

Abstract
Metabolism consists of a series of reactions that occur within cells of living organisms to sustain life. The process of metabolism involves many interconnected cellular pathways to ultimately provide cells with the energy required to carry out their function. The importance and the evolutionary advantage of these pathways can be seen as many remain unchanged by animals, plants, fungi, and bacteria. In eukaryotes, the metabolic pathways occur within the cytosol and mitochondria of cells with the utilisation of glucose or fatty acids providing the majority of cellular energy in animals. Metabolism is organised into distinct metabolic pathways to either maximise the capture of energy or minimise its use. Metabolism can be split into a series of chemical reactions that comprise both the synthesis and degradation of complex macromolecules known as anabolism or catabolism, respectively. The basic principles of energy consumption and production are discussed, alongside the biochemical pathways that make up fundamental metabolic processes for life.
Keywords: biochemistry, glycolysis, metabolism
Introduction
The basics of metabolism
When many people think about metabolism, they think of food and drink or the huge metabolic pathway diagram with thousands of connections. However, understanding metabolism is key to understanding life and this has been a subject of fascination with biochemists for more than 150 years. The great Nobel Prize-winning scientist Hans Krebs was inspired to study metabolism by his university professor Prof France Knoop (who discovered β-oxidation of fatty acids). He unpicked and described both the citric acid cycle and the urea cycle which lie as fundamental processes of metabolism. Prof Franz Knoop said:
“The final goal of physiological chemistry/(metabolism)” is to “present a scheme that puts together an unbroken series of equations of all of the reactions from the food stuffs which continuously supply to the organism its energy needs, all the way to the slag that again leaves the organism as energyless final oxidation products.” Prof Franz Knoop 1931 - Hans Krebs: The formation of a scientific life 1900–1933 by F.L. Holmes.
Whilst it can be daunting to think about every metabolic pathway that is occurring, we can break it down and understand its smaller aspects. Knoop’s words underpin the true meaning of metabolism and one of its central roles in biochemistry and physiological chemistry. Metabolism is derived from the Greek word, metabolē meaning ‘to change’ and comprises the total of all chemical reactions that take place in the cell that are essential for life. These chemical reactions comprise both the synthesis and degradation of complex macromolecules and can be divided into either catabolism or anabolism (Figure 1 – catabolism vs anabolism). Catabolism is the degradation of complex macromolecules into simpler molecules such as carbon dioxide, water, and ammonia. Anabolism is the biosynthetic pathways that generate complex macromolecules such as nucleic acids, proteins, polysaccharides, and lipids.
Figure 1. Coupling of anabolic and catabolic pathways.

Anabolism utilises energy to make macromolecules and biomolecular polymers. Catabolism releases energy when these are broken down into simpler molecules.
To maintain cellular and whole-body function, living organisms require energy continuously. Energy is required for mechanical work (contraction and cellular movement), active transport of ions/substrates (i.e. K+, Mg2+, and Ca2+, for example in cardiac contraction) and the biosynthesis of complex macromolecules (such as glycogen).
This review will focus on the basics of metabolism within mammals, with mentions of other organisms too. The aim is to provide you with an understanding of the metabolic pathways that are present in animals, how energy is derived from these systems, and how they are controlled. Finally, we will touch on the exciting elements of research in metabolism, including how understanding metabolism could help with treating cancer, how it can be used in biotechnology to generate bioethanol, and how metabolic diseases make up several key inherited conditions.
Why pathways?
Metabolic pathways are vital in capturing useful energy. This is in contrast with uncontrolled combustion, where energy is rapidly released into the environment, as heat and light, which would be unsustainable for life. Metabolism is organised into distinct metabolic pathways to either maximise the capture of energy or minimise its use. In catabolism, metabolic pathways are organised such that energy is released slowly in discrete quanta of energy, which is captured by the synthesis of adenosine triphosphate (ATP), guanosine triphosphate (GTP), NAD(P)H (nicotinamide adenine nucleotide (phosphate)) or by the electron transport chain (ETC). In anabolism, metabolic pathways use these discrete quanta of energy in the form of ATP and NADPH to perform work, such as the synthesis of biomolecules.
The action of metabolic pathways in the cell is particularly impressive with the ability to organise several hundred metabolic reactions occurring simultaneously within the cell and occurring at a relatively low temperature. Most of this is achieved by specific enzymes and compartmentalisation of reactions and enzymes. Sometimes this compartmentalisation is achieved by separating reactions into different organelles, or by coupling reactions together, to prevent uncontrolled combustion. Enzymes allow discrete reactions to occur, which when combined give the same overall effect as combustion, but in a controlled fashion. In this review, we will discuss the energy of reactions, the role of metabolic enzymes, key metabolic pathways, and then the vital organelles for energy generation.
Energy within the system
One of the most important biomolecules in the cell is the nucleotide; ATP. ATP has a linear triphosphate structure which provides four negative charges and therefore it exists as a highly charged molecule. Due to this negative charge, these bonds can store a large amount of energy, which can be liberated easily at the site of work. Along with ATP, NAD+ also acts as a store for energy in its reduced form, NADH + H. NAD+ acts as a universal electron carrier in the cell, transporting electrons from catabolism site to the ETC.
Émilie du Châtelet proposed the law of conservation, which stated that energy can be neither created nor destroyed; rather, it can only be transformed from one form into another. This is linked to the first law of thermodynamics and it helps us to explain how energy flows through biological systems. The laws of thermodynamics also help us to predict if a reaction is possible and how much energy is required or released in the process. This brings us to an important concept: the role of chemical equilibria, where a reaction can be reversible (see Figure 2). At equilibrium, there is no net reaction as both the forward and reverse reactions are moving equally fast. The system aims to bring reactions to equilibrium. However, true equilibrium is not compatible with life, as there is no longer a flow of energy.
Figure 2. Reaching equilibrium.

If you increase the concentration of A and B, this pushes the reaction to make more C and D. If you do the opposite and add more C and D, then the reverse reaction occurs. The aim is to bring the reaction back to equilibrium.
The direction of the reaction can be governed by many aspects, including the concentration of substrate or products, the energy released or required for the reaction, and in the case of metabolic pathways, the activity of the enzyme. Figure 2 shows that simple equilibria can be influenced by the change in substrates and products and that the direction of the reaction is governed by which has a higher concentration. However, the fulcrum of the metaphorical seesaw is not always in the centre and this is described by the equilibrium constant or standard free energy change.
Gibbs free energy is used to describe whether our reaction will run in one direction or the other and is termed as the energy available to do work. This is different from total energy, which is the energy obtained from combustion. Standard free energy change is a special form of the Gibbs free energy and provides a constant energy change during the standard state – pH 7.0 at 25°C and 1 atm, when the concentration of substrates is 1.0 M. This is termed as ΔG°′ and has a characteristic constant value for a given reaction and tells us where the balancing point is. However, we still want to know if the reaction is going in one direction or the other. Change in free energy (ΔG) is a much more realistic measure of the energy available to the system under normal cellular environments. This is because in mammals internal body temperatures are not at 25°C and metabolic substrates are not at 1 M. Whilst ΔG°′ has a characteristic constant value for a given reaction, ΔG is not a constant as it is dependent on ΔG°′, the concentration of substrates, and the temperature. ΔG can be described by the following (eqn 1):
| (1) |
Enthalpy (H) = total energy; Entropy (S) = disorder of the system; T = temperature (measured in Kelvin)
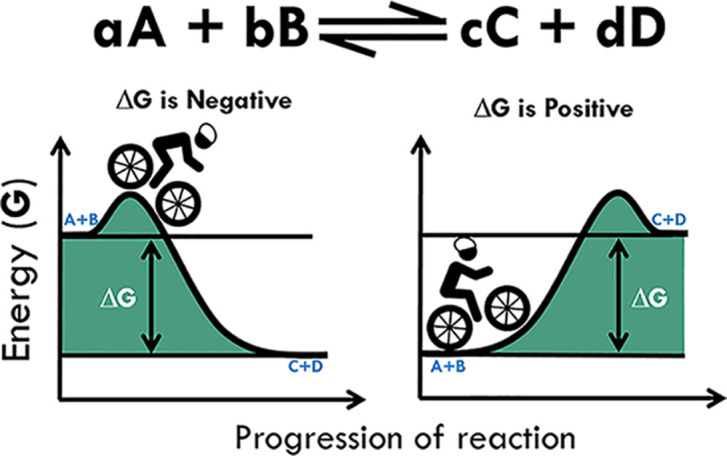
The second law of thermodynamics states that for a process (reaction) to take place, there must be an increase in entropy in the universe (or system). This means that for a reaction to take place spontaneously there needs to be an increase in entropy and therefore a negative change in free energy (ΔG). We can now consider what this means in terms of reaction kinetics and how ΔG can dictate the direction of the reaction, using the analogy of bicycling (Figure 3). If the ΔG is negative, a reaction proceeds spontaneously and with a loss of free energy. Using our bike analogy, this would be like the energy released as you cycle from the top of a hill down to the bottom. This kind of reaction is termed EXERGONIC, or if it releases heat, exothermic. If on the other hand, ΔG is positive, and you are looking up at the hill, then the reaction is unfavourable or not spontaneous, i.e. the reaction is ENDERGONIC, or in the case of heat, endothermic. As reactions can be reversible, if the forward reaction has a positive ΔG, then the reverse reaction will be negative, therefore the reaction could occur in reverse.
Figure 3. The effect of changes in ΔG on the reaction A+B and C+D.
If we put this into practical terms to consider the second reaction in glycolysis catalysed by glucose-6-phosphate (G6P) isomerase (G6PI) (eqn 2):
| (2) |
The ΔG°′ for this reaction is +1.7 kJ.mol−1, which is close to equilibrium, would be endogenic and therefore not spontaneous. However ΔG°′ describes the free energy available under standard state conditions. If we look at a more physiological condition, i.e. at 37°C, then if the concentration of G6P increases, this decreases the ΔG into negative and drives the forward reaction. The counter is true, so if fructose-6-phosphate (F6P) accumulates the forward reaction is inhibited (due to ΔG now becoming positive) then the reverse reaction is favoured (as the ΔG for this is negative). This is an example of one of the freely reversible reactions in glycolysis.
Another thing to take into account is that some reactions will not be reversible under physiological conditions. For example, if the ΔG is very negative, then changing the amount of product or substrate will not affect, especially as the temperature can very rarely be altered to any real degree. One example of where this is particularly seen is in one of the key regulatory steps in glycolysis – the reaction catalysed by phosphofructokinase 1 (PFK1), shown in (eqn 3). The ΔG°′ is −14.2 kJ.mol−1, meaning that under physiological conditions the ΔG is very negative, and therefore even a build-up of fructose-1-phosphate is not sufficient to reverse the reaction. This reaction is effectively irreversible under physiological conditions; we will discuss this in more detail later.
| (3) |
One important caveat about ΔG and reactions is that ΔG does not predict the rate of reaction. The rate of reaction is governed by the activation energy and is catalysed by enzymes. If ΔG is negative, then the reaction should occur spontaneously, but there is still a need to overcome the initial activation energy. Reactions that need catalysis can be controlled by the activity of the enzyme. For example, whilst the ΔG for (eqn 3) is very negative, PFK1 is regulated by feedback inhibition and activation. Increasing concentrations of either phosphoenolpyruvate (PEP; the penultimate intermediate in glycolysis) or ATP inhibit PFK1, as they act as a signal for sufficient energy demand. Conversely, an increase in the concentration of adenosine monophosphate (AMP) signals an energy deficit in the cell and therefore activates PFK1. For further information on enzyme inhibition and feedback, then look at the Enzymes: principles and biotechnological applications (Essays in Biochemistry (2015), 59, 1–41; DOI: 10.1042/bse0590001).
Coupled reactions and pathways
There is an intrinsic value to coupling reactions together to form complex pathways. As we can see from Table 1, several reactions have a positive ΔG and so in theory, should not be spontaneous nor go in reverse. However, by linking the reactions together, the product of one reaction becomes the substrate for the next. Thus, by coupling reactions together, substrates can build up and products can be rapidly removed, changing the equilibrium seesaw (Figure 2) making the ΔG negative (for reversible reactions). We will discuss glycolysis in more detail later and how regulation can control key steps and the other fates of G6P.
Table 1. Standard free energy and Gibbs free energy of each step in glycolysis.
| Enzyme | ΔG°′ in kJ.mol−1 | ΔG in kJ.mol−1 |
|---|---|---|
| Hexokinase | −16.7 | −33.5 |
| G6PI | +1.7 | −2.5 |
| PFK | −14.2 | −22.2 |
| Aldolase | +23.8 | −1.3 |
| Triose phosphate isomerase | +7.5 | +2.5 |
| Glyceraldehyde 3 phosphate dehydrogenase | +6.3 | −1.7 |
| Phosphoglycerate kinase | −18.8 | +1.3 |
| Phosphoglycerate mutase | +4.6 | +0.8 |
| Enolase | +1.7 | −3.3 |
| Pyruvate kinase | −31.4 | −16.7 |
Finally, ATP can be used to drive reactions forward by coupling the release of energy from its hydrolysis to overcome a positive ΔG. As we have previously seen, ATP stores a large amount of energy in the phosphate bonds and cleavage of one phosphate liberates −31 kJ.mol−1. This can be used to overcome endergonic reactions such as the first reaction in glycolysis:
The native reaction, the addition of inorganic phosphate, is endergonic and requires +14 kJ.mol−1. However, because the hydrolysis of ATP liberates Pi and Pi is a substrate for the first reaction, these reactions can be coupled and the energy of hydrolysis of ATP is used to drive the reaction. Note this coupling only works because Pi is a shared intermediate in both reactions.
In summary, reactions are favoured when they have a negative ΔG. Coupling reactions together in pathways enable unfavourable reactions to occur. Metabolism is organised into distinct metabolic pathways to maximise the capture of energy and prevent uncontrolled combustion.
Metabolism from thin air
Organisms fall into two distinct groups, either heterotrophs or autotrophs. Autotrophs derived from the words ‘auto’ for ‘self’ and ‘-trophs’ for ‘food’ are a set of organisms that can obtain energy from sunlight and inorganic nutrients from their environment to fix carbon into complex macromolecules. Heterotrophs represent a much larger group of organisms, including most microorganisms and animal cells. In contrast with autotrophs, they are not able to fix their own carbon, but instead use the carbon fixed by autotrophs to grow.
Autotrophs can generally be split into two groups, chemoautotrophs, and photoautotrophs. These organisms use the energy from inorganic chemical reactions (chemoautotrophs, i.e. Nitrobacter) or light (photoautotrophs, i.e. plants) to fix carbon dioxide as a usable energy source. One example of a key group of chemoautotrophs are the diazotrophs, which are nitrogen-fixing bacteria and archaea. They are responsible for fixing nitrogen from the air (N2) into inorganic nitrogen compounds such as ammonia (NH4+), nitrite (NO2−), and nitrate (NO3−). They are also capable of using these compounds to obtain energy for biosynthesis of macromolecules. Ammonia-oxidising (AOB) and nitrite-oxidising (NOB) bacteria take advantage of processes similar to the eukaryotic ETC (in mitochondria) to generate their ATP (Figure 4). Oxidation of inorganic nitrogen compounds, such as NH4+ and NO2− release electrons that are captured and transferred by copper-containing enzymes to a final electron acceptor. The transfer of these electrons drive protons (H+) to be pumped out to generate a proton gradient, also known as the proton motive force. These protons then flow back down their concentration gradient through an ATP synthase, to generate ATP. This ATP can then be utilised in biosynthetic processes such as the Calvin cycle, which captures CO2 and converts it into carbohydrates.
Figure 4. The role of nitrogen in generating macromolecules in chemoautotrophs – AOB, NOB, NH4+, NO2−, and NO3−.

Photoautotrophs represent the major class of autotrophs, which utilise light as an energy source to fix carbon dioxide (Figure 5). Multicellular plants and unicellular organisms (algae and cyanobacteria) are perhaps the best examples of this class, using the process of photosynthesis to turn sunlight into ATP and NADPH. Matthew Johnson wrote an excellent review on Photosynthesis for Essays In Biochemistry ((2016) 60, 255–273; DOI: 10.1042/EBC20160016). In brief, these organisms use photons of light to obtain electrons from water and produce oxygen as a by-product. These electrons are again used to drive ATP formation, which in turn is used by pathways, such as the Calvin cycle to generate carbohydrates. In eukaryotic organisms such as green plants and algae, this process of capturing photons is achieved by the chloroplast. Cyanobacteria lack these organelles, instead photosynthesising using folded membranes. Genome analysis data suggest that an ancient cyanobacterium might have provided the ancestor of the chloroplast in eukaryotic cells (for more information on this topic, the recent work of Sánchez-Baracaldo et al. (2017) describes their work to identify the early photosynthetic eukaryotes).
Figure 5. Basic pathway of photosynthesis and biosynthesis in green plants.

Animal metabolism
As we have already seen, animals are heterotrophs and rely on organic chemical nutrients to produce physiologically useful energy. Animals, therefore, need a dietary intake of carbohydrate, fat, and protein for energy, along with vitamins and ions. Although the recommended dietary intake for humans varies from country to country, in the U.K. the NHS recommends a diet comprising 68.5% carbohydrate, 18.5% fat, and 13% protein. However, within the general population these numbers are shifting. There is a much greater reliance on fat and sugar within the diet. The increased overconsumption of fats and sugar are potentially fuelling an obesity epidemic, with the greatest driving force being sugar intake. Therefore, a greater understanding of metabolic pathways and the processes that govern weight loss and gain is required. There are five major sources of metabolic fuel used by animal tissues, these include glucose, fatty acids, ketones, amino acids, and lactate. Glucose, fatty acids, and amino acids are generally derived from dietary sources, although some can be synthesised by specific organs. Glucose and fatty acids can be stored within the body in polymers such as glycogen and triglycerides (TAGs), respectively, and during times of starvation, amino acids can be liberated from proteins. Ketones and lactate are only transiently produced in the body and are not stored.
Many tissues are flexible with how they can derive ATP. Under times of fasting or starvation (fasting for longer than 24 h), our body prioritises breakdown of fuel sources so that the least important for tissue survival is used first. Over the first 24 h, the body uses liver glycogen stores to maintain blood glucose concentrations, but as can be seen in Table 2, there are limited stores of glucose in the body. The brain requires between 100 and 120 g of glucose per day to maintain normal function, meaning that glycogen levels alone would not be sufficient. There is therefore a need to increase glucose production to maintain normal blood glucose at close to 5.5 mmol.l-1 (with blood glucose not normally dropping below 3.5 mmol.l-1 in extreme conditions). As the body progresses through this first 24 h, it starts to increase the synthesis of ‘new’ glucose from other sources and by day 2, gluconeogenesis is the predominant source of glucose. To maintain normal blood glucose levels, some tissues, such as the heart and muscle, increase fatty acid usage to minimise their glucose utilisation. As we enter this longer period of fasting and starvation the body starts to breakdown ‘mobilisable’ protein in skeletal muscle (proteolysis), to provide substrates for gluconeogenesis (in the liver). These ‘mobilisable’ proteins are initially proteins that are deemed expendable or whose action is energy demanding; however, as starvation progresses this expendable protein breakdown is followed by the breakdown of less expendable proteins. If this protein breakdown continues, it can lead to muscle wasting and eventually compromised physiological function, damage to the diaphragm, and death.
Table 2. Mass of fuel reserves in tissue and total in a typical 70-kg man (adapted from G.F., Jr, Cahill (1976) Clin. Endocrinol. Metab. 5, 398).
| Tissue | Glucose or glycogen (g) | Mobilisable proteins (g) | TAGs (g) |
|---|---|---|---|
| Adipose tissue | 20 | 10 | 15000 |
| Blood | 15 | 0 | 5 |
| Brain | 2 | 0 | 0 |
| Liver | 100 | 100 | 50 |
| Skeletal muscle | 300 | 6000 | 50 |
| Total (g) | 437 | 6110 | 15105 |
Metabolic fuels are selectively metabolised
The tissues of the body show differing abilities to utilise various metabolic fuels, and most show flexibility in fuel selection. For several tissues, glucose is the main fuel, but others, including the heart, can use a wide range of fuels. The human brain relies almost completely on glucose to meet the high energy demand, with a small amount of ketones used during starvation. It is estimated that at rest the brain uses nearly two-thirds of glucose consumption, with the erythrocytes (red blood cells) and skeletal muscle using most of the rest. Erythrocytes are solely dependent on glucose as they lack mitochondria, therefore can only undergo anaerobic glycolysis, highlighting the importance of maintaining normal blood glucose levels. During exercise, skeletal muscle will increase its demand for glucose, breaking down the extensive glycogen stores (∼300 g) to maintain peak output.
Fatty acids are a major source of energy for humans and most terrestrial animals, due to their ability to densely pack into TAGs, meaning mol for mol they generate more ATP than glucose. TAGs are hydrophobic and allow efficient storage of fatty acids in lipid droplets, such as in adipose tissue. Upon lipolysis (the breakdown of TAGs), free fatty acids are released into the blood, where they can be oxidised by several tissues. At rest the heart derives 60–70% of its energy demand from fatty acids, highlighting the importance of this fuel source. However, elevated levels of fatty acids are linked to conditions such as type 2 diabetes (which is covered in more detail later). The liver uses fatty acids, both as a fuel source and, in times of fasting and starvation, for the generation of ketones (also called ketone bodies). The liver can generate the two major ketones, acetoacetate and β-hydroxybutyrate, using acetyl Coenzyme A (CoA) from β-oxidation in a process known as ketogenesis. The liver lacks the enzyme succinyl CoA transferase to use ketones as a fuel source and therefore releases it for other tissues, such as the heart, skeletal muscle, and the brain. Ketones can pass through the blood–brain barrier and can offer an additional source of energy to glucose in the brain, although they cannot fully replace glucose. Ketones are oxidised in these tissues in a process known as ketolysis, to generate 20 and 22.5 ATP (acetoacetate and β-hydroxybutyrate, respectively). In the blood, ketones exist in an acidic form and at low concentrations (normally under 0.6 mmol.l-1) are unlikely to cause the body any harm. However, during diabetic ketoacidosis (an acute complication of type 1 diabetes), there is a rapid and uncontrolled overproduction of ketones (≥3.1 mmol.l-1) resulting in a decrease in blood pH and potentially leading to a coma. An overview of tissue interaction in starvation can be seen in Figure 6.
Figure 6. Summary of metabolic pathways active during starvation.

During starvation, there is an increase in fatty acid utilisation in the muscle (not shown here for simplicity) and a breakdown of proteins into amino acids. Intermediates (in black), tissues (in green), and pathways (in red).
From Figure 6 it can be seen that there is a requirement for cross-talk between tissues to survive during the time of starvation. The same is true for the fed state, where regulation and metabolic integration of tissues is vital to maintain normal function. Each organ is responsible for carrying out a specific range of metabolic transformations and processing of molecules at each stage. This is important to reduce the chances of futile cycles, whereby a tissue synthesises and breaks down a metabolite at the same time, leading to a net loss of energy.
There are three main interorgan pathways in order to either regenerate glucose or to control the use of glucose in the muscle. The Cori cycle (Figure 7) is activated under strenuous exercise when the skeletal muscle (or ischaemic heart) are contracting using anaerobic glycolysis, which leads to an accumulation of lactate. Lactate is transported to the liver where it regenerates glucose (gluconeogenesis), which can then be used by the exercising muscle again. As you will see later, whilst the use of anaerobic glycolysis generates far less ATP than the oxidation of glucose, this process does not require oxygen, which can be limited in strenuous exercise.
Figure 7. The two key pathways to recycle lactate or alanine from muscle and regenerate glucose in the liver.

Intermediates (in black), tissues (in green), and pathways (in red).
During times of starvation, the glucose-alanine cycle can regenerate glucose and remove excess nitrogen formed in the breakdown of amino acids (Figure 7). During proteolysis, amino acids that are liberated can provide carbon skeletons to top up different pathways, but must dispose of the amino group. This amino group is transferred to pyruvate by alanine aminotransferase to form alanine. Alanine is the predominant amino acid released by the muscle. In the cycle, glucose taken up by the muscle is used to generate the pyruvate, thereby aiding in proteolysis, without a net loss of glucose. The alanine is released by the muscle and taken up by the liver, where it is converted into pyruvate, and back into glucose to start the cycle again. Finally, the amino group liberated by the conversion of alanine back into pyruvate enters the urea cycle for disposal.
Hormonal control of metabolism
In mammals, metabolism can also be controlled by an interplay between the small peptide hormones: insulin and glucagon. Both of these are produced within the islets of Langerhans within the pancreas, insulin within pancreatic β-cells and glucagon within pancreatic α-cells. Both insulin and glucagon are held within vesicles in their respective cells, awaiting a signal for release into the bloodstream. Upon blood glucose levels rising, for example after eating, insulin is released from its vesicles into the blood. The effects of insulin are widespread. Along with regulating metabolic function in the liver, it is also able to significantly increase glucose uptake in peripheral tissue. Insulin binds to the insulin receptor (a tyrosine kinase receptor) on the cell surface which autophosphorylates and recruits the insulin receptor substrate (IRS). IRS then initiates the signalling transduction pathway, which eventually leads to the phosphorylation of AKT (also known as PKB), the protein that mediates or directs insulin actions.
Insulin has both short- and long-term effects, depending on the metabolic state of the organism. In skeletal muscle, heart, and adipose tissue, insulin signalling causes the translocation of the glucose transporter (GLUT4) to the plasma membrane, from internal vesicle stores. This significantly increases the potential for glucose uptake into these cells. As discussed briefly later, insulin is also able to regulate gene expression to increase glucose ultilisation, storage as glycogen, fatty acid uptake, and storage as TAGs. Insulin’s action in the adipose tissue is also to increase de novo lipogenesis, or the formation of new fatty acids from glucose.
When blood glucose levels decrease, glucagon is released from the α-cells. Insulin and glucagon are in a delicate balance, and the ratio of the two is important in determining the metabolic pathways active at specific times. A lack of insulin, in comparison with the effect of glucagon, can be a powerful signal. For example, insulin inhibits the action of the hormone-sensitive lipase (HSL) in the adipose tissue. The role of HSL is to stimulate lipolysis (breakdown of TAGs) by hydrolysing a fatty acid from the TAG, but this is inhibited by the continued signalling by insulin. When blood glucose levels decrease, this inhibitory signal is removed and HSL allows the process of lipolysis to occur. Glucagon’s main area of action is on the liver, with limited glucagon receptors found on other tissues such as adipose tissue. Glucagon works via a G-protein coupled receptor, which regulates adenylate cyclase and causes an increase in cyclic AMP. Glucagon turns the liver from an importer of glucose, into a net exporter by stimulating the formation of new glucose in gluconeogenesis and suppressing glucose usage in glycolysis and storage as glycogen.
As you progress through this review, you will see the action of insulin and glucagon. Other hormones are also at play in controlling metabolism, these include adrenaline (during the fight or flight response), thyroid hormones, cortisol, and the incretin hormones. For further reading on metabolic regulation, the role of hormones and metabolism in humans, we would recommend Human Metabolism: A Regulatory Perspective by Rhys Evans and Keith N. Frayn.
Glucose metabolism
Central metabolism and G6P
Glucose is transported into a cell through GLUTs and sodium glucose cotransporters (SGLTs) via facilitated diffusion. These transporters can move glucose into and out of cells. To ensure that glucose remains within the cell, it is quickly ‘trapped’ and phosphorylated to form G6P. This phosphorylation occurs via a kinase enzyme called hexokinase or glucokinase, which catalyses the transfer of a phosphoryl group from an ATP molecule to an acceptor molecule. In this case, G6P is a highly negative, polar molecule meaning it is unable to diffuse across the cell membrane. Furthermore, the addition of the phosphate group renders G6P too large to escape back out of the cell through GLUT transporters. By trapping glucose in cells as G6P, the gradient of glucose between the cytosol and the extracellular space increases, resulting in a net movement of glucose into cells. Glucose holds a high osmotic potential, and so by removing glucose, the movement of water out of the cell is reduced. This reaction, therefore, ensures the fate of glucose as G6P to facilitate the initiation of further metabolic processes.
G6P is the central molecule of metabolism. It is a ‘crossroad’ marker and holds many possible fates within a cell, dependent on its conditions and metabolic needs (Figure 8). G6P lies at the centre of four metabolic pathways:
Glycolysis – The formation of pyruvate and lactate.
Gluconeogenesis – G6P is converted by glucose-6-phosphatase during gluconeogenesis to form glucose. Glucose-6-phosphatase is primarily expressed in the liver but also in the kidney cortex at times of starvation. It has further been found to be expressed in the β-cells of pancreatic islets and human intestinal mucosa in starved and diabetic states.
Glycogenesis – Storage as glycogen. G6P is converted via glycogen synthase into glycogen for storage.
The pentose phosphate pathway (PPP) – The generation of NADPH molecules allows fatty acid synthesis. The formation of ribose-5-phosphate to synthesise nucleotides. The PPP regenerates the intermediates of glycolysis such as F6P and glyceraldehyde-3-phosphate (GAP).
Figure 8. The metabolic crossroad and fate of G6P.

G6P links four key metabolic processes; glycolysis, gluconeogenesis, glycogenesis/glycogenolysis, and the PPP.
The first step of these pathways is tightly controlled and acts as control points, to ensure the fate of the cell is established.
The importance of glycolysis
Glycolysis represents the first stage of glucose catabolism in organisms that perform cellular respiration. The glycolytic pathway involves the breakdown of glucose to two pyruvate molecules in ten sequential enzymatic reactions within the cytosol (Figure 9).
Figure 9. The pathway is split into an initial ‘investment’ phase, where ATP is used and then the ‘payout’ phase, where ATP is regenerated.

Intermediates (in black), by-products (in green), and enzymes (in red).
Glycolysis occurs in most living cells and can succeed in the absence of oxygen. However, the fate of its end product depends on the anaerobic or aerobic environment of the cell following glycolysis.
Glycolysis relies on NAD+ to accept electrons from glucose forming NADH and H+. NAD+ can be re-oxidised from NADH in order to ensure a cyclic effect of glycolysis in all cells. This can be accomplished under both aerobic and anaerobic conditions. However, in the presence of oxygen, NADH passes its electrons into the ETC, allowing the complete oxidation of glucose. This yields a net production of 30–32 ATP molecules. Under anaerobic conditions, fermentation occurs and NADH donates its electrons to regenerate NAD+, and no further ATP is formed.
Despite glycolysis only yielding two ATP molecules, the process is vital. As previously mentioned, the mammalian erythrocytes rely entirely on the ATP generated through glycolysis as its energy source because they lack mitochondria. Furthermore, within the liver, glucose regulation is vital to ensure glucose homoeostasis in the body. Here, glycolysis can be tightly regulated. Under states of fasting, hepatic glucose production can be elevated, making the liver the main source of glucose production at this time. Here, pyruvate can also be used to form precursors for the synthesis of fats, cholesterol, bile, and plasma proteins. For microorganisms, the glycolytic pathway ensures a source of energy for respiration and bacterial photosynthesis, along with necessary biosynthetic precursors.
The glycolytic pathway
The enzymatic process of glycolysis occurs within the cytosol and can be divided into two definitive stages of energy investment and energy recovery (Figure 9).
Stage I energy investment phase
The first reaction of glycolysis is catalysed by hexokinase (or glucokinase in the liver and pancreas), involving the transfer of a phosphoryl group from ATP to glucose, forming G6P. G6P is isomerised to F6P by G6PI and is then further phosphorylated by PFK, to form fructose-1,6-bisphosphate (FBP). This phosphorylation step is irreversible and utilises the second ATP molecule in glycolysis. Aldolase catalyses the cleavage of FBP (6-carbon molecule) to form two 3-carbon molecules GAP and dihydroxyacetone phosphate (DHAP). Another isomerase, triose phosphate isomerase (TIM), catalyses the interconversion between DHAP and GAP, allowing DHAP to convert into GAP, and proceed along the glycolytic pathway. As a result, for every glucose molecule, two molecules of GAP are produced. Therefore, from this stage onwards, all intermediates and by-products are doubled in production.
Stage II energy payout
GAP is oxidised and phosphorylated by NAD+ and Pi to form 1,3-bisphosphoglycerate (1,3-BPG), catalysed by GAP dehydrogenase (GAPDH). Intermediates NADH and H+ are produced alongside 1,3-BPG. The conversion of 1,3-BPG into 3-phosphoglycerate (3PG) is catalysed by phosphoglycerate kinase (PGK) and signifies the first step in glycolysis to generate ATP molecules through phosphorylation of adenosine diphosphate (ADP). 3PG can be interconverted by phosphoglycerate mutase (PGM), to form 2-phosphoglycerate (2PG) that is dehydrated to PEP by enolase. The final reaction of glycolysis generates the final ATP molecule alongside pyruvate in a cleavage reaction catalysed by pyruvate kinase (PK).
The initial energy investment in the form of two ATP molecules is doubly repaid in the later stage of glycolysis due to the formation of two 3-carbon GAP molecules, which are each transformed to pyruvate and ATP. Therefore, generating four molecules of ATP, a net gain of 2 ATP.
Overall, glycolysis holds a negative ΔG value of −310 kJ.mol−1. The reaction is as follows:
Regulation of glycolysis
Glycolysis is regulated by three rate-limiting steps. These are slower, regulated stages and therefore determine the overall rate of the pathway. Within the glycolytic pathway, these rate-limiting steps are coupled with the hydrolysis of ATP or the phosphorylation of ADP. This ensures these steps are energetically favourable, i.e. holding a very negative ΔG value and are therefore irreversible under physiological conditions.
Hexokinase/glucokinase
Hexokinase and glucokinase are the first regulatory enzymes within the glycolytic pathway. Hexokinase exists in abundance within tissues in our body. It holds a low Km value, thus, ensuring its high affinity for glucose. Due to its low Km it means that hexokinase is more useful in a state of hypoglycaemia, where glucose levels are low. Hexokinase is feedback-inhibited by its own product, meaning a build-up of G6P can inhibit hexokinase and therefore, the phosphorylation of glucose. Hexokinase does ensure the irreversible formation of G6P. In mammalian skeletal muscle, where the major source of energy is glycogen and not glucose, this step is ultimately overcome. Within pancreatic islets, hexokinase allows the control of insulin and glucagon release in the β- and α-cells, respectively.
Glucokinase is an isoenzyme of hexokinase that exists in the liver and pancreatic β-cells. Contrary to hexokinase, glucokinase holds a high Km value and therefore a high Vmax. This means that glucokinase exists with a low affinity for glucose. As a result of this, glucokinase is utilised in a state of hyperglycaemia or a post-prandial state. Within pancreatic β-cells, glucokinase acts as a sensor to control the rate of entry of glucose into the glycolytic pathway by phosphorylation. Within the liver, it ensures that glucose is synthesised into glycogen or fatty acids post-prandially, when glucose levels are high. Unlike hexokinase, glucokinase is not inhibited by high levels of G6P and can therefore remain active to ensure glucose is stored as glycogen when glucose levels are high. The low affinity of glucokinase for glucose ensures that within a state of low glucose, peripheral tissue hexokinase can phosphorylate glucose to G6P for glycolysis and the liver and β-cells stop the uptake of glucose.
PFK
PFK is another enzyme that acts as a key regulator of glycolysis. It has a highly negative ΔG value, thus ensuring that the reaction will still occur despite accumulation of FBP. PFK holds two conformational states that exist in equilibrium, with, ATP acting as both an activator and inhibitor of both the states. When ATP levels are high (e.g. in the liver), ATP acts as an allosteric inhibitor of PFK, shifting its equilibrium and decreasing its affinity for F6P. However, where levels of ATP are low, PFK is activated, shifting its equilibrium and affinity for F6P to form FBP.
PK
PK ensures the fate of PEP to form pyruvate in the last step of glycolysis. Pyruvate is an essential intermediate building block for many further metabolic pathways such as fatty acid synthesis, the tricarboxylic acid (TCA) cycle or, under anaerobic conditions, converted into lactic acid or ethanol (in yeast). Therefore, PK is noted as the most important regulator of glycolysis.
Gluconeogenesis
The importance of gluconeogenesis
Gluconeogenesis is an anabolic process whereby glucose is formed from non-carbohydrate carbon precursors including pyruvate. The gluconeogenic pathway largely occurs within the liver and kidneys to maintain blood glucose levels following glycogen depletion, and in the renal cortex during starvation. Gluconeogenesis has also been found to occur within the β-cells of the islets of Langerhans and intestinal mucosa in starved and diabetic states.
Gluconeogenesis aims to do the reverse of glycolysis; however, due to the presence of irreversible steps within the glycolytic pathway, gluconeogenesis is not simply a reversal of glycolysis. These irreversible steps are overcome in gluconeogenesis, using additional enzymes than those present in the glycolytic pathway. It is crucial that gluconeogenesis is not just the reverse of glycolysis. This is because the last step of glycolysis (Figure 9) involves the irreversible and highly energetically favourable formation of pyruvate. To bypass this, gluconeogenesis is split into a two-step process with specific steps occurring within the mitochondria and the cytosol. Within the mitochondria, pyruvate is converted into oxaloacetate which, in turn, converts into malate to transport out of the mitochondria into the cytosol. Once here, it is immediately converted back into oxaloacetate and then to PEP by PEP carboxykinase (PEPCK). This not only overcomes the irreversible step within glycolysis but also avoids the cell undergoing a futile cycle whereby pyruvate is immediately converted back into PEP. Furthermore, during steps 1 and 3 of glycolysis (Figure 9) ATP is invested in order to phosphorylate the product formed. Therefore, if gluconeogenesis were the reverse of glycolysis, it would essentially mean that gluconeogenesis would need to regenerate ATP, a process that is not possible. Gluconeogenesis is instead ATP-dependent and therefore requires additional enzymes to bypass steps 1 and 3, where ATP is not regenerated.
The importance of gluconeogenesis lies in the fact the brain and erythrocytes rely almost entirely on glucose as a form of energy and, therefore, it is essential that glucose ultimately depleted in glycolysis is restored by gluconeogenesis in a cyclic fashion.
Gluconeogenic pathway
The formation of oxaloacetate from pyruvate
Pyruvate is carboxylated by pyruvate carboxylase (PC) to oxaloacetate at the expense of 1 ATP molecule. This reaction occurs inside the mitochondria. PC is activated through increased concentration of acetyl CoA and inhibited in the presence of glucose and ADP.
Oxaloacetate is reduced to malate in the presence of NADH, to be transported over the mitochondrial membrane and into the cytosol. Malate crosses the mitochondrial membranes via the malate-aspartate shuttle, where it is re-oxidised to oxaloacetate.
At the expense of one GTP molecule, oxaloacetate is decarboxylated and phosphorylated by PEPCK.
Formation of F6P
A hydrolysis reaction occurs in a phosphate ester located at carbon 1 of fructose-1,6-bisphosphate, facilitated by fructose-1,6-bisphosphatase (F16BPase).
G6P and free glucose formation
F6P is readily converted into G6P by G6PI.
In many scenarios, G6P is utilised to generate glycogen, ending gluconeogenesis. Alternatively, it can be dephosphorylated to form free glucose molecules.
The site for the formation of glucose
During the final step of gluconeogenesis, glucose is formed. This occurs within the lumen of the endoplasmic reticulum. The glucose formed is ultimately shuttled into the cytosol by GLUTs, which are readily available and located in the endoplasmic reticulum.
Regulation of glucose metabolism by gene expression
One of the actions of insulin is to increase glycolysis, whilst suppressing gluconeogenesis (in the liver). In response to increased insulin and glucose, the mammalian liver, muscle, and peripheral tissue increases the expression of GLUT1–4, hexokinase/glucokinase and key glycolytic genes: GAP dehydrogenase, PK and the bifunctional enzyme (which stimulates PFK activity). To suppress gluconeogenesis in the liver, insulin decreases the expression of glucose-6-phosphatase, fructose-1,6-bisphosphatase, and EP carboxylase. This alteration in gene expression pattern, increases glucose utilised in the cells, and maintains glycolytic activity. In the mammalian liver, glucose can increase the expression of PK via the transcription factor known as carbohydrate-responsive element binding protein (ChREBP). Interestingly, the action of glucagon on the liver suppresses this transcription factor, thereby reducing the expression of PK.
Glycogen
What is glycogen?
Glycogen is a large, multibranched polysaccharide of glucose. It contains α-1,4-glycosidic bonds between adjacent glucose molecules and α-1,6-glycosidic bonds at branching points at every tenth residue within the chain. It is essentially the storage form of glucose in animals, fungi, and bacteria. It is also the storage molecule of glucose within the body and can be broken down to yield glucose when energy is required. Glycogen is stored within muscle and liver in the body. Within the muscle, the breakdown of glycogen serves to supply energy to that muscle, whereas within the liver it is degraded to maintain blood glucose levels in the body. It is present within these sites as granules within the cytosol that are up to 40 nm in size.
The importance of glycogen
Glycogen can be degraded to supply energy to the body. This is specifically important as cells within the brain rely almost entirely on glucose for energy and therefore glucose released from liver cells can help supply this. Within periods of sudden activity, such as sprinting, the glucose obtained from glycogen degradation can produce enough energy when no oxygen is initially available.
Furthermore, the question ‘why cannot all excess fuels in the human body be stored as fatty acids?’ is usually probed. There are two main reasons as to why glycogen storage is beneficial over fatty acid storage. Firstly, glycogen is readily mobilised to glucose and therefore can be utilised quickly in situations where glucose is needed immediately. Secondly, the breakdown of glycogen is highly controlled. Therefore, the subsequent release of glucose is also controlled to help raise or maintain blood glucose levels.
Synthesis and degradation
The synthesis of glycogen requires an activated form of glucose called uridine diphosphate glucose (UDP-glucose). This is formed by the addition of UTP to glucose-1-phosphate. UDP-glucose is added to the non-reducing end of glycogen, expanding its size.
The degradation of glycogen requires the release of glucose-1-phosphate from glycogen and the remodelling of glycogen substrates to warrant further degradation. Glucose-1-phosphate is then converted into G6P which has several fates within metabolism.
PPP
The importance of the PPP and its intermediates
The PPP is an essential biochemical process that occurs within the cytosol of living organisms (Figure 10). This pathway runs parallel to glycolysis in the cytosol, as it utilises some similar components of this pathway for its own use. It is known to have several important roles.
- The production of nicotinamide adenine dinucleotide phosphate (NADPH). NADPH is a crucial reducing agent which is used in:
- Fatty acid synthesis
- Cholesterol biosynthesis
- Nucleotide synthesis
- Neurotransmitter synthesis.
- It synthesises pentose sugars which are precursors for nucleotide synthesis
- DNA, RNA, FADH2, ATP, NADH and CoA.
It establishes a way to breakdown 5-carbon sugars which are consumed within the diet.
It also provides a way to synthesise and break 4- and 7-carbon sugars which are less popular within the body.
Figure 10. PPP is split into the oxidative and non-oxidative phases.

The oxidative phase represents the conversion of G6P into ribulose-5-phosphtase which generates NADPH molecules. The non-oxidative phase shows the generation of ribose-5-phosphate and also glycolysis pathway intermediates. Intermediates (in black), by-products (in green), and enzymes (in red).
The PPP
The PPP consists of two major phases: the oxidative phase, which produces NAPDH molecules, and the non-oxidative phase, which produces the ribose-5-phosphate molecules for nucleotide synthesis.
During the PPP, at various points, the intermediates of glycolysis are available (highlighted in Figure 10). Therefore, this pathway is shown to occur in parallel with glycolysis. This ensures that sufficient amounts of NADPH and pentose sugars are produced for subsequent events such as electron transfer within the electron transfer chain.
The fate of pyruvate and acetyl CoA
Pyruvate is the end product of glycolysis and is a key intermediate in numerous metabolic pathways. Its fate is dependent on the organism in which it has been synthesised and also the oxygen conditions within the cell.
Anaerobic utilisation of pyruvate
The NADH and H+ molecules that are ultimately generated during glycolysis are re-oxidised to form NAD+ molecules. The recycling of these is a fundamental process that allows glycolysis to continue in a cyclic fashion.
The fate of pyruvate and NADH is dependent on the conditions within the cell. In the presence of oxygen, pyruvate is oxidised completely at the mitochondria, to form carbon dioxide and water to yield ATP molecules. However, where oxygen is absent, anaerobic respiration occurs. In animal tissues, such as muscle, pyruvate is reduced to lactate by homolactic fermentation due to lactate dehydrogenase (LDH). This regenerates NAD+ molecules for the continuation of glycolysis and the subsequent formation of 2 ATP molecules. Anaerobic respiration therefore only synthesises 2 ATP molecules which, in comparison with the 30–32 ATP molecules yielded in aerobic respiration, is far less efficient. Therefore, energy from anaerobic respiration is not sustainable for whole organism use (in mammals) but is instead required for individual cell survival. For example, erythrocytes lack mitochondria and so rely solely on anaerobic respiration for energy. In the case of erythrocytes, this is highly advantageous as it means that they do not use the oxygen which they carry. Instead, they use the energy supplied from anaerobic respiration to transport the oxygen to other cells in the body.
In yeast, alcoholic fermentation produces NAD+ and ethanol. This occurs as pyruvate is decarboxylated to carbon dioxide and acetaldehyde, which is reduced to NADH to ultimately form NAD+ by yeast alcohol dehydrogenase (YADH).
Aerobic fate of pyruvate
Following glycolysis, under aerobic conditions, pyruvate is oxidised to form acetyl CoA, which then enters the TCA cycle to further cellular respiration in cells. This reaction is catalysed by pyruvate dehydrogenase (PDH) and is a crucial convergence point between the TCA cycle and glycolysis, lipid, and amino acid metabolic pathways. PDH is regulated based on the demand of the cell for the use of carbohydrates as energy. Where carbohydrate stores are depleted, PDH activity is down-regulated to diminish the use of glucose via oxidative phosphorylation. Therefore, other sources of energy, such as fatty acids and ketone bodies, can be used in various tissue types such as the heart and muscle.
Structurally, PDH exists as three subunits: E1, E2, and E3. Regulation of PDH occurs at serine residues within subunit E1, where its activity is inhibited through reversible phosphorylation at these sites. PDH kinases (PDK 1–4) catalyse this phosphorylation reaction and therefore inhibits PDH activity. On the other hand, PDH phosphatases (PDP1 and PDP2) catalyse the reverse dephosphorylation reaction to restore PDH activity. The kinases and phosphatases are respectively differentially expressed in a multitude of tissues within the body. The activity of these enzymes is tightly controlled, kinases are stimulated by increased NADH and acetyl CoA concentrations (indicative of high energy production) and transcriptionally by peroxisome proliferator-activated receptor α (PPARα – increased fatty acid uptake), however inhibited by increased pyruvate concentrations. Whereas phosphatases are stimulated by increased levels of insulin, Mg2+, and Ca2+ (in the case of the heart and muscle). Increasing levels of magnesium are linked to the breakdown of ATP to ADP, as magnesium ions are found coordinated around ATP, to decrease its highly negative charge. Calcium on the other hand comes from contraction, which is a highly energy-dependent process, and therefore requires glucose to be fully oxidised.
CoA and acetyl CoA
CoA is a ubiquitous, indispensable cofactor that is present in all living organisms. CoA functions to carry acyl groups and is a carbonyl-activating group carrier, which is essential for many metabolic processes such as fatty acid oxidation and the TCA cycle. CoA naturally derives from pantothenic acid, also known as vitamin B5, in a series of steps that require ATP. Pantothenate is synthesised de novo in bacteria and plants and is found in foods such as cereals, meat, and potatoes. Pantothenate undergoes phosphorylation, its product is then condensed with a cysteine molecule followed by a decarboxylation reaction. AMP is added to form dephospho-CoA, which is then phosphorylated to yield CoA. This pathway is regulated by end-product inhibition as CoA is a competitive inhibitor of pantothenate kinase, the first enzyme involved in the phosphorylation of pantothenate.
Acetyl CoA is a molecule that lies at the hub of carbohydrate and fatty acid metabolism. Its main function is to deliver its acetyl group to the TCA cycle for energy production. Here, acetyl CoA readily combines with oxaloacetate to form citrate and begin the TCA cycle. Also, acetyl CoA is formed via fatty acid β-oxidation and therefore acts as an intermediate molecule for the TCA cycle, fatty acid metabolism, and glycolysis. It is known that acetyl CoA is central to maintain the balance between carbohydrate and fatty acid metabolism for a source of energy. As acetyl CoA can inhibit PDH, an increase in fatty acid uptake into muscle (and heart) causes a build-up of β-oxidation-derived acetyl CoA and inhibition of glucose oxidation. This is part of the glucose-fatty acid cycle, also known as the Randle cycle. In the adipose tissue, a counter-reaction occurs, whereby a build-up of glucose (used for making new fatty acids) inhibits lipolysis and reduces fatty acid release from this tissue.
Fatty acids
Structure, function, properties of lipids
Lipids are chemically defined as substances that are insoluble in water but are soluble in nonpolar solvents such as acetone. Their insolubility in water is due to the presence of a long hydrophobic, hydrocarbon chain which can be either saturated or unsaturated. A free fatty acid, made up of lipids, consists of a carboxyl group (–COOH) linked to a straight chain of carbon atoms bound with hydrogen. The carbon chain, which can be up to 24 carbons in length, may be either saturated or unsaturated based on the carbon–carbon bonds they hold (see Figure 11) and may contain functional groups. If the carbon chain holds a double bond, the fatty acid is unsaturated and can exist in either a cis or trans form.
Figure 11. The chemical structural differences amongst saturated, monosaturated, and polyunsaturated fatty acids.

Saturated fatty acids hold no double bond within their structure meaning the carbon atoms are fully ‘saturated’ with hydrogens. Monounsaturated fats have one carbon–carbon double bond in their structure and polyunsaturated hold two or more.
Lipids can exist as TAGs, an efficient storage solution. TAGs are composed of a glycerol molecule, where the three hydrogen atoms are esterified by fatty acid chains. These TAGs function as energy storage in adipose tissues and are a major form of energy in both animals and plants. A major function of lipids is to provide an alternative energy source to carbohydrates by the hydrolysis of ester bonds between TAGs.
Biologically, lipids are essential components of cellular membranes and the nervous system. Lipids make up adipose tissue, where its role is to protect internal organs and provide insulation. In terms of metabolism, lipids are stored as TAGs for use as energy. TAGs are stored due to their high energy value, providing more energy per gram than carbohydrates and proteins alone even though carbohydrates are the preferable source of energy in animals.
Where do animals obtain fatty acids from?
Fatty acids are the essential building blocks of fat within our bodies. During digestion, the fats that we consume within our diet are broken down into fatty acid molecules to aid absorption into the blood. Fatty acids are usually formed in groups of three to form TAGs. These reside in the bloodstream to reach capillary beds, which eventually allow diffusion to muscles where they can be oxidised to form ATP molecules.
There are various sources from where fats can be obtained, as stated below:
-
Diet
Mammals consume TAGs within our diet. As they are consumed, the small intestine packages these fats into protein carrier molecules called chylomicrons. These are eventually released into the lymphatic system where they reach the bloodstream.
-
Adipose cells
Adipose cells are specialised cells that can store large amounts of fat. A few hours after the consumption of a meal, insulin levels decrease. This in turn also diminishes the levels of amylin, a molecule that is secreted with insulin to inhibit glucagon secretion. Due to diminished levels of amylin, glucagon secretion rises. It is at this point, where insulin levels are reduced, where the adipose tissues release the stored fatty acids into the bloodstream. Due to its hydrophobic nature, fats usually bind with proteins within the blood such as albumin.
-
Liver synthesis
The liver is the main site of fatty acid synthesis. Here, excess glucose that has not been used for ATP synthesis or glycogen, is synthesised into fatty acids. These are packaged in the liver into TAGs alongside cholesterol, to form very low-density lipoproteins (VLDLs) which can be transported within the bloodstream.
The yin and yang of fatty acids
As listed above, we consume fats within our diet. Fats exist here as either saturated or unsaturated. Unsaturated fats can be further divided into monounsaturated or polyunsaturated. The difference between these three groups of fats is based on their chemical structure, which ultimately determines whether they hold beneficial or harmful effects within our body. The structure of fats is ultimately a long hydrocarbon chain bonded to a glycerol backbone.
Saturated fats, such as palmitic acid, are harmful to our body. It is often found in butter, lard, and cheese. Saturated fats tip the balance between low-density lipoproteins (LDLs) and high-density lipoproteins (HDLs) to favour LDL concentration, which is harmful. Consuming large amounts of saturated fat within the diet is associated with an increased risk of heart disease, stroke, and type 2 diabetes. Within their structure, they contain a long single-bonded carbon chain with lots of hydrogen atoms as shown in Figure 11.
In opposition, unsaturated fats are beneficial when consumed. They are found within vegetables, nuts, and fish and are liquid at room temperature. Their chemical structure contains less hydrogen to carbon bonds due to the presence of double bonds between carbon atoms within their tail chain. Monounsaturated fats found within olive oil, peanuts, and avocados contain one carbon-to-carbon double bond within their structures. Whereas polyunsaturated fats, such as sunflower oil and those found within salmon, contain two or more carbon double bonds within their structure (Figure 11). These fats can increase levels of HDLs within humans, reducing the chance of heart disease, stroke, and diabetes. Some studies claimed that increasing these fats can treat some of the listed diseases above.
The yin and yang of fatty acids are apparent. Fats live within a balance in the body. As you eat more saturated fats, this diminishes the availability of HDLs within the body, causing harm. The opposite effect is seen when unsaturated fats are consumed. Therefore, keeping a balance between the two is key to staying healthy and diminishing harsh side-effects associated with the overconsumption of saturated fatty acids. One example of this is with the onset of type 2 diabetes. It is known that the ratio of palmitic acid:oleic acid impacts diabetes risk in humans. In humans, the increased consumption of saturated fatty acids within the diet, such as palmitic acid, alongside the over consumption of carbohydrates, could eventually cause obesity. Chronic obesity and increased visceral fat can cause insulin resistance in insulin target tissues over time, which can manifest as type 2 diabetes. In contrast, the consumption of monounsaturated fatty acids such as oleic acid, appears to not only diminish the ability for an individual to develop diabetes but, in diabetic patients, can help to reduce or reverse the disease.
Fatty acid uptake into the cell and activation by acyl synthetase
The breakdown of TAGs provide twice as much energy per gram compared with the utilisation of carbohydrates and proteins. The heart, the most energy-expensive organ in the body, utilises fatty acids for 50–70% of its energy.
Fats are taken up into the cytosol from the bloodstream, either diffusing across the membrane, or actively by specific transporters. However, the first step for fatty acid oxidation occurs within the mitochondria.
Fatty acids are initially ‘activated’ in the cytosol and then transported over the outer and inner mitochondrial membranes for fatty acid oxidation to proceed. The activation of fatty acids begins with the reaction of fatty acids with CoA to create Acyl CoA, a reaction catalysed by acyl synthetase (thiokinase). This reaction is coupled to a hydrolysis reaction utilising 1 ATP molecule to form AMP and PPi (inorganic pyrophosphate group), which is rapidly hydrolysed (due to it being unstable in aqueous solution) to inorganic phosphate (PO43_). The reverse reaction to form pyrophosphates from this would require heating phosphates. Therefore, the rapid hydrolysis of PPi to inorganic phosphate renders the cleavage of ATP to AMP and PPi irreversible and thus, the reaction coupled to this hydrolysis, too.
The long-chain fatty-acyl CoA cannot readily pass through the outer mitochondrial membrane. To overcome this, the acyl group is transferred to a carnitine molecule, releasing the CoA group, a reaction catalysed by carnitine palmitoyl transferase I (CPT1). The acyl-carnitine can readily diffuse through pores in the outer mitochondrial membrane into the intermembrane space.
Acylcarnitine is then transported via a protein carrier on the inner mitochondrial membrane called the acyl carnitine translocase, into the mitochondrial matrix. Here, carnitine is substituted for a CoA molecule from the mitochondrial matrix, forming acyl CoA and carnitine molecules.
Here the carnitine is transported back through the carnitine carrier protein to the cytosol and the remaining acyl group is transferred to a CoA molecule from the mitochondrial pool of CoA. The acyl carnitine translocase protein pump is efficient in that, for every acyl carnitine it pumps into the mitochondrial matrix, it exchanges it for one molecule of carnitine. This can then be recycled in the cytosol. The production of long-chain fatty acyl CoA within the matrix of the mitochondria marks the start of β-oxidation.
Regulation of fatty acid utilisation
Fatty acid transport is regulated by CPT1. This allows the formation of acylcarnitine in the cytosol to readily diffuse across the outer mitochondrial membrane, for subsequent transportation to the matrix. CPTI is a rate-limiting step, thus making it the slowest step in the pathway. Malonyl CoA is an allosteric inhibitor of CPT1 and is formed by carboxylating acetyl CoA. Therefore, providing a direct relationship with the synthesis of fatty acids and the utilisation of fatty acids for oxidation. If fatty acid synthesis is increased (more malonyl CoA), then we do not need to break down fats. Therefore, inhibiting the rate-limiting step to ensure a net production of fatty acids. It can be deemed that the processes of fatty acid synthesis and breakdown are essentially exclusive and limiting to one another.
This is also controlled on a gene level in the mammalian liver and peripheral tissue, where increased fatty acid uptake into cells causes fatty acid binding to the transcription factor PPARα. The PPARα–fatty acid complex forms a heterodimer with retinoid X receptor (RXR), which binds to PPAR response elements and leads to the increased expression of CPT1, liver fatty acid binding protein (FABP) and fatty acid β-oxidation genes. As mentioned earlier, PPARα also potentially decreases glucose oxidation by increasing the expression of the PDH inhibitor, PDK4. The process of PPARα activation by fatty acids, means that an increase in fatty acid availability is met by an increase in metabolism of fatty acids.
Fatty acid β-oxidation
Fatty acid β-oxidation is the mitochondrial aerobic process of breaking down a fatty acid into acetyl CoA, NADH, and FADH2. As we have just seen, fatty acids are simple lipids and usually have a long hydrocarbon chain with a terminal carboxyl group. Fatty acid β-oxidation involves the break down of long-chain fatty acids by two carbons at a time, starting from the carboxylic acid end. The product formed by its breakdown ultimately feeds into the TCA acid cycle.
Fatty acid β-oxidation occurs within the mitochondrial matrix (Figure 12). Initially, fatty acyl CoA is oxidised by FAD to form trans-enoyl CoA, where a dehydrogenation reaction removes two hydrogen molecules between carbon 2 and 3 of the fatty acid chain. Next, the hydration step adds a water molecule across the double bond forming hydroxyacyl CoA. The next NAD-dependent dehydrogenation step generates an NADH molecule and ketoacyl CoA. Eventually, a thiolytic cleavage reaction forms an acetyl CoA molecule and acyl CoA that is 2 carbons shorter in length. This acyl CoA can be recycled and reused cyclically for β-oxidation of fatty acids.
Figure 12. β-oxidation process of fatty acids including the enzymes involved.

The process results in the formation of acetyl CoA and acyl CoA molecules from the oxidation, hydration, and cleavage of fatty acyl CoA. Intermediates (in black), by-products (in green), enzymes (in red), and black boxes summarise the steps.
Fatty acid oxidation can also occur within peroxisomes. Peroxisomal oxidation of fatty acids occurs on fats that the mitochondria are unable to utilise, such as very long chain fatty acids, pristanic acid, and bile intermediates. Here, fatty acid oxidation proceeds via a similar mechanism; however, enzymes and regulation can differ.
The NADH and FADH2 formed within the β-oxidation steps are utilised during the ETC.
Transport of acetyl CoA for fatty acid synthesis
De novo lipogenesis, or fatty acid synthesis, takes place in the liver and adipocytes, where glucose is ultimately formed into fatty acids. Glycolysis takes place within the cytosol yielding pyruvate, which is transported into the mitochondrial matrix. Here, pyruvate undergoes an oxidative decarboxylation reaction catalysed by PDH to produce acetyl CoA, the initial precursor for the TCA cycle.
The enzymes required for fatty acid synthesis reside in the cytosol. Therefore, acetyl CoA must be exported from the mitochondria to allow fatty acid synthesis to occur. However, due to unavailable protein shuttles, acetyl CoA cannot readily cross the mitochondrial membrane. Instead, acetyl CoA combines with oxaloacetate forming citrate, which readily crosses the mitochondrial membrane into the cytosol. Here, citrate is converted back into acetyl CoA and oxaloacetate via ATP citrate lyase (ACLY) in order to re-form acetyl CoA for the initiation of fatty acid synthesis. Oxaloacetate is recycled to form pyruvate, forming NADH and carbon dioxide.
Fatty acid synthesis is an anabolic reaction, where a monomer (acetyl CoA) forms a polymer (fatty acid), meaning it holds a positive ΔG value, therefore it is coupled to the hydrolysis of ATP.
Initially, acetyl CoA is carboxylated to form malonyl CoA, a reaction that is catalysed by acetyl CoA carboxylase. As this reaction holds a positive ΔG value, to make this more favourable, it is coupled to the hydrolysis of ATP (ΔG < 0). Malonyl CoA then undergoes polymerisation to form the long-chain fatty acid, catalysed by fatty acid synthase (FAS). Here CO2 and H2O are released, alongside the synthesis of 2 NADPH molecules. Two NADPH are used to break carbon double bonds for fatty acid synthesis. The amount of CO2, H2O, and NADPH that is utilised depends on how long the fatty acid end product is required.
Example: To form 16 carbon palmitic acid from a 2-carbon acetyl CoA molecule, the following reaction occurs.
The fatty acid is esterified into TAGs and packaged to VLDLs to enter the bloodstream to be delivered to the rest of our tissues in our body.
The FAS complex
FAS is the enzyme complex that catalyses the formation of long-chain fatty acids via fatty acid synthesis of palmitate (C16:0). It is a large dimerised complex with seven catalytic sites. In the centre of the complex lies the acyl carrier protein (ACP) which contains a prosthetic 4′ phosphopantetheine group. FAS consists of two identical polypeptides which exist in a yin-yang formation. FAS is known to dimerise this way due to cysteine cross-linking between the KS domain in one FAS monomer to the prosthetic group in ACP of the other monomer.
This reaction occurs in the presence of acetyl CoA, malonyl CoA (as a 2-carbon donor), and NADPH. In humans and animals, the seven catalytic groups (Figure 13) of the FAS and ACP are linked covalently in a single polypeptide chain. The seven catalytic sites are separated into three domains: domain I, II, and III.
Figure 13. FAS domains and their respective catalytic sites.

At the first two catalytic sites where acetyl transacylase (AT) and malonyl transacylase (MT) are present, they both transfer their respective acetyl and malonyl groups to the ACPs prosthetic group, forming malonyl ACP and acetyl ACP, respectively. The condensing enzyme (CE), also known as acyl-malonyl ACP condensing enzyme, forms a ketoacyl ACP molecule by combining the acetyl and malonyl groups. The first stage of condensation also occurs at this catalytic site. The initial phase of this process is termed as the elongation process. Following on from this β-ketoacyl ACP reducatase (KR), β-hydroxyacyl ACP dehydratase (DH), and enoyl ACP reductase (ER) carry out the next steps of reduction, dehydration and the second reduction step for fatty acid synthesis by reducing the β-keto group to a fully saturated carbon chain. Thioesterase (TE) cleaves the thioester bond between palmitate and the phosphopantetheine group within ACP, upon reaching a length of C16. Palmitate is released from the fatty synthase complex.
Regulation of fatty acid synthesis
The enzyme acetyl CoA carboxylase, which catalyses the reaction of acetyl CoA to malonyl CoA, is the rate-limiting step of fatty acid synthesis. It is regulated allosterically and hormonally. Allosterically, citrate can bind as an activator, whereas long chain fatty acids bind as inhibitors. This is ideal because as cytosolic concentrations of citrate increase, fatty acid synthesis should be activated to form long-chain fatty acids. However, where too many fatty acids are being formed, this step needs to be regulated to inhibit this process and activate fatty acid oxidation. The regulation of acetyl CoA carboxylase in this manner prevents the possibility of a futile cycle. If a futile cycle were to occur the formation and oxidation of fatty acids would occur simultaneously, keeping concentrations the same rather than allowing the favourable reaction to take place.
Malonyl CoA is also an important inhibitor of fatty acid β-oxidation, by preventing the uptake of fatty acids into the mitochondria via inhibition of CPT1. Whilst tissues like the heart and muscle lack functional FAS, they still undergo the first steps of the process to generate malonyl CoA. In this case, the malonyl CoA is used as a CPT1 inhibitor and acts to regulate β-oxidation. This shifts the heart and muscle to store fatty acids as TAGs for future use.
Hormonally, insulin can activate ACC where glucagon inhibits. Insulin has also been shown to increase the expression of both ACC and FAS in the mammalian liver and adipose tissue. This is because once a meal is consumed, insulin levels rise as glucose levels rise. Increased glucose means activation of glycolysis and thus increased production of acetyl CoA. Increased insulin concentrations will allow the utilisation of the acetyl CoA to form fatty acids. Glucagon aims to increase blood glucose levels several hours after a meal. Where there is not excess glucose, fatty acid oxidation occurs instead.
Amino acid metabolism
What are amino acids and where do they come from?
Amino acids are organic compounds that are composed of nitrogen, carbon, hydrogen, and oxygen, along with a variable side chain. Amino acids are the individual monomers that make up proteins, nucleotide bases (used for DNA, RNA, and ATP synthesis) and other nitrogenous products. As we have already seen in times of starvation, amino acid metabolism can be vital to maintain glucose levels and provide alternative carbon sources. Amino acids can be consumed from dietary sources or synthesised within our bodies and are categorised on this basis, respectively (Figure 14).
Figure 14. The categorisation of amino acids into their essential, conditional and non-essential groups in humans.

Conditional amino acids are not usually essential amino acids, only in times of illness and stress. Essential amino acids are not produced naturally by the body and must come from dietary intake whereas non-essential amino acids are produced by our body.
Amino acid transamination
Excess amino acids that are not required within the body are excreted as they cannot be stored. The metabolism of amino acids occurs predominantly within the liver however, the kidney, muscles, and adipose tissues also carry out amino acid metabolism. This consists of a two-step process. A transamination step and an oxidative deamination step. The first step of amino acid catabolism involves the transfer of an α-amino group from the original amino acid to a carrier molecule, an α-keto acid such as α-ketoglutarate. This transamination step forms another α-keto acid and amino acid such as glutamate. This step is catalysed by the enzyme aminotransferase, which is found within cell cytosols and is abundant in liver cells, along with the kidney, intestines, and the muscle.
Aminotransferases exist in many forms, two of which are: alanine aminotransferase and aspartate aminotransferase. All aminotransferases require a co-enzyme pyridoxal phosphate (vitamin B6) which acts to initially accept the nitrogen-containing group from the α-amino acid before the transfer to form the α-keto acid. Alanine aminotransferase transfers the α-amino group from alanine to the α-keto acid, α-ketoglutarate, forming glutamate and pyruvate. However, aspartate aminotransferase transfers an α-amino group from aspartate to α-ketoglutarate yielding oxaloacetate, the α-keto acid and glutamate.
The transamination step exists as a reversible step. Therefore, it is important to note that when amino acid concentrations are high, they are broken down, however, where their concentrations are low they can be formed from other amino acids and α-keto acids. The formed amino acid, in this case, glutamate, must continue to undergo oxidative deamination to form ammonia (see Urea cycle later). The formed α-keto acid can be utilised as a form of energy as ATP molecules.
Oxidative deamination
Oxidative deamination consists of two steps: a dehydrogenation and a hydrolysis step. The deamination step removes the amino group from glutamate to form an intermediate molecule. The intermediate undergoes a hydrolysis reaction where the amino group forms ammonium ions (NH4+) and regeneration of α-ketoglutarate.
Glutamate dehydrogenase is predominantly found within the liver and the kidneys, inside mitochondria, for this reaction to occur. This reaction occurs within the mitochondria to ensure that the toxic ammonium yielded does not cause cytotoxicity within the cell. It utilises two co-enzymes; NAD+ and NADP+. These co-enzymes are respectively used based on the conditions for the cell. When driving the forward reaction, in conditions where amino acid concentrations are high, for example after consuming a protein-rich meal, NAD+ is used as the co-enzyme. However, where amino acid or glutamate levels are low, the reverse reaction occurs to form more glutamate which can aid the synthesis of other non-essential amino acids. ADP and guanosine diphosphate (GDP) are both allosteric activators of glutamate dehydrogenase. Therefore, when energy levels are low (high ADP, GDP but low ATP and GTP), this reaction occurs to breakdown glutamate and amino acids for energy production.
Following amino acid metabolism within the liver, due to its toxicity, the synthesised ammonia, cannot be simply transported in the bloodstream. Due to this, the ammonia is transformed into a non-toxic compound, glutamine. Glutamine is transported to the kidneys by the enzyme glutamine synthetase which is present in peripheral tissues.
Glutamine is transported via the bloodstream and travels to the liver. Within the liver it is converted back into glutamate and ammonia by the enzyme glutaminase. In terrestrial vertebrates, ammonia is converted into urea which is excreted (see the section on Urea later).
Within the kidneys, the proximal tubule is the primary site for ammoniagenesis of predominantly glutamine metabolism. Ammonia produced here is excreted directly into the urine or returned to the systemic circulation.
Single-step catabolism of amino acids
Some particular amino acids only undergo a single step deamination process. These include serine and threonine. The one step process is catalysed by the enzyme dehydratase.
During these reactions, a dehydration reaction occurs to form an unstable, high energy, intermediate molecule such as aminoaceylate. This readily converts into a final product and yields ammonium. The ammonium carries into the urea cycle whereas the carbon skeletons formed, such as pyruvate, can be used for energy purposes.
The TCA cycle
The importance of the TCA cycle and its discovery
The TCA cycle was initially known as the citric acid cycle by Hans A. Krebs, a German biochemist, in 1937. It is more commonly known as the Krebs cycle, in recognition of Hans Krebs’ discovery. Krebs’ work was in collaboration with William A. Johnson at the University of Sheffield. However, before their end discovery, a succession of experiments took place by many other scientists.
Stern carried out his experiments with minced animal tissue. Here, he found that these tissues contained specific substances that could transfer H+ atoms from several intracellular organic acids. In particular, fumarate, malate, succinate, and citrate. He studied this using Methylene Blue, where this transfer of H+ atoms resulted in the reduction of the dye to a colourless form. This was found to occur within the presence of oxygen at high rates, knowing that active enzymes existed here. In the 1920s, Thunberg described a respiratory cycle that was present to oxidise acetate when particular tissue dehydrogenases were available.
Albert Szent-Gyorgyi later described the sequence of events of succinate oxidation. He also found that by adding a small amount of either malate or oxaloacetate stimulated their complete oxidation. This was indicated by an excessive amount of oxygen being converted into an oxidised form. Therefore, he concluded that this must cause the oxidation of an endogenous substance such as glycogen, which resides within tissues.
Martius and Knoop furthered Szent-Gyorgyis’s work by discovering a further piece of the sequence.
Krebs found that certain organic acids were readily oxidised by muscle, whereas the oxidation of carbohydrates and pyruvate was stimulated by the presence of specific organic acids. These acids happened to be the intermediates that are present throughout the TCA cycle.
Furthermore, using malonate, an inhibitor of succinate dehydrogenase, in muscle suspension experiments, Krebs found an accumulation of succinate in the presence of citrate isocitrate, α-ketoglutarate, fumarate, malate, and oxaloacetate. He, therefore, deemed a cyclic nature of all of these reactions to lead to succinate. This was proposed by Krebs in 1937 for which he won the Nobel Prize in Physiology or Medicine in 1953.
The TCA cycle is the predominant central driver of ATP synthesis and biosynthesis in most cells (Figure 15). In eukaryotes, the TCA cycle takes place in the matrix of the mitochondria following the biosynthesis of acetyl CoA via the oxidation of pyruvate. In prokaryotes, these steps occur within the cytosol.
Figure 15. The TCA cycle with all intermediates (in black), by-products (in green), and enzymes (in red).

The TCA cycle proceeds after the formation of acetyl CoA from either pyruvate during aerobic respiration following glycolysis or β-oxidation. The TCA cycle forms carbon dioxide, NADH, GTP, and FADH2 molecules.
The pathway
The cycle is formed of eight major steps, see Table 3.
Table 3. The reactions of the TCA cycle alongside the standard free energy changes and physiological free energy changes of the reactions.
| Step | Reaction | Enzyme | Type of reaction | ΔG°′ kJ.mol −1 | ΔG kJ.mol −1 |
|---|---|---|---|---|---|
| 1 | Citrate synthase | Condensation | −31.5 | Negative | |
| 2 | Aconitase | Isomerisation | ∼5 | ∼0 | |
| 3 | Isocitrate dehydrogenase | Oxidative decarboxylation | −21 | Negative | |
| 4 | α-ketoglutarate dehydrogenase | Oxidative decarboxylation | −33 | Negative | |
| 5 | Succinyl CoA synthase | Substrate level phosphorylation | −2.1 | ∼ 0 | |
| 6 | Succinate dehydrogenase | Dehydrogenation | +6 | ∼ 0 | |
| 7 | Fumarase | Hydration | −3.4 | ∼ 0 | |
| 8 | Malate dehydrogenase | Dehydrogenation | +29.7 | ∼0 |
Important steps of TCA
Step 1 – The combination of 2-carbon acetyl CoA and 4-carbon oxaloacetate forms 6-carbon citrate. Here, citrate can either move into the cytosol to initiate fatty acid synthesis or is destined to carry through the oxidation steps involved in the TCA cycle. This reaction is irreversible and highly regulated with a highly negative ΔG.
Step 3 – This step is highly regulated and allows the commitment of citric acid to the TCA cycle instead of fatty acid synthesis. This step is activated and stimulated by NAD+ and ADP but inhibited by NADH and ATP. Here, the first NADH molecules are formed along with CO2.
Step 5 – This reaction is coupled to the phosphorylation of GDP to GTP, making it the only reaction in the TCA cycle to generate a high energy phosphate. Following the formation of GTP, this can either be converted to ATP or used in protein synthesis.
Balance sheet
Glucose
Following glycolysis, 2 pyruvate, 2 ATP, and 2 NADH are formed. The pyruvate molecules are broken down by PDH to 2 acetyl CoA, 2 NADH, and 2 CO2.
These acetyl CoA molecules enter the TCA cycle and generate: 1 molecule of GTP (interchangeable with ATP), 3 NADH, and 1 FADH2 co-enzymes. These are doubled as two molecules of acetyl CoA are generated per glucose.
Palmitate (C16)
From β-oxidation: 7 NADH, 7 FADH2, and 8 acetyl CoA
From TCA cycle: 24 NADH, 8 FADH2, 8 GTP/ATP
Anaplerosis and cataplerosis
The importance
Anaplerosis is the act of replenishing intermediates of the TCA cycle that have been used up for biosynthesis. These anions must be replaced to retain the function and cyclic fashion of the TCA cycle. However, to ensure that these intermediates do not over-supply the TCA cycle, anaplerosis is coupled with the exit of intermediates from the TCA cycle, called cataplerosis (Figure 16).
Figure 16. The anaplerotic and cataplerotic reactions within the TCA cycle.

The major reactions are illustrated here including the entry of amino acids, formation and breakdown of oxaloacetate and the link to gluconeogenesis.
The NADH that is generated from the TCA cycle is used by the cell in the form of ATP. Even though the TCA cycle forms over 70% of the ATP that is used by a cell, it also provides the intermediate building blocks for other processes. As a result of this, the subsequent intermediates within the TCA cycle can be used up excessively and these must be replenished through the process of anaplerosis.
Intermediates and their use:
Citrate – Used during fatty acid synthesis.
α-ketoglutarate – Amino acid and purine nitrogenous base synthesis.
Succinyl CoA – Haem groups, porphyrin synthesis.
Oxaloacetate – Glucose, amino acids, purine, pyrimidine bases synthesis.
To keep up with the energy demands of the cell, intermediate concentrations such as those listed above, need to be maintained at a minimal level.
Anaplerotic reactions
Oxaloacetate can be formed directly from pyruvate (as discussed in gluconeogenesis), which in turn replenishes the other intermediates within the cycle.
This is a controlled step within the process where pyruvate decarboxylase is the archetypical anaplerotic enzyme. As the energy demand of a cell increases, oxaloacetate is formed at a higher rate to act as a building block for the formation of amino acids, purine and pyrimidine bases and therefore needs to be replenished at a higher rate. However, where the energy demands are low, oxaloacetate is used to drive the TCA cycle for the formation of NADH molecules to increase ATP concentrations.
Oxaloacetate can also be formed via an irreversible reaction from aspartate, which is catalysed by aspartate transaminase.
Upon the oxidation of fatty acids, succinyl CoA is formed. Therefore, the process of β-oxidation of fats can allow the formation of succinyl CoA molecules required in the TCA cycle.
α-ketoglutarate is regenerated from glutamate by glutamate dehydrogenase.
Fumarate is regenerated from adenylsuccinate during purine synthesis.
Cataplerosis
During the catabolism of amino acids, 4- to 5-carbon intermediates are formed, which ultimately enter the TCA cycle. The TCA cycle is unable to fully oxidise these and therefore are removed by the process of cataplerosis. Cataplerosis aims to remove intermediates, thus ensuring that there is no accumulation of anions in the mitochondrial matrix. Three main cataplerotic enzymes exist; PEPCK, aspartate aminotransferase, and glutamate dehydrogenase. The reactions involved within cataplerosis, as described below, form a product that essentially removes specific intermediates. For example, within the liver and kidney, PEPCK forms PEP which can be utilised for gluconeogenesis. However, within the muscle, PEP can be decarboxylated to form acetyl CoA for oxidation within the TCA cycle.
PEPCK
Aspartate aminotransferase
Glutamate dehydrogenase
The synthesis of carbohydrates from fat
The glyoxylate cycle is a variation of the TCA cycle that does not exist in animals. In animals, carbohydrates are readily converted into fat; however, the reverse process cannot occur. This follows on from the nature of the TCA cycle and the irreversible conversion of pyruvate into acetyl CoA.
The glyoxylate cycle allows the synthesis of carbohydrates from fat within plants, bacteria, fungi, algae, and protozoa which grow on acetate as their carbon source for energy and cell components. This is especially important for seeds rich in oil such as peanuts, olives, and sunflowers when they are germinating. The fatty acids stored within the seeds are broken down to form glucose to be used as energy for germination until photosynthesis is established.
The glyoxylate cycle is an anabolic reaction which bypasses the CO2 forming steps of the TCA cycle (isocitrate to succinate), conserving a glyoxylate molecule to form malate. It occurs within specialised peroxisomes called glyoxysomes.
As represented in Figure 17, within this cycle, isocitrate is formed as per the TCA cycle; however, this is broken down by isocitrate lyase into glyoxylate (2-carbon molecule) and succinate. Glyoxylate is then combined with acetyl CoA forming malate, a reaction catalysed by malate synthase. Malate forms oxaloacetate, which proceeds to generate energy in the form of glucose from gluconeogenesis.
Figure 17. The glyoxylate shunt (green) coexisting amongst the TCA cycle (black).

The glyoxylate shunt converts fatty acids into carbohydrates by bypassing decarboxylation steps of the TCA cycle.
The carbon skeletons that enter the TCA cycle as acetyl CoA are lost during the decarboxylation steps of the cycle. As oxaloacetate undergoes cataplerosis to make glucose, there is no oxaloacetate remaining for the TCA cycle to continue. Due to this, fats cannot produce glucose at a net rate. The glyoxylate cycle bypasses the decarboxylation steps, creating a compound that can form glucose without depleting the starting compound of the TCA cycle.
Energy production by organelles
There are several common features of the organelles responsible for energy production in eukaryotes – the mitochondria and chloroplast. Firstly, both organelles rely on the presence of a highly folded membrane, within a ‘protective’ outer membrane, which significantly increases surface area for energy generation. Secondly, these folded membranes are studded with enzymes which use Cu and Fe ions, and ubiquinone for the transfer of electrons. Thirdly, these electrons are used to pump proton (H+) ions against a concentration gradient, generating a proton motive force. This proton motive force is then used to generate ATP via ATP synthase, as protons move back down their concentration gradient. Finally, both organelles appear to have a prokaryotic origin and one theory is that they have developed an endosymbiotic relationship with an ancient pre-eukaryotic cell, to generate modern-day eukaryotes.
Organisation of organelles
We have already seen that plants are autotrophic and use chloroplasts to capture the energy from sunlight to fix CO2 and synthesise useful macromolecules. Chloroplast use their heavily folded membranes to significantly increase the amount of sunlight that can be captured and thus maximise energy generation. As well as enzymes in the membranes, the enzymes responsible for CO2 fixation, known as the Calvin cycle, are found within the stroma (large central space) which is surrounded by the inner membrane. Unlike mitochondria, chloroplast pump protons into the thylakoids, rather than the intermembrane space, which acts to generate the proton motive force required. For a lot more detailed understanding of the chloroplast and the process of photosynthesis as a whole, please refer to the Understanding Biochemistry review: Photosynthesis, Matthew P. Johnson (2016), Essays In Biochemistry, 60, 255–273; DOI: 10.1042/EBC20160016.
Unlike chloroplasts, mitochondria use organic chemical nutrients to obtain physiological energy in the form of ATP. We have already seen that ATP can be generated in glycolysis, but per glucose molecule that enters, only two ATP are generated. To capitalise on the energy held in glucose, it needs to be fully oxidised through the TCA cycle and then FAD and NADH enter the ETC. This then liberates 30–32 ATP per glucose molecule, a 15–16-fold increase in energy return. These last two pathways, TCA cycle and ETC, require functioning mitochondria and oxygen to be present. In the lumen (known as the matrix), enzymes for the TCA cycle, PDH, and β oxidation reside (Figure 18). Embedded into the cristae (folds in the inner membrane) are the enzymes for the ETC.
Figure 18. Basic structure of the mitochondrion and chloroplast, showing the membrane structures.

One enzyme crosses over between the TCA cycle and the ETC, which is succinate dehydrogenase. This enzyme catalyses the reaction succinate to fumarate and generates FADH2. Succinate dehydrogenase is integral to complex 2 and passes electrons from FADH2, directly into the ETC. Generally speaking, the greater the number of cristae, the higher the respiratory demand of the tissue. This is seen in the heart, where there are a high number of cristae and a large number of mitochondria present. Several studies have shown that one of the consequences of heart failure is mitochondrial dysfunction, where cristae superstructure breaks down and the mitochondria become less able to produce ATP.
Mitochondria are dynamic
Mitochondria are often thought of as the static structure presented in Figure 18, but in reality, they can exist in different forms. Fluorescently tagged neuronal mitochondria have been visualised moving from the neuronal body, along the cytoskeleton of the axon and to the synapse where energy demand is high. Whilst mitochondria can be present as single mitochondrion, they can also form long connected networks similar to chains seen in some prokaryotes (e.g. cyanobacteria). In these networks, mitochondria can undergo fission (splitting) and then re-join (fusion), depending on energetic demand and mitochondrial health. This act of fusion is thought to help recycle or refresh damaged mitochondria and allow mutations in mitochondrial DNA and damage to enzymes to be diluted throughout the network. In fact defects in genes regulating fission and fusion (for example PINK1 and Parkin), have been reported in neurological diseases such as Parkinson’s disease. Mutations in these genes (and others) lead to mitochondrial undergoing higher levels of fission and are less able to fuse, thereby not recycling damaged mtDNA or metabolic enzymes, affecting neuronal health and viability.
Oxidative phosphorylation in the mitochondria
Oxidative phosphorylation encompasses both the ETC and ATP synthase. This process occurs within the inner mitochondria membrane, where the complexes for the ETC and ATP synthase are present (Figure 19). The role of the ETC is, as its name suggests, to transport electrons through a series of complexes to the final electron acceptor: oxygen. As electrons flow along this chain, they start with a high energy potential losing energy as they reduce electron carriers as they travel through the chain (Figure 20). The complexes use this energy to pump protons from the matrix, into the intermembrane space. As the inner membrane is impermeable to protons, a concentration and pH gradient develops across the membrane due to the positive charge of the protons. The matrix becomes negatively charged due to the pumping out of protons and this generates an electrical potential across the membrane, equivalent to ∼150–200 mV. The electrochemical gradient is used to drive ATP synthase to produce ATP. Along with the complexes, there are two electron carriers coenzyme Q (also known as quinone and CoQ) and cytochrome c, which are responsible for passing electrons from complex to complex (Figure 19).
Figure 19. Oxidative phosphorylation – ETC and ATP synthase.

Drawn from PDB and OPM codes: 5XTD (Complex I), 1ZOY (Complex II), 1L0L (Complex III), 2DYR (Complex IV) and 6J5I (ATP synthase).
Figure 20. As electrons (e-) are passed along the chain, they reduce the next carrier and decrease their energy levels, until they reach the final electron acceptor – oxygen.

Complex I (also known as NADH dehydrogenase) and complex II (also known as succinate dehydrogenase) are flavoproteins, which means that they contain either FAD or FMN and use a series of iron–sulphur clusters to accept two electrons and two protons from NADH + H+ or succinate (eqn 4). The electrons are then passed via coenzyme Q to complex III (Q-cytochrome c oxidoreductase) which is the first cytochrome is the pathway. Complex III, IV, and cytochrome c are all cytochrome proteins, meaning that they use both haem prosthetic groups and iron–sulphur clusters to transfer electrons. Within the haem group, the iron ion alternates between a reduced Fe2+ (ferrous) state and oxidised Fe3+ (ferric) state as electrons are transferred. At complex IV, the electrons are finally passed to oxygen, which generates water. As the electrons pump through complex I, III, and IV they cause the release of 4H+, 4H+, and 2H+ into the intermembrane space, respectively (eqn 4).
Summary of equations for the four complexes of the ETC. CoQ = coenzyme Q, Cyt c = cytochrome c.
| (4) |
There are two entry points into the ETC, this is either via NADH + H+ and complex I or succinate/FADH2 and complex II. Ultimately, both pathways deliver electrons into the ETC and generate ATP at the end. However, the amount of ATP generated varies depending on which entry point, complex I or complex II, is used. This difference is due to the number of protons pumped out into the intermembrane space by the complexes. As can be seen in Figure 19, the different complexes pump protons from the matrix into the intermembrane space, thereby generating a proton gradient. Roughly four protons are needed for the generation of one ATP molecule, approximately three protons go through ATP synthase and one is used for the nucleotide transport into the mitochondria (to import Pi and ADP). Complex I can pump four protons, whilst complex II does not directly pump any protons out, therefore NADH + H+, via complex I, can generate one more ATP than FADH2, via complex II. In total, 1 NADH + H+ generates 2.5 ATP and 1 FADH2 generates 1.5 ATP. After a breakdown of the ETC process, we will return to the balance sheets that we saw earlier for the number of NADH + H+ and FADH2 generated from the oxidation of glucose and palmate.
ATP synthase
The ATP synthase multiprotein complex is a molecular motor powered of protons passing through it. In humans, ATP synthase is made up of 29 subunits of 18 different types that are, interestingly, encoded for by different genomes: some genes are nuclear whilst others are mtDNA. These subunits form the three main structures in ATP synthase: the FO (rotor), F1 (catalytic head and stalk), and FO-stalk arm. The FO is a transmembrane protein complex, which is the main rotor, accepting protons and turning the F1 head. Protons enter a channel on the intermembrane space side of FO, these protons then bind into a pocket in the FO and the rotor turns one space. This turning releases a proton on the matrix side via another channel. The complex can hold ten protons and a single proton turns just under 360° as it passes from the intermembrane space to the matrix. In the F1 subunit, there are 3 catalytic domains each containing binding sites for ADP and Pi. Within the catalytic domain, ADP and Pi react to form ATP, the action of turning the F1 releases one ATP into the matrix at a time. Therefore, a complete turn of the ATP synthase transfers ten protons from the intermembrane space to the matrix and generates three ATP molecules.
Final balance sheets
Note these numbers are estimation and values differ in different textbooks, depending on if the proton lost for the input of ADP and Pi is counted, or if you count the number of ATP produced as either 3 or 3.3 protons. There is also a difference in the number of protons that can pass through ATP synthase in different organisms. The c-ring, which is the subunit responsible for binding protons in FO, can vary in the number of subunits and therefore the number of protons that it can bind. It has been seen that this can be between 8 and 17, therefore altering the potential number of protons needed to make a 360° transition of the ATP synthase complex. The values we have used here account for the import of ADP and Pi and use the value of three protons per ATP, therefore 1 NADH = 2.5 ATP and 1 FADH2 = 1.5 ATP.
Glucose
NADHcytosol needs to be transported into the mitochondria to be used in the ETC. As NADH cannot be directly transported, the electrons and protons are transferred to malate, which via the malate-aspartate shuttle, regenerates NADH in the matrix. The use of the malate-aspartate shuttle however transports one proton from the intermembrane space, into the matrix, reducing the effective ATP production from 2.5 to 2.25.
20 ATP + 4.5 ATP + 4 ATP + 3 ATP = 31.5 ATP per glucose
Palmitate (C16)
From β-oxidation: 7 NADH, 7 FADH2, and 8 acetyl CoA
From TCA cycle: 24 NADH, 8 FADH2, 8 GTP/ATP
77.5 ATP (from NADH) + 22.5 ATP (from FADH2) + 8 ATP = 108 ATP
Even chained fatty acid with n number of carbons (Cn)
Whilst this looks scary, if you input the number of carbons in the fatty acids, then you will get the number of ATP produced when oxidised.
From β-oxidation: (Cn−2/2) NADH, (Cn−2/2) FADH2, and Cn acetyl CoA
From TCA cycle: 3 (Cn/2) NADH, (Cn/2) FADH2, (Cn/2) GTP/ATP
Or more simply for any given fatty acid with a chain length of Cn, this is the number of ATP produced via complete oxidation:
Detoxification and urea biosynthesis (waste products)
Detoxification and waste products
During their lifetime, most organisms will be exposed to harmful and hazardous substances from the environment. These substances are transformed into non-toxic products in a process of detoxification, which is carried out in a series of steps by a range of enzymes. Some toxic molecules can also arise from cellular metabolism in the form of ammonia. Ammonia is a by-product of protein metabolism through the transamination of amino acids and the deamination of l-glutamate. Ammonia is also naturally produced by gut flora.
Elevated blood concentrations of ammonia (hyperammonemia) is toxic to the central nervous system and can manifest as cerebral edema, seizures, and asterixis. Therefore, ammonia is rapidly detoxified in the body to form non-toxic urea for disposal. In mammals, the liver is the main site of metabolism and detoxification of pharmacological compounds and food. The liver also acts to maintain low levels of ammonia in the blood and, therefore, it is the main site of ammonia detoxification in mammals.
Many terrestrial animals such as humans and frogs, alongside marine mammals such as whales and seals, synthesise urea as their nitrogenous waste product and are therefore termed as ureotelic. Urea is formed from ammonia through a metabolic process called the ornithine (or urea) cycle. Detoxification occurs mostly in hepatocytes in the liver and, to a lesser extent, the kidneys.
The discovery of the urea cycle
The urea cycle was discovered by Hans Krebs and Kurt Henseleit in 1932 and was the first metabolic cycle to be elucidated.
Initially, it was found that, in liver slices, urea synthesis occurred at high rates in the presence of ornithine and ammonium ions. Even though Krebs and Henseleit were sceptical of this, it was deemed that there may be a potential link with the presence of arginase within the liver. Arginase is an enzyme that converted arginine into ornithine and urea. Later experiments found that ornithine was recycled and regenerated once urea was made and simply acted as a catalyst. Following this finding, the search for intermediates began and the cycle was formed.
The urea cycle
The main function of the urea cycle is to detoxify excess ammonium (NH4+) into a non-toxic compound called urea (Figure 21). The first stages of the cycle occur within the mitochondrial matrix of hepatocytes. Ammonia travels to the liver in the bloodstream in the form of alanine and glutamine or via the hepatic portal vein from the gut. The initial reaction includes the formation of a carbomoyl phosphate molecule which then combines with ornithine to form citrulline. For this reaction to occur, ornithine moves into the mitochondrial matrix from the cytosol via an ornithine transporter. Once formed, citrulline moves into the cytosol via a citrulline transporter in exchange for cytosolic ornithine, which ultimately begins the cycle.
Figure 21. The urea cycle intermediates and by-products.

Ornithine is imported into the mitochondria, where it combines with carbomoyl phosphate to generate citrulline. Citriuline is exported to the cytosol, where the cycle continues, until the release of urea and the regeneration of ornithine.
In the cytosol, citrulline is condensed with aspartate to form argininosuccinate driven by the hydrolysis of ATP. Argininosuccinate is cleaved to arginine and fumarate, a reaction that is catalysed by argininosuccinate lyase. In a state of fasting when gluconeogenesis occurs, fumarate can be used to form malate which can then go on to form glucose or oxaloacetate. Arginine, on the other hand, proceeds in the ornithine cycle and is cleaved to form urea and regenerate ornithine molecules via arginase.
The overall reaction of the urea cycle is as follows:
The first step of the urea cycle is a rate-limiting step. The enzyme carbomoyl phosphate synthetase I requires an activator, N-acetyl-glutamate (NAG). NAG is formed by the addition of glutamate and acetyl CoA occurring under the influence of NAG synthase, which is regulated by the presence of arginine.
Ammonotelic, uricotelic, and the in-between
Aquatic animals such as fish are termed ammonotelic. Ammonotelic organisms excrete ammonia directly as nitrogenous waste products. Ammonia is a highly soluble substance and requires large volumes of water to dissolve. Therefore, aquatic animals like fish use their gills to excrete over 80% of ammonia directly into surrounding waters.
Terrestrial organisms such as lizards, snakes, insects, and birds are termed uricotelic organisms. These excrete uric acid as their waste product instead of urea. The complex pathway ultimately stems from the metabolic breakdown of purine nucleotides. The metabolic pathway has a higher energy cost, in comparison with the synthesis of urea, but can reduce water loss in these animals.
In some cases, animals can shift from utilising one form of waste product mechanism to another. In the metamorphosis of amphibians, you can see the development of the urea cycle as they adapt to terrestrial life. Tadpoles excrete ammonia as their primary waste product before metamorphosis. However, during its development to becoming a frog and the ultimate invasion of land, an irreversible change to ureotelism occurs. This, therefore, means that urea is the excretory end-product in comparison.
The reverse is seen in earthworms, which are primarily ammonotelic. Earthworms possess the ability to carry out the urea cycle in full to synthesise arginine. However, during times of starvation, earthworms shift to become ureotelic where they then utilise the urea cycle to detoxify ammonia producing urea. Earthworms can shift between these two states depending on their state of starvation.
The role of metabolism in inherited diseases
Metabolic processes that have been discussed extensively within this review largely oversimplify and underestimate the pervasive role of metabolism in all living organisms. The discovery and breakdown of individual metabolic pathways have been at the forefront of scientific research for many years up until the 1960s. Since then research into the role of metabolism has been largely driven by the recognition of metabolic perturbations and their manifestation in human diseases. As metabolism is essential for life, disruptions or dysregulation in metabolic processes can be detrimental. Certain mutations in metabolic enzymes can be embryonically lethal in mouse knockout studies, demonstrating the curial role that metabolic pathways play in life. Metabolic perturbations underlie many human diseases and here we will briefly discuss metabolic processes that occur within inborn errors of metabolism, which lead to inherited diseases.
Inborn errors of metabolism
In the U.K. at 6 days of age, a newborn baby will receive a heel prick test (Guthrie test). This test screens for diseases such as sickle cell aneamia, congenital hypothyroidism, and inherited metabolic diseases. These metabolic diseases, listed in Table 4, can manifest as serious illnesses that can cause severe developmental problems and can be detrimental to life.
Table 4. A list of the six common inherited metabolic diseases that are screened for in newborn babies, alongside their respective metabolic disruptions.
| Metabolic disease | Metabolic disruption |
|---|---|
| Phenylketonuria (PKU) | Amino acid disorder (Phenylalanine) |
| Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) | Fatty acid oxidation disorder (Octanonyl carnitine) |
| Maple syrup urine disease (MSUD) | Amino acid disorder |
| Isovaleric acidaemia (IVA) | Amino acid disorder (Leucine) |
| Glutaric aciduria type 1 (GA1) | Amino acid disorder (lysine, hydroxylysine, and tryptophan) |
| Homocystinuria (pyridoxine unresponsive) (HCU) | Amino acid disorder (methionine) |
Phenylketonuria (PKU) and medium-chain acyl-CoA dehydrogenase deficiency (MCADD) are the two most commonly inherited metabolic disorders, which affects approximately 1 in 10000 newborns in the U.K.
PKU is an amino acid disorder that is caused by the deficiency of the enzyme phenylalanine hydroxylase causing an enzymatic block. This results in the decreased metabolism of the amino acid phenylalanine causing an increased accumulation in the blood and brain. If left untreated in newborns, this can cause developmental delay or brain damage. Treatment begins early with a low protein diet, supplemented with an amino acid mixture with phenylalanine removed. However, a small proportion of individuals diagnosed with PKU, do not respond to this suggested treatment type. These individuals usually present with defects in dihydropteridine reductase or biopterin synthesis, causing a defect in phenylalanine hydroxylase function. These individuals also usually present with defects in tyrosine hydroxylase too, which can lead to a deficiency of neurotransmitters. These patients then require additional supplementation with neurotransmitters as well as a low phenylalanine-based diet.
MCADD is a lifelong condition that occurs due to a mutation in the medium-chain acyl-CoA dehydrogenase (MCAD) of fatty acid β-oxidation. This mutation impairs the breakdown of medium-chain fatty acids in acetyl CoA. A loss or insufficiency of MCAD reduces the oxidation of fatty acyl-CoAs that contain more than six carbons, as the first dehydrogenation step of β-oxidation cannot occur. Using tandem mass spectrometry can be seen that the blood fatty acid profile in MCADD shows an accumulation of C6, C8, and C10:1. MCADD is a major cause of hypoketotic hypoglycaemia and can cause liver dysfunction with metabolic acidosis, hyperammonemia, and sudden death. MCADD is particularly dangerous during a fasting state, where the body utilises glycogen stores and free fatty acids are released from adipose tissue for energy. The reduced ability to metabolise medium fatty acids significantly reduces the availability of substrates for ketogenesis, ATP synthesis, and the TCA cycle, at a time of low energy. The accumulation of fatty acid intermediates inhibits gluconeogenesis, exacerbating hypoglycaemia. This accumulation might also contribute to cardiovascular and neurological complications found in these conditions. Treatment for patients with MCADD includes the intake of high sugar drinks and avoidance of long fasted periods.
The final inherited metabolic disorder we are discussing is much rarer, occurring in 1 in 100000 or 150000 newborns. Maple syrup urine disease (MSUD) occurs due to the deficiency or decreased function of the branched-chain α-keto acid dehydrogenase complex (BCKAD). This results in an accumulation of branched-chain amino acids (BCAAs) such as leucine, isoleucine, and valine in the blood and urine. The name of the condition comes from the maple syrup smell of the urine, due to the excess BCAA. BCAAs are consumed within protein rich diets in foods such as meat, fish, eggs, and milk. Normally excess amino acids are broken down through branched-chain aminotransferases (BCATs) into α-ketoacids within the mitochondria. In the second step of catabolism, the BCKAD complex initiates oxidative decarboxylation of the α-ketoacids resulting in the formation of acetoacetate, acetyl CoA and succinyl CoA. The normal functioning of amino acid catabolism is essential for protein synthesis, cell signalling, and glucose metabolism. BCKAD is formed of four subunits. Mutations within the catalytic components of BCKAD, decrease its activity and therefore increase BCAA levels manifesting as MSUD and causing dysfunction of the immune system, skeletal muscle, and central nervous system. As the accumulation of toxic metabolites, such as lactic acid and ammonia occurs, immune cell function is inhibited causing its dysregulation. Skeletal muscle is impaired as shown in studies that found diminished muscle fiber diameters and myofiber lesions in MSUD rats, however, its mechanism is not fully understood. Nervous system dysregulation, in particular, brain damage, was associated with the accumulation of toxic metabolites. However, studies have shown that the generation of nitrogen reactive species in MSUD patients can induce morphological changes in C6 glioma cells. Also, markers of protein, DNA, and lipid oxidative damage are found in MSUD patients potentially as a result of free radical production.
The role of metabolism in cancer
Cancer metabolism
In 1931 Warburg won the Nobel Prize in Physiology or Medicine for his ‘Discovery of the nature and mode of action of the respiratory enzyme’. Warburg’s work focused on the metabolic reprogramming of cancer cells, which is now deemed as one of the hallmarks of cancer as put forward by Douglas Hanahan and Robert Weinberg in Hallmarks of Cancer: The Next Generation 2011 (Figure 22).
Figure 22. The hallmarks of cancer development put forward by Hanahan and Weinberg in 2011.

Deregulated cellular energetics is included in these hallmarks as a driver of cancer progression.
In the presence of oxygen and glucose, the mammalian quiescent cell will uptake glucose for respiration. Here the glucose molecule will undergo the reactions of glycolysis to form pyruvate and then enter the TCA cycle and the ETC for oxidative phosphorylation to yield 30–32 ATP molecules. In anaerobic conditions, glucose partakes in glycolysis yielding two ATP molecules and lactic acid. Warburg found that tumour slices and cancer cells, even in the presence of oxygen, do not continue with oxidative phosphorylation but instead increase lactate production. This cellular metabolic adaptation in cancer cells is referred to as aerobic glycolysis or the Warburg effect (Figure 23). Similar to what is seen in red blood cells. The resulting lactate overproduction would provoke a state of metabolic acidosis within the tumour environment. But also, lactate would be taken up by surrounding cells to support tumour growth and inhibiting apoptosis. By increasing the gene expression of GLUTs, GLUT1, there is an increased uptake of glucose into the cell. Along with the reprogramming of several glycolytic genes, this increased glucose drives an increase in glycolytic flux. This increase in glycolytic flux allows the formation of glycolytic intermediates for biosynthesis and fulfils the proliferative hallmark of cancer cells, i.e. increased nucleotide formation. Cancer or proliferating cells uptake large amounts of nutrients such as glucose and glutamine that are used to support cellular growth to help generate proteins, lipids, and nucleic acids necessary to aid cell division as summarised in Figure 24.
Figure 23. The Warburg effect.

A cell with no oxygen supply generates lactate as it undergoes glycolysis anaerobically. In aerobic conditions, with oxygen, a cell proceeds with oxidative phosphorylation yielding a higher number of ATP molecules. However, in a cancer tumour microenvironment, the ability to carry out oxidative phosphorylation is disrupted, despite oxygen presence. Instead, lactate is formed, and a cell is suggested to undergo aerobic glycolysis.
Figure 24. Increased glucose and glutamine uptake and utilisation drive the increased synthesis of nucleotide, protein, and lipid synthesis in cancer cells.
The up-regulation of glycolysis increases cellular lactate concentrations.
The increased uptake of glucose is furthered in cancer or proliferating cells due to the increased expression of GLUT transporters alongside the up-regulation of metabolic enzymes such as PFKFB3 to increase glycolytic flux. In cancer, hypoxia-inducible factor (HIF)-1α is activated, even in the presence of oxygen. HIF-1α is suggested to be activated due to mutations in isocitrate dehydrogenase (IDH) enzymes which also increase cellular glycolysis within the cells and halts oxidative phosphorylation. Glutamine uptake and catabolism are also increased within these cells to ensure a supply of nitrogen for nucleotide biosynthesis and the formation of α-ketoglutarate for TCA cycle progression. Intermediates of both glycolysis and the TCA cycle both generate major macromolecules required for growth; ribose-5-phosphate for nucleotide synthesis, serine, and glycine to fuel protein synthesis and support nucleotide synthesis, and citrate which is generated to be exported into the cytoplasm providing acetyl CoA for fatty acid synthesis.
Targeting metabolism in cancer cell drug discovery
As one of the hallmarks of cancer, targeting metabolic pathways is critical in the development of new therapeutics, and might serve as useful targets to inhibit cancer cell growth. As cancer cells are rapidly dividing, any disruption in their ability to obtain energy will slow their proliferation. There are two main strategies in targeting glucose metabolism in cancer cells, this is to either inhibit anaerobic glycolysis, thereby starving the cells, or by increasing glucose oxidation, increasing the usage of mitochondria and therefore apoptosis.
Examples of targeting glycolysis are found in inhibitors of hexokinase (2-deoxyglucose (2DG)) or enolase (phosphonoacetohydroxamate (PHAH) or SF-2312). These compounds aim to reduce the flow of glucose within cells that have a high glycolytic flux, i.e. cancer cells. This would, therefore, reduce the energy available to the cell and slow rapid cell proliferation. The problem is that these compounds might have additional effects in red blood cells, which are solely reliant on glycolysis.
Another way of reducing anaerobic glycolysis is to inhibit the export of lactic acid from the cell. By targeting the monocarboxylate transporters (MCTs) (AZD3965 or syrosingopine), this increases the intracellular concentration of lactic acid and either increases the acidity of the cell and/or decreases the activity of LDH. Decreased LDH activity, leads to an accumulation of NADH and pyruvate, which both lead to inhibition of glycolysis by feedback mechanisms discussed earlier.
To target glucose oxidation, the drug dichloroacetate (DCA) inhibits PDK, which causes the activation of PDH and an increase in acetyl CoA. PDH activation allows mitochondrial oxidation of pyruvate to occur, disrupting the metabolic glycolytic advantage of cancer cells. Furthermore, DCA acts to counter the acidosis caused by increased lactate production by decreasing flow through LDH, inhibiting the progression and development of the tumour microenvironment. Therefore, drug therapies have shown the ability to target the altered cellular metabolic pathways in cancer cells to slow the uptake of glucose by disrupting and inhibiting glycolysis and the production of lactate.
Imaging metabolism in cancer
Along with targeting metabolism for treating cancer, it is also possible to hijack the importance of metabolism in cancer to find and identify tumours. Several techniques take advantage of changes in metabolic state to image where a tumour is and also, its response to treatment. Two techniques: positron emission tomography and magnetic resonance spectroscopy (MRS) or imaging (MRI) have demonstrated their ability in this field.
FDG–PET
2-deoxy-2-[18F]fluoro-D-glucose (FDG) is a radiotracer that acts similarly to glucose, by being taken up by glucose transporters, i.e. GLUTs and SGLT. Once inside the cell, FDG goes through the first step in glycolysis and is phosphorylated to [18F]FDG-6-phosphate, which unlike G6P is unable to continue through glycolysis. Outside of the liver (and fasted kidney), the lack of glucose-6-phosphatase traps [18F]FDG-6-phosphate inside the cells. As this molecule accumulates, the 18F radiotracer allows the molecule to be detected using PET, which is overlaid onto an anatomical image generated by computed tomography (CT) or MRI. This tracer has been useful in the detection and sizing of tumours, as an increase in [18F]FDG-6-phosphate detection compared with the surrounding tissue, is indicative of high glucose uptake and rapidly proliferating cells and possibly cancer. This is particularly useful, as metabolic imaging of cancerous cells as the potential to detect tumours, before they are visible using conventional imaging, such as X-ray, CT, or MRI. However, there are still limits to the sensitivity of this technique for the detection of tumours, it can also be difficult to detect changes in the brain (where there is a high background glucose uptake) and it only measures glucose uptake and not directly glycolysis.
Hyperpolarised 13C MRI/MRS
Whilst MRI and MRS have been around for many years, monitoring real-time metabolism using hyperpolarised carbon 13 (13C) is relatively new. 13C MRS generates a set of peaks depending on the molecular environment that the 13C molecule is found in, this is called a spectrum. The new technique relies on an external machine (a polariser) to significantly enhance the signal from a 13C containing compound. This enhanced state is temporary (between 1 and 5 min), so the compound needs to be rapidly injected into a patient inside an MRI machine to enable detection. Unlike PET, 13C tracers are non-radioactive and can also monitor real-time metabolism. The most common tracer has been [1-13C]pyruvate, which can be rapidly taken up by cells and has three possible fates; through PDH, ALT, or LDH. When [1-13C]pyruvate is metabolised by one of these three enzymes, it produces a new 13C compound, which has a different peak on the spectrum. Meaning you can trace the conversion of pyruvate into CO2/bicarbonate (acetyl-CoA is not seen as a carbon is lost as CO2), alanine, and lactate, respectively. These can be visualised on a spectrum as a series of peaks, with a new spectrum being produced every 0.1–1 s, thereby generating a time course to work out enzyme kinetics. Or the peaks can have spatial information added and an image of where metabolism is occurring can be produced for a specific tissue.
The first in man trial for using this technique in patients with prostate cancer was only completed in 2013. They monitored the conversion of [1-13C]pyruvate into [1-13C]lactate and produced images of where this conversion occurred in the human prostate. This was used as a marker of elevated glycolytic flux (although it only measures the last step in anaerobic glycolysis) and showed cancerous tissue amongst healthy tissue. In rodent models of cancer this technique can also monitor the effectiveness of treatment in tumours. This technique has also been applied to the study of cardiac metabolism, where it was able to image real-time metabolism in the human heart.
Both PET and hyperpolarised 13C MRS/MRI have their limitations, these are mainly due to the injection of tracers. These tracers only provide you with a snapshot of the processes occurring, as they only make up a very small percentage of the endogenous metabolites. They also only show you a limited number of steps within metabolic pathways and several assumptions are required to enable analysis and understanding of the pathways at work. Having said that, these are exciting areas and have shown an ability to visualise metabolism occurring in real-time, within the human body, which takes us from perfused animal organ and tissue slices of last century to live metabolic imaging of the human body.
The importance of metabolism in biotechnology
The potential of metabolism
Along with drug discovery and diagnosis of human diseases, metabolism plays an exciting role in the development of biotechnology. Whether that is bio-engineering of Escherichia coli to meet the growing demand for bioethanol or manipulating plants to generate better defence against pests, there are lots of possibilities. Advancements of knowledge in these particular fields have allowed the development of more specific biotechnological advancements. These advancements allow us to overcome hurdles that once caused issues with yield and even resistance. These ideas are discussed below as how biotechnology has ensured efficient and elaborate systems to target particular areas of metabolism.
Bioethanol production from E. coli
Ethanol holds a variety of applications in the food, chemical, medical, and health industry and is commonly used in beverages, fuels, and paint. Ethanol is largely produced from coal and petroleum using non-renewable processes; however, ethanol can be produced by fermentation of sugars and starch. Microorganisms hold the capacity to produce ethanol via fermentation and due to this, have been identified and studied to produce ethanol in high capacity. Microorganisms can be metabolically engineered to redirect carbon sources to desired fuel products. One of these microorganisms is E. coli.
E. coli can naturally utilise a variety of carbon sources such as sugars and sugar alcohols, under both aerobic and anaerobic conditions. All biofuels that are derived from E. coli are derived from the modification of central carbon catabolism including the conversion of pentose/hexose sugar molecules into diatomic carbon.
E. coli can produce ethanol under anaerobic conditions to produce 1 mole of ethanol for every 1 mole of glucose used, alongside formate and acetate. The reduction reaction that occurs to synthesise these, utilises two NADH molecules but the initial stages of glycolysis only yield one. This causes a redox imbalance. This redox imbalance is overcome by E. coli by balancing the production of ethanol by oxidation of acetyl CoA into acetate, which requires no NADH. These E. coli were engineered to express pyruvate decarboxylase and alcohol dehydrogenase II genes of Zymomonas mobilis. This pathway produces 95% ethanol, without a redox imbalance as it only requires 1 NADH molecule.
Modification of metabolism in plants
Plant metabolism is highly organised and controlled by innate regulations alongside homoeostatic mechanisms. Their metabolic procedures are highly compartmentalised where each organelle carries out a specific metabolic function.
Within nature, plants are constantly challenged by insect herbivores. In response to this, as a defence to reduce insect-related damage to the plant, they produce toxins and small deterrent molecules for protection. Some of these defence responses are inducible where these are related to the up-regulation or down-regulation of metabolites and proteins in response to an attack.
For example, the tobacco plant naturally synthesises nicotine to deter insects from eating them. However, if an attack does occur on a tobacco plant, this response is up-regulated. Here, the roots synthesise large amounts of nicotine, and this is transported to the shoot. This response is regulated by jasmonic acid and auxin, both signals that are triggered by wounding. This increase in nicotine production is to avoid further herbivore attack.
In addition to defences such as those described above, plants can also induce changes in their metabolism in response to attacks. These include elevating or decreasing their photosynthetic efficiency, remobilisation of carbon and nitrogen resources alongside altering their plant growth.
Amino acids are the form of nitrogen found in plants. They can regulate plant growth and are precursors for defence-related metabolites. When plants are infested by herbivore insects, studies found that amino acid production increased to synthesise defensive metabolites to ensure a second attack does not occur. This increase in amino acid production occurs as genes involved in amino acid synthesis are up-regulated and sulphur assimilation, which is ultimately required for the biosynthesis of cysteine and methionine, are all up-regulated upon caterpillar feeding on Arabidopsis thaliana. Furthering this, a similar study further found that the increased expression of genes involved in amino acid biosynthesis, in particular, related to methionine and tryptophan synthesis, led to a greater accumulation of glucosinolates. These are defence-related molecules that are derived from methionine and tryptophan in A. thaliana. Interestingly, this was also found in tomatoes. Upon attack, tomato plants accumulate tryptophan within their stems and apex of tomatoes, to serve as a precursor for the production of defensive metabolites.
Plant biotechnology is a set of techniques that are used to adapt plants for survival and defence. One of the most effective ways to manage plant pathogenicity is to use genetic modification alongside genome editing. This is particularly useful in the agriculture industry to ensure increased food supply. Globally, up to 30% of food is damaged by pathogens and disease pests. Therefore, the adaptation of plants to pathogens and pests is imperative for survival. Gene modification and genome editing can help manage plant pathogens to ensure plant cell survival in times of attack, these solutions currently include bacterial, viral, fungal, and oomycete pathogens.
As knowledge regarding the plant immune system has expanded, it has been shown that plants can distinguish between ‘self’ and ‘non-self’ through monitoring the extracellular and intracellular environment. However, pathogens can overcome this system by producing proteins and effectors to suppress plant host immunity and colonise it. A bacterial pathogen can hijack the coronatine-insensitive protein 1 jasmonate receptor, altering the plant’s defence response and activating jasmonate responses and suppressing salicylic acid defence pathways. Manipulation like these has allowed the development of biotechnology to intervene at the point of pathogen perception. These can be created by introducing receptors from other plants with novel recognition specificity, reactivation of genes disabled in NOD-like receptors, and modification of domains in NOD-like receptors that are targeted by pathogens. This knowledge reveals which components of the host can be manipulated to promote a disease state. Therefore, by removing these vulnerable points or replacing them with variants that are immune to specific effectors, the natural functionality of the plant can be retained. Bacterial pathogens, that express the transcription activate like effectors that activate susceptibility genes within the host, can be overcome by the deletion of TAL DNA-binding sites in the promoter. Another approach includes engineering resistance to bacterial pathogens by adding TAL effector binding sites to an executor gene.
Another way to regulate plant pathogen perception is by the intervention of the defence signalling pathways. The major hormones involved in defence signalling are salicylic acid, jasmonate, and ethylene. In tomato plants, a loss of function allele was engineered for Downy mildew resistance 6 (DMR6) to ensure resistance to biotrophic pathogens. DMR6 is widely conserved and encodes a salicylate-5-hydrozylase enzyme around infection sites.
Biotechnology advancements regarding plants and plant metabolism help ensure defence and healthy plant growth. In conclusion, biotechnology has allowed the intervention of defence signalling and regulation, pathogen recognition, effector recognition, and allowed targeting of genes associated with plant pathogenicity.
Conclusion
At least 25 Nobel Prizes have been awarded for work related to metabolism, with the most recent for the oxygen-sensing role of HIF1α in 2019. This review has introduced the basic metabolic pathways of life and demonstrated how a series of reactions can combine to sustain life. Over the last 150–200 years, the understanding of metabolism has moved from individual enzymes and metabolites to the complicated network we see today. This has allowed us to visualise metabolism occurring in real-time within the human body, to help us understand metabolic alterations in human disease. Manipulation of metabolic pathways in both microorganisms and plants has also led the way in the development of new biotechnological techniques.
Acknowledgements
We would like to thank Jennifer Greaves (Coventry University) for her useful discussions on this article and A.R. Teal (Faculty of Science and Technology, North East Surrey College of Technology) for the original Metabolism BASC article (1994) on which this essay is partly based.
Abbreviations
- ACP
acyl carrier protein
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- BCAA
branched-chain amino acid
- BCAT
branched-chain aminotransferase
- BCKAD
branched-chain α-keto acid dehydrogenase complex
- CoA
coenzyme A
- CPT1
carnitine palmitoyl transferase I
- CT
computed tomography
- DCA
dichloroacetate
- DHAP
dihydroxyacetone phosphate
- DMR6
Downy mildew resistance 6
- ER
enoyl ACP reductase
- ETC
electron transport chain
- FAS
fatty acid synthase
- FDG
2-deoxy-2-[18F]fluoro-D-glucose
- F6P
fructose-6-phosphate
- GAP
glyceraldehyde-3-phosphate
- GDP
guanosine diphosphate
- GLUT
glucose transporter
- GTP
guanine triphosphate
- G6P
glucose-6-phosphate
- G6PI
G6P isomerase
- HDL
high-density lipoprotein
- HIF
hypoxia-inducible factor
- HSL
hormone-sensitive lipase
- IRS
insulin receptor substrate
- LDH
lactate dehydrogenase
- LDL
low-density lipoprotein
- MCAD
medium-chain acyl-CoA dehydrogenase
- MCADD
medium-chain acyl-CoA dehydrogenase deficiency
- MRI
magnetic resonance imaging
- MRS
magnetic resonance spectroscopy
- MSUD
maple syrup urine disease
- NAD(P)H
nicotinamide adenine nucleotide (phosphate)
- NAG
N-acetyl-glutamate
- NH4+
ammonium ions
- NO2−
nitrite
- PC
pyruvate carboxylase
- PDH
pyruvate dehydrogenase
- PEP
phosphoenolpyruvate
- PEPCK
PEP carboxykinase
- PFK
phosphofructokinase
- PK
pyruvate kinase
- PKU
phenylketonuria
- PPi
inorganic pyrophosphate group
- PPARα
peroxisome proliferator-activated receptor α
- PPP
pentose phosphate pathway
- SGLT
sodium glucose cotransporter
- TAG
triglyceride
- TCA
tricarboxylic acid
- UDP-glucose
uridine diphosphate glucose
- VLDL
very low-density lipoprotein
- 1,3-BPG
1,3-bisphosphoglycerate
- 3PG
3-phosphoglycerate
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Funding
Ayesha Judge is funded by a PhD studentship from Coventry University with additional funding and support from the University Alliance Doctoral Training Alliance. Michael Dodd is funded by Coventry University and the Centre for Sport, Exercise and Life Sciences.
References and further reading
- Albrecht T. and Baron S. (1996) Medical Microbiology, University of Texas Medical Branch at Galveston, Dept. of Microbiology & Immunology, Galveston, TX, U.S.A., Chapter 4 [Google Scholar]
- Ameer F., Scandiuzzi L., Hasnain S., Kalbacher H. and Zaidi N. (2014) De novo lipogenesis in health and disease. Metabolism 63, 895–902 10.1016/j.metabol.2014.04.003 [DOI] [PubMed] [Google Scholar]
- Berg J., Tymoczko J. and Stryer L. (2002) The first step in amino acid degradation is the removal of nitrogen. https://www.ncbi.nlm.nih.gov/books/NBK22475 [Google Scholar]
- Bishop S. and Campbell J. (1965) Arginine and urea biosynthesis in the earthworm Lumbricus terrestris. Comp. Biochem. Physiol. 15, 51–71 10.1016/0010-406X(65)90240-9 [DOI] [PubMed] [Google Scholar]
- Brown G., Brown W. and Cohen P. (1962) Comparative biochemistry of urea synthesis IV. [14C]Urea synthesis by liver slices of the metamorphosing tadpole. Biochim. Biophys. Acta 60, 185–186 10.1016/0006-3002(62)90387-6 [DOI] [Google Scholar]
- Chirala S., Jayakumar A., Gu Z. and Wakil S. (2001) Human fatty acid synthase: Role of interdomain in the formation of catalytically active synthase dimer. Proc. Natl. Acad. Sci. U.S.A. 98, 3104–3108 10.1073/pnas.051635998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis R. and Chandel N. (2016) Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esse H., Reuber T. and Does D. (2019) Genetic modification to improve disease resistance in crops. New Phytol. 225, 70–86 10.1111/nph.15967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayn K. and Evans R. (2018) Human Metabolism: A Regulatory Perspective, 4thedn, Wiley Blackwell [Google Scholar]
- Gartner V., McGuire P. and Lee P. (2015) Child Neurology: medium-chain acyl-coenzyme A dehydrogenase deficiency. Neurology 85, e37–e40 10.1212/WNL.0000000000001786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibala M., Young M. and Taegtmeyer H. (2000) Anaplerosis of the citric acid cycle: role in energy metabolism of heart and skeletal muscle. Acta Physiol. Scand. 168, 657–665 10.1046/j.1365-201x.2000.00717.x [DOI] [PubMed] [Google Scholar]
- Guo X., Li H., Xu H., Woo S., Dong H., Lu F. et al. (2012) Glycolysis in the control of blood glucose homeostasis. Acta Pharm. Sin. B 2, 358–367 10.1016/j.apsb.2012.06.002 [DOI] [Google Scholar]
- Hanahan D. and Weinberg R. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- He J., Ford H., Carroll J., Douglas C., Gonzales E., Ding S. et al. (2018) Assembly of the membrane domain of ATP synthase in human mitochondria. Proc. Natl. Acad. Sci. U.S.A. 115, 2988–2993 10.1073/pnas.1722086115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz M. and Stein L. (2008) Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol. Lett. 278, 146–156 10.1111/j.1574-6968.2007.00970.x [DOI] [PubMed] [Google Scholar]
- Koppolu V. and Vasigala V. (2016) Role of Escherichia coli in biofuel production. Microbiol. Insights 9, 29–35 10.4137/MBI.S10878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg H. and Beevers H. (1957) A mechanism of conversion of fat to carbohydrate in castor beans. Nature 180, 35–36 10.1038/180035a0 [DOI] [PubMed] [Google Scholar]
- Krebs H. (1973) The discovery of the ornithine cycle of urea synthesis. Biochem. Educ. 1, 19–23 10.1016/0307-4412(73)90048-4 [DOI] [Google Scholar]
- Leonardi R., Zhang Y., Rock C. and Jackowski S. (2005) Coenzyme A: back in action. Prog. Lipid Res. 44, 125–153 10.1016/j.plipres.2005.04.001 [DOI] [PubMed] [Google Scholar]
- Li C., Teng W., Shi Q. and Zhang F. (2007) Multiple signals regulate nicotine synthesis in tobacco plant. Plant Signal. Behav. 2, 280–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navale A. and Paranjape A. (2016) Glucose transporters: physiological and pathological roles. Biophys. Rev. 8, 5–9 10.1007/s12551-015-0186-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S., Kurhanewicz J., Vigneron D., Larson P., Harzstark A., Ferrone M. et al. (2013) Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med. 5, 198.ra108 10.1126/scitranslmed.3006070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen O., Kalhan S. and Hanson R. (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412 10.1074/jbc.R200006200 [DOI] [PubMed] [Google Scholar]
- Palomer X., Pizarro-Delgado J., Barroso E. and Vázquez-Carrera M. (2018) Palmitic and oleic acid: the yin and yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol. Metab. 29, 178–190 [DOI] [PubMed] [Google Scholar]
- Park J., Davis R. and Sue C. (2018) Mitochondrial dysfunction in Parkinson’s disease: new mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 18, 10.1007/s11910-018-0829-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider O., Apps A., Miller J., Lau J., Lewis A., Peterzan M. et al. (2020) Noninvasive in vivo assessment of cardiac metabolism in the healthy and diabetic human heart using hyperpolarized 13 C MRI. Circ. Res. 126, 725–736 10.1161/CIRCRESAHA.119.316260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P. (2015) Enzymes: principles and biotechnological applications. Essays Biochem. 59, 1–41 10.1042/bse0590001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger A., Muñoz-Gómez S. and Kamikawa R. (2017) The origin and diversification of mitochondria. Curr. Biol. 27, R1177–R1192 10.1016/j.cub.2017.09.015 [DOI] [PubMed] [Google Scholar]
- Sánchez-Baracaldo P., Raven J.A., Pisani D. and Knoll A.H. (2017) Early photosynthetic eukaryotes inhabited low-salinity habitats. Proc. Natl. Acad. Sci. U.S.A. 114, E7737–E7745 10.1073/pnas.1620089114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz O., Brock B. and Rungby J. (2004) Amylin agonists: a novel approach in the treatment of diabetes. Diabetes 53, S233–S238 10.2337/diabetes.53.suppl_3.S233 [DOI] [PubMed] [Google Scholar]
- Sitta A., Ribas G., Mescka C., Barschak A., Wajner M. and Vargas C. (2013) Neurological damage in MSUD: the role of oxidative stress. Cell. Mol. Neurobiol. 34, 157–165 10.1007/s10571-013-0002-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnet D., O’Leary M., Gutierrez A., Nguyen S.M., Mateen S., Hsu Y. et al. (2016) Metformin inhibits branched chain amino acid (BCAA) derived ketoacidosis and promotes metabolic homeostasis in MSUD. Sci. Rep. 6, 10.1038/srep28775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf P., Conn E., Miflin B. and Lea P. (1987) The Biochemistry of Plants, 11thedn, pp. 29–30, Academic Press, San Diego [Google Scholar]
- Voet D., Voet J. and Pratt C. (2013) Principles of Biochemistry 4thedn, (Hoboken N.J., ed.), John Wiley & Sons [Google Scholar]
- Wang Z., Ohliger M., Larson P., Gordon J., Bok R., Slater J. et al. (2019) Hyperpolarized 13C MRI: state of the art and future directions. Radiology 291, 273–284 10.1148/radiol.2019182391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner I. and Verlander J. (2013) Renal ammonia metabolism and transport. Compr. Physiol. 3(1), 201–220 10.1002/cphy.c120010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B., Schisler J. and Willis M. (2010) Sir Hans Adolf Krebs: architect of metabolic cycles. Lab. Med. 41, 377–380 10.1309/LMZ5ZLAC85GFMGHU [DOI] [Google Scholar]
- Yudkoff M. (1999) Urea cycle. https://www.ncbi.nlm.nih.gov/books/NBK27982 [Google Scholar]
- Zhang S., Hulver M., McMillan R., Cline M. and Gilbert E. (2014) The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Lou Y., Tzin V. and Jander G. (2015) Alteration of plant primary metabolism in response to insect herbivory. Plant Physiol. 169(3), 1488–98 10.1104/pp.15.01405 [DOI] [PMC free article] [PubMed] [Google Scholar]