

Abstract
Nanopore sequencing enables direct measurement of RNA molecules without conversion to cDNA, thus opening the gates to a new era for RNA biology. However, the lack of molecular barcoding of direct RNA nanopore sequencing data sets severely affects the applicability of this technology to biological samples, where RNA availability is often limited. Here, we provide the first experimental protocol and associated algorithm to barcode and demultiplex direct RNA nanopore sequencing data sets. Specifically, we present a novel and robust approach to accurately classify raw nanopore signal data by transforming current intensities into images or arrays of pixels, followed by classification using a deep learning algorithm. We demonstrate the power of this strategy by developing the first experimental protocol for barcoding and demultiplexing direct RNA sequencing libraries. Our method, DeePlexiCon, can classify 93% of reads with 95.1% accuracy or 60% of reads with 99.9% accuracy. The availability of an efficient and simple multiplexing strategy for native RNA sequencing will improve the cost-effectiveness of this technology, as well as facilitate the analysis of lower-input biological samples. Overall, our work exemplifies the power, simplicity, and robustness of signal-to-image conversion for nanopore data analysis using deep learning.
The emergence of third-generation sequencing (TGS) technologies has revolutionized our ability to sequence genomes and transcriptomes (Ardui et al. 2018; Pollard et al. 2018). Compared to second-generation sequencing technologies, TGS has the ability to produce long sequencing reads, avoiding the hassle of fragmenting the RNA or DNA molecules into smaller pieces to then reassemble them back together. Furthermore, TGS technologies have the ability to sequence DNA and RNA without a PCR amplification step, thus allowing direct detection of DNA and RNA modifications, with single nucleotide resolution and in individual molecules.
Direct sequencing of native RNA molecules (dRNA-seq) can be achieved using the platform offered by Oxford Nanopore Technologies (ONT). This platform relies on the use of protein nanopores embedded in a membrane that are subjected to an electric field. Characteristic disruptions in the electric current are measured as the RNA molecule passes through the pore, enabling the observation of single molecules. Low translocation velocity of the RNA molecule is achieved through the association of motor proteins that regulate the translocation of the nucleic acid molecule, and the resulting current intensity measurements can, in turn, be converted into sequence information using previously trained base-calling algorithms (Rang et al. 2018).
The first direct RNA sequencing protocol developed by ONT (SQK-RNA001) became commercially available in 2017 and was designed to sequence mRNAs (Garalde et al. 2018), although later efforts have shown that this protocol can be adapted to sequence non-poly(A)-tailed RNAs, such as ribosomal RNAs (Smith et al. 2019). The current ONT dRNA-seq library preparation protocol comprises three main steps: (1) ligation of a double-stranded, pre-annealed DNA RT Adapter (RTA), which contains an oligo-dT overhang to anneal to poly(A)+ mRNAs; (2) optional reverse transcription, which linearizes the RNA molecule into an RNA-DNA duplex; and (3) ligation of the RNA sequencing adapter (RMX), which contains the motor protein that directs RNA molecules to the pores and regulates their translocation (Fig. 1A). Currently, there are no manufacturer-provided protocols for molecular barcoding of direct RNA sequencing data sets, which greatly improve the cost-effectiveness of certain dRNA-seq applications by combining multiple samples on the same consumable flow cell. Multiplexing would greatly benefit sequencing designs in which the required number of reads per sample is low or transcriptomes are of low complexity, such as in vitro–transcribed RNA sequences, ribosomal RNAs, or viral RNA genomes.
Figure 1.

Schematic overview of the direct RNA barcoding and demultiplexing strategy. (A) Overview of Oxford Nanopore library preparation protocol for native RNA sequencing. (B) Adaptation of A to include custom DNA barcodes. (C) Barcode segmentation and transformation, where the electric current associated with a barcode adapter (highlighted in red) is extracted and converted into an image using GASF transformation. (D) Deep learning is used to classify the segmented and GASF-transformed squiggle signals into their corresponding bins, without the need of base-calling the underlying sequence. The convolution architecture of the final residual neural network classifier (ResNet-20) described in this work: FC = fully connected layer.
To overcome these limitations, here, we provide a novel experimental protocol and associated algorithm to barcode and demultiplex direct RNA nanopore sequencing data sets, which consists of classifying barcode current intensity data into images or arrays of pixels, followed by classification using a deep learning algorithm.
Results
Barcoding in vitro–transcribed RNAs with shuffled DNA adapters
We designed three custom DNA barcode adapters by shuffling the double-stranded sequence of the default ONT RTA (Fig. 1B). The three custom barcodes, together with the standard ONT RTA, were individually ligated to four distinct in vitro–transcribed RNA sequences (see Methods and Supplemental Table S1). In total, we performed five sequencing runs with the RTA and custom adapters: replicates 1 and 3 contained four unique Sequins transcripts (Hardwick et al. 2016), while replicates 2, 4, and 5 contained four unique Sequins and four unique Curlcake sequences (Liu et al. 2019), each of them ligated to a distinct barcoded adapter (Table 1). In addition, replicate 3 was spiked-in with the manufacturer-provided yeast ENO2 control strand (RCS). Each run produced between 600,000 and 1,000,000 reads, which were base-called and uniquely aligned to the reference sequences (Table 1; see also Supplemental Table S2). The reference alignments were used to empirically demultiplex the sequences, thus establishing a truth set to train the barcode classifier.
Table 1.
Mapping statistics from direct RNA sequencing runs

Extraction of barcode signals from raw FAST5 reads
Raw nanopore barcode signal data, consisting of a time series of electric current values, were extracted from the files corresponding to the uniquely mapped reads. Atomic structural differences between DNA and RNA produce conspicuously different mean current signal intensities, which can effectively be used to identify the boundaries of the proximal DNA adapter in the raw signal—a process henceforth referred to as barcode segmentation. We modified the Segmenter utility of SquiggleKit (Ferguson and Smith 2019) to create an automated workflow for barcode segmentation (termed B_roll) that targets the lower average current level of the DNA barcodes by comparing the current of a given window to the average current of the read using a sliding window. We also tested a barcode segmentation strategy that uses raw current signal smoothing followed by convolutional transformation of the data (termed B_conv) to identify major current intensity change points along the read (see Methods). We found that B_roll extracted signal at an average speed of 0.013 sec per read, while B_conv extracted signal at an average speed of 0.3 sec per read. The two methods showed high agreement in the extracted regions, with a median overlap of segmented signals of 89% (Supplemental Fig. S1A–C). Although both methods proved sufficient for training a classifier (Supplemental Fig. S1D), the B_roll method for barcode segmentation was chosen for subsequent analyses given its greater speed and recovery.
Transformation of segmented barcode signals into 2D images
We reasoned that conveying raw current signal into a higher dimension could facilitate the recognition of similar patterns in the data by employing deep learning strategies for the downstream classification. Indeed, supervised machine learning using deep convolutional neural networks (CNNs) and, in particular, deep residual neural networks (ResNets) has been shown to perform optimally for the classification of images (He et al. 2016; Pak and Kim 2017). To leverage the power of ResNet classifiers, we converted the raw signal corresponding to the extracted barcodes into an array of pixels (Fig. 1C) and used diverse image transformation strategies previously shown to be effective for subsequent CNN training and classification, including recurrence plots (RPs) (Eckmann et al. 1987), Markov Transition Fields (MTFs), Gramian Angular Difference Fields (GADFs), and Gramian Angular Summation Fields (GASFs) (Wang and Oates 2015). An example of the different image transformations for a given raw signal segment can be found in Supplemental Figure S2. GASF transformation was retained as it was found to be substantially faster at computing images than the other methods (Table 2). Furthermore, the symmetrical images that GASF produces generated slightly more accurate results than the nonsymmetrical GADF images or any of the other image transformation methods tested (Table 2). Figure 2 illustrates the conversion of segmented nanopore dRNA-seq barcode signals into GASF images that were subsequently used for deep learning.
Table 2.
Accuracy and average speed of signal to image conversions from 1000 runs

Figure 2.

Barcode segmentation and signal transformation. A randomly selected example of barcode signal segmentation (red outline) for each of the four barcodes is shown with its corresponding GASF image below. An additional five randomly selected segmented barcode signals and their corresponding GASF images are shown for each of the four barcodes. Sequencing reads were drawn from replicate 2. (GASF) Gramian Angular Summation Field.
Deep residual networks accurately classify raw signal barcodes
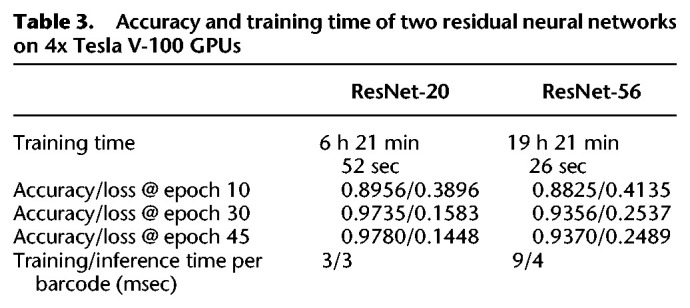
We combined sequencing data from replicates 2, 3, and 4 to train different CNN architectures using the GASF images generated from the segmented barcodes (Fig. 1C), which were previously disambiguated by aligning the base-called sequences of the ligated RNA sequenced to the reference sequence of their unique ligation templates. A total of 240k images were divided into three groups of four barcodes for training, testing, and validation at a ratio of 4:1:1, respectively (160K training:40K testing:40K withheld for validation). We compared a ResNet V2 implementation with 20 layers (ResNet-20) (see Fig. 1D) to a ResNet V2 with 56 layers (ResNet-56) for classification of transformed images corresponding to barcode signals. We found that ResNet-20 performed slightly better than ResNet-56 while being one third smaller and three times faster (Table 3).
Table 3.
Accuracy and training time of two residual neural networks on 4x Tesla V-100 GPUs
The resulting ResNet-20 model was applied to the withheld validation set to assess its accuracy. Receiving operator characteristic (ROC) analysis revealed an area under the curve of 0.998, a sensitivity of 98.9% and a false positive rate of 0.3% at maximal accuracy (99.4%) (Table 4; see also Fig. 3A–C), suggesting that the ResNet-20 model is highly accurate but might be potentially overfitted to the input, despite the latter being composed of three independent sequencing data sets.
Table 4.
Accuracy and recovery of ResNet-20 on the testing set, validation set, and two independent replicates
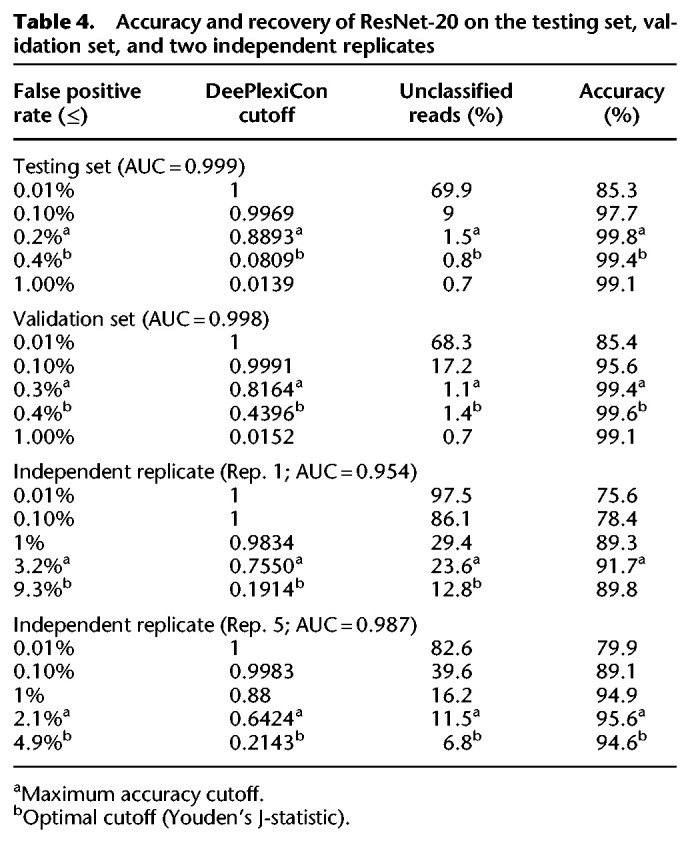
Figure 3.

Performance of 2D convolutional neural network barcode classifier. (A) Receiving operator characteristic (ROC) analysis and area under the curve (AUC) metrics of the final model on three evaluation sets: (1) Replicates 2–4 validation set (left column), which was generated from the same sequencing runs used to train the model but were withheld from training; (2) Replicate 1 set (middle column), composed of reads generated using the RNA001 library kit; and (3) Replicate 5 set (right column), derived from an independent sequencing run using the RNA002 kit. Optimal Youlden index (J statistic) is marked as a black cross on the ROC curve. (B) The associated precision recall curves on the three test sets. (C) Accuracy (black) and percentage of reads recovered (blue) in function of the scoring threshold (cut-off) emitted by the trained model, for three different data sets presented in A.
To further evaluate the model's accuracy and assess potential overfitting, we applied the model onto two additional independent biological replicates (Table 4; see also Supplemental Table S3), not used during algorithm training or testing. The global accuracy of demultiplexing was slightly lower than the other replicates, with AUC values of 0.954 and 0.987, respectively (Fig. 3A). These decreased AUC values suggest that the ResNet-20 model may indeed be slightly overfitted to the sequencing data used for training but nonetheless remains highly accurate at classifying reads from independent sequencing runs generated with different chemistries (RNA001 and RNA002; see Discussion).
Discussion
In the last decade, third-generation sequencing technologies have emerged as powerful methods to comprehensively study the (epi)transcriptome (van Dijk et al. 2018). In contrast to second-generation sequencing technologies, TGS is not limited by read length, and consequently, does not require prior fragmentation of the RNA or cDNA molecules, providing transcriptome-wide maps of full-length molecules.
In 2017, the direct RNA sequencing technology appeared, making it possible for the first time to sequence native RNA molecules. Moreover, this technology could also identify chemical RNA modifications present in the native RNA molecules (Garalde et al. 2018; Leger et al. 2019; Liu et al. 2019; Smith et al. 2019), as well as estimations for their poly(A)-tail lengths (Krause et al. 2019; Workman et al. 2019). However, two major caveats of dRNA-seq are the lack of multiplexing options and the large amount of poly(A)-selected RNA material that is needed, that is, typically 500 ng of poly(A)+ RNA. In this regard, pooling samples via multiplexing in the same flow cell would allow this technology to be applied to situations where the amount of input RNA is limiting, and it would decrease the sequencing cost per sample.
In contrast to dRNA-seq libraries, ONT does offer barcoding strategies for cDNA libraries, which rely on direct ligation of DNA adapters to the cDNA sequences. In this scenario, both the barcode and the cDNA sequence can be easily base-called under a DNA model. However, this is not possible in the context of RNA sequencing kits, as the adapter is DNA and, therefore, the DNA adapter cannot be properly base-called under an RNA model. Alternatively, one could base-call the DNA adapter using the DNA model; however, this is not straightforward because the translocation speed of RNA reads (70 bp/sec) differs from that of DNA reads (450 bp/sec).
Here, we propose a novel strategy to barcode and efficiently demultiplex dRNA-seq data (Fig. 1B). Our strategy does not require additional ligation steps compared to the standard direct RNA sequencing library preparation, as it relies on the use of shuffled DNA oligonucleotides that are incorporated during the first ligation step. To demultiplex the dRNA-seq libraries, we employ deep convolutional neural networks which are able to demultiplex dRNA-seq reads without the need of base-calling. Specifically, our strategy relies on conversion of the barcoded DNA adapter region into images, which are fed onto the trained CNNs to determine the underlying barcode. The DNA barcodes do not appear in the base-called FASTA sequence, but their electronic signal is present in the raw FAST5 sequencing data, which is used as input for our demultiplexing algorithm. Thus, demultiplexing is performed via a two-step process: (1) the transformation of raw FAST5 signals into images using Gramian Angular Summation Field (GASF), followed by (2) classification using a deep residual neural network learning model (Fig. 1; He et al. 2016). We demonstrate that our proposed methodology and algorithm is a highly effective strategy to multiplex direct RNA sequencing reads, yielding 99.9% specificity, while recovering 60% of the reads, or 95.1% specificity with 93% of read recovery, if enhanced recovery is preferred. Lower accuracy but increased recovery may be desired for specific applications; the user can choose to increase the recovery rate at the expense of accuracy if desired (Table 4; see also Supplemental Table S3).
CNNs have been widely used in signal and time-series analysis problems, including speech recognition and electrical and optical signal coding-decoding (Ismail Fawaz et al. 2019). Compounding this fact, many of the recently developed DNA base-callers for nanopore signals rely on the use of CNNs, such as DeepNano (Boža et al. 2017), DeepSignal (Ni et al. 2019), or Chiron (Teng et al. 2019). Similarly, previous efforts have shown that nanopore DNA barcodes can be correctly classified using 1D CNNs, using a tool called DeepBinner (Wick et al. 2018). Here, we employ 2D CNNs, which are widely used in computer vision and pattern recognition (LeCun et al. 2015), for direct classification of raw current intensity signals. Using this strategy, we correctly classified 84% of reads at 99% specificity (Table 3), which corresponds to 96.5% precision (positive predictive value) and 94.9% accuracy. The performance of DeePlexiCon is comparable to the signal-based DNA demultiplexing algorithm DeepBinner, which displays slightly higher sensitivity and precision (92% and 98.5%, respectively) (Wick et al. 2018). In an attempt to compare DeepBinner (1D CNN) to DeePlexiCon (2D CNN), we recreated the code from the GitHub DeepBinner repository (Wick et al. 2018) and trained this network on our dRNA-seq data. Using the same training and test set data (see Methods), we found that DeepBinner's 1D CNN achieved 61.4% accuracy, whereas DeePlexiCon achieved 94.2% accuracy (Supplemental Fig. S3). Thus, we conclude that 2D CNNs are best suited for the classification of barcodes from dRNA-seq runs; however, it is possible that future solutions better than ours will rely on the use of 1D CNNs.
Although DeePlexiCon is well-suited for multiplexing up to four samples on the same flowcell, there is room for future improvement. Firstly, barcodes could be increased in length, which may improve the accuracy of the algorithm due to a larger amount of discriminative information. Here we designed the barcodes by shuffling the paired region of oligoA and oligoB (20 bp), with some additional constraints to minimize ligation bias across barcodes (see Methods). One possible improvement would be to increase the length of the barcode, for example, up to 40 bp, in a similar fashion to the longer barcodes that are typically employed in ONT DNA multiplexing. Secondly, barcode sequences could be redesigned to maximize the differences between current intensity signals derived from the barcodes. Lastly, it is possible to train new models with additional barcodes to increase the level of multiplexing using the methodology and DeePlexiCon software described herein.
The barcodes used in this work were designed such that: (1) the same nucleotide was maintained in the 5′ end in all four barcodes to minimize ligation efficiency differences across barcodes, and (2) the nucleotide content of the annealed region between A and B was maintained across the four barcodes to ensure that the melting temperature of the four oligonucleotides was the same (see Methods). Biases in ligation efficiency are known to be sequence-dependent and are heavily affected by the identity of the 3′ and 5′ nucleotides that are being ligated. We tried to alleviate this known bias by designing barcodes that had the same 5′ nucleotide as the original ONT adapter (in this case, “G”). Thus, all sequences ligated have an “A” (from the poly(A) tail) at their 3′ end and a “G” at their 5′ end (the barcode). We should note, however, that this design will alleviate ligation bias, but we cannot rule out its existence. Nevertheless, even in a scenario with bias in ligation efficiency, this will only lead to a slight difference in the proportion of reads represented by each barcode but not in the sequencing results per se.
We should note that, in the library preparation, replicate 1, which was one of the two data sets used for independent validation of the demultiplexing accuracy (Table 4), was loaded onto a R9.5 flowcell, which bears a modified nanopore protein optimized for rapid adapter uptake, whereas the remaining replicates were loaded onto R9.4 flowcells (Supplemental Table S2). Although observed sporadically in other sequencing runs, replicate 1 revealed an increased frequency of spurious (equal barcode assignment probabilities), chimera (multimapping reads), and dual barcode ligations (false-false positive assignments evidenced by visual and algorithmic confirmation of dual barcodes in the raw signal), which may explain the lower—yet reasonable—accuracy for this sample (Supplemental Fig. S4). The presence of multiple barcodes in a read might occur due to free-floating adapters in solution in conjunction with minimal time between the first adapter/barcode passage, and the next, with a true read attached. However, this may also be due to the lack of clear open pore signal, causing MinKNOW to miss the segmentation and thus produce a single FAST5 file with both events included. Nonetheless, DeePlexiCon was able to demultiplex the sample with respectable accuracy (92%–96%), demonstrating the power of deep learning for disentangling noisy data. The ability to barcode and accurately demultiplex direct RNA sequencing reads opens new avenues to enable nanopore native RNA sequencing of samples with limited RNA availability. Moreover, it improves the cost-effectiveness of sequencing low diversity samples, such as target-enriched or in vitro–transcribed libraries.
In the last few years, deep learning has gained a lot of attention for the analysis of biomedical data. Here, we exploit the fact that residual neural networks (a type of convolutional neural network) excel at image classification by converting raw signal into abstract images. We describe the conversion of raw nanopore current intensities into images for pattern recognition and apply it for the purpose of dRNA-seq demultiplexing. The resulting model is sufficiently powerful that it can classify reads from distinct library preparation chemistries. The ability to extract biologically meaningful information from nanopore raw signals without base calling can be employed to tackle a large variety of biological problems, such as rapid binning of metagenomic samples. We hope our work will provide grounds for the development of base-calling-free classification of DNA or RNA sequences produced using nanopore sequencing.
Methods
Synthetic sequences
Curlcake sequences (Liu et al. 2019) were ordered from General Biosystems. Curlcake plasmids were double-digested overnight with EcoRV-BamHI-HF. Sequin plasmid constructs (R2_117_1, R2_63_3, R1_103_1 and R1_81_2), used commercially for RNA sequencing experiments as a spike-in control (Hardwick et al. 2016) were a kind gift from Dr. Tim Mercer (https://www.sequinstandards.com/). Sequin plasmids were digested overnight with EcoRI-HF. After digestion, DNA was extracted with phenol-chloroform followed by ethanol precipitation. Plasmid digestion was confirmed by agarose gel (Supplemental Fig. S5A). Digestion product quality was assessed with NanoDrop before proceeding to in vitro transcription.
Barcode design
To incorporate barcodes into the direct RNA sequencing library preparation without additional ligation steps, we redesigned oligonucleotide A (5′-/5Phos/GGCTTCTTCTTGCTCTTAGGTAGTAGGTTC-3′) and oligonucleotide B (5′-GAGGCGAGCGGTCAATTTTCCTAAGAGCAAGAAGAAGCCTTTTTTTTTT-3′), which are employed in the first ligation step of the direct RNA sequencing protocol. The barcodes were designed by shuffling the underlined regions of oligoA such that (1) the same nucleotide was maintained in the 5′ end—in this case, G—in all four barcodes, to minimize ligation efficiency differences across barcodes, and (2) the nucleotide content of the annealed region between A and B (underlined above) was maintained across the four barcodes, to ensure that the melting temperature of the four oligonucleotides was the same. Similarly, we chose to not change the barcode length of the RMX adapters to ensure that the clean-up steps would work as efficiently as in the original direct RNA sequencing protocol. The FASTA sequences corresponding to each barcoded oligonucleotide A and B pairs used in this work to obtain barcoded direct RNA sequencing libraries can be found in Supplemental Table S1.
In vitro transcription, capping, and polyadenylation
Using 1 µg of purified digestion product as starting material, Curlcake in vitro–transcribed (IVT) sequences were produced using the AmpliScribe T7-Flash Transcription kit (Lucigen ASF3507). Sequin IVT sequences were produced using SP6 Polymerase (NEB M0207S), following the manufacturer's recommendations. Each IVT reaction was incubated for 4 h at 42°C for Curlcake sequences and at 40°C for Sequin sequences. In vitro–transcribed RNA was then incubated with Turbo DNase (Lucigen) for 15 min, followed by purification using the RNeasy Mini kit (Qiagen 74104). Correct IVT product lengths for Sequins were confirmed using Bioanalyzer (Supplemental Fig. S5B). Each IVT product was 5′-capped using Vaccinia Capping Enzyme (NEB M2080S) following the manufacturer's recommendations. The capping reaction was incubated for 30 min at 37°C. Capped IVT products were purified using RNA Clean XP beads (Beckman Coulter A66514). Curlcake IVT products were poly(A)-tailed using the Escherichia coli Poly(A) Polymerase kit (NEB M0276S), following the manufacturer's recommendations. Poly(A)-tailed RNAs were purified using RNA Clean XP beads. Correct IVT product lengths for Curlcakes were confirmed using TapeStation (Supplemental Fig. S5C). Concentration of IVT products was determined using Qubit Fluorometric Quantitation and purity was measured with a NanoDrop 2000 Spectrophotometer (Supplemental Table S4).
Direct RNA library preparation and sequencing
Custom RT adaptors (IDT) were annealed in following conditions. Oligo A and B were mixed in annealing buffer (0.01 M Tris-HCl at pH 7.5, 0.05 M NaCl) to the final concentration of 1.4 µM each in a total volume of 75 µL. The mixture was incubated at 94°C for 5 min and slowly cooled down (−0.1°C/sec) to room temperature. An RNA library for direct RNA sequencing (SQK-RNA001 for replicates 1 and 2; SQK-RNA002 for replicates 3, 4, and 5) was prepared following the ONT Direct RNA Sequencing protocol (version DRS_9026_v1_revP_15Dec2016 for replicates 1 and 2; DRS_9080_v2_revI_14Aug2019 for replicates 3, 4, and 5).
For replicates 2, 3, 4, and 5, 500 ng total of each IVT product (four Curlcakes and/or four Sequins, as described in Table 1) were individually ligated to pre-annealed custom RT adaptors (Supplemental Table S2) in four separate eppendorfs, using concentrated T4 DNA Ligase (NEB M0202T) and were reverse-transcribed using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific 18080044). The products were purified using 1.8X Agencourt RNAClean XP beads (Thermo Fisher Scientific NC0068576), washing with 70% freshly prepared ethanol. In total, 50 ng of reverse-transcribed RNA from each reaction was pooled, and RNA Adapter (RMX), composed of sequencing adapters with motor protein, was ligated onto the RNA:DNA hybrid. The mix was purified using 1X Agencourt RNAClean XP beads, washing with wash buffer twice. The sample was then eluted in elution buffer and mixed with RNA running buffer prior to loading onto a primed R9.4.1 flowcell (replicates 2,3,4, and 5) or R9.5 flowcell (replicate 1); the samples were run on either a GridION (replicates 1 and 3) or MinION (replicates 2, 4, and 5) sequencer for 48 h or less (until all pores were inactive).
For replicate 1, library preparation steps were mainly performed as described above but with slight variations. Specifically, the pooling of barcoded samples was performed after the ligation step with pre-annealed custom RT adaptors, prior to reverse-transcription. This strategy was discarded for the subsequent replicates, as we considered that there could be potential cross-ligation of barcodes and IVT products if the pooling was performed prior to clean-up.
Base calling, mapping, and organization of sequencing data
Reads were base-called with Guppy version 3.1.5 on a GPU-enabled Sun Grid Engine high-performance computing server (parameters “‐‐chunks_per_runner 1500 ‐‐gpu_runners_per_device 1 –cpu_threads_per_caller 4 -x “cuda:0 cuda:1 cuda:2 cuda:3” -r” and configuration “rna_r9.4.1_70bps_hac.cfg”. Base-called reads (FASTQ) were aligned to Sequin transcripts (R2_117_1, R2_63_3, R1_103_1 and R1_81_2) (Hardwick et al. 2016) in replicate 1, and to both Sequin and ‘Curlcake’ constructs (CC1, CC2, CC3, and CC4) in replicate 2, using minimap2 (Li 2018) with v.2.17-r943-dirty with parameters “-k 14 ‐‐secondary = no”. Reference FASTA sequences used to map both Sequin and Curlcake reads can be found in Supplemental File S1. Mapped reads were filtered for unique targets and mapping quality (MAPQ=60), quantified, and binned into four groups based on the ligated sequence against which they mapped, and the associated raw signal data was extracted using the fast5_fetcher and SquigglePull modules from the SquiggleKit package (Ferguson and Smith 2019). The resulting tab delimited files were used as input for barcode segmentation, that is, identifying and extruding the signal associated with DNA adapter barcodes.
Extraction (segmentation) of raw signal associated with barcodes
Barcode segmentation from raw signal was performed using two strategies. The first strategy, which we term B_roll, calculates the global mean of the signal over a rolling window (2000 signal points) and identifies DNA barcode edges by setting a threshold of the mean, relative to the standard deviation. This strategy was performed by running the dRNA_segmenter.py script from SquiggleKit, with default parameters (Ferguson and Smith 2019). The second strategy, which we term B_conv, consisted in applying the discrete convolution operation of the numpy Python package (van der Walt et al. 2011) to smooth the unidimensional signal data and manifest large shifts in the data, which facilitates the identification of boundaries delimiting the different sections of the sequencing read. The second derivative of the convolved signal was calculated using a rolling window of 1001 points by applying the Savitzky-Golay filter (Savitzky and Golay 1964). Maximal absolute values of derivatives were considered as the most likely location of boundary signal points, that is, adapter start and end points. Mean and standard deviation of the current intensities were considered to further refine the boundaries. The raw signal comprised between the two boundary points, identified by either strategy, was used as input for the following steps. The efficiency and accuracy of both methods was assessed by visually inspecting 100 start and stop sites in the segmentation output of both methods. A comparative analysis of the segments obtained using either B_conv or B_roll is shown in Supplemental Figure S1. We also examined the robustness of our algorithm toward inaccuracies in the segmentation. We found that small shifts, trims, or extensions (150 data points) of the barcode segment did not significantly affect the accuracy and/or recovery of the algorithm, with only a very slight decrease (1%) in accuracy in the case of trimming the signal (Supplemental Fig. S6).
Signal transformation and deep learning
The extracted raw signals were converted into 2D images using the Python PyTS package (https://zenodo.org/record/2561773). We implemented a model training method in Python that employs Tensorflow, Keras, Scikit, Pandas, PyCM, and PyTS libraries (Supplemental Table S5; Hunter 2007; McKinney 2010; Pedregosa et al. 2011; van der Walt et al. 2011; Abadi et al. 2016; Gulli and Pal 2017; Haghighi et al. 2018). Keras implementations of ResNet-20 and ResNet-56 were slightly modified to support multi-GPU training, to adjust the learning rate scheduler, and to limit the channels to one and outputs to four classes (see Jupyter notebook in git repository v1.0.0 release source code). To drastically increase the speed of training, we employed Keras multi-GPU processing with Tensorflow-1.32. A Jupyter notebook presenting all commands used for the ResNet training protocol is available in the accompanying GitHub repository (release v1.0.0). Training was performed on a server with 4x NVIDIA V100 GPUs with 16GB memory each using NVLink. Statistics on the demultiplexing accuracy for each barcode separately can be found in Supplemental Table S3.
Comparison of image transformation methods
To compare how the choice of image transformation method affects the accuracy of prediction of the algorithm, we trained the ResNet-56 with 32,000 barcodes from replicates 2 to 4 as a training set and 8000 barcodes from replicates 2 to 4 as a test set. Specifically, we compared the following 2D image transformation methods: (1) recurrence plot; (2) Markov Transition Field; (3) Gramian Angular Summation Field; and (4) Gramian Angular Difference Field. Comparative performance of the accuracy of the algorithm using distinct image transformation methods can be found in Table 2. Comparative results of per-image processing time required by each image transformation method can also be found in Table 2.
Implementation of DeepBinner for RNA barcodes and comparison to DeePlexiCon
To compare the performance of a 1D CNN to a 2D CNN for classification of barcodes in dRNA-seq data sets, we have recreated the DeepBinner (Wick et al. 2018) code from the DeepBinner repository (https://github.com/rrwick/Deepbinner) and trained this network using our data. Comparative performance using the same training and test set data can be found in Supplemental Figure S3.
Performance evaluation
ROC and precision metrics were computed using the ROCit package in R (R Core Team 2017). Code for performance evaluation is accessible at GitHub (https://github.com/Psy-Fer/deeplexicon/blob/master/benchmarking/metrics_plots.R). We should note that performance of demultiplexing can only be assessed from those reads that have been mapped. While barcodes can be predicted from raw FAST5 reads—not only from mapped reads—these would not be useful to assess the accuracy of the method, because unless the read is mapped, it is not possible to know whether the barcode has been correctly or incorrectly predicted.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA545820. The individual accession numbers for each MinION run are: SRR10584784 (replicate 1), SRR10584783 (replicate 2), SRR10584782 (replicate 3), SRR10584781 (replicate 4), SRR10584780 (replicate 5). Code, models, and scripts used to demultiplex direct RNA reads, including benchmarking scripts, can be found at GitHub (https://github.com/Psy-Fer/deeplexicon). Additional documentation on how to use DeePlexiCon and build barcoded libraries can be found in a GitHub Page (https://psy-fer.github.io/deeplexicon). All code, models, and scripts used in this work can be found in the stable release at GitHub (https://github.com/Psy-Fer/deeplexicon/releases/tag/v1.1.0), as well as in Supplemental Code.
Competing interest statement
M.A.S., J.M.F., and E.M.N. have received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences. Otherwise, the authors declare that the submitted work was carried out in the absence of any professional or financial relationships that could potentially be construed as a conflict of interest.
Supplementary Material
Acknowledgments
We thank Dr. Tim Mercer for providing us with the sequin plasmids that have been used in this work. O.B. is supported by an international PhD fellowship (University International Postgraduate Award) from the University of New South Wales. M.C.L. is supported by Centre for Genomic Regulation (CRG) International PhD Fellowships Programme. E.M.N. was supported by a Discovery Early Career Researcher Award fellowship from the Australian Research Council (DE170100506) and is currently supported by CRG Severo Ochoa Funding. This work was funded by the Australian Research Council (DP180103571). We acknowledge the support of the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) to the European Molecular Biology Laboratory partnership, Centro de Excelencia Severo Ochoa and ‘Centres de Recerca de Catalunya’ Programme/Generalitat de Catalunya.
Author contributions: M.A.S., T.E., J.M.F., and H.L. performed the bioinformatic analysis of the data and developed demultiplexing pipelines. T.E. designed and performed all deep learning models. M.C.L., O.B., and L.B. prepared the synthetic RNAs. M.C.L., O.B., L.B., and K.B. prepared the direct RNA libraries and ran the sequencing. M.A.S. and E.M.N. conceived the project. M.A.S. and E.M.N. supervised the project. M.A.S. and E.M.N. wrote the manuscript with assistance from all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.260836.120.
References
- Abadi M, Barham P, Chen J, Chen Z, Davis A, Dean J, Devin M, Ghemawat S, Irving G, Isard M, et al. 2016. Tensorflow: asystem for large-scale machine learning. In Proc. of the Twelfth USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), pp. 265–283, Savannah, GA. [Google Scholar]
- Ardui S, Ameur A, Vermeesch JR, Hestand MS. 2018. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res 46: 2159–2168. 10.1093/nar/gky066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boža V, Brejová B, Vinař T. 2017. DeepNano: deep recurrent neural networks for base calling in MinION nanopore reads. PLoS One 12: e0178751 10.1371/journal.pone.0178751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann J-P, Oliffson Kamphorst S, Ruelle D. 1987. Recurrence plots of dynamical systems. Europhysics Letters (EPL) 4: 973–977. 10.1209/0295-5075/4/9/004 [DOI] [Google Scholar]
- Ferguson JM, Smith MA. 2019. SquiggleKit: a toolkit for manipulating nanopore signal data. Bioinformatics 35: 5372–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A, et al. 2018. Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods 15: 201–206. 10.1038/nmeth.4577 [DOI] [PubMed] [Google Scholar]
- Gulli A, Pal S. 2017. Deep learning with Keras. Packt Publishing Ltd, Birmingham, UK. [Google Scholar]
- Haghighi S, Jasemi M, Hessabi S, Zolanvari A. 2018. PyCM: multiclass confusion matrix library in Python. JOSS 3: 729 10.21105/joss.00729 [DOI] [Google Scholar]
- Hardwick SA, Chen WY, Wong T, Deveson IW, Blackburn J, Andersen SB, Nielsen LK, Mattick JS, Mercer TR. 2016. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat Methods 13: 792–798. 10.1038/nmeth.3958 [DOI] [PubMed] [Google Scholar]
- He K, Zhang X, Ren S, Sun J. 2016. Deep residual learning for image recognition. In 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV 10.1109/cvpr.2016.90 [DOI] [Google Scholar]
- Hunter JD. 2007. Matplotlib: a 2D graphics environment. Comput Sci Eng 9: 90–95. 10.1109/MCSE.2007.55 [DOI] [Google Scholar]
- Ismail Fawaz H, Forestier G, Weber J, Idoumghar L, Muller P-A. 2019. Deep learning for time series classification: a review. Data Min Knowl Discov 33: 917–963. 10.1007/s10618-019-00619-1 [DOI] [Google Scholar]
- Krause M, Niazi AM, Labun K, Torres Cleuren YN, Müller FS, Valen E. 2019. tailfindr: alignment-free poly(A) length measurement for Oxford Nanopore RNA and DNA sequencing. RNA 25: 1229–1241. 10.1261/rna.071332.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCun Y, Bengio Y, Hinton G. 2015. Deep learning. Nature 521: 436–444. 10.1038/nature14539 [DOI] [PubMed] [Google Scholar]
- Leger A, Amaral PP, Pandolfini L, Capitanchik C, Carprao F, Barbieri I, Migliori V, Luscombe NM, Enright AJ, Tzelepis K, et al. 2019. RNA modifications detection by comparative Nanopore direct RNA sequencing. bioRxiv 10.1101/843136v1 [DOI] [PMC free article] [PubMed]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Begik O, Lucas MC, Ramirez JM, Mason CE, Wiener D, Schwartz S, Mattick JS, Smith MA, Novoa EM. 2019. Accurate detection of m6A RNA modifications in native RNA sequences. Nat Commun 10: 4079 10.1038/s41467-019-11713-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney W. 2010. Data structures for statistical computing in Python. In Proceedings of the Ninth Python in Science Conference, Vol. 445, pp. 51–56, Austin, TX. [Google Scholar]
- Ni P, Huang N, Zhang Z, Wang D-P, Liang F, Miao Y, Xiao C-L, Luo F, Wang J. 2019. DeepSignal: detecting DNA methylation state from Nanopore sequencing reads using deep-learning. Bioinformatics 35: 4586–4595 10.1093/bioinformatics/btz276 [DOI] [PubMed] [Google Scholar]
- Pak M, Kim S. 2017. A review of deep learning in image recognition. In 2017 Fourth International Conference on Computer Applications and Information Processing Technology (CAIPT), pp. 1–3, Kuta Bali, Indonesia. [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. 2011. Scikit-learn: machine learning in Python. J Mach Learn Res 12: 2825–2830. [Google Scholar]
- Pollard MO, Gurdasani D, Mentzer AJ, Porter T, Sandhu MS. 2018. Long reads: their purpose and place. Hum Mol Genet 27: R234–R241. 10.1093/hmg/ddy177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang FJ, Kloosterman WP, de Ridder J. 2018. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol 19: 90 10.1186/s13059-018-1462-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Savitzky A, Golay MJE. 1964. Smoothing and differentiation of data by simplified least squares procedures. Anal Chem 36: 1627–1639. 10.1021/ac60214a047 [DOI] [Google Scholar]
- Smith AM, Jain M, Mulroney L, Garalde DR, Akeson M. 2019. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS One 14: e0216709 10.1371/journal.pone.0216709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Cao MD, Hall MB, Duarte T, Wang S, Coin LJM. 2019. Correction to: Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning. Gigascience 8: giz049 10.1093/gigascience/giz049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S, Colbert SC, Varoquaux G. 2011. The NumPy array: a structure for efficient numerical computation. Comput Sci Eng 13: 22–30. 10.1109/MCSE.2011.37 [DOI] [Google Scholar]
- van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. 2018. The third revolution in sequencing technology. Trends Genet 34: 666–681. 10.1016/j.tig.2018.05.008 [DOI] [PubMed] [Google Scholar]
- Wang Z, Oates T. 2015. Encoding time series as images for visual inspection and classification using tiled convolutional neural networks. In Workshops at the Twenty-Ninth AAAI Conference on Artificial Intelligence, Austin, TX https://aaai.org/ocs/index.php/WS/AAAIW15/paper/viewPaper/10179 [Google Scholar]
- Wick RR, Judd LM, Holt KE. 2018. Deepbinner: demultiplexing barcoded Oxford Nanopore reads with deep convolutional neural networks. PLoS Comput Biol 14: e1006583 10.1371/journal.pcbi.1006583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman RE, Tang AD, Tang PS, Jain M, Tyson JR. 2019. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nature Method 16: 1297–1305. 10.1038/s41592-019-0617-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.