with a vehicle control (Figure S14). We then determined that the frequency of myeloid cells was reduced after treatment with lymphodepleting chemotherapy followed by infusion with CD90.1+ pmel T cells (Figure 7F). Specifically, M-MDSCs and PMN-MDSCs were reduced in the blood and spleens of LD IL-6KO recipient mice compared to WT recipient mice (Figures 7G–7I). Furthermore, the ratio of PMN-MDSCs to T cells was reduced in the spleens of IL-6KO recipient mice in comparison to WT recipient mice (Figure 7J). Taken together, these data indicated that IL-6 regulates MDSC survival and Fas expression after lymphodepletion treatment.
Figure 7.

IL-6 Reduces Fas Expression in MDSCs That Expand after Lymphodepletion
(A) Annexin V and PI staining among MDSCs cultured for 48 h. (B) Frequency of live MDSCs from LD mice after 24 h of co-culture with T cells with or without FasL blockade. (C and D) Expression of Fas in PMN-MDSCs. Compiled data from three independent experiments with biological replicates (C) with representative histogram (D) are shown. (E) Frequency of live PMN-MDSCs cultured for 24 h with isotype antibodies or αFas. (F) Experimental design for (G)–(J). (G) Representative dot plots showing donor CD90.1+ pmel T cells and reduced CD11b+ cells in IL-6KO recipients 24 h after infusion. (H and I) Frequency of MDSCs in blood (H) and spleen (I) 24 h after ACT. (J) PMN-MDSC/T cell ratio in the spleens of mice 24 h after ACT. Data are reflective of two to three independent experiments. p values were determined by a two-tailed t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. In (C), error bars represent SD. Error bars represent SEM in remaining figures.
IL-6 Signaling during Progenitor Differentiation to MDSCs Is Essential for the Regulation of Fas Expression and Resistance to Apoptosis
In cells from IL-6R conditional knockout mice, we observed that the rescue of IL-6R signaling did not promote the immunosuppressive capacity of MDSCs (Figure S11). Moreover, exogenous IL-6 did not enhance the ability of human PMN-MDSCs to suppress autologous TILs (Figure 8A; Figure S15). Hence, we postulated that IL-6 may be most relevant during the differentiation of MDSCs from mobilized progenitors. To test our hypothesis, we collected mobilized HSPCs from LD mice and differentiated these cells to MDSCs in vitro. MDSCs were generated in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) alone or GM-CSF in combination with IL-6 for 4 days (Figure 8B). MDSCs that were differentiated from HSPCs in the presence of GM-CSF and IL-6 exhibited reduced Fas expression compared to MDSCs differentiated with GM-CSF alone (Figures 8C and 8D). Next, we cultured the MDSCs collected on day 4 for an additional 24 h in media containing IL-6. Intriguingly, the addition of IL-6 failed to reduce Fas expression in MDSCs generated from HSPCs in the presence of GM-CSF alone or GM-CSF and IL-6, suggesting that the expression of Fas in MDSCs may be inherited from parental cells (Figure 8E). In parallel, we cultured the MDSCs collected on day 4 and exposed them to Fas agonistic antibodies with or without IL-6. Consistent with the reduction of Fas expression in MDSCs generated with GM-CSF and IL-6 observed in Figures 8C and 8D, survival was superior in response to Fas agonism compared to MDSCs generated with GM-CSF alone (Figure 8F). Notably, the addition of IL-6 to MDSCs in this post-differentiation culture only had a modest effect on the viability of cells or promoting any resistance to Fas agonism in cells differentiated in either GM-CSF alone or the combination of GM-CSF with IL-6 (Figure 8F). We validated these results by differentiating mobilized HSPCs from WT and IL-6KO mice that were treated with lymphodepleting chemotherapy with the same protocol as in Figure 8B. GM-CSF-generated MDSCs from mobilized IL-6KO HSPCs exhibited a reduced viability in T cell co-cultures in the presence of an isotype antibody and αFasL compared to MDSCs derived from WT mice (Figure 8G). In contrast, MDSC viability was similar between cells differentiated from WT and IL-6KO HSPCs with the addition of GM-CSF and IL-6. This suggests that the IL-6 signal received during differentiation conferred resistance to apoptosis even in IL-6 naive cells that were collected from IL-6KO mice (Figure 8H). Additionally, Fas expression was reduced in GM-CSF+IL-6-generated MDSCs compared to MDSCs generated with GM-CSF alone from both WT and IL-6KO progenitors (Figure 8I). Importantly, the suppressive capacity of MDSCs generated from mobilized IL-6KO HSPCs was impaired compared to MDSCs differentiated from WT HSPCs. When T cells were cultured alone, the blockade of FasL enhanced T cell proliferation. Despite this enhancement of T cell proliferation, the suppressive capacity of MDSCs was amplified upon FasL blockade. Specifically, weakly suppressive MDSCs generated from IL-6KO HSPCs became highly suppressive upon FasL blockade, indicating that the ability of MDSCs to survive during contact with T cells was critical to mediate immunosuppressive functions (Figure 8J). Taken together, these data demonstrate that IL-6 differentiation signals regulate the survival and Fas expression patterns of post-lymphodepletion MDSCs that are necessary to resist apoptotic signals from T cells and mount immunosuppressive functions.
Figure 8.

IL-6 Differentiation Signals Reduce Fas Expression and Increase Survival in MDSCs
(A) 40 ng/mL IL-6 was added to co-cultures of TILs and PMN-MDSCs collected from a melanoma patient. TIL proliferation was measured by 3H thymidine incorporation after 72 h of culture. (B) Experimental design for (C)–(F). MDSCs were differentiated with GM-CSF or GM-CSF+IL-6 from Lin−c-kit+ HSPCs collected from the spleens of LD B16 tumor-bearing mice. (C and D) Fas expression in MDSCs measured on day 4 of differentiation protocol. (C) Representative histogram showing Fas expression and (D) summarized data. (E) Differentiated MDSCs were collected on day 4 and then cultured for an additional 24 h with or without IL-6. Fas expression is shown. (F) Annexin V and PI staining was used to determine the ratio of dead/live cells after differentiated MDSCs were cultured for 24 h in the indicated conditions. (G–J) MDSCs were differentiated from Lin−c-kit+ cells as in (B). WT or IL-6KO mice were treated as in (B). (G and H) Percentage of viableMDSCs generated with (G) GMCSF or (H) GMCSF+IL-6 after 24 h of co-culture with T cells in combination with isotype antibodies for αFasL. (I) Fas expression measured in MDSCs differentiated from WT or IL-6KO progenitors in respective conditions. (J) T cell proliferation at 72 h in co-cultures with in vitro-generated MDSCs at a 1:8 ratio as in with/without FasL blockade. Dotted lines represent the mean of pmel T cell proliferation upon culture with cognate peptide with isotype antibodies (black) or FasL blocking antibodies (red). Data are reflective of two to three independent experiments. In (D)–(F), each data point is representative of cells pooled from 5 mice; 20 mice total. In (I)-(J), biological replicates are shown. p values were determined by a two-tailed t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Error bars represent SEM.
Discussion
Melanoma patient responses to ACT with TILs have been associated with T cell-intrinsic differences among patients, including the number of TILs infused, the expression of B and T lymphocyte attenuator (BTLA), in vivo T cell persistence, and the magnitude of IFN-γ production by TILs.8,28,34 Our data suggest that lymphodepleting chemotherapy induces the rapid expansion of MDSCs that ultimately impacts the function of adoptively transferred T cells, leading to diminished therapeutic efficacy and reduced TIL persistence. Importantly, the overlap of lymphodepleting regimens in patients treated with ACT with TILs, CAR-T cells, and T cells with transgenic TCRs is highly suggestive that similar mechanisms of MDSC-mediated suppression may take place in most ACT settings.1,7,35 We show that the repertoire of T cell clonotypes was diminished in patients who had high frequencies of CD11b+ myeloid cells, which was ultimately associated with poor long-term survival (Figures 2G–2K). It is feasible that the most abundant adoptively transferred TIL clones have a higher probability of interacting with and being suppressed by MDSCs. However, we cannot rule out that characteristic differences among individual TIL clones, such as the prevalence of tumor-reactive clones or phenotypic memory characteristics, could make certain T cells more susceptible to MDSC suppression.36,37 Hence, a deeper understanding of the interactions between MDSCs and adoptively transferred T cells is necessary to guide strategies to render T cells more resistant to myeloid-mediated immune suppression and promote in vivo T cell persistence.
Cyclophosphamide, a common chemotherapeutic drug used in lymphodepleting regimens, has been described to have immunomodulatory effects that promote immunosuppressive myeloid cell accumulation.10,38 In breast cancer patients, the frequency of Lin−HLA-DR−CD11b+CD33+ MDSCs increased after treatment with a doxorubicin-cyclophosphamide chemotherapy regimen,39 In mouse models, lymphodepleting doses of total body irradiation9 and cessation of treatment with Gr-1-depleting antibodies40 promoted the subsequent accumulation of myeloid cells. Hence, it is likely that non-myeloablative immunodepleting methods can lead to “reactive myelopoiesis.”19 While previous studies have shown that cyclophosphamide leads to the accumulation of MDSCs, little is known about the mechanisms that drive the expansion of MDSCs during the recovery phase after lymphodepletion treatment, particularly in human patients. Moreover, the immunosuppressive capacity of granulocytes and monocytes that accumulate in non-malignant pathological settings of emergency myelopoiesis have not been examined.20,21 Nearly all patients in our study exhibited a significant elevation of myeloid cells after lymphodepletion and ACT, which potently suppressed donor T cells and autologous TILs (Figure 1). It is known that during tumor progression, myeloid cells acquire immunosuppressive characteristics in part by a skewing that occurs during myeloid progenitor commitment.41,42 Intriguingly, we observed a strong myelopoietic bias when HSPCs were transferred to LD recipients (Figures 4I and 4J), which suggests that host-derived factors potentiate myeloid differentiation in response to lymphodepleting chemotherapy. Tumor-derived factors such as retinoic acid can skew the differentiation of M-MDSCs to macrophages at the expense of generating dendritic cells, and the administration of all-trans retinoic acid (ATRA) can reverse this effect.43,44 However, early precursors such as GMPs and CMPs need to be targeted to skew the development of terminally differentiated PMN-MDSCs.42,45 CMPs and GMPs are rare populations of cells often restricted to the BM space, which may make it a challenge to manipulate differentiation signals specific to these cell populations.46 Interestingly, the rapid increase of myeloid progenitors that we observed shortly after lymphodepleting chemotherapy treatment may provide a therapeutic window to target the differentiation of myeloid cells when myelopoietic signals are most relevant (Figures 4 and 8). Hence, the manipulation of differentiation signals and the exploitation of progenitor cell plasticity to modulate the immunosuppressive capacity of daughter cells, including MDSCs, during ACT regimens is an attractive therapeutic strategy and warrants further investigation.
IL-6 and STAT3 are known to promote the function, differentiation, and survival of MDSCs.14,31 In this study, we show that lymphodepletion prompted BM progenitor mobilization and that IL-6 is required for promoting survival signals during differentiation. Likewise, IL-6 has been reported to simultaneously drive the expansion of HSPCs, enhance myelopoiesis, and block lymphopoiesis.47,48 Notably, we describe that the role of IL-6 goes beyond providing an enhancement of immunosuppressive capabilities during the differentiation from progenitor cells.31 Our functional studies in murine models and RNA sequencing indicated that post-lymphodepletion MDSCs exhibited distinct biological and transcriptional characteristics compared to MDSCs from NT mice (Figures 7 and 8; Figures S10 and S13). The inhibition of IL-6 signals in murine models improved the efficacy of ACT by causing the dysregulation of survival signals and the sensitization of MDSCs to Fas-induced apoptosis (Figures 5, 7, and 8). In the setting of an acute induction of lymphopenia, we show that a cytokine signal, such as IL-6, is critical during progenitor differentiation. IL-6 imparted an improved survival capacity and reduced Fas expression in daughter cells during the differentiation from progenitors (Figures 8C–8F). We show that the addition of IL-6 to already differentiated MDSCs failed to improve the viability of cells or reduce Fas expression (Figure 8A). Taken together, these data indicated that survival patterns can be inherited from parental hematopoietic cells, which impact downstream immunosuppressive capabilities of MDSCs. Hence, MDSCs have the potential to be targeted uniquely in the setting of ACT by targeting factors specifically induced by lymphodepleting regimens, which can impact the differentiation trajectory of mobilized HSPCs.
We showed that lymphodepleting chemotherapy enhances IL-6 production in BM-derived myeloid cells and HSPCs early after treatment (Figures 6G and 6H). Subsequently, the IL-6 differentiation signal was critical for promoting MDSC resistance to T cell-mediated apoptosis (Figure 8J). We found that a single infusion of MDSCs along with T cells was sufficient to accelerate tumor growth in comparison to mice that received T cells alone. Importantly, the transfer of IL-6 naive MDSCs taken from IL-6KO donor mice failed to negatively impact the efficacy of ACT in IL-6 competent recipients (Figures 5L and 5M). This provides further support that IL-6 impacts MDSC function indirectly during their differentiation from progenitors, rather than on cells in a post-differentiation state. In addition to IL-6, our RNA sequencing experiments revealed that several upstream regulators, including tumor necrosis factor alpha (TNF-α) and GM-CSF (CSF2), may drive the expansion and function of MDSCs that expand after treatment with lymphodepleting chemotherapy (Figures 5A and 5B). TNF-α can promote the survival of HSPCs and promote the accumulation of myeloid cells in response to stimulation with polyinosinic:polycytidylic acid (poly I:C), LPS, or treatment with 5-fluorouracil (5-FU).49 Meanwhile, GM-CSF has been shown to promote extramedullary myelopoiesis within inflamed joints of mice with experimental spondyloarthritis.22 Interestingly, TNF-α and GM-CSF promoted a myelopoietic bias in these experimental settings, which aligns with the differentiation bias of HSPCs that we observed in LD mice (Figure 5I). While we did not examine other factors in this current study, we are actively investigating the role of other cytokines and growth factors in the regulation of lymphodepletion-driven myelopoiesis. Collectively, the determination of key host-factor signals and the characterization of myelopoietic niches during lymphodepletion-driven myelopoiesis could identify new therapeutic targets with the potential to enhance patient responses to ACT.
In patients receiving CAR-T cells, the onset of cytokine release syndrome (CRS) was attributed to the production of IL-6 and IL-1β by monocytes after CAR-T cell infusion and that the symptoms of CRS could be ameliorated by the administration of IL-6R blocking antibodies and/or IL-1 receptor antagonists.50 However, it is unclear which host factors induced by lymphodepleting chemotherapy, including IL-6 and IL-1β, are responsible for the induction of myeloid-mediated immunosuppression during ACT regimens. Notably, patients who receive ACT with TILs do not exhibit symptoms of CRS, but rather they endure toxicities related to lymphodepleting chemotherapy and/or bolus high-dose IL-2 administration.51 Thus, the role of IL-6 beyond its role in the induction of CRS remains unclear in a variety of ACT settings. However, the ongoing clinical use of IL-6R blocking antibodies in patients receiving CD19-directed CAR-T cells provides feasibility for this treatment to be used prophylactically to reduce myeloid-mediated immunosuppression in patients receiving any modality of ACT in combination with lymphodepleting chemotherapy.52 We acknowledge that immunological abnormalities associated with pre-existing cytopenias, late-onset neutropenia, and a history of HSCT, which are frequently observed in patients with hematological malignancies, could have an impact on myelopoiesis in patients receiving CD19-directed CAR-T cells.53,54 Thus, differences associated with lymphodepletion-driven myelopoiesis between patients with solid tumors and hematological malignancies remain unclear. Moreover, the impact of MDSCs on patient outcomes after CAR-T cell infusion has not been thoroughly evaluated and is an active point of investigation for our group.
This study is characterized by important limitations. For example, the small patient sample size limits the statistical power of our study. Hence, future prospective studies that analyze MDSC accumulation and its impact on the survival of patients receiving ACT are necessary. Importantly, we focused on peripheral immune suppression during lymphodepletion recovery in this study. It is well established that tumor-derived factors can enhance the suppressive capacity of MDSCs.55 We also demonstrated this by generating immunosuppressive myeloid cells from a patient’s CD34+ cells (Figures 4M and 4N; Figure S9). However, the lack of patient tumor biopsies after TIL infusion limited our ability to evaluate therapy-related changes in the tumor microenvironment. Hence, future studies in murine models and patient specimens will evaluate the differentiation and infiltration of HSPCs and myeloid cells in the tumor microenvironment after treatment with lymphodepleting chemotherapy and its ensuing impact on the efficacy of ACT.
While the therapeutic efficacy of ACT and the persistence of infused T cells are associated with lymphodepletion-induced increases of T cell homeostatic cytokines,4,6,56 we highlight an opposing mechanism in that lymphodepleting chemotherapy regimens prompt the mobilization of HSPCs followed by dramatic expansion of immunosuppressive myeloid cells. With the evidence provided in our study, we propose that the full benefits of pre-conditioning lymphodepletion regimens may be achieved by inhibiting counter-immunosuppressive reactions, potentially through skewing the differentiation of progenitor cells. Accordingly, we report that the modulation of MDSCs by blocking IL-6 differentiation signals is a feasible approach to enhance therapeutic outcomes in patients receiving ACT with TILs and provides support for future clinical trials.
Materials and Methods
Study Design
The primary research objective was to evaluate mechanisms that drive the expansion and function of immunosuppressive myeloid cells in patients receiving ACT and relevant murine models. For human specimen analysis, the data shown include all acquired data for the patient cohorts for this study. For murine experiments, animals were randomized after tumor inoculation and experiments were repeated between two and four times as noted.
Patient Samples
Patient lymphodepletion was carried out via administration of cyclophosphamide (60 mg/kg/day) and mesna (20 mg/kg), which were given intravenously (i.v.) on day −7 and day −6 relative to the anticipated TIL infusion date. Fludarabine (25 mg/m2) was given daily intravenously from day −5 to day −1. The TIL infusion was administered on day 0, and a 720,000 IU/kg intravenous bolus of IL-2 (aldesleukin, Prometheus Laboratories, San Diego, CA, USA) was given every 8–16 h for up to 15 doses, beginning 12–16 h after TIL infusion. Preparation of TIL was performed as previously described.28 Briefly, surgically resected tumors were minced to 1-mm pieces and placed into individual wells of a 24-well plate containing 6,000 IU/mL IL-2. TILs were expanded for up to 5 weeks and then tested for IFN-γ production in co-cultures with autologous tumor cell lines or cryopreserved tumor digest cell suspensions. IFN-γ+ TILs underwent a rapid expansion protocol (REP).28 For TIL-MDSC co-cultures, cryopreserved post-REP TILs were thawed and rested for 48–72 h in AIM-V (Thermo Fisher Scientific) containing 3,000 IU/mL IL-2 prior to co-culture. Myeloid cells were sorted from fresh PBMCs collected from melanoma or NSCLC patients. Sorted MDSCs were cultured with autologous TILs or donor T cells as indicated below.
Myeloid Cell Isolation for Functional Assays
For human specimens, PMN-MDSCs were purified from fresh PBMCs using CD15 microbeads (Miltenyi Biotec). M-MDSCs were purified by the negative selection PBMCs by labeling with biotinylated antibodies for CD3, CD19, CD56, and HLA-DR. The negative fraction was then labeled with CD14 microbeads (Miltenyi Biotec). MDSCs isolated from human specimens were cultured with donor T cells or autologous TILs. T cells were stimulated using Dynabeads human T-activator CD3/CD28 for T cell expansion and activation (Thermo Fisher Scientific, 11132D). When available, TILs were co-cultured with autologous tumor cell lines and varying concentrations of MDSCs. For murine specimens, MDSCs were purified from spleens using anti-Gr-1 biotinylated antibodies, anti-biotin microbeads, or streptavidin microbeads (Miltenyi Biotec) with a purity >90% after elution through magnetic columns. CD8+ T cells were purified from the spleens of pmel or OT-I mice using an EasySep mouse CD8+ T cell isolation kit (STEMCELL Technologies). Gr-1+ cells were co-cultured with pmel or OT-I T cells for 72 h in round-bottom 96-well plates. Co-cultures were incubated in media containing 1 μg/mL of cognate peptide, glycoprotein (gp)10025–33 or ovalbumin (OVA)SIINFEKL (both from AnaSpec). T cell proliferation was assessed by CellTrace Violet dilution (Thermo Fisher Scientific) or 3H-thymidine incorporation. For 3H-thymidine incorporation, 3H-thymidine was added at the final 18 h of culture and cells were harvested at 72 h. For detection of IFN-γ, supernatants were collected after 72 h of culture and concentrations were measured by ELISA (BD Biosciences or R&D Systems).
Generation of Human Melanoma Cell Line and TCM
A surgically resected melanoma tumor was subjected to a TIL expansion protocol as described above. The remaining tumor was digested in media containing collagenase (type II and type IV), hyaluronidase, and DNAase (Fisher Scientific, Pittsburgh, PA, USA) for 1 h at 37°C and mechanical dissociation by gentleMACS (Miltenyi Biotec). After digestion, the cell suspension was filtered to remove undigested tumor and connective tissue to generate a single-cell suspension. Cells were suspended in complete media (CM) containing RPMI 1640 media supplemented with 10% heat-inactivated fetal bovine serum (FBS), 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 2 mM fresh l-glutamine, 100 mg/mL streptomycin, 100 U/mL penicillin, 50 mg/mL gentamicin, 0.5 mg/mL Fungizone (all from Life Technologies, Rockville, MD, USA), and 0.05 mM 2-mercaptoethanol (2-ME) (Sigma-Aldrich, St. Louis, MO, USA) and grown to confluency. Adherent cells were passaged multiple times. To generate TCM, tumor cells were grown to confluency in CM and dissociated using enzyme-free cell dissociation buffer with PBS (Thermo Fisher Scientific). Cells were pelleted and washed twice with PBS before suspending in serum-free RPMI 1640 at 1e6 cells/mL and cultured in a 24-well plate for 24 h. Cell-free supernatant was harvested and stored at −80°C until ready for use. Cytokine concentrations were determined by LEGENDplex human (HU) essential immune response panel (13-plex) (BioLegend).
MDSC Differentiation
For murine specimens, Lin−c-kit+ cells were collected from the spleens of LD mice. 1 × 106 cells were cultured in six-well plates for 4 days in 4 mL of media containing 40 ng/mL recombinant murine GM-CSF (PeproTech) or GM-CSF in combination with 40 ng/mL recombinant murine IL-6 (PeproTech). At the end of culture, Gr-1+ cells were isolated and used for functional analysis. For human specimens, CD34+ cells were purified from cryopreserved patient PBMCs using a CD34 microbead kit, human (Miltenyi Biotec) with a purity >95%. 5 × 104 CD34+ cells were cultured in six-well plates in StemSpan serum-free expansion medium II (SFEM II) supplemented with StemSpan CC110 (STEMCELL Technologies), 100 mg/mL streptomycin, and 100 U/mL penicillin. SFEMII+CC110 media were supplemented with 40 ng/mL recombinant human G-CSF (PeproTech). CD34+ cells were cultured for the first 7 days in media containing cytokines and CC110 diluted 1:100 per the manufacturer’s recommendation. For the final 7 days of culture, CC110 was diluted 1:1,000 from the manufacturer’s stock concentration. Media were refreshed every 4 days by addition of fresh media and cytokines to each well. For the final 4 days of culture, media were refreshed containing cytokines plus 30% RPMI 1640 (vehicle) or TCM. At the end of the culture (14 days total), cells were harvested and co-cultured with TILs for functional analysis.
Mouse Models and Treatment
Female C57BL/6 mice (6–8 weeks old) were purchased from Charles River Laboratories (Wilmington, MA, USA). B6.129S2-Il6tm1Kopf/J (IL-6KO) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). To generate a conditional IL-6R knockout, B6;SJL-Il6ratm1.1Drew/J mice (The Jackson Laboratory) were bred with B6.129P2-Lyz2tm1(cre)Ifo/J mice (The Jackson Laboratory). Il6ra+/fl heterozygotes, hemizygous for Lyz2-cre, were bred with homozygous Il6rafl/fl mice to generate mice that were Il6rafl/flLyz2cre (denoted as IL-6RM-KO; M indicates myeloid). Cre− littermates (Il6rafl/fl) were used as control mice. Female B6.SJL-Ptprca Pepcb/BoyJ CD45.1+ (6–8 weeks old) mice were purchased from The Jackson Laboratory. Pmel and OT-I mice were bred and housed at the Animal Research Facility of the H. Lee Moffitt Cancer Center and Research Institute. Mice were humanely euthanized by CO2 inhalation according to the American Veterinary Medical Association guidelines. Mice were observed daily and were humanely euthanized if a solitary subcutaneous tumor exceeded 400 cm2 in area or mice showed signs referable to metastatic cancer.
Murine Cell Lines
B16 melanoma (obtained from ATCC) and Panc02 pancreatic cancer (obtained from ATCC) cell lines were cultured in CM: RPMI 1640 media supplemented with 10% heat-inactivated FBS, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 2 mM fresh l-glutamine, 100 mg/mL streptomycin, 100 U/mL penicillin, 50 mg/mL gentamicin, 0.5 mg/mL Fungizone (all from Life Technologies, Rockville, MD, USA), and 0.05 mM 2-ME (Sigma-Aldrich, St. Louis, MO, USA). The cell lines tested negative for mycoplasma contamination. All cell lines were passaged fewer than 10 times after initial revival from frozen stocks. All cell lines were validated in core facilities prior to use.
In Vitro T Cell Culture, Lymphodepletion, Adoptive Transfer, and In Vivo Treatment
T cells were isolated from the spleens of mice using a pmel EasySep mouse CD8+ T cell isolation kit (STEMCELL Technologies). Pmel T cells were cultured for 3 days in CM containing 10 IU/mL IL-2 and 5 μg/mL gp10025–33 peptide. Recipient mice with established tumors were lymphodeleted by intraperitoneal (i.p.) injection of 200 mg/kg cyclophosphamide (Baxter) followed by 100 mg/kg fludarabine (Sagent Pharmaceuticals) 24 h after cyclophosphamide injection. Adoptive transfer of 2.5 × 106 activated pmel T cells were infused intravenously via tail vein injection 24 h after fludarabine administration. For the adoptive transfer of MDSCs, B16 tumor-bearing mice with established tumors were given lymphodepleting chemotherapy. Seven days after treatment, recipient mice were lymphodeleted and MDSCs were isolated from the spleens of mice and co-transferred at a 1:1 ratio with activated pmel T cells 24 h after fludarabine administration. IL-2 (2.5e5 IU) was given intraperitoneally following T cell injection, continuing every 12 h for 3 days, for a total of six injections. Following this treatment, tumor size was measured and recorded every 3–4 days.
For the adoptive transfer of HSPCs, CD45.1+ mice with established B16 tumors were given lymphodepleting treatment. Seven days after treatment, HSPCs were purified from spleens by depleting Lin+ cells using direct lineage depletion kit (Miltenyi Biotec) followed by positive selection using CD117 microbeads (Miltenyi Biotec). Purity of Lin−c-kit+ cells was >90%. 5 × 106 CD45.1+Lin−c-kit+ cells were infused intravenously via tail vein injection to CD45.2+ C57BL/6 mice.
For IL-6R blockade, mice were administered via intraperitoneal injection 1.0 mg of anti-IL-6R antibody (15A7, Bio X Cell) 1 day prior to cyclophosphamide injection, followed by 0.5 mg of anti-IL-6R antibody every 5 days for the duration of the experiment. For JAK2/STAT3 inhibition, LD tumor-bearing mice were administered 1 mg/kg JSI-124 (Cayman Chemical) by intraperitoneal injection once daily starting 1 day prior to cyclophosphamide injection. DMSO was the carrier and was used as a vehicle control.
Flow Cytometry
Spleens and BM were harvested under sterile conditions. Spleens were homogenized by applying pressure to tissue on 100-μm cell strainers. BM was harvested by flushing media with a needle and syringe through femurs and tibias. Bones were then crushed and the resulting BM was collected. Single-cell suspensions were prepared, and red blood cells were removed using red blood cell lysis buffer (BioLegend). The resulting suspension was passed through a 70-μm cell strainer and washed once with PBS. Cells were resuspended in to a concentration of 0.5–1 × 106 cells/mL for flow cytometric analysis in fluorescence-activated cell sorting (FACS) buffer containing PBS, 5% FBS, 1 mM ethylenediaminetetraacetate (EDTA) (Sigma-Aldrich), and 0.1% sodium azide (Sigma-Aldrich). Cell viability was measured by staining cell suspensions with Zombie NIR (BioLegend). Prior to surface staining, cells were incubated with Fc Shield (Tonbo Biosciences) for murine specimens and Fc Blocker (Miltenyi Biotec) for human specimens. For surface staining of murine specimens, cells were stained in FACS buffer with the following antibodies: CD3 (145-2C11), CD4 (GK1.5), CD8 (53-6.7), CD19 (1D3), NK1.1 (PK136), CD11b (M1/70), Ly6G (1A8), Ly6C (HK1.4), F4/80 (BM8), c-kit (2B8), Sca-1 (D7), CD16/32 (93), IL-7R (A7R34), IL-6R (D7715A7) (all from BioLegend), Fas (Jo2) (BD Biosciences), and lineage cocktail (Tonbo Biosciences). For human specimens, cell surface staining was conducted with the following antibodies: CD3 (145-2C11), CD4 (RPA-T4), CD8 (RPA-T8), CD19 (HIB19), CD56 (B159), CD11c (Bly6), CD14 (MoP9), CD15 (HI98), CD11b (ICRF44), CD33 (P67-6), HLA-DR (G46-6), IL-6R (M5), CD34 (581), CD38 (HIT2), CD45RA (HI100), CD90 (5E10) (all from BD Biosciences), and LOX-1 (15C4) and PD-L1 (29E-2A3) (from BioLegend). Cells were acquired by an LSR II or FACSCelesta (BD Biosciences), and the data were analyzed with FlowJo (Tree Star).
IL-6 Detection and In Vitro Stimulation of IL-6 Signaling
IL-6 was measured in plasma samples collected from melanoma patients who received ACT with TILs at the Moffitt Cancer Center using a human IL-6 Quantikine ELISA kit (R&D Systems). For human specimens, 40 ng/mL recombinant human IL-6 (PeproTech) was added to T cell/TIL co-cultures with patient-derived MDSCs. For murine specimens, 40 ng/mL recombinant murine IL-6 (PeproTech) was added to MDSC/T cell co-cultures. Where indicated, 200 ng/mL recombinant mouse IL-6/IL-6R alpha protein chimera (R&D Systems) was added to co-cultures containing MDSCs collected from IL-6RM-KO mice. For intracellular IL-6 staining, BM cells were incubated in CM with and without 1 mg/mL LPS for 18 h in 24-well plates at a concentration of 5 × 105 cells per well. During the final 6 h of culture, 500 ng/mL brefeldin A solution (BioLegend) was added to each well. After 6 h of incubation with brefeldin A, non-adherent cells were collected. Adherent cells were collected after incubation with 1 mM EDTA solution and gentle scraping. Adherent cells were pooled with non-adherent cells. Cells were stained with Zombie NIR, Fc Shield, and cell surface markers followed by fixation and permeabilization via a Cytofix/Cytoperm kit (BD Biosciences). Cells were stained with anti-IL-6 antibodies, washed twice, and then data were acquired immediately by FACSCelesta (BD Biosciences). For pSTAT3 (pY705) staining, cells were incubated in serum-free RPMI 1640 for 30 min at 37°C. To induce STAT3 phosphorylation, cells were incubated with 100 ng/mL IL-6 (PeproTech) or 200 ng/mL recombinant mouse IL-6/IL-6R alpha protein chimera (R&D Systems). Cells were washed and stained for cells surface markers. BD Phosflow lyse/fix buffer was added to each sample for fixation followed by permeabilizaton with BD Phosflow perm buffer III. Permeabilized cells were incubated with antibodies specific for pSTAT3 for 30 min at room temperature. Cells were washed three times and then data were acquired immediately for flow cytometric analysis.
MDSC Apoptosis
Purified MDSCs were cultured for 24–48 h in 24-well plates at 5 × 105–1 × 106 cells per well in media containing 1 μg/mL purified hamster anti-mouse Fas (CD95) (Jo2, BD Biosciences) or isotype Armenian hamster immunoglobulin (Ig)G2 (BD Biosciences). In MDSC co-cultures with activated T cells, cells were incubated with 10 μg/mL purified anti-mouse CD178 (FasL) (MFL3, BioLegend) or isotype mouse IgG1 (BioLegend). Apoptosis was measured by annexin V and propidium iodide (PI) staining (Thermo Fisher Scientific) and assessed by flow cytometric analysis.
TCRβ Sequencing and Analysis
DNA extraction was performed using the DNeasy Blood & Tissue kit (QIAGEN) on post-REP TILs, apheresis samples collected 6 weeks post-TIL infusion, and patient PBMCs. Samples were subjected to TCR clonotyping, and TCRβ CDR3 regions were analyzed using the immunoSEQ analyzer (Adaptive Biotechnologies). TIL frequency was determined by identifying unique V-beta and J-beta genes identified within each sample and calculating the sum frequency of productive rearrangements among the total detected in both post-REP TILs and post-TIL infusion PBMCs. A persistent TIL clone in post-TIL infusion PBMCs or apheresis products were determined by the detection of the same rearrangements initially identified in post-REP TILs. The fold change of TIL frequency was determined by identifying overlapping unique rearrangements detected in post-REP TILs and post-TIL infusion PBMCs and calculating their respective proportion of the total detected rearrangements. Fold change was calculated between the sum productive frequency of overlapping rearrangements detected at week 6 post-TIL infusion by the frequency of the same rearrangements detected in post-REP TILs and correlated with CD11b+ cell frequency.
RNA Sequencing
M-MDSCs (CD11b+Ly6C+Ly6G−) and PMN-MDSCs (CD11b+Ly6C+Ly6G+) were sorted by a FACSAria special order research product (SORP) (BD Biosciences) with a purity >99%. Sorted cells were washed and stored as dry pellets. RNA was extracted with an RNeasy mini kit (QIAGEN). Paired-end RNA sequencing reads were subjected to adaptor trimming and quality assessment before being aligned to mouse reference genome mm10 using STAR v2.5.3a (PMID: 23104886). Quantification of read counts aligned to the region associated with each gene was performed using HTSeq v0.6.1 (PMID: 25260700) based on the RefSeq gene model. Read counts of all samples were normalized based on library size estimation using the R/Bioconductor package DESeq2 v1.6.3 (PMID: 25516281). Differential gene expression between different conditions was performed by serial dispersion estimation and statistical model fitting procedures implemented in DESeq2. Genes with a Benjamini-Hochberg corrected p value of less than 0.05 were considered to be significantly differentially expressed. Significant genes affected by at least 2-fold were analyzed for enrichment of upstream regulators using QIAGEN’s IPA software (https://www.qiagen.com/ingenuity, “canonical pathways,” “diseases and functions,” and “upstream regulators” options). For upstream regulator analysis, cytokine upstream regulators were filtered and sorted by p value for the dataset overlap between molecules known to be regulated by that given cytokine. GEO: GSE136574; GEO: GSE136639.
Statistical Analysis
Graphs were generated using GraphPad Prism software. Graphs represent mean values with SEM. p values were calculated in each respective Figure where statistical tests are indicated. Retrospective analysis for patient survival was performed. Mantel-Cox p values are shown on each respective Kaplan-Meier plot. Patient groups for Kaplan-Meier survival plots were established by determining the median cutoff by frequency distribution analysis. The median follow-up for survival analysis was determined by reverse Kaplan-Meier analysis (median follow-up, 22 months). p values and R2 values were determined by a two-tailed Pearson r correlation test. For mouse-tumor growth studies, tumor growth curves are shown as mean with SEM, and significance was determined by a two-way ANOVA and Sidak’s multiple comparisons test. Mice were randomized after tumor cell implantation into respective treatment groups. For all other experiments, data were compared using either an unpaired two-tailed Student’s t test corrected for multiple comparisons by a Bonferroni adjustment or Welch’s correction. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Study Approval
All animal experiments were approved by the University of South Florida Institutional Animal Care and Use Committee and performed in accordance with the US Public Health Service policy and National Research Council guidelines. Studies were performed under approved Institutional Review Board (IRB) laboratory protocols at the H. Lee Moffitt Cancer Center (Tampa, FL, USA). TILs, PBMCs, and autologous tumors were collected from melanoma patients or PBMCs from NSCLC tumor patients as part of TIL ACT clinical trials. All samples were de-identified prior to use in research studies. All patients signed approved consent forms. Specimens were obtained from patients who were enrolled in the following clinical trials: Vemurafenib with lymphodepletion plus adoptive cell transfer & high dose IL-2 metastatic melanoma, MCC-16992, ClinicalTrials.gov: NCT01659151, https://clinicaltrials.gov/ct2/show/NCT01659151?term=16992&draw=2&rank=4; Ipilimumab with lymphodepletion plus adoptive cell transfer and high dose IL-2 in melanoma mets pts, MCC-17057, ClinicalTrials.gov: NCT01701674, https://clinicaltrials.gov/ct2/show/NCT01701674?term=17057&draw=2&rank=4; Combining PD-1 blockade, CD137 agonism and adoptive cell therapy for metastatic melanoma, MCC-18377, ClinicalTrials.gov: NCT02652455, https://clinicaltrials.gov/ct2/show/NCT02652455?term=18377&draw=2&rank=2; Nivolumab and tumor infiltrating lymphocytes (TIL) in advanced non-small cell lung cancer, MCC-19122, ClinicalTrials.gov: NCT03215810, https://clinicaltrials.gov/ct2/show/NCT03215810?term=19122&draw=2&rank=1.
Author Contributions
P.I. designed and performed experiments, analyzed data, and wrote the manuscript. K.K. designed and performed experiments. B.S., M.H., D.W, L.N., S.A, and A.M.. provided technical and material support. A.M.. provided administrative support. M.B. performed data interpretation. B.C.C. and A.A.S. provided patient samples. A.A.S. performed patient data acquisition, methodology development, and designed experiments. S.P.-T. performed experimental design, study supervision, and manuscript revision.
Conflicts of Interest
The H. Lee Moffitt Cancer Center and Research Institute has licensed intellectual property related to the proliferation and expansion of tumor-infiltrating lymphocytes (TILs) to Iovance Biotherapeutics. S.P.-T., A.A.S., and M.H. are inventors on such intellectual property. S.P.-T. receives salary support on sponsored research agreements between Moffitt Cancer Center and Iovance Biotherapeutics, Myst Therapeutics, Intellia Therapeutics, and Provectus Biopharmaceuticals. A.A.S. is a paid consultant for Iovance Biotherapeutics and has undertaken sponsored travel. None of these organizations provided funding for this study. B.C.C. is an advisor or consultant with E.R. Squibb & Sons LLC, AbbVie Inc., GlaxoSmithKline plc, Celgene Corp., Gilead Sciences Inc., and Achilles plc.; is on the Speaker’s Bureau for Hoffman-LaRoche AG, E.R. Squibb & Sons LLC, AstraZeneca LLC, Takeda Pharmaceutical Company Ltd, and Foundation Medicine Inc.; and has a contracted/support research grant from Prometheus Inc., Iovance Biotherapeutics Inc., and Adaptive Biotechnologies Corp. The remaining authors declare no competing interests.
Acknowledgments
We would like to thank Autumn Joerger, Patrick Verdugo, Jennifer Morse, and Scott Kidd for technical assistance. We also would like to thank Dr. Michael Schell of the H. Lee Moffitt Cancer Center and Research Institute Biostatistics and Bioinformatics Core Facility for advice and statistical expertise. This work was supported in part by the Flow Cytometry Core Facility, Biostatistics and Bioinformatics Core Facility, Cell Therapies Core Facility, Molecular Genomics Core Facility, and Tissue Core Facility of the H. Lee Moffitt Cancer Center and Research Institute and in part by the H. Lee Moffitt Cancer Center Support Grant P30 CA076292 from the National Institutes of Health. This work was also supported by a Bankhead Coley Cancer Research Grant for the Florida Department of Health, a Miles for Moffitt Award, and Swim Across America. A.A.S. was supported by NCI grant 5K23CA178083. We also thank the Comparative Medicine Department of the University of South Florida.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.06.025.
Supplemental Information
References
- 1.Turtle C.J., Hanafi L.A., Berger C., Hudecek M., Pender B., Robinson E., Hawkins R., Chaney C., Cherian S., Chen X. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016;8 doi: 10.1126/scitranslmed.aaf8621. 355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee D.W., Stetler-Stevenson M., Yuan C.M., Shah N.N., Delbrook C., Yates B., Zhang H., Zhang L., Kochenderfer J.N., Rosenberg S.A. Long-term outcomes following CD19 CAR T cell therapy for B-ALL are superior in patients receiving a fludarabine/cyclophosphamide preparative regimen and post-CAR hematopoietic stem cell transplantation. Blood. 2016;128:218. [Google Scholar]
- 3.Gattinoni L., Finkelstein S.E., Klebanoff C.A., Antony P.A., Palmer D.C., Spiess P.J., Hwang L.N., Yu Z., Wrzesinski C., Heimann D.M. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kochenderfer J.N., Somerville R.P.T., Lu T., Shi V., Bot A., Rossi J., Xue A., Goff S.L., Yang J.C., Sherry R.M. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J. Clin. Oncol. 2017;35:1803–1813. doi: 10.1200/JCO.2016.71.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dudley M.E., Wunderlich J.R., Robbins P.F., Yang J.C., Hwu P., Schwartzentruber D.J., Topalian S.L., Sherry R., Restifo N.P., Hubicki A.M. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dudley M.E., Yang J.C., Sherry R., Hughes M.S., Royal R., Kammula U., Robbins P.F., Huang J., Citrin D.E., Leitman S.F. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pilon-Thomas S., Kuhn L., Ellwanger S., Janssen W., Royster E., Marzban S., Kudchadkar R., Zager J., Gibney G., Sondak V.K. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J. Immunother. 2012;35:615–620. doi: 10.1097/CJI.0b013e31826e8f5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forget M.A., Haymaker C., Hess K.R., Meng Y.J., Creasy C., Karpinets T., Fulbright O.J., Roszik J., Woodman S.E., Kim Y.U. Prospective analysis of adoptive TIL therapy in patients with metastatic melanoma: response, impact of anti-CTLA4, and biomarkers to predict clinical outcome. Clin. Cancer Res. 2018;24:4416–4428. doi: 10.1158/1078-0432.CCR-17-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kodumudi K.N., Weber A., Sarnaik A.A., Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J. Immunol. 2012;189:5147–5154. doi: 10.4049/jimmunol.1200274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding Z.C., Lu X., Yu M., Lemos H., Huang L., Chandler P., Liu K., Walters M., Krasinski A., Mack M. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014;74:3441–3453. doi: 10.1158/0008-5472.CAN-13-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pawelec G., Verschoor C.P., Ostrand-Rosenberg S. Myeloid-derived suppressor cells: not only in tumor immunity. Front. Immunol. 2019;10:1099. doi: 10.3389/fimmu.2019.01099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solito S., Marigo I., Pinton L., Damuzzo V., Mandruzzato S., Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N Y Acad. Sci. 2014;1319:47–65. doi: 10.1111/nyas.12469. [DOI] [PubMed] [Google Scholar]
- 13.Condamine T., Ramachandran I., Youn J.I., Gabrilovich D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015;66:97–110. doi: 10.1146/annurev-med-051013-052304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabrilovich D.I., Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Condamine T., Dominguez G.A., Youn J.-I., Kossenkov A.V., Mony S., Alicea-Torres K., Tcyganov E., Hashimoto A., Nefedova Y., Lin C. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016;1:aaf8943. doi: 10.1126/sciimmunol.aaf8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mollee P., Pereira D., Nagy T., Song K., Saragosa R., Keating A., Crump M. Cyclophosphamide, etoposide and G-CSF to mobilize peripheral blood stem cells for autologous stem cell transplantation in patients with lymphoma. Bone Marrow Transplant. 2002;30:273–278. doi: 10.1038/sj.bmt.1703653. [DOI] [PubMed] [Google Scholar]
- 17.Lévesque J.-P., Hendy J., Takamatsu Y., Williams B., Winkler I.G., Simmons P.J. Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp. Hematol. 2002;30:440–449. doi: 10.1016/s0301-472x(02)00788-9. [DOI] [PubMed] [Google Scholar]
- 18.Lévesque J.P., Takamatsu Y., Nilsson S.K., Haylock D.N., Simmons P.J. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood. 2001;98:1289–1297. doi: 10.1182/blood.v98.5.1289. [DOI] [PubMed] [Google Scholar]
- 19.Manz M.G., Boettcher S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014;14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H., Nguyen-Jackson H., Panopoulos A.D., Li H.S., Murray P.J., Watowich S.S. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010;116:2462–2471. doi: 10.1182/blood-2009-12-259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwak H.J., Liu P., Bajrami B., Xu Y., Park S.Y., Nombela-Arrieta C., Mondal S., Sun Y., Zhu H., Chai L. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity. 2015;42:159–171. doi: 10.1016/j.immuni.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Regan-Komito D., Swann J.W., Demetriou P., Cohen E.S., Horwood N.J., Sansom S.N., Griseri T. GM-CSF drives dysregulated hematopoietic stem cell activity and pathogenic extramedullary myelopoiesis in experimental spondyloarthritis. Nat. Commun. 2020;11:155. doi: 10.1038/s41467-019-13853-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guillamot M., Ouazia D., Dolgalev I., Yeung S.T., Kourtis N., Dai Y., Corrigan K., Zea-Redondo L., Saraf A., Florens L. The E3 ubiquitin ligase SPOP controls resolution of systemic inflammation by triggering MYD88 degradation. Nat. Immunol. 2019;20:1196–1207. doi: 10.1038/s41590-019-0454-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapuis A.G., Egan D.N., Bar M., Schmitt T.M., McAfee M.S., Paulson K.G., Voillet V., Gottardo R., Ragnarsson G.B., Bleakley M. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019;25:1064–1072. doi: 10.1038/s41591-019-0472-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goff S.L., Dudley M.E., Citrin D.E., Somerville R.P., Wunderlich J.R., Danforth D.N., Zlott D.A., Yang J.C., Sherry R.M., Kammula U.S. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J. Clin. Oncol. 2016;34:2389–2397. doi: 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenberg S.A., Yang J.C., Sherry R.M., Kammula U.S., Hughes M.S., Phan G.Q., Citrin D.E., Restifo N.P., Robbins P.F., Wunderlich J.R. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullinax J.E., Hall M., Prabhakaran S., Weber J., Khushalani N., Eroglu Z., Brohl A.S., Markowitz J., Royster E., Richards A. Combination of ipilimumab and adoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic melanoma. Front. Oncol. 2018;8:44. doi: 10.3389/fonc.2018.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wrzesinski C., Paulos C.M., Kaiser A., Muranski P., Palmer D.C., Gattinoni L., Yu Z., Rosenberg S.A., Restifo N.P. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J. Immunother. 2010;33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wright D.E., Cheshier S.H., Wagers A.J., Randall T.D., Christensen J.L., Weissman I.L. Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood. 2001;97:2278–2285. doi: 10.1182/blood.v97.8.2278. [DOI] [PubMed] [Google Scholar]
- 31.Marigo I., Bosio E., Solito S., Mesa C., Fernandez A., Dolcetti L., Ugel S., Sonda N., Bicciato S., Falisi E. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Condamine T., Gabrilovich D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25. doi: 10.1016/j.it.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinha P., Chornoguz O., Clements V.K., Artemenko K.A., Zubarev R.A., Ostrand-Rosenberg S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood. 2011;117:5381–5390. doi: 10.1182/blood-2010-11-321752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chandran S.S., Somerville R.P.T., Yang J.C., Sherry R.M., Klebanoff C.A., Goff S.L., Wunderlich J.R., Danforth D.N., Zlott D., Paria B.C. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: a single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017;18:792–802. doi: 10.1016/S1470-2045(17)30251-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chapuis A.G., Desmarais C., Emerson R., Schmitt T.M., Shibuya K., Lai I., Wagener F., Chou J., Roberts I.M., Coffey D.G. Tracking the fate and origin of clinically relevant adoptively transferred CD8+ T cells in vivo. Sci. Immunol. 2017;2 doi: 10.1126/sciimmunol.aal2568. eaal2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raber P.L., Sierra R.A., Thevenot P.T., Shuzhong Z., Wyczechowska D.D., Kumai T., Celis E., Rodriguez P.C. T cells conditioned with MDSC show an increased anti-tumor activity after adoptive T cell based immunotherapy. Oncotarget. 2016;7:17565–17578. doi: 10.18632/oncotarget.8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 38.Angulo I., de las Heras F.G., García-Bustos J.F., Gargallo D., Muñoz-Fernández M.A., Fresno M. Nitric oxide-producing CD11b+Ly-6G(Gr-1)+CD31(ER-MP12)+ cells in the spleen of cyclophosphamide-treated mice: implications for T-cell responses in immunosuppressed mice. Blood. 2000;95:212–220. [PubMed] [Google Scholar]
- 39.Diaz-Montero C.M., Salem M.L., Nishimura M.I., Garrett-Mayer E., Cole D.J., Montero A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Condamine T., Kumar V., Ramachandran I.R., Youn J.I., Celis E., Finnberg N., El-Deiry W.S., Winograd R., Vonderheide R.H., English N.R. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Invest. 2014;124:2626–2639. doi: 10.1172/JCI74056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel S., Fu S., Mastio J., Dominguez G.A., Purohit A., Kossenkov A., Lin C., Alicea-Torres K., Sehgal M., Nefedova Y. Unique pattern of neutrophil migration and function during tumor progression. Nat. Immunol. 2018;19:1236–1247. doi: 10.1038/s41590-018-0229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strauss L., Sangaletti S., Consonni F.M., Szebeni G., Morlacchi S., Totaro M.G., Porta C., Anselmo A., Tartari S., Doni A. RORC1 regulates tumor-promoting “emergency” granulo-monocytopoiesis. Cancer Cell. 2015;28:253–269. doi: 10.1016/j.ccell.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Devalaraja S., To T.K.J., Folkert I.W., Natesan R., Alam M.Z., Li M., Tada Y., Budagyan K., Dang M.T., Zhai L. Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell. 2020;180:1098–1114.e16. doi: 10.1016/j.cell.2020.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tobin R.P., Jordan K.R., Robinson W.A., Davis D., Borges V.F., Gonzalez R., Lewis K.D., McCarter M.D. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int. Immunopharmacol. 2018;63:282–291. doi: 10.1016/j.intimp.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Youn J.I., Kumar V., Collazo M., Nefedova Y., Condamine T., Cheng P., Villagra A., Antonia S., McCaffrey J.C., Fishman M. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat. Immunol. 2013;14:211–220. doi: 10.1038/ni.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pang W.W., Price E.A., Sahoo D., Beerman I., Maloney W.J., Rossi D.J., Schrier S.L., Weissman I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maeda K., Baba Y., Nagai Y., Miyazaki K., Malykhin A., Nakamura K., Kincade P.W., Sakaguchi N., Coggeshall K.M. IL-6 blocks a discrete early step in lymphopoiesis. Blood. 2005;106:879–885. doi: 10.1182/blood-2005-02-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maeda K., Malykhin A., Teague-Weber B.N., Sun X.H., Farris A.D., Coggeshall K.M. Interleukin-6 aborts lymphopoiesis and elevates production of myeloid cells in systemic lupus erythematosus-prone B6.Sle1.Yaa animals. Blood. 2009;113:4534–4540. doi: 10.1182/blood-2008-12-192559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamashita M., Passegué E. TNF-α coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. 2019;25:357–372.e7. doi: 10.1016/j.stem.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Norelli M., Camisa B., Barbiera G., Falcone L., Purevdorj A., Genua M., Sanvito F., Ponzoni M., Doglioni C., Cristofori P. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 51.Besser M.J., Itzhaki O., Ben-Betzalel G., Zippel D.B., Zikich D., Kubi A., Brezinger K., Nissani A., Levi M., Zeltzer L.A. Comprehensive single institute experience with melanoma TIL: long term clinical results, toxicity profile, and prognostic factors of response. Mol. Carcinog. 2020;59:736–744. doi: 10.1002/mc.23193. [DOI] [PubMed] [Google Scholar]
- 52.Le R.Q., Li L., Yuan W., Shord S.S., Nie L., Habtemariam B.A., Przepiorka D., Farrell A.T., Pazdur R. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist. 2018;23:943–947. doi: 10.1634/theoncologist.2018-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fried S., Avigdor A., Bielorai B., Meir A., Besser M.J., Schachter J., Shimoni A., Nagler A., Toren A., Jacoby E. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019;54:1643–1650. doi: 10.1038/s41409-019-0487-3. [DOI] [PubMed] [Google Scholar]
- 54.Juluri K.R., Hirayama A.V., Mullane E., Cleary N., Wu Q.V., Maloney D.G., Turtle C.J., Bar M., Gauthier J. Severe cytokine release syndrome is associated with impaired hematopoietic recovery after CD19-targeted CAR-T cell therapy. Blood. 2019;134:3229. [Google Scholar]
- 55.Thevenot P.T., Sierra R.A., Raber P.L., Al-Khami A.A., Trillo-Tinoco J., Zarreii P., Ochoa A.C., Cui Y., Del Valle L., Rodriguez P.C. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity. 2014;41:389–401. doi: 10.1016/j.immuni.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson C.B., Riesenberg B.P., May B.R., Gilreath S.C., Li G., Staveley-O’Carroll K.F., Garrett-Mayer E., Mehrotra S., Cole D.J., Rubinstein M.P. Effector CD8+ T-cell engraftment and antitumor immunity in lymphodepleted hosts is IL7Rα dependent. Cancer Immunol. Res. 2015;3:1364–1374. doi: 10.1158/2326-6066.CIR-15-0087-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.