

Abstract
The precise genetic diagnosis of dystrophinopathies can be challenging, largely due to rare deep intronic variants and more complex structural variants (SVs). We report on the genetic characterization of a dystrophinopathy patient. He remained without a genetic diagnosis after routine genetic testing, dystrophin protein and mRNA analysis, and short‐ and long‐read whole DMD gene sequencing. We finally identified a novel complex SV in DMD via long‐read whole‐genome sequencing. The variant consists of a large‐scale (~1Mb) inversion/deletion‐insertion rearrangement mediated by LINE‐1s. Our study shows that long‐read whole‐genome sequencing can serve as a clinical diagnostic tool for genetically unsolved dystrophinopathies.
Introduction
Duchenne muscular dystrophy (DMD) and its two allelic forms, Becker muscular dystrophy and X‐linked dilated cardiomyopathy, are caused by loss‐of‐function variants in the DMD gene. The broad spectrum of pathogenic DMD variants, ranging from single nucleotide variants to large chromosomal events, are well‐described in the literature. 1 , 2 , 3 , 4 As small pathogenic variants and large deletions/duplications almost all occur in coding and/or adjacent non‐coding regions of DMD, most DMD variants can be detected by routine genetic testing for dystrophinopathies, 3 which consists of multiplex ligation‐dependent probe amplification (MLPA) and/or short‐read sequencing of all exons and flanking regions of DMD. 3
Most undetected DMD variants after routine testing are deep intronic variants, 5 which can be identified by mRNA analysis and genomic sequencing approaches, such as Sanger sequencing and short‐read sequencing. However, some patients remain without a genetic diagnosis after the use of these methods due to the presence of very rare and more complex structural variants (SVs). These complex SVs can evade detection by short‐read sequencing or even by long‐read sequencing that lacks the read length to cover the SVs. Newly emerged genetic approaches, including next‐generation mapping 2 and long‐read sequencing, 6 have high sensitivity to detect large SVs. Here, we performed long‐read whole‐genome sequencing in a DMD patient who remained without a genetic diagnosis after routine testing, dystrophin protein and mRNA analysis, and short‐ and long‐read whole DMD gene sequencing. Finally, we identified a novel pathogenic complex SV in DMD by long‐read whole‐genome sequencing, a large‐scale inversion/deletion‐insertion rearrangement (almost 1Mb), which was mediated by long interspersed nuclear element‐1 (LINE‐1) retrotransposons. This study is a continuation of our previous work, 7 which includes a stepwise application of various genetic approaches to genetically diagnose dystrophinopathies, with the exception of the application of long‐read whole‐genome sequencing.
Methods and Results
Patient and routine genetic testing
This study was approved by the Ethics Committee at Peking University First Hospital. The patient analyzed in this study is a 9.5‐year‐old boy with clinical characteristics compatible with a DMD phenotype. He presented to Peking University First Hospital at the age of 6.5 years because of delayed motor milestones and obvious proximal muscle weakness since the age of 2 years. Physical examination confirmed that he had limb‐girdle muscle weakness, a positive Gowers’ sign, calf hypertrophy, and mild bilateral tendon contractures at 6.5‐years of age. Currently, at the age of 9.5 years, he is still able to independently walk but presents with obvious tendon contractures, toe walking, and waddling gait. Serum creatine kinase was markedly elevated in every test (range 6,510–11,896 IU/L; normal 25–170 IU/L). His muscle MRI examination showed a distinctive muscle involvement pattern, the trefoil with single fruit sign at proximal thigh level, which is highly specific for a dystrophinopathy. 8 On the basis of his DMD phenotype, we initiated routine genetic testing for DMD variants, including MLPA‐based deletion/duplication analysis of DMD and a short‐read sequencing‐based gene panel. 9 However, routine testing did not identify any causal variants. Variants in DMD were described in relation to genomic reference sequence NC_000023.10 (genome build GRCh37/hg19), coding DNA reference sequence NM_004006.2, and protein reference sequence NP_003997.1.
Dystrophin protein and mRNA analysis
As no genetic diagnosis could be confirmed by routine testing, a muscle biopsy was performed and revealed a dystrophic pattern and absent expression of dystrophin (Fig. 1), which established the molecular diagnosis of DMD in this patient. Therefore, we performed a dystrophin mRNA analysis to detect possible aberrant transcripts. We isolated mRNA from the remaining muscle tissue and amplified 22 overlapping cDNA fragments of the entire dystrophin mRNA using RT‐PCR (Supplementary Figure S1). 7 Gel electrophoresis analysis of the cDNA products showed that the 2nd–15th cDNA fragments were absent (Supplementary Figure S1C), suggestive of a possible skipping of exons 8/9/10 to 49/50/51. Further cDNA analysis confirmed the skipping of exons 8–51 from the mature mRNA (Supplementary Figure S1D), indicating a possible variant involving exons 8–51 at the genomic level. This aberrant transcript (r.650_7542del) was predicted to create a frameshift and premature termination codon occurring 33 codons downstream of exon 52 (p.Asp217Glyfs*33), which was consistent with the absent expression of dystrophin due to nonsense‐mediated decay.
Figure 1.
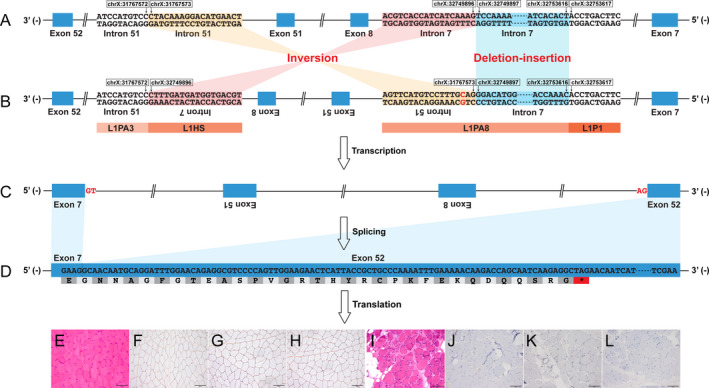
Graphic representation of the aberrant splicing of DMD caused by the complex structural variant. The complex structural variant, 982,323bp inversion flanked by 3,719bp deletion‐insertion, caused the skipping of exons 8–51 from the mature mRNA. The aberrant transcript was predicted to create a frameshift and premature termination codon, which was consistent with the absent expression of dystrophin observed on immunostaining. (A) reference genome; (B) patient genome (NC_000023.10); (C) dystrophin pre‐mRNA; (D) dystrophin mRNA (NM_004006.2). (E) and (I) hematoxylin and eosin staining (×20); (F) and (J) immunohistochemical staining for dystrophin‐N (×20); (G) and (K) dystrophin‐C (×20); (H) and (L) dystrophin‐R (×20). (E)–(H), a healthy control; (I)–(L), the patient. L1, LINE‐1.
Short‐ and long‐read sequencing of the whole DMD gene
Genomic DNA was isolated from peripheral blood and paired‐end short‐read whole DMD gene sequencing was performed as previously described 7 to search for variants that could cause the exon skipping. No potentially causative intronic variants were identified. Only one pair of reads (length 150bp) were found indicating a possible inversion involving exons 8–51, but they were not sufficiently informative for further validation (Supplementary Figure S2A). We then performed the long‐read sequencing of the DMD gene (average read length 1377bp; average read depth 191X) using a previously described protocol. 7 Ten reads with an average length of 1938bp indicated a possible inversion involving exons 8–51, g.(31767103‐31767576)_(32749380‐32749897)inv (Supplementary Figure S2B), suggesting that its breakpoints would be involved in LINE‐1s as both the genomic regions chrX:32749016‐32755046 and chrX:31767278‐31768677 were classified as a LINE‐1 annotated by RepeatMasker (http://www.repeatmasker.org/cgi‐bin/WEBRepeatMasker). However, we could only successfully validate the 3′ breakpoint region (Fig. 2C) according to the information provided by those 10 reads. The low coverage and depth of the 5′ breakpoint region was not informative enough for further Sanger validation, due in part to a possible large deletion event around the 5′ breakpoint region (Supplementary Figure S2B).
Figure 2.
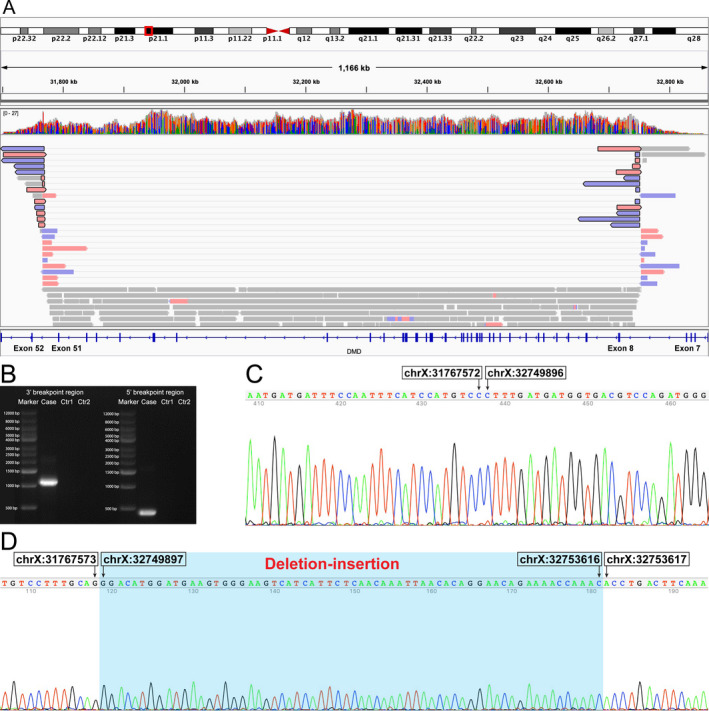
Identification and validation of the complex structural variant in DMD. (A) An integrative genomics viewer screenshot of long‐read whole‐genome sequencing showing that 13 reads indicating an inversion and a deletion event around the 5′ breakpoint region (indicated in reads with black frame). (B) Gel electrophoresis analysis of the PCR products of the genomic breakpoint regions using primers designed for the complex structural variant in the patient (positive) and healthy control (negative). Successful Sanger validation of the 3′ (C) and 5′ (D) breakpoint regions. Ctr1, a healthy control; Ctr2, a reagent control (blank).
Long‐read whole‐genome sequencing
For further precise the identification and validation of the possible inversion, we performed whole‐genome long‐read sequencing using the Nanopore PromethION (Oxford Nanopore, Oxford, UK) sequencer. Reads (average length 21,173bp; average depth 15X) were aligned to the human genome using minimap2. Small variants and SVs were called using Clairvoyante and Sniffles, respectively. 6 We found that 13 reads with the average length of 62,738bp not only indicated the possible inversion but also a deletion event around the 5′ breakpoint region (Fig. 2A). After Sanger sequencing, we finally confirmed the exact sequence of the 5′ breakpoint region and discovered that it was a deletion‐insertion event (Fig. 2D). Therefore, this complex SV, 982,323bp inversion flanked by 3,719bp deletion‐insertion, was eventually described as follows (Fig. 1): g.[31767573_32749896inv;31767575A>G;32749897_32753616delinsGGACATGGATGAAGTGGGAAGTCATCATTCTCAACAAATTAACACAGGAACAGAAAACCAAAC]; c.[650‐36206_650‐32487delinsGTTTGGTTTTCTGTTCCTGTGTTAATTTGTTGAGAATGATGACTTCCCACTTCATCCATGTCC;7543‐19710T>C;650‐32486_7543‐19708inv]. The inserted sequence at the 5′ breakpoint was completely homologous to a region in LINC02343 (BLAT homology search, chr13:34940968‐34941030, GRCh37/hg19). Both of the 400bp sequences around the 5′ and 3′ breakpoint regions were classified as a LINE‐1 annotated by RepeatMasker (Fig. 1B). This novel complex SV was absent from a healthy control (Fig. 2B and Supplementary Figure S2) and several databases as well, including the Database of Genomic Variants (DGV; http://dgv.tcag.ca/dgv/app/home), Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and Leiden Open Variation Database (LOVD; https://databases.lovd.nl/shared/genes/DMD).
Discussion
The DMD gene, spanning over 2.5Mb, is the largest gene described in the human genome, more than 99% of which is the intronic region. 10 Transposable elements that can mediate gross genomic rearrangements are relatively frequent in the intronic region of DMD. 11 , 12 The above structural complexity of DMD underlies the complex spectrum of pathogenic DMD variants. Thus, the identification of the full spectrum of DMD variants demands the application of various genetic approaches in addition to the routine testing. 1 , 3 , 4 , 11
It has been increasingly reported that deep intronic variants, the main source of the undetected variants after routine testing, can be identified via dystrophin mRNA analysis. 5 Hence, we performed dystrophin mRNA analysis in a DMD patient without a pathogenic variant identified through routine testing and found an informative aberrant splicing event. Then short‐ and long‐read whole DMD gene sequencing were performed to search for possible causal variants, which revealed a potential large‐scale inversion in DMD. However, as both breakpoints of the complex SV locate in LINE‐1s, they are hard to study via short reads, and sometimes even difficult to study via long reads that lack the read length to cover the SV and its breakpoints sufficiently. Therefore, we could not further reconstruct the complex SV according to the information provided by long‐read whole DMD gene sequencing. We ultimately validated the complex SV using longer reads obtained from long‐read whole‐genome sequencing, highlighting the significance of long‐read whole‐genome sequencing in the identification and reconstruction of large‐scale complex SVs in DMD. It is imperative to improve the detection rate and accurate reconstruction of pathogenic DMD variants for various reasons, including genetic counseling, prenatal diagnosis, and disease management in dystrophinopathies.
LINE‐1s are very common retrotransposons, typically of ~6 kb in length and occupying ~17% of the human genome. 13 If a de novo insertion of a LINE‐1 has a deleterious effect on its host gene, the LINE‐1 may be inactivated via silencing effects and accumulation of mutations. 13 Furthermore, LINE‐1s are hard to detect with the commonly used short‐read sequencing. Hence, diseases related to LINE‐1 insertions have been rarely reported. Only seven cases with dystrophinopathies caused by pathogenic LINE‐1 insertions in DMD have been reported. Four of the insertions are in exons causing dystrophinopathies by exonic disruptions, 14 , 15 , 16 , 17 one in 5′ untranslated region of DMD causing dystrophinopathies by affecting the transcription process or the stability of mature mRNA, 18 and two in introns causing dystrophinopathies by partial exonization of themselves. 7 , 11 The mutational event related to LINE‐1 identified in our case is a genomic rearrangement mediated by the retrotransposition activity of two LINE‐1s in deep intronic regions. To our knowledge, this is the first report of a LINE‐1‐meditated large‐scale complex SV in DMD shown to cause a dystrophinopathy, expanding the genetic spectrum of dystrophinopathies.
Conflict of Interest
None.
Supporting information
Figure S1. cDNA analysis of the DMD muscle transcript. (A) The entire dystrophin mRNA (NM_004006.2) was divided into 22 overlapping cDNA fragments for RT‐PCR amplification. (B) Gel electrophoresis analysis showed that the 22 overlapping cDNA fragments were successfully amplified in healthy control. (C) Gel electrophoresis analysis showed that the 2nd–15th cDNA fragments were absent in the patient, suggestive of skipping of exons 8/9/10 to 49/50/51 from the mature mRNA. (D) Further cDNA analysis confirmed the skipping of exons 8–51 from the mature mRNA in the patient. F, fragment.
Figure S2. Indications for the complex structural variant in DMD found in short‐ and long‐read whole DMD gene sequencing. (A) An integrative genomics viewer (IGV) screenshot of short‐read whole DMD gene sequencing showing that there was one pair of reads indicating a possible inversion involving exons 8–51 (indicated in the red pair‐reads; patient track). No short reads indicating possible disease‐causing structural variants were found in a healthy control track. (B) An IGV screenshot of long‐read whole DMD gene sequencing showing 10 reads (not fully presented in the IGV screenshot) indicating a possible inversion involving exons 8–51 (indicated in the colorful reads with black frame; patient track). No long reads indicating possible disease‐causing structural variants were found in a healthy control track.
Acknowledgments
The authors thank Miss Li Wang and Chao Zhou (Science and Technology, PrecisionMDx Inc., Beijing, China) for genetic data support of short‐read sequencing. This study was supported by a grant from the Beijing Municipal Science and Technology Commission (grant number Z191100006619034).
Funding Statement
This work was funded by Beijing Municipal Science and Technology Commission grant Z191100006619034.
Contributor Information
Isabelle Schrauwen, Email: is2632@cumc.columbia.edu, Email: sml3@cumc.columbia.edu, Email: drwangzx@163.com, Email: yuanyun2002@126.com.
Suzanne M. Leal, Email: is2632@cumc.columbia.edu, Email: sml3@cumc.columbia.edu, Email: drwangzx@163.com, Email: yuanyun2002@126.com.
Zhaoxia Wang, Email: drwangzx@163.com.
Yun Yuan, Email: yuanyun2002@126.com.
References
- 1. Bladen CL, Salgado D, Monges S, et al. The TREAT‐NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum Mutat 2015;36(4):395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barseghyan H, Tang W, Wang RT, et al. Next‐generation mapping: A novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med 2017;9(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kong X, Zhong X, Liu L, et al. Genetic analysis of 1051 Chinese families with Duchenne/Becker Muscular Dystrophy. BMC Med Genet 2019;20(1):4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aartsma‐Rus A, Van Deutekom JCT, Fokkema IF, et al. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading‐frame rule. Muscle Nerve 2006;34(2):135–144. [DOI] [PubMed] [Google Scholar]
- 5. Gonorazky H, Liang M, Cummings B, et al. RNAseq analysis for the diagnosis of muscular dystrophy. Ann Clin Transl Neurol 2016;3(1):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deng J, Gu M, Miao Y, et al. Long‐read sequencing identified repeat expansions in the 5′UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet 2019;758–764. [DOI] [PubMed] [Google Scholar]
- 7. Xie Z, Sun C, Liu Y, et al. Practical approach to the genetic diagnosis of unsolved dystrophinopathies: a stepwise strategy in the genomic era. J Med Genet 2020. (Accept and in press). [DOI] [PubMed] [Google Scholar]
- 8. Xie Z, Xie Z, Yu M, et al. Value of muscle magnetic resonance imaging in the differential diagnosis of muscular dystrophies related to the dystrophin‐glycoprotein complex. Orphanet J Rare Dis 2019;14(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xie Z, Hou Y, Yu M, et al. Clinical and genetic spectrum of sarcoglycanopathies in a large cohort of Chinese patients. Orphanet J Rare Dis 2019;14(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2(12):731–740. [DOI] [PubMed] [Google Scholar]
- 11. Gonçalves A, Oliveira J, Coelho T, et al. Exonization of an intronic LINE‐1 element causing becker muscular dystrophy as a novel mutational mechanism in dystrophin gene. Genes (Basel) 2017;8(10):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McNaughton JC, Hughes G, Jones WA, et al. The evolution of an intron: Analysis of a long, deletion‐prone intron in the human dystrophin gene. Genomics 1997;40(2):294–304. [DOI] [PubMed] [Google Scholar]
- 13. Kim Y‐J, Lee J, Han K. Transposable Elements: No More “Junk DNA”. Genomics Inform 2012;10(4):226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Narita N, Nishio H, Kitoh Y, et al. Insertion of a 5’ truncated L1 element into the 3’ end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J Clin Invest, 1993;91(5):1862–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Musova Z, Hedvicakova P, Mohrmann M, et al. A novel insertion of a rearranged L1 element in exon 44 of the dystrophin gene: Further evidence for possible bias in retroposon integration. Biochem Biophys Res Commun 2006;347(1):145–149. [DOI] [PubMed] [Google Scholar]
- 16. Awano H, Malueka RG, Yagi M, et al. Contemporary retrotransposition of a novel non‐coding gene induces exon‐skipping in dystrophin mRNA. J Hum Genet 2010;55(12):785–790. [DOI] [PubMed] [Google Scholar]
- 17. Holmes SE, Dombroski BA, Krebs CM, et al. A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat Genet 1994;7(2):143–148. [DOI] [PubMed] [Google Scholar]
- 18. Yoshida K, Nakamura A, Yazaki M, et al. Insertional mutation by transposable element, L1, in the DMD gene results in X‐linked dilated cardiomyopathy. Hum Mol Genet 1998;7(7):1129–1132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. cDNA analysis of the DMD muscle transcript. (A) The entire dystrophin mRNA (NM_004006.2) was divided into 22 overlapping cDNA fragments for RT‐PCR amplification. (B) Gel electrophoresis analysis showed that the 22 overlapping cDNA fragments were successfully amplified in healthy control. (C) Gel electrophoresis analysis showed that the 2nd–15th cDNA fragments were absent in the patient, suggestive of skipping of exons 8/9/10 to 49/50/51 from the mature mRNA. (D) Further cDNA analysis confirmed the skipping of exons 8–51 from the mature mRNA in the patient. F, fragment.
Figure S2. Indications for the complex structural variant in DMD found in short‐ and long‐read whole DMD gene sequencing. (A) An integrative genomics viewer (IGV) screenshot of short‐read whole DMD gene sequencing showing that there was one pair of reads indicating a possible inversion involving exons 8–51 (indicated in the red pair‐reads; patient track). No short reads indicating possible disease‐causing structural variants were found in a healthy control track. (B) An IGV screenshot of long‐read whole DMD gene sequencing showing 10 reads (not fully presented in the IGV screenshot) indicating a possible inversion involving exons 8–51 (indicated in the colorful reads with black frame; patient track). No long reads indicating possible disease‐causing structural variants were found in a healthy control track.