

Abstract
Rhodopsin-mediated autosomal dominant retinitis pigmentosa (RHO-adRP) is a hereditary degenerative disorder in which mutations in the gene encoding RHO, the light-sensitive G protein-coupled receptor involved in phototransduction in rods, lead to progressive loss of rods and subsequently cones in the retina. Clinical phenotypes are diverse, ranging from mild night blindness to severe visual impairments. There is currently no cure for RHO-adRP. Although there have been significant advances in gene therapy for inherited retinal diseases, treating RHO-adRP presents a unique challenge since it is an autosomal dominant disease caused by more than 150 gain-of-function mutations in the RHO gene, rendering the established gene supplementation strategy inadequate. This review provides an update on RNA therapeutics and therapeutic editing genome surgery strategies and ongoing clinical trials for RHO-adRP, discussing mechanisms of action, preclinical data, current state of development, as well as risk and benefit considerations. Potential outcome measures useful for future clinical trials are also addressed.
Keywords: retinitis pigmentosa, autosomal dominant retinitis pigmentosa, gene therapy, CRISPR, genome editing
Graphical Abstract
There have been recent advances in gene replacement therapies for autosomal recessive and X-linked inherited retinal disorders, but treating autosomal dominant diseases remains challenging. Tsang and colleagues highlight recent developments in gene therapy for rhodopsin-mediated autosomal dominant retinitis pigmentosa. The therapeutic strategies discussed are applicable to all autosomal dominant disorders.
Main Text
Retinitis pigmentosa (RP) is a group of rare inherited disorders with phenotypes ranging from mild night blindness (nyctalopia) to total blindness, affecting 1 in 3,000–7,000 individuals.1,2 Aberrations in photoreceptors (rods and cones) and the retinal pigment epithelium cause progressive vision loss. In the initial stages of the disease process, rod photoreceptors start to die, and this causes night blindness. Affected individuals begin to experience difficulty seeing in dim light and adapting to changes in light sensitivity, leading to difficulties with driving at night or entering darkened rooms. Following the loss of rod photoreceptors, cone photoreceptors (which produce high acuity, bright light vision) are progressively lost. The cone degeneration in RP is considered secondary to rod death, possibly due to neighbor effects of decreased trophic factors, nutrient shortage, and oxidative stress.3,4 The progression of RP is slow and involves the continuous loss of photoreceptors, leading to loss of peripheral vision termed “tunnel vision,” whereby only the central vision is preserved. RP patients also report experiencing continuous flashes of light (photopsia). In late stages of the disease central vision is lost, resulting in total blindness. Patients may lose up to 90% of rod cells before vision changes are detected, resulting in initial diagnosis at advanced stages of disease.5 An electroretinogram (ERG) evaluating rod and cone functions can detect changes in rod function during initial stages of the disease before significant visual dysfunctions occur.6,7
The rhodopsin (RHO) gene was the first identified gene causing RP.8,9 Human RHO contains five exons and is 6.7 kb, and it is located on chromosome 3q22.1.10 More than 150 different mutations in RHO are associated with 25% of autosomal dominantly inherited RP (adRP) cases. Patients with RHO-mediated adRP have discernible differences in the pattern of retinal dysfunction between families with different mutations.11 Two classes of RHO-adRP have been described in the literature based on clinical observations. Class A patients (possessing mutations R135G, R135L, R135W, V345L, and P347L) lose rod function over the entirety of the retina and experience onset of night blindness earlier in life.12 There is a catastrophic loss of rod function, which may not be corrected, and therefore therapies should be focused on cone preservation in these patients. Class B patients demonstrate a milder phenotype, including normal rod activation kinetics and preserved rod outer segment length with abnormalities in the rod visual cycle that are mutation specific. Among subclass B1 (T17M, P23H, T58R, V87D, G106R, and D190G) patients, photoreceptor degeneration is heterogeneous and patients show an inferior to superior disease progression. Subclass B2 patients (G51A, Q64ter, and Q344ter) show no regional retinal predisposition for disease. In class B patients, rods have the potential to be rescued, and rod preservation should be a target in order to protect cones. Of note, the P23H mutation is the most prevalent RHO mutation in North America, accounting for 10% of adRP cases because of a founder effect. P23H is not found elsewhere, including Europe and Asia. Individuals possessing the P23H mutation have significantly better visual acuity and larger electroretinographic amplitudes.13
Multiple mechanisms underlie RHO-mediated retinal degeneration.14 RHO is the visual pigment of retinal rods, which facilitates vision in dim light and absorbs light at 495 nm.15 It is a 348-aa G protein-coupled receptor protein with seven transmembrane domains, with a luminal N terminus and a cytoplasmic C terminus.16 The cytoplasmic face of RHO is made of three loops with catalytic sites that prompt guanosine triphosphate (GTP)-guanosine diphosphate GDP exchange by transducing GNAT1 and sites for light-dependent phosphorylation by RHO kinase. It also contains sites sites for N-glycosylation, and the site lys296 is where retinal attachment occurs. The vast majority of pathogenic mutations in RHO cause retinal degeneration by way of gain-of-function mutations leading to adRP. Of note, a few RHO mutations have been associated with autosomal recessive RP, but they are relatively uncommon.14 Further details regarding the molecular and cellular basis of RHO-mediated RP are beyond the scope of this review and can be found in the excellent review by Athanasiou et al.14
There is no universally effective treatment or cure for RHO-RP, and multiple approaches are being studied. Stem cell or retinal tissue transplantation, nutritional supplementations, retinal implants, as well as targeted and non-targeted gene therapies have been proposed and tested. Transplantation of stem cells or retinal tissue by the use of retinal progenitor cells may provide beneficial effects. In studies that tested the effect of transplanted newly born rods, responses to dim light were restored in blind mice.17 Nutritional supplementation for RP with nutrients such as vitamin A, docosahexaenoic acid (DHA), and lutein have produced limited and controversial results.18 Retinal implants and prostheses have been investigated as potential interventions in the treatment of advanced RP, including the Argus II retinal prosthesis system and the Alpha IMS from Retina Implant. Clinical trials showed improvement in various visual function tests but several serious adverse events.19,20 Optogenetics delivered as non-targeted gene therapy for advanced RP are also being tested. Channelrhodopsins (ChRs), when expressed in the retina, depolarize in response to light-generating signals, which are then transmitted to the brain. There are two ongoing current clinical trials using optogenetics in RP patients, RST-001 (ClinicalTrials.gov: NCT02556736) and GS030 (ClinicalTrials.gov: NCT03326336). Targeted gene therapy holds great promise for RHO-adRP and is the focus of this review.
Gene Therapy for RHO-adRP
There have been recent advances in gene replacement therapies for autosomal recessive and X-linked inherited retinal disorders. In these cases, since the genes of interest have loss-of-function mutations (both copies in autosomal recessive disorders and the only copy in X-linked disorders), a straightforward replacement approach by gene supplementation is appropriate. The first, and currently only, US Food and Drug Administration (FDA)-approved gene therapy for a retinal disease is voretigene neparvovec-rzyl (Luxturna), a gene replacement therapy targeting RPE65 enzyme deficiency in Leber congenital amaurosis (LCA) and RP. Subretinal injection of the adeno-associated viruses (AAVs)-hRPE65 vector expressing RPE65 restores its production in the transduced RPE cells, resulting in functional visual improvements measured by navigational ability and light sensitivity.21 In addition, there are many ongoing clinical trials with gene supplementation therapies for X-linked RP caused by RP GTPase regulator (RPGR) mutations, autosomal recessive RP caused by PDE6B and MERTK mutations, autosomal recessive achromatopsia caused by CNGA3 and CNGB3 mutations, X-linked choroideremia caused by REP1 mutations, as well as X-linked retinoschisis caused by RS1 mutations (https://clinicaltrials.gov/).
In contrast, treating autosomal dominant diseases with gain-of-function mutations such as RHO-adRP requires a substantially different approach and has been traditionally difficult. The goal for targeted gene therapy of RHO-mediated adRP is to inhibit expression of the mutant RHO protein and to increase the ratio of wild-type (WT)-to-mutant RHO in order to reduce the rate of retinal degeneration. One approach is to design therapeutics with high specificity that can differentiate between a single point mutation between the diseased and WT RHO alleles in order to selectively decrease expression of toxic protein from mutant RHO. Although it is preferable to leave the WT RHO intact, gene therapy strategies that offer such a high level of allele selectivity have to be designed specifically for each of the 150+ mutations in RHO, making it impractical. An alternative approach involves disrupting both copies of endogenous RHO genes, mutant and WT, and replacing them with an exogenous RHO gene. Such a strategy can be mutation-independent and bypass the need to design unique therapy for each RHO mutant, offering a simplified treatment strategy for all RHO-adRP patients.
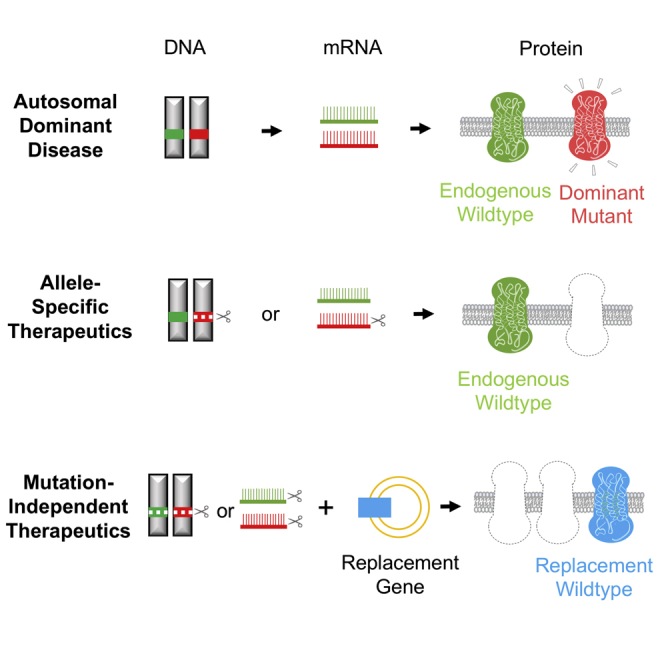
Herein, we provide a review of both mutation-specific and mutation-independent gene therapy strategies currently under development for RHO-adRP (Figure 1; Table 1). For the purpose of this review, gene therapy is defined as delivering nucleic acids in vivo and covers both RNA and DNA targeting therapeutics.
Figure 1.

Gene Therapy Strategies for Autosomal Dominant Diseases
Top: in autosomal dominant disorders such as RHO-adRP, one mutated copy of gene encodes the abnormal mRNA and protein (red) sufficient to cause disease. The wild-type copy is shown in green. Middle: allele-specific therapeutics target the mutated RHO gene or its mRNA product without affecting the wild-type copy. Bottom: mutation-independent therapeutics disrupt both the mutated and wild-type RHO genes or their RNA products and replace them with an exogenous copy of the codon-modified CRISPR or RNAi resistant RHO gene (blue).
Table 1.
Comparison of Gene Therapy Candidates Currently under Development for RHO-adRP
| Developer | Drug Name | Targeted Mutation | Mechanism of Action | Preclinical Model and Efficacy Data | Route of Administration/ Frequency | Current Stage of Development | |
|---|---|---|---|---|---|---|---|
| RNA targeting | ProQR Therapeutics/Ionis Pharmaceuticals | PR1123/ION357 | P23H | antisense oligonucleotides targeting mutated RHO mRNA | • murine/rat | • intravitreal injections | • phase 1/2 started October 2019 |
| • 40% knockdown of RHO expression; ONL 18% thicker; ERG amplitude 181% higher (scotopic a-wave) | • expected to need repeat injections | • target to enroll 35 patients | |||||
| • expected to conclude in October 2021 | |||||||
| IVERIC Bio | IC-100 | mutation-independent | shRNA suppression of endogenous RHO expression + replacement with codon-modified shRNA-resistant RHO delivered by a single AAV vector | • canine | single subretinal injection | plan to initiate a phase 1/2 clinical trial during the fourth quarter of 2020 | |
| • >97% knockdown of RHO expression; qualitative ONL and ERG improvement | |||||||
| • estimated equivalent human years of useful vision gain: 8a | |||||||
| Roche/Spark Therapeutics | RhoNova | mutation-independent | shRNA suppression of endogenous RHO expression + replacement with codon-modified shRNA-resistant RHO delivered by dual AAV vectors | • murine | single subretinal injection | • preclinical | |
| • 68% knockdown of RHO expression; ONL 35% thicker; ERG amplitude 244%–429% higher (rod-isolated response) | • received orphan drug designations in Europe (2010) and in the US (2013) | ||||||
| • estimated equivalent human years of useful vision gain: 11a | |||||||
| Alnylam/Regeneron | – | – | RNAi molecule targeting RHO | – | – | • preclinical | |
| Therapeutic editing genome surgery | Editas Medicine | – | mutation-independent | CRISPR-based knockout of endogenous RHO genes + replacement RHO cDNA delivered by dual AAV vectors | • in vitro (preliminary) | single subretinal injection (presumed) | • preclinical |
| • 70% knockdown of RHO expression | • declared development candidates May 2020 | ||||||
| Precision Bio | – | P23H | ARCUS meganuclease-medicated knockout of mutant RHO delivered by a single AAV vector | • swine (preliminary) | single subretinal injection | • preclinical | |
| • qualitative rescue of outer retinal morphology and ERG rod response | |||||||
| • estimated equivalent human years of useful vision gain: 14.5a |
Estimation based on extrapolation of original data from preclinical studies. There is significant variability in the models and outcome measures used across studies.
RNA Targeting Therapeutics
Antisense Oligonucleotides (ASOs)-Based Therapy
ASOs are single-stranded DNA molecules complementary to mRNA targets. Upon hybridization with target RNA through specific nucleotide pairing, ASOs induce target RNA degradation by recruiting cellular enzyme ribonuclease H1 (RNase H1), which cleaves the target RNA.22 ASOs remain intact through this process and therefore can be active for additional targets. This approach has been routinely used in basic research to achieve downregulation of gene expression. Due to recent advances in ASO technology, it is now possible for second-generation ASOs to selectively target a mutant allele with a single base pair difference from the WT allele, significantly expanding their potential as disease-modifying therapeutics for autosomal dominant genetic disorders in vivo.23 Three ASO medications have been successfully commercialized to date, including Spinraza/nusinersen for spinal muscular atrophy, Tegsedi/inotersen for hereditary transthyretin-mediated amyloidosis, and Waylivra/volanesorsen for familial chylomicronemia syndrome. Dozens more ASO drug candidates are currently under clinical development covering a broad range of disease areas.
PR1123 (previously named ION357) is an ASO drug in a gapmer configuration targeting the P23H mutation in the human RHO gene. It has been shown to knock down expression of P23H mutant mRNA specifically without affecting the WT RHO RNA expression in cell lines and mouse models.24 Unilateral intravitreal injection of PR1123 caused a 40% reduction in the RHO mRNA level compared to the contralateral eye in P23H mice while no significant change in RHO level was observed in WT mice. Moreover, PR1123 treatment led to ERG improvement and structural preservation in P23H mouse and rat models.24,25 Improved scotopic a-wave response amplitude at all stimulus intensities were observed via ERG on P23H rats following treatment. Treated eyes of P23H mice and rats had increased outer nuclear layer (ONL) thickness, a measurement of photoreceptor cell number. Additionally, a safety study was performed in primates that showed that QR-1123 did not affect levels of WT RHO mRNA in cynomolgus monkeys after single intravitreal injections.
Initially developed by Ionis Pharmaceuticals, PR1123 was in-licensed to ProQR Therapeutics in 2018. It received orphan drug and fast track designations for P23H-adRP by the FDA in 2019. ProQR Therapeutics is currently sponsoring a phase 1/2 study that started recruitment in October 2019 (ClinicalTrials.gov: NCT04123626). The study intends to enroll up to 35 adult P23H-adRP patients followed for 12 months. An open label single-dose cohort as well as a double-masked multiple-dose escalation cohort are planned with repeat unilateral intravitreal injection of QR-1123 every 3 months compared to unilateral sham procedures. In addition to evaluating safety and tolerability as primary outcome measures, multiple modalities will be used to evaluate efficacy by measuring structural and functional improvements. The phase 1/2 study is scheduled to conclude in October 2021.
Short Hairpin RNA (shRNA)-Based Therapy
shRNAs are short RNA molecules that spontaneously form hairpin structures. shRNAs are recognized by the intracellular RNA interference (RNAi) pathway, such as RNase III enzyme Dicer, and are processed to active small interfering RNAs (siRNAs). Subsequently, the siRNAs bind to target mRNAs through base pairing facilitated by the RNA-induced silencing complex, resulting in cleavage and degradation of target mRNAs. shRNAs are expressed by DNA vectors in contrast to their synthetic siRNA counterparts.26 In vivo delivery of DNA vectors expressing shRNAs often requires viral vectors such as AAV and lentiviruses.
A dual AAV vector strategy of suppression with shRNA and replacement was tested in a murine model of RHO-adRP, Rhop347s/+, by Millington-Ward et al.27 in a proof-of-concept study.28 One AAV2/5 vector, AAV-shRNA, expresses an shRNA targeting human RHO gene at nucleotides 254–274, a site independent of known mutations. Therefore, it is designed to knock down expression of all endogenous RHO genes, mutant and WT, in a mutation-independent way.29 An shRNA-resistant human RHO with codon modification at wobble nucleotides is expressed by a second AAV2/5 vector as replacement (AAV-RHO). Of note, the endogenous murine Rho gene is naturally resistant to the shRNA due to mismatches at the RNAi target site between the two species and was continuously expressed in this model. Both vectors were delivered together through subretinal injections to Rhop347s/+ mice. An AAV vector expressing non-targeting control RNAi was used as a control. A 68% ± 2.4% reduction in the ratio of RHO/Rho mRNA was observed in transduced retinal cells after AAV-shRNA injection, suggesting an efficient knockdown of RHO gene expression. As for replacement, AAV-RHO injection in WT mice resulted in RHO/Rho ratio of 31% ± 5% at the mRNA level. The authors argued that since only 40% of the retina was transduced but the whole retina was analyzed, the true expression level of AAV-RHO in transduced cells could be much higher. To evaluate functional improvement, rod-mediated ERG responses were analyzed. Rod b-wave amplitudes were 184.5 ± 65.4 μV in treated eyes at 6 weeks post-injection (wpi) compared to control eyes at 34.9 ± 16.8 μV. The improvement was still significant at 20 wpi (58.1 ± 19.8 μV in the dual vector group versus 16.9 ± 12.6 μV in the control vector group). The control single vector injections did not result in improvement in rod responses. Histologically, ONL thickness was 17.9 ± 3.4 μm in dual vector-injected eyes compared to 13.3 ± 2.0 μm in sections from control eyes at 6 wpi. At 20 wpi, ONL thickness was 8.9 ± 1.2 μm in treated eyes, whereas in control eyes the ONL had almost completely disappeared and was no longer measurable due to disease progression. Additionally, electron microscopy revealed preservation of rod photoreceptor outer segments with correctly formed membrane disks in treated eyes compared to control eyes, which exhibited degenerating outer segments and disorganized membrane disks. This dual vector shRNA suppression and replacement therapeutic strategy for RHO-adRP was named RhoNova and received orphan drug designation in Europe in 2010 and in the US in 2013. Genable Technologies, the original owner of RhoNova, was acquired by Spark Therapeutics in 2016, which was subsequently acquired by Roche in 2019. There has been no publicly available update on RhoNova’s clinical development.
More recently, a gene therapy candidate for RHO-adRP composed of a single AAV2/5 vector expressing both an shRNA targeting human RHO and a healthy copy of the RHO gene modified to be resistant to the shRNA has been developed.30 The shRNA targets a part of human RHO unaffected by any known mutations, similar to RhoNova. A codon-modified human RHO gene resistant to shRNA is expressed by the same vector as replacement. Cideciyan et al.30 performed preclinical studies in a naturally occurring canine model of RHO-adRP (RHOT4R/+) sensitive to acute light-induced retinal degeneration with single subretinal injections. One of the shRNA candidates, shRNA820, caused a near-complete knockdown of RHO mRNA and protein in WT and RHO mutant retinas. When the shRNA-resistant human RHO gene, named RHO820, was supplemented in addition to shRNA820 by a single AAV2/5 viral vector, the resistant RHO was expressed at 118%–132% at the mRNA level and 31%–33% at the protein level compared to untreated retinas. There was some evidence that this suppression and replacement strategy could rescue the light-induced retinal degeneration in RHO mutant retina. ERG measurements showed greater rod and cone responses in AAV-shRNA820-RHO820-injected retinas compared to those injected with balanced salt solution. There were also qualitative data suggesting retention of ONL thickness, photoreceptor cell bodies, and rod and cone outer segments in AAV-shRNA820-RHO820-injected retinas. More recently, a separate study showed that subretinal injection of the same vector preserved the ERG response and the ONL thickness in P23H Rho transgenic mice.31
In 2018, IVERIC bio licensed the right to develop and commercialize this gene therapy product candidate for RHO-adRP, now named IC-100, from the University of Florida Research Foundation and the University of Pennsylvania. Currently, further preclinical studies and investigational new drug (IND)-enabling activities for IC-100 are being conducted by IVERIC bio, with a phase 1/2 clinical trial expected to begin in the near future.
Other siRNA-Based Therapies
As discussed above, siRNAs are synthetic RNA molecules that are recognized by cellular RNAi pathways and cause degradation of target mRNAs. Recently, siRNAs have been successfully developed into medications for the treatment of genetic diseases. Onpattro/patisiran, an RNAi therapeutic developed by Alnylam Pharmaceuticals, is the first in class and received FDA approval in 2018 for the treatment of hereditary transthyretin amyloidosis with polyneuropathy by targeting transthyretin mRNA. Another RNAi therapeutic Givlaari/givosiran was approved by the FDA in 2019 for adult acute hepatic porphyria, and several others RNAi therapeutics are in late-stage development for other hepatic diseases. Alnylam Pharmaceuticals has since expanded its clinical programs to extra-hepatic diseases and signaled its interest in developing RNAi therapeutics for eye diseases, including RHO-adRP, in collaboration with Regeneron. No preclinical data have been published to date, and no development timeline is publicly available.
Genome Editing Therapeutics
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Based Therapies
CRISPR-based genome editing technology has rapidly advanced since the first demonstration of its use.32,33 Short RNA sequences are used to guide CRISPR-associated (Cas) nucleases to their DNA targets through base pairing, leading to increased specificity compared to previous DNA editing techniques.34 After a targeted double-stranded break is created, DNA sequence alterations can be introduced through cellular DNA repair mechanisms such as homology-directed repair (HDR) and non-homologous end joining (NHEJ). The development of the homology-independent targeted integration (HITI) strategy was a milestone in the advancement of CRISPR-based technology since it enables gene editing in post-mitotic cells by utilizing NHEJ rather than HDR.35 In the landmark study the HITI method was used to correct the genetic defect in the Mertk gene in a rat model of human RP leading to anatomical and functional visual improvements.35 More recent developments in CRISPR technology include prime and base editing techniques that enable changing single nucleotides in a predictable manner.34,36 Prime and base editors as well as other CRISPR strategies with improved specificity for target sites can potentially discriminate between mutant and WT alleles of target genes. Updated sequencing analysis tools such as CRISPResso2 have since been developed with improved alignment algorithms for validation and characterization of accurate indel analysis and precise genomic changes.37 CRISPR-based gene editing technologies are now increasingly being developed as therapeutics for inherited genetic diseases to permanently correct the underlying mutations in adult non-dividing cells in vivo.
Mutation-Independent CRISPR Strategies for RHO-adRP
In 2018, Tsai et al.38 used dual AAV vectors to deliver CRISPR-based ablate-and-replace gene therapy in mouse models of RHO-adRP. One vector, AAV-Cas, encodes the codon-optimized Streptococcus pyogenes Cas9 (SpCas9). The second vector encodes two guide RNAs (gRNAs) targeting murine Rho (mRho) gene exon 1, as well as a copy of human RHO cDNA as replacement (AAV-GR). Delivering the gRNAs in the same vector as the replacement RHO gene instead of in the AAV-Cas vector means that gene disruption will occur only in cells receiving the replacement gene, which is a significant safety feature. This mutation-independent CRIPSR strategy aims to disrupt all endogenous murine Rho genes, including both the mutated and WT copy.
In vitro and in vivo studies showed effective ablation of mRho genes with this CRISPR-based gene therapy.38 The double gRNA strategy led to 90% ± 7.8% ablation of the mRho gene due to truncations in vitro, whereas the traditional single gRNA strategy resulted in 60.0% ± 9.9% ablation of the mRho gene. Subretinal injection of the dual vectors (AAV-Cas+AAV-GR) in WT mice also resulted in effective ablation of mRho. Whole-retina mRho mRNA analysis revealed a 25% decrease in mRho mRNA and detectable xenogeneic human RHO mRNA expression. Historical analysis revealed retention of ONL layers in all adRP mouse models examined (RhoP23H/P23H, RhoD190N/+) at 3 months after treatment. AAVs-Cas9+GR treatment resulted in six- to eight-layer-thick ONLs compared to three to four layers for the control PBS treatment in RhoP23H/+ mice. In addition, the ERG of maximal responses with mixed rod-cone components revealed significant increases in a- and b-wave amplitudes after AAVs-Cas9+GR treatment.
A similar dual vector CRISPR-based knockout and replacement strategy for RHO-adRP has been proposed by Editas Medicine,39 a company with expertise in application of CRISPR-based gene editing technologies in ocular diseases that is currently conducting a phase 1/2 study of first-in-human CRISPR-based therapeutic EDIT-101 in another hereditary retinal disease, LCA-10. The first AAV5 vector encodes Staphylococcus aureus Cas9 (SaCas9) cRNA under a minimal RHO promoter. The second vector encodes gRNAs as well as codon-optimized RHO cRNA under the minimal RHO promoter. In preclinical studies presented in May 2020, lead gRNA candidates were shown to knock down RHO expression in cell lines in a dose-dependent fashion and were highly specific based on sequencing assays. In human retinal explants, the two lead gRNA candidates predominantly generated frameshifting deletions. Additionally, the optimized RHO replacement vector was shown to express RHO mRNA 8-fold higher than the previously published one from Cideciyan et al.,30 and the expression level was comparable to endogenous RHO expression in human retinal explants.
Allele-Specific CRISPR Strategies for RHO-adRP
Allele-specific CRISPR gene therapies for RHO-adRP have been explored by several preclinical studies in vitro and in vivo aiming to disrupt the dominant negative allele while leaving the WT allele intact. In a RhoS334/+ rat model of RHO-adRP, Bakondi et al.40 used Cas9 and a gRNA complementary to a protospacer-adjacent motif (PAM) site unique to the RhoS334 locus delivered by electroporation, resulting in cleavage of only the mutant allele. Morphological preservations of photoreceptors and their synapses were also observed. Additionally, preferential editing of the P23H allele over the WT Rho allele in P23H mouse models was demonstrated using either a variant SpCas9 paired with truncated single gRNA37,41 or SaCas9 with conventional 20-nt gRNA.42 Similarly, specific knockdown of the P347S RHO mutant with little effect on WT RHO using SpCas9 or its high-fidelity variant SpCas9 VQR (D1135V/R1335Q/T1337R) paired with specific gRNAs was shown in cell lines.43
Meganuclease Gene Editing Technique
Meganuclease is an I-CreI (chloroplast-encoded homing endonuclease I)-based endonuclease originally discovered in Chlamydomonas reinhardtii. WT I-CreI recognizes a specific 22-bp DNA sequence within the chloroplast ribosomal RNA23S gene and causes a double-strand break at this site.44 Meganucleases are developed by engineering the I-CreI DNA-recognition surface using structure-based redesign in order to alter the target DNA sequences.
The potential of meganuclease as in vivo gene editing therapeutics was first demonstrated in 2018 when the PCSK9 gene was successfully targeted and knocked out in the livers of rhesus macaques by an engineered meganuclease expressed by the AAV vector leading to sustained reduced levels of low-density lipoprotein (LDL) cholesterol.45 This genome editing technology, named ARCUS by Precision BioSciences, has since been applied in many preclinical studies of different liver diseases. A potential cure of hepatitis B infection by permanently eliminating hepatitis B viral genome from liver cells is currently being co-developed between ARCUS and Gilead with a targeted IND submission in 2021.
Therapeutic efficacy was recently reported in a transgenic swine model of human P23H RHO-adRP using ARCUS meganuclease.46 A single subretinal injection of an AAV5 vector expressing P23H-RHO-targeting ARCUS meganuclease was performed in unilateral eyes. Sustained reduction in mutant RHO expression was observed. Additionally, ERG showed modest improvements in rod responses of up to 42 wpi as well as preserved cone function after 42 wpi compared to no rod responses in untreated eyes. Furthermore, the outer retinal morphology including rod photoreceptors and cone morphology were rescued by the treatment.
Discussion
Risk versus Benefit Comparison of Gene Therapy Strategies
Mutation-Specific versus Mutation-Independent
By design, ASO therapeutic QR1123 and ARCUS meganuclease target the specific P23H mutation in RHO-adRP, limiting their use to the approximately 10% of all RHO-adRP cases in North America.8,47 Similarly, the emerging allele-specific CRISPR strategy targets P347S RHO, which comprises an even smaller subpopulation of RHO-adRP.48
In contrast, IC-100, RhoNova and the ablation strategy using CRISPR from Editas Medicine are mutation-independent, making them suitable for all RHO-adRP patients. However, because the mutation-independent strategies inevitably disrupt the endogenous WT allele, an exogenous RHO replacement has to be provided to maintain sufficient levels of RHO expression in the treated retinas. It is difficult to compare the RHO replacement strategies and attempt to predict their efficiency in human retina given different ratios of mutant transgene to endogenous RHO copy numbers used in various preclinical models. The optimized RHO replacement vector from Editas Medicine appears to have the strongest preclinical data so far, expressing at the highest mRNA level comparable to endogenous expression in human retinal explants. The expression level of exogenous RHO should be carefully titrated in future studies since excessive expression above the physiological level could also be harmful.
RNAi versus Genome Editing
RNAi therapeutics such as QR1123, IC-100, and RhoNova target the defective mRNA products produced by mutant DNA, effectively causing a knockdown rather than a complete knockout. This could limit their efficacy since some level of the mutant DNA is still expressed, causing a toxic effect. A highly effective candidate that is constantly expressed can potentially overcome the limit of knockdown instead of the knockout effect of RNAi. For example, IC-100 caused a near-complete knockdown of RHO mRNA and protein in WT and RHO mutant retinas in the canine model of RHO-adRP, compared to the 40% knockdown by QR1123 and 68% knockdown by RhoNova in RHO mRNA levels in murine models.
Genome editing technologies such as CRISPR-Cas9 and ARCUS meganuclease permanently disrupt mutant (and WT) RHO genes at the DNA level, leading to knockout of mutant RHO and promising a potential cure. Clinical safety is a concern for applying genome editing technologies and requires careful risk and benefit considerations.49 There is ongoing discussion in the literature on the frequency of off-target effects in vivo by CRISPR-Cas9, including translocation events, off-target effects caused by AAV vector insertion versus CRISPR-Cas9 by itself, immunogenicity of Cas9 protein, and the best strategies to decrease these effects moving forward.50, 51, 52, 53, 54, 55 The National Institute of Standards and Technology has set the limit for an acceptable CRISPR off-targeting rate in patients at 25%.56, 57, 58, 59 The increasing use of high-fidelity CRISPR-Cas9 variants such as SpCas9-HF1, -HF2, or -HF4 further increases the specificity of genome editing and reduces the off-target rate.60 Additionally, a wide variety of on-target editing events can occur and are difficult to predict from preclinical studies. Long-term follow-up after administration of gene therapy products past the active follow-up period of a clinical trial has been recommended by the FDA to monitor long-term safety and delayed adverse events of gene therapy products.57
Delivery System, Route of Administration, and Frequency of Administration
ASO therapeutic QR1123 is synthetic, does not require a separate delivery vehicle, and can be delivered directly to the retina via intravitreal injections as routine office procedures. It was developed on the same prolific ASO platform that has resulted in several FDA-approved therapeutics with excellent safety profiles. Eliminating subretinal injections for drug delivery reduces the risk of associated surgical complications. However, given that defective RHO mRNA is constantly expressed, it is likely that QR1123 needs to be given chronically over a lifetime in the form of repetitive intravitreal injections. This will not only pose significant financial and logistical hurdles for patients receiving treatments but also increase risks for side effects, most notably infection risk associated with repeated intravitreal injections.
All of the other gene therapy candidates require AAV as a delivery vector and subretinal injections as a delivery method. AAV-mediated gene delivery in vivo is an attractive choice and is widely used given the vector’s ability to infect non-dividing cells, non-pathogenicity, low host immune response, and long-term stable expression.34 The packaging capacity of 4.7 kb has been a long-standing limitation of AAV viral vectors. Given that Cas9 genes themselves are approaching the size limit of AAV vectors, usage of long promoters to increase specificity and expression level is prohibited. gRNAs and replacement genes have to be delivered via a second vector in the ablate-and-replace strategies developed by Tsai et al.38 and Editas Medicine, potentially limiting gene editing efficacy. Although lentivirus vectors have a larger packaging capacity approaching 9.7 kb, present-day lentivirus vectors do not infect photoreceptor cells efficiently, limiting their clinical use. ARCUS meganuclease is much smaller (∼300 aa) and does not require gRNAs, making it easier to deliver via a single AAV vector. Another concern with AAV-mediated gene delivery is the unknown long-term effect of persistent expression of a gene editing system, including xenogeneic Cas9. Inducible AAV expression, self-cleaving AAV-CRISPR, and other non-viral delivery methods are currently being explored.
Potential Outcome Measures for Clinical Trials of RHO-adRP
The only ongoing clinical trial for RHO-adRP evaluating QR-1123 has proposed outcome measures of best-corrected visual acuity (BCVA), low-luminance visual acuity, dark-adapted chromatic perimetry, static visual field, microperimetry, spectral-domain optical coherence tomography (SD-OCT), full-field light sensitivity threshold (FST), full-field ERG, fundus autofluorescence, contrast sensitivity, and color vision, all of which are mentioned in this section. Since traditional outcomes measures for eye diseases are not always helpful in quantifying severely impaired vision,61 we also discuss new outcome measures developed to evaluate changes in low vision as well as tests that assess functional changes in vision rather than structural or anatomic changes in order to evaluate clinically relevant parameters.
BCVA is a commonly reported endpoint in clinical trials of ocular interventions. However, this methodology may not be sensitive enough for inherited retinal diseases such as RHO-adRP given that the disease primarily affects rods and peripheral vision with late effects on visual acuity.62 BCVA assigning the logarithm of the minimum angle of resolution for off-chart visual measurements should be performed. Low-luminance visual acuity may be a better alternative to evaluate reduced visual acuity associated with dim lighting.63 It is measured by placing a neutral density filter over the best correction for each eye and having the subject read the normally illuminated Early Treatment Diabetic Retinopathy Study (ETDRS) chart. Low-luminance visual acuity is a stable measure over time and is simple and rapid to perform.
The multi-luminance mobility test (MLMT) was developed specifically to assess functional vision in patients with inherited retinal dystrophies.64 MLMT objectively evaluates functional vision in patients with low vision and nyctalopia by measuring their ability to follow a marked path while avoiding obstacles, handling raised steps, and identifying a door at different illumination levels. Patients may be evaluated by MLMT at up to nine different standardized light levels. Positive changes in MLMT scores indicate that subjects have passed the test at lower light levels. Although visual acuity is typically used as a primary endpoint in clinical trials of ocular interventions, MLMT was used successfully in clinical trials on inherited retinal diseases primarily affecting rods and peripheral vision with late effects on visual acuity.62 This test of functional vision and the ability to carry out vision-dependent daily life activities is important because such activities are linked to quality of life. MLMT performance also correlates with visual acuity and visual field assessments.
FST testing was developed to evaluate clinical efficacy of gene therapy in patients with LCA due to RPE65 mutations since it was challenging to use traditional efficacy outcomes to quantify the severely impaired vision with nystagmus in LCA patients.61 FST utilizes white light stimuli to determine the lowest illumination perceived, or light sensitivity, over the complete visual field. FST measures underlying physiological functions of the rod photoreceptors that are primarily affected by RP. FST is a particularly useful tool, as results are unaffected by nystagmus and the test may be used over variable levels of visual impairment among RP patients.
Visual field testing should be performed to assess the functional area of the light-sensitive retina. In a phase 3 study of voretigene neparvovec-rzyl, in subjects who could see, cumulative visual field calculations were performed across 24 meridians in both eyes separately. Higher sum total degrees correlated to a greater area of functional light-sensitive retina, indicating a greater field of vision for the subject. In order to monitor changes in visual function over time, visual field sensitivity or microperimetry are more accessible and accurate tests. Microperimetry is fundus-tracked perimetry that measures luminance increment sensitivity at specific retinal locations by using a laser ophthalmoscope to visualize the retina through the entirety of the examination.65 Microperimetry has been used more frequently as an outcome measure in clinical trials and is being used to a greater degree in observational studies as well.
Dark-adapted chromatic perimetry quantifies rod-mediated vision and addresses the lack of rod-focused outcome measures.66 This perimeter utilizes a protocol involving a commercial static perimeter under dark-adapted conditions with a stimulus wavelength near the peak of scotopic sensitivity. This technique records rod sensitivity to a cyan wavelength stimulus through comparisons with the maximum theoretical sensitivity of cones at testing points throughout the visual field. Another technique quantifies rod sensitivity through two-color perimetry where sensitivity to cyan is compared to sensitivity to a red stimulus in the same location. The usefulness of dark-adapted chromatic perimetry in clinical trials involving RP patients is still being determined.
Contrast sensitivity is the ability to distinguish an object from its background and may be evaluated using the Pelli-Robson charts at 2 m with the assessment in letters and converted to LogMar (logarithm of the minimum angle of resolution).67 Color vision may be quantitatively measured using the computerized Cambridge color test and the low vision color Cambridge color test, which are currently been investigated as possible outcome measures in inherited retinal degeneration (ClinicalTrials.gov: NCT01878032).
Full-field ERGs may be conducted to assess intervention outcomes, as they provide a global assessment of photoreceptor function.68 An ERG was completed in the phase 1 portion of the clinical trial for voretigene neparvovec-rzyl, but it did not show significant change. Due to the difficulties performing the test in pediatric patients as well as patient discomfort, ERGs were not performed in later trials. Of note, some RHO mutations such as T17M lead to increased susceptibility to light.69 In these patients, bright light flashes may be damaging and, as a result, ERGs at high light intensity (and fundus photography) should be avoided if possible.
Changes in retinal structures may also be assessed from the baseline during clinical trials. SD-OCT has been used to visualize retinal layers and evaluate the integrity of the external limiting membrane, ellipsoid zone, and ONL thickness.70 SD-OCT may also be used to detect cystoid macular edema. Fundus autofluorescence using blue-light excitation may provide information on the metabolic activity of the RPE by observing fluorophores such as A2E in photoreceptor outer segments and lipofuscin granules that aggregate in the RPE as a byproduct of phagocytosis of shed photoreceptor outer segments. SD-OCT and fundus photography were used as inclusion criteria in the RPE65 trials to determine whether patients had sufficient viable retinal cells as determined by retinal thickness, but they were not used in a comparative manner to draw meaningful conclusions on treatment efficiency.
Novel outcome measures are being developed to more easily assess functional vision changes in subjects with severe visual impairment following treatment. As an example, the Institut de la Vision is developing a set of new objective behavioral measures using an artificial street named “StreetLab.”71 A motion capture system with passive markers, eye-tracking, inertial sensors, and surveillance cameras is used to evaluate motion behaviors. The evaluation of locomotion (obstacle avoidance, walking among others) in RP patients under different lighting conditions may be performed. Additionally, gaze behavior may be measured using eye-head coordination, fixation location, and gaze sampling strategy. StreetLab diverges from classical, highly precise, and standardized clinical tests. It provides real world evaluations of a therapeutic intervention’s ability to make a difference in the patients’ lives with performance-based tests.
Conclusions
There are many promising gene therapy candidates for RHO-adRP, with the first-in-human phase 1/2 trial initiated in late 2019, another clinical trial expected to enroll later in 2020, and several other candidates in early development. Well-designed clinical studies with outcome measures suitable to detect moderate changes in low functional vision are critical for the successful development of novel treatments for RHO-adRP. Although only gene therapy candidates for RHO-adRP are covered in this review, the therapeutic strategies discussed herein are applicable to all autosomal dominant ophthalmic disorders and potentially autosomal dominant disorders in other organ systems.
Author Contributions
D.M. and S.D.R. wrote the manuscript. S.H.T. provided essential advice and edited the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We are grateful to Dr. Tarun Sharma and Megan Soucy for insightful comments and the imaging facilities provided by Jonas Children’s Vision Care. Jonas Children’s Vision Care and the Barbara & Donald Jonas Laboratory of Regenerative Medicine and Bernard & Shirlee Brown Glaucoma Laboratory are supported by the National Institutes of Health (5P30CA013696, U01 EY030580, U54OD020351, R24EY028758, R24EY027285, 5P30EY019007, R01EY018213, R01EY024698, R01EY026682, R21AG050437); the Schneeweiss Stem Cell Fund, New York State (DHDOH01-C32590GG-3450000); the Foundation Fighting Blindness New York Regional Research Center Grant (PPA-1218-0751-COLU); Nancy & Kobi Karp; the Crowley Family Funds; the Rosenbaum Family Foundation; the Alcon Research Institute; Abeona Therapeutics; the Gebroe Family Foundation; the Research to Prevent Blindness (RPB) Physician-Scientist Award; and by unrestricted funds from RPB, New York, NY, USA.
References
- 1.Tsang S.H., Sharma T. Retinitis pigmentosa (non-syndromic) Adv. Exp. Med. Biol. 2018;1085:125–130. doi: 10.1007/978-3-319-95046-4_25. [DOI] [PubMed] [Google Scholar]
- 2.Verbakel S.K., van Huet R.A.C., Boon C.J.F., den Hollander A.I., Collin R.W.J., Klaver C.C.W., Hoyng C.B., Roepman R., Klevering B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018;66:157–186. doi: 10.1016/j.preteyeres.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Narayan D.S., Wood J.P., Chidlow G., Casson R.J. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016;94:748–754. doi: 10.1111/aos.13141. [DOI] [PubMed] [Google Scholar]
- 4.Caruso S., Ryu J., Quinn P.M., Tsang S.H. Precision metabolome reprogramming for imprecision therapeutics in retinitis pigmentosa. J. Clin. Invest. 2020;130:3971–3973. doi: 10.1172/JCI139239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malanson K.M., Lem J. Rhodopsin-mediated retinitis pigmentosa. Prog. Mol. Biol. Transl. Sci. 2009;88:1–31. doi: 10.1016/S1877-1173(09)88001-0. [DOI] [PubMed] [Google Scholar]
- 6.Gouras P., Carr R.E. Electrophysiological studies in early retinitis pigmentosa. Arch. Ophthalmol. 1964;72:104–110. doi: 10.1001/archopht.1964.00970020106022. [DOI] [PubMed] [Google Scholar]
- 7.Berson E.L. Retinitis pigmentosa and allied diseases: applications of electroretinographic testing. Int. Ophthalmol. 1981;4:7–22. doi: 10.1007/BF00139576. [DOI] [PubMed] [Google Scholar]
- 8.Dryja T.P., McGee T.L., Reichel E., Hahn L.B., Cowley G.S., Yandell D.W., Sandberg M.A., Berson E.L. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- 9.Dryja T.P., McGee T.L., Hahn L.B., Cowley G.S., Olsson J.E., Reichel E., Sandberg M.A., Berson E.L. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 1990;323:1302–1307. doi: 10.1056/NEJM199011083231903. [DOI] [PubMed] [Google Scholar]
- 10.National Center for Biotechnology Information . 2020. Gene. RHO rhodopsin [Homo sapiens (human)]https://www.ncbi.nlm.nih.gov/gene/6010 [Google Scholar]
- 11.Jacobson S.G., Kemp C.M., Sung C.H., Nathans J. Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am. J. Ophthalmol. 1991;112:256–271. doi: 10.1016/s0002-9394(14)76726-1. [DOI] [PubMed] [Google Scholar]
- 12.Cideciyan A.V., Hood D.C., Huang Y., Banin E., Li Z.Y., Stone E.M., Milam A.H., Jacobson S.G. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc. Natl. Acad. Sci. USA. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berson E.L., Rosner B., Sandberg M.A., Dryja T.P. Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His) Arch. Ophthalmol. 1991;109:92–101. doi: 10.1001/archopht.1991.01080010094039. [DOI] [PubMed] [Google Scholar]
- 14.Athanasiou D., Aguila M., Bellingham J., Li W., McCulley C., Reeves P.J., Cheetham M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018;62:1–23. doi: 10.1016/j.preteyeres.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nathans J., Piantanida T.P., Eddy R.L., Shows T.B., Hogness D.S. Molecular genetics of inherited variation in human color vision. Science. 1986;232:203–210. doi: 10.1126/science.3485310. [DOI] [PubMed] [Google Scholar]
- 16.Nathans J., Hogness D.S. Isolation and nucleotide sequence of the gene encoding human rhodopsin. Proc. Natl. Acad. Sci. USA. 1984;81:4851–4855. doi: 10.1073/pnas.81.15.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacLaren R.E., Pearson R.A., MacNeil A., Douglas R.H., Salt T.E., Akimoto M., Swaroop A., Sowden J.C., Ali R.R. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006;444:203–207. doi: 10.1038/nature05161. [DOI] [PubMed] [Google Scholar]
- 18.Berson E.L., Rosner B., Sandberg M.A., Hayes K.C., Nicholson B.W., Weigel-DiFranco C., Willett W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch. Ophthalmol. 1993;111:761–772. doi: 10.1001/archopht.1993.01090060049022. [DOI] [PubMed] [Google Scholar]
- 19.da Cruz L., Dorn J.D., Humayun M.S., Dagnelie G., Handa J., Barale P.O., Sahel J.A., Stanga P.E., Hafezi F., Safran A.B., Argus II Study Group Five-year safety and performance results from the Argus II Retinal Prosthesis System clinical trial. Ophthalmology. 2016;123:2248–2254. doi: 10.1016/j.ophtha.2016.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang A.L., Knight D.K., Vu T.T., Mehta M.C. Retinitis pigmentosa: review of current treatment. Int. Ophthalmol. Clin. 2019;59:263–280. doi: 10.1097/IIO.0000000000000256. [DOI] [PubMed] [Google Scholar]
- 21.Maguire A.M., Russell S., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., Marshall K.A. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: results of phase 1 and 3 trials. Ophthalmology. 2019;126:1273–1285. doi: 10.1016/j.ophtha.2019.06.017. [DOI] [PubMed] [Google Scholar]
- 22.Crooke S.T. Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Ther. 2017;27:70–77. doi: 10.1089/nat.2016.0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scoles D.R., Minikel E.V., Pulst S.M. Antisense oligonucleotides: a primer. Neurol. Genet. 2019;5:e323. doi: 10.1212/NXG.0000000000000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biasutto P., Adamson P.S., Dulla K., Murray S., Monia B., McCaleb M. Allele specific knock-down of human P23H rhodopsin mRNA and prevention of retinal degeneration in humanized P23H rhodopsin knock-in mouse, following treatment with an intravitreal GAPmer antisense oligonucleotide (QR-1123) Invest. Ophthalmol. Vis. Sci. 2019;60:5719. [Google Scholar]
- 25.Murray S.F., Jazayeri A., Matthes M.T., Yasumura D., Yang H., Peralta R., Watt A., Freier S., Hung G., Adamson P.S. Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest. Ophthalmol. Vis. Sci. 2015;56:6362–6375. doi: 10.1167/iovs.15-16400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambeth L.S., Smith C.A. Short hairpin RNA-mediated gene silencing. Methods Mol. Biol. 2013;942:205–232. doi: 10.1007/978-1-62703-119-6_12. [DOI] [PubMed] [Google Scholar]
- 27.Millington-Ward S., Chadderton N., O’Reilly M., Palfi A., Goldmann T., Kilty C., Humphries M., Wolfrum U., Bennett J., Humphries P. Suppression and replacement gene therapy for autosomal dominant disease in a murine model of dominant retinitis pigmentosa. Mol. Ther. 2011;19:642–649. doi: 10.1038/mt.2010.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiang A.S., Palfi A., Ader M., Kenna P.F., Millington-Ward S., Clark G., Kennan A., O’reilly M., Tam L.C., Aherne A. Toward a gene therapy for dominant disease: validation of an RNA interference-based mutation-independent approach. Mol. Ther. 2005;12:555–561. doi: 10.1016/j.ymthe.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 29.O’Reilly M., Palfi A., Chadderton N., Millington-Ward S., Ader M., Cronin T., Tuohy T., Auricchio A., Hildinger M., Tivnan A. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am. J. Hum. Genet. 2007;81:127–135. doi: 10.1086/519025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cideciyan A.V., Sudharsan R., Dufour V.L., Massengill M.T., Iwabe S., Swider M., Lisi B., Sumaroka A., Marinho L.F., Appelbaum T. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA. 2018;115:E8547–E8556. doi: 10.1073/pnas.1805055115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed C.M., Massengill M.T., Ildefonso C.J., Zhu P., Li H., Hauswirth W.W. AAV mediated RNA replacement as therapy in a mouse model of autosomal dominant retinitis pigmentosa. Mol. Ther. 2020;28(4 Suppl 1):320. [Google Scholar]
- 32.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang D., Zhang F., Gao G. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell. 2020;181:136–150. doi: 10.1016/j.cell.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai Y.-T., Wu W.-H., Lee T.-T., Wu W.-P., Xu C.L., Park K.S., Cui X., Justus S., Lin C.S., Jauregui R. Clustered regularly interspaced short palindromic repeats-based genome surgery for the treatment of autosomal dominant retinitis pigmentosa. Ophthalmology. 2018;125:1421–1430. doi: 10.1016/j.ophtha.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diner B.A., Dass A., Nayak R., Flinkstrom Z., Tallo T., DaSilva J. Dual AAV-based “knock-out-and-replace” of RHO as a therapeutic approach to treat RHO-associated autosomal dominant retinitis pigmentosa (RHO adRP) Mol. Ther. 2020;28(4 Suppl 1):108–109. [Google Scholar]
- 40.Bakondi B., Lv W., Lu B., Jones M.K., Tsai Y., Kim K.J., Levy R., Akhtar A.A., Breunig J.J., Svendsen C.N., Wang S. In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol. Ther. 2016;24:556–563. doi: 10.1038/mt.2015.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li P., Kleinstiver B.P., Leon M.Y., Prew M.S., Navarro-Gomez D., Greenwald S.H., Pierce E.A., Joung J.K., Liu Q. Allele-specific CRISPR-Cas9 genome editing of the single-base P23H mutation for rhodopsin-associated dominant retinitis pigmentosa. CRISPR J. 2018;1:55–64. doi: 10.1089/crispr.2017.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D’Amico A., Butcher R., Kantardzhieva A., Takeuchi R., Lukason M., Pierce E. Allele-specific CRISPR-Cas9 via dual AAV-mediated gene editing rescue retinal degeneration in a humanized RHO-P23H mouse model. Mol. Ther. 2020;28(4 Suppl 1):454. [Google Scholar]
- 43.Patrizi C., Llado M., Benati D., Guarascio R., Cheetham M., Auricchio A. Specific knock-down of P347S dominant mutation in rhodopsin gene by CRISPR/Cas9 system. Mol. Ther. 2020;28(4 Suppl 1):286–287. [Google Scholar]
- 44.Dürrenberger F., Thompson A.J., Herrin D.L., Rochaix J.-D. Double strand break-induced recombination in Chlamydomonas reinhardtii chloroplasts. Nucleic Acids Res. 1996;24:3323–3331. doi: 10.1093/nar/24.17.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L., Smith J., Breton C., Clark P., Zhang J., Ying L., Che Y., Lape J., Bell P., Calcedo R. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat. Biotechnol. 2018;36:717–725. doi: 10.1038/nbt.4182. [DOI] [PubMed] [Google Scholar]
- 46.McCall M.A., Jalligampala A., Fransen J., Noel J., Wang W., Fleissig E. Therapeutic efficacy of ARCUS meganuclease gene editing—arrest of rod degeneration and restoration of rod function in a transgenic pig model of autosomal dominant retinitis pigmentosa. Mol. Ther. 2020;28(4 Suppl 1):1. [Google Scholar]
- 47.Farrar G.J., Kenna P., Redmond R., McWilliam P., Bradley D.G., Humphries M.M., Sharp E.M., Inglehearn C.F., Bashir R., Jay M. Autosomal dominant retinitis pigmentosa: absence of the rhodopsin proline—histidine substitution (codon 23) in pedigrees from Europe. Am. J. Hum. Genet. 1990;47:941–945. [PMC free article] [PubMed] [Google Scholar]
- 48.Dryja T.P., Hahn L.B., Cowley G.S., McGee T.L., Berson E.L. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. USA. 1991;88:9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teboul L., Herault Y., Wells S., Qasim W., Pavlovic G. Variability in genome editing outcomes: challenges for research reproducibility and clinical safety. Mol. Ther. 2020;28:1422–1431. doi: 10.1016/j.ymthe.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 51.Ma H., Marti-Gutierrez N., Park S.W., Wu J., Hayama T., Darby H., Van Dyken C., Li Y., Koski A., Liang D. Ma et al. reply. Nature. 2018;560:E10–E23. doi: 10.1038/s41586-018-0381-y. [DOI] [PubMed] [Google Scholar]
- 52.Cullot G., Boutin J., Toutain J., Prat F., Pennamen P., Rooryck C., Teichmann M., Rousseau E., Lamrissi-Garcia I., Guyonnet-Duperat V. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019;10:1136. doi: 10.1038/s41467-019-09006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Enache O.M., Rendo V., Abdusamad M., Lam D., Davison D., Pal S., Currimjee N., Hess J., Pantel S., Nag A. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020;52:662–668. doi: 10.1038/s41588-020-0623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cho G.Y., Schaefer K.A., Bassuk A.G., Tsang S.H., Mahajan V.B. CRISPR genome surgery in the retina in light of off-targeting. Retina. 2018;38:1443–1455. doi: 10.1097/IAE.0000000000002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maragh S.K.N., Tona A., Eskandari T. 2019. NIST Genome Editing Consortium.https://www.nist.gov/programs-projects/nist-genome-editing-consortium [Google Scholar]
- 57.US Food and Drug Administration . 2020. Human gene therapy for retinal disorders: guidance for industry.https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-retinal-disorders [Google Scholar]
- 58.Wang H.X., Song Z., Lao Y.H., Xu X., Gong J., Cheng D., Chakraborty S., Park J.S., Li M., Huang D. Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc. Natl. Acad. Sci. USA. 2018;115:4903–4908. doi: 10.1073/pnas.1712963115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.US Food and Drug Administration. 2020. Long term follow-up after administration of human gene therapy products: guidance for industry.https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-administration-human-gene-therapy-products [Google Scholar]
- 60.Akcakaya P., Bobbin M.L., Guo J.A., Malagon-Lopez J., Clement K., Garcia S.P., Fellows M.D., Porritt M.J., Firth M.A., Carreras A. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018;561:416–419. doi: 10.1038/s41586-018-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garafalo A.V., Cideciyan A.V., Héon E., Sheplock R., Pearson A., WeiYang Yu C., Sumaroka A., Aguirre G.D., Jacobson S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020 doi: 10.1016/j.preteyeres.2019.100827. Published online December 30, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ciulla T.A., Hussain R.M., Berrocal A.M., Nagiel A. Voretigene neparvovec-rzyl for treatment of RPE65-mediated inherited retinal diseases: a model for ocular gene therapy development. Expert Opin. Biol. Ther. 2020;20:565–578. doi: 10.1080/14712598.2020.1740676. [DOI] [PubMed] [Google Scholar]
- 63.Sunness J.S., Rubin G.S., Broman A., Applegate C.A., Bressler N.M., Hawkins B.S. Low luminance visual dysfunction as a predictor of subsequent visual acuity loss from geographic atrophy in age-related macular degeneration. Ophthalmology. 2008;115:1480–1488.e2. doi: 10.1016/j.ophtha.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chung D.C., McCague S., Yu Z.F., Thill S., DiStefano-Pappas J., Bennett J., Cross D., Marshall K., Wellman J., High K.A. Novel mobility test to assess functional vision in patients with inherited retinal dystrophies. Clin. Exp. Ophthalmol. 2018;46:247–259. doi: 10.1111/ceo.13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu Z., Cimetta R., Caruso E., Guymer R.H. Performance of a defect-mapping microperimetry approach for characterizing progressive changes in deep scotomas. Transl. Vis. Sci. Technol. 2019;8:16. doi: 10.1167/tvst.8.4.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bennett L.D., Klein M., Locke K.G., Kiser K., Birch D.G. Dark-adapted chromatic perimetry for measuring rod visual fields in patients with retinitis pigmentosa. Transl. Vis. Sci. Technol. 2017;6:15. doi: 10.1167/tvst.6.4.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Verdina T., Piaggi S., Peschiera R., Russolillo V., Ferraro V., Chester J., Mastropasqua R., Cavallini G.M. Biofeedback low vision rehabilitation with Retimax vision trainer in patients with advanced age-related macular degeneration: a pilot study. Semin. Ophthalmol. 2020;35:164–169. doi: 10.1080/08820538.2020.1774624. [DOI] [PubMed] [Google Scholar]
- 68.Menghini M., Cehajic-Kapetanovic J., MacLaren R.E. Monitoring progression of retinitis pigmentosa: current recommendations and recent advances. Expert Opin. Orphan Drugs. 2020;8:67–78. doi: 10.1080/21678707.2020.1735352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.White D.A., Fritz J.J., Hauswirth W.W., Kaushal S., Lewin A.S. Increased sensitivity to light-induced damage in a mouse model of autosomal dominant retinal disease. Invest. Ophthalmol. Vis. Sci. 2007;48:1942–1951. doi: 10.1167/iovs.06-1131. [DOI] [PubMed] [Google Scholar]
- 70.Hood D.C., Lin C.E., Lazow M.A., Locke K.G., Zhang X., Birch D.G. Thickness of receptor and post-receptor retinal layers in patients with retinitis pigmentosa measured with frequency-domain optical coherence tomography. Invest. Ophthalmol. Vis. Sci. 2009;50:2328–2336. doi: 10.1167/iovs.08-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.StreeLab . 2020. Artificial Street.https://www.streetlab-vision.com/rue-artificielle/?lang=en [Google Scholar]