

Abstract
Objective
To investigate the impact of rare variants underlying neurodegenerative‐related genes to familial Alzheimer’s disease (AD).
Methods
We performed targeted sequencing of 277 neurodegenerative‐related genes on probands from 75 Chinese AD families non‐carrying causative mutation of dementia genes. Rare coding variants segregated in families were tested for association in an independent cohort of 506 patients with sporadic AD and 498 cognitively normal controls. East Asians data from the Exome Aggregation Consortium (ExAC) were used as a reference control.
Results
A novel rare variant, P410S of PLD3 was found in an early‐onset AD family. LRRK2 I2012T, a causative mutation of Parkinson’s disease, was identified in another early‐onset AD family. Missense variants in ABCA7 (P143S and A1507T) and CR1(T239M) were significantly associated with familial AD (P = 0.005437, 0.001383, 0.000549), a missense variant in TREM2(S183C) was significantly associated with AD (P = 0.000396) when compared with the East Asian controls in ExAC database. A non‐frameshift variant in FUS (G223del) was frequent in AD cases and significantly associated with familial AD (P = 0.008).
Interpretation
Multiple rare coding variants of causal and risk neurodegenerative genes were presented in clinically diagnosed AD families that may confer risk of AD. Our data supported that the clinical, pathological, and genetic architectures of AD, PD, and FTD/ALS may overlapping. We propose that targeted sequencing on neurodegenerative‐related genes is necessary for genetically unclear AD families.
Introduction
Alzheimer’s disease (AD) is a kind of neurodegenerative disease characterized by progressive episodic memory loss and other multiple cognitive decline including learning, attention, orientation, and calculation. AD is the most common form of dementia, followed by vascular dementia (VD), dementia with Lewy body (DLB), and frontotemporal dementia (FTD). 1 , 2 , 3 According to the World Alzheimer Report 2018, 4 50 million people were living with dementia worldwide in 2018 and the number will more than triple to 152 million by 2050. The circumstance is more severe in low‐ and middle‐income countries where 66% of the people with dementia live. That 66% is set to rise to 71 or 72% by 2050. AD, together with other forms of dementia, has become a major social problem that seriously affects human health, especially for low‐ and middle‐income countries. AD cannot yet be cured, however, knowledge of the factors that cause AD or modify the risk of AD to delay its onset would have a massive global impact on the severe social problem.
AD can be divided into familial AD (FAD) and sporadic AD (SAD) depending on whether there is family aggregation. FAD was thought to be caused by a certain causal gene inherited inside the family following Mendel's law. Presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein (APP) involved in amyloidogenic processing have been identified as the causal genes of FAD. 5 , 6 To date, a small portion of AD families (about 500 families to date) has been identified as carrying mutations of the three causal genes (http://www.molgen.ua.ac.be/ADMutations/), leaving the genetic factors of the rest unknown. The etiology of SAD is complicated and multifactorial, including aging, sex, education, and risk genetic factors. APOE, together with more than 20 loci identified by genome‐wide association studies (GWAS), is recognized as a risk gene for SAD. 7 , 8 Those loci are common variants (MAF > 5%), and their effect on AD is assumed to be low to moderate as each locus accounts for only a small proportion of the variance in AD susceptibility, thus, they do not lead to family aggregation.
To elucidate the causative or high‐risk genetic factors of AD, studies on AD families were performed using multiple sequencing technique such as whole‐exome sequencing (WES), whole‐genome sequencing (WGS) and targeted sequencing. To date, no novel causal gene has been identified; however, a series of rare coding variants from risk genes that segregated in large AD families were identified to be causal to FAD, including R47H in TREM2, 9 rare variants in SORL1, 10 , 11 and ABCA7. 12 The rare variants above are mostly located in the functional regions that lead to amino acid changes, researchers tend to consider them as high risk that is enough to cause family aggregation of AD patients. Thus, we propose that rare damaging variants of GWAS loci and risk genes may account for a portion of FAD.
Neurodegenerative diseases are a series of diseases with insidious onset and slow progression, characterized by aggregation of abnormal proteins in neurons and other cells, including AD, Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), DLB, and FTD etc. Neurodegenerative diseases have some neuropathology, clinical and/or genetic crossover. 13 , 14 For example, Parkinson’s disease dementia (PDD) has both α‐synuclein and Aβ deposits in the brain that may co‐cause the disease onset. 15 , 16 ALS and FTD share some common causal genes such as C9orf72. 17 , 18 Recent sequencing studies on subjects of European, American, and Thailand ancestry found that pathogenic and potential mutations of FTD‐ and PD‐related genes were presented in clinically diagnosed AD patients and considered to be probably/possibly pathogenic and risk. 19 , 20 Whether mutations of genes related to neurodegenerative diseases, such as other types of dementia, PD, and ALS, contribute to risk and family aggregation of AD remains to be further investigated.
To elucidate the underlying genetic factors of familial AD non‐carrying known causative mutations, we performed targeted sequencing covering 277 candidate genes involved in neurodegenerative diseases (Table S1) in a discovery set of 75 Chinese AD families, including causative genes, previously reported risk loci and genes functional associated with AD, FTD, DLB, PD, ALS and other dementia‐related genes, such as PRNP for prion disease, NOTCH3 for VD, CSF1R for hereditary diffuse leukoencephalopathy with neuroaxonal spheroids (HDLS). Candidate causal and risk variants were further genotyped in an independent sporadic case‐control dataset. Our study will help to deepen the insight on the genetic basis of AD and provide genetic characteristics of Chinese FAD patients.
Materials and Methods
Sample selection
A total of 75 AD families, 506 sporadic AD patients and 498 cognitively normal controls from mainland China were recruited in this study (Table 1). All families selected for targeted sequencing and all sporadic AD patients for follow‐up genotyping were referred to the outpatient neurology clinics of Xiangya Hospital. For each patient, probable or definite AD diagnosis was established using the National Institute of Aging‐Alzheimer's Association (NIA‐AA) Criteria and the International Working Group (IWG‐2) criteria by two experienced neurologists. Each selected family had at least two patients suffering from AD in two generations. All families involved in this study were given a ‘Goldman score’ between 1 and 4 following Goldman’s category for pedigrees 21 where 1 is autosomal dominant inheritance that there were at least three patients in two generations with one person being a first‐degree relative of the other two, 2 is familial aggregation of three of more family members with dementia, 3 is single affected first‐degree family member with dementia (modified to give a score of 3 only if there is a history of early onset dementia within the family, i.e., <65, and 3.5 if onset above 65) and 4 is no or unknown family history. Families with known mutations in APP, PSEN1, PSEN2, GRN, MAPT, or C9orf72 were excluded from the study. Cognitively normal controls were unrelated individuals of matched geographical ancestry, age and sex, recruited from communities and the health examination center. All control individuals had scores indicative of no cognitive impairment (>26/30) on the Mini‐Mental State Examination (MMSE). The study was approved by the Ethics Committee of Xiangya Hospital, Central South University in China (equivalent to an Institutional Review Board) and performed in accordance with the approved guidelines and regulations. Written informed consent was signed by each subject.
Table 1.
Demographics of all individuals recruited in this study.
| Status | N | Mean age at onset (y) or last examination (y) | Female No. (%) | APOE ε4+(%) | |
|---|---|---|---|---|---|
| Targeted sequencing | FAD | 75 | 32% | ||
| EOAD | 40 | 51.62 ± 8.34 | 0.47 | 16% | |
| LOAD | 35 | 71.87 ± 5.72 | 0.61 | 51% | |
| Follow‐up genotyping | SAD | 506 | 72.33 ± 7.12 | 0.62 | 41% |
| NC | 498 | 70.65 ± 5.33 | 0.56 | 20% |
Targeted sequencing of neurodegenerative disease related genes in AD families
Targeted sequencing was performed in probands from 75 AD families, targeting all exonic sequence of genes including 119 dementia‐related genes, 133 PD related‐genes and 25 ALS‐related genes (the gene list can be found in Table S1).
Genomic DNA was extracted from peripheral blood leukocytes using a QIAGEN kit following the supplier’s instructions. We used NanoDrop (Thermo Fisher Scientific, Waltham, MA) and Qubit 3.0 to detect the concentration and purity of DNA. Qualified genomic DNA was then prepared for establishing a sequencing library through the following procedure including genomic DNA break by Bioruptor Pico, end‐filling, A‐tail add, joint link, and pre‐PCR reactions. The length of the DNA library fragment was measured between 220 and 320 bp by QSEP 100. The targeted DNA area was captured using the Dynabeads MyOne Streptavidin T1 magnetic beads, and then enriched through a post‐PCR reaction. Sequencing of all DNA samples occurred on Illumina HiSeq 2500. The sequencing data were then processed using the Illumina Sequence Control Software to assess the quality.
Variants that passed quality control were analyzed and selected based on the following standards: (1) rare variants with minor allele frequency <5% in the Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium (ExAc); (2) heterozygous variants located in the exon and splice‐site; (3) functional variants (missense, nonsense and frameshift). Multiple lines of computational evidence (including SIFT, Polyphen2, and Mutationtaster) were additional reference standards. Predicted pathogenic variants and rare, damaging variants shared by at least two families were validated by Sanger sequencing and then genotyped in sporadic AD cases and cognitively normal controls to determine its population frequencies and risk. We also used the East Asians data from ExAC as a reference control to compare the frequencies of the variants found in FAD.
Follow‐up genotyping in an independent case‐control dataset
A total of six rare, damaging variants with overrepresentation in AD families were genotyped in sporadic AD cases (N = 506) and controls (N = 498) using Multiplex Snapshot SNP assay. Samples were first purified and then amplified using extension primers, designed adjacent to the six variants sites, and labeled with different single fluorescent dideoxyribonucleoside triphosphate (ddNTP) added to the polymorphic site. The products were then sequenced on an ABI3730XL capillary electrophoresis instrument. GeneMapper 4.0 software (Applied Biosystems Co., Ltd., USA) was used for data analysis.
Statistics
Frequency comparisons between the case and control were made using Pearson’s χ 2 test. When the SNP numbers were fewer than 5 in a group, Fisher’s test was adopted. For repeated measures, P < 0.05/n (n = total number of SNPs in comparison) was taken as statistically significant after the Bonferroni correction. All data are presented as mean + standard deviation (SD). Statistical analysis was conducted using GraphPad Prism version 5.0.
Results
A total of 75 AD families underwent targeted sequencing. Forty of them were early‐onset AD (EOAD) with an onset age younger than 65 years, and 35 of them were late‐onset AD (LOAD) with an onset age older than 65. According to the Goldman’s criteria for pedigree, 21 45 probands had a Goldman score of 1, 17 probands had a Goldman score of 3 and 13 probands had a Goldman score of 3.5. Their APOE status is summarized in Table 1. On average, a higher frequency for the APOE ε4 allele was observed in the LOAD families and LOAD sporadic cases compared with normal controls(P < 0.05). In the targeted sequencing, a total of 263 rare coding variants were sequenced in 67 families (including 36 EOAD families and 31 LOAD families). All these variants had a high sequencing quality score with a 20× coverage of 96.86%. The average sequencing depth was 256.86. The prevalence of rare coding variants in families was not statistically associated with age at onset and Goldman score.
Identification of a novel rare variant, P410S of PLD3 gene in an early‐onset AD family
A novel rare variant c. 1228C > T, p.P410S of PLD3 gene was found in an early‐onset AD family (Fig. 1). PLD3 gene is a risk gene for AD first identified by Cruchaga et al. in 2014. 22 P410S identified in our study was absent in the Exome Sequencing Project, 1000 Genomes Project, ExAC database, and our case‐control cohort. The variant P410S was detected in an affected individual (II:3) and a subjective cognitive decline (SCD) individual (II:5), absent in two unaffected siblings (II:7 and II:8). P410S was found in the conserved region of the PLD3 protein. Pathogenicity predictions revealed that P410S was damaging using SIFT, Polyphen2, and Mutationtaster. According to the American College of Medical Genetics and Genomics (ACMG) guidelines for sequence variants, 23 p.P410S of PLD3 gene was divided as variant of uncertain significance.
Figure 1.
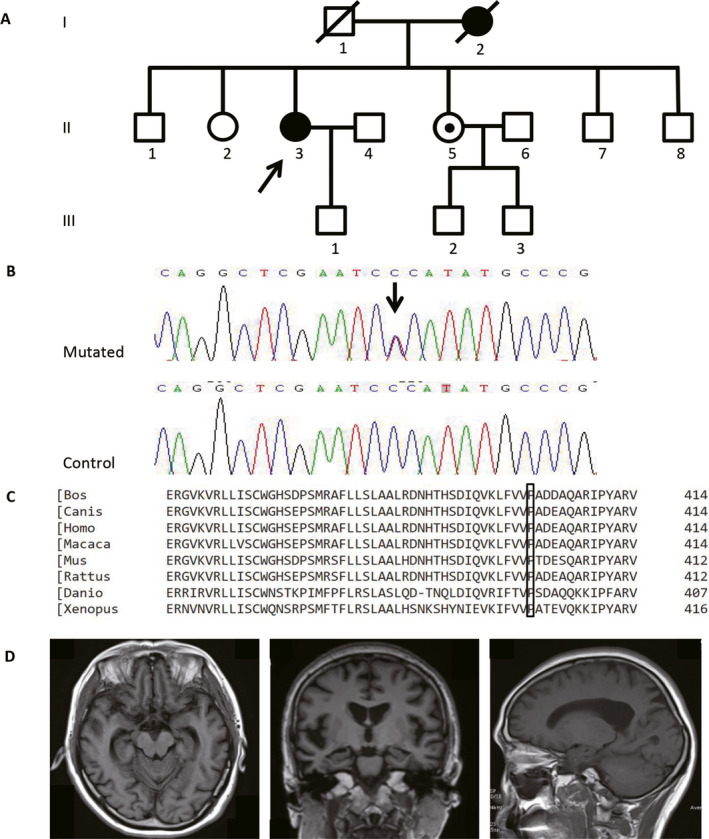
EOAD family carrying p.P410S of PLD3 gene. Note: (A) the pedigree of the EOAD family carrying p.P410S of PLD3 gene, the black arrow shows the proband. (B) sanger sequencing diagram of p.P410S variant, and the black arrow shows the mutated site. (C) conservatism of the 410th amino acid of PLD3 protein detected by ClustalW2 website. (D) brain MRI of the II:3 case showed asymmetrical atrophy in the bilateral hippocampus and mild atrophy of parietal areas.
The proband (II:3) was a 56‐year‐old woman who complained of memory impairment and personality change since the age of 53. Her neuropsychological assessment revealed MMSE 12/30 points, Montreal Cognitive Assessment (MoCA) 7/30 points, Clinical Dementia Rating (CDR) 0.5 points, Alzheimer’s Disease Assessment Scale‐Cognitive section (ADAS‐Cog) 31 points, Neuropsychiatric Inventory (NPI) 1 point and Hachinski Inchemic Score (HIS) 1 point, indicating impairment of short‐ and long‐term memory, as well as deficit of attention and executive functions. Her APOE genotype was 3/3. Cerebrospinal fluid Aβ1–42 level was 434.2 pg/mL (control values > 651 pg/mL), Aβ1–42/Aβ1–41 ratio was 0.07 (control ratio > 0.1), total tau value was 491.43 pg/mL (control values < 290 pg/mL). The brain Magnetic resonance imaging (MRI) showed asymmetrical atrophy in the bilateral hippocampus and mild atrophy of parietal areas (Fig. 1). She was diagnosed as typical AD according to IWG‐2 criteria. Her sister (II:5) was 50 years old. She complained of episodic memory loss but presented normally upon neuropsychological assessment and cranial MRI scanning. Her APOE genotype was 3/3. The status of the carrier (II:5) was uncertain now. Based on the research criteria for SCD established by the Subjective Cognitive Decline Initiative (SCD‐I), 24 we classified her as SCD that may occur at the preclinical stage of AD. We will take follow‐up observation on the phenotype of the patient.
Identification of a known causal mutation I2012T of the LRRK2 gene in an early‐onset AD family
We identified a missense mutation p.I2012T of the LRRK2 gene in an early‐onset AD family (Fig. 2). LRRK2 I2012T mutation was a causative mutation of PD, previously reported in patients with PD or FTD with parkinsonism. To the best of our knowledge, this is the first dementia case but without parkinsonism of LRRK2 I2012T mutation.
Figure 2.
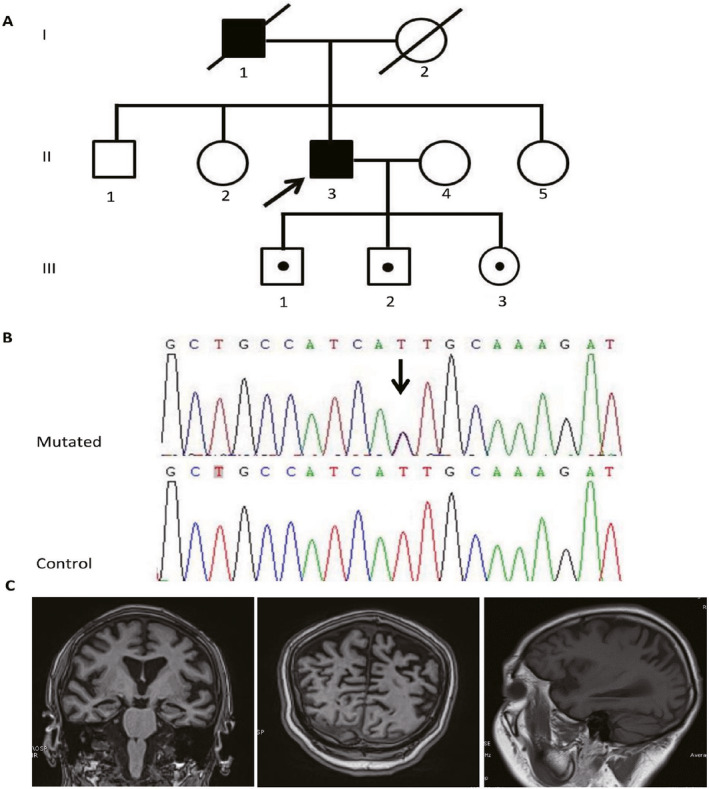
EOAD family carrying p.I2012T mutation in LRRK2 gene. Note: (A) the pedigree of the EOAD family carrying p.I2012T in LRRK2 gene, the black arrow shows the proband. (B) sanger sequencing diagram of p.I2012T mutation, and the black arrow shows the mutated site. (C) Brain MRI of the II:3 case showed mild to moderate atrophy in the bilateral hippocampus, temporal and parietal areas.
The index LRRK2 I2012T mutation carrier was a 59‐year‐old man. At 53 years of age, he presented initial symptoms with episodic memory loss and disorientation. He could not tell the name of his family members and his home address. As the dementia progressed, he got lost frequently when leaving home alone. Two years later, he became progressive apathy and fewer words. His neuropsychological assessment revealed 2/30 in MMSE, 1/30 in MoCA, 3 points in CDR and 14 points in NPI, indicating a dementia clinical course with some psychiatric symptoms. He could not finish the ADAS‐cog test. Physical examination revealed no resting tremor, rigidity, bradykinesia or postural instability. Brain MRI showed mild to moderate atrophy mainly in the bilateral hippocampus, temporal and parietal areas. His APOE genotype was 3/4. He was clinically diagnosed as probable AD according to NIA‐AA criteria. His father died of heart attack at the age of 78. The caregiver recalled that he had cognitive decline during his last 5 years. No brothers or sisters of the proband complained of dementia or parkinsonism. We further examined the p.I2012T status of the three offspring (III:1, III:2, and III:3), and found that all the three children of the proband were p.I2012T carriers. Their ages were 38, 36, and 32, respectively. The neuropsychological assessment and physical examination were all normal. We will take follow‐up observation on the three p.I2012T carriers.
Variants of AD risk genes and follow‐up genotyping
Fifty‐one rare variants of the established AD risk genes were detected in 36 probands of EOAD families and 22 probands of LOAD families (Table S2), including 17 missense variants in ABCA7, 11 missense variants in SORL1, 1 frameshift and 4 missense variants in ABCA1, 3 missense variants in CD33, 1 frameshift and 3 missense variants in CR1, 2 missense variants in BIN1 and PTK2B, and 1 missense variant in FERMT2, MS4A6A, TREM2 and CLU, respectively. Sixteen probands had a co‐occurrence of 2 or more rare variants. The prevalence of rare variants in risk genes tended to be higher in EOAD families (90%) than LOAD families (63%). No significant association was observed in co‐occurrence of rare variants with age at onset and Goldman score.
A total of six rare variants in four genes were present in two or more unrelated families, including one missense variant each in TREM2, and ABCA1, and two missense variants in CR1 and ABCA7, respectively (Table 2). We tested their association with risk of AD in an independent sporadic AD case‐control cohort. However, none of the enriched variants presented significant P‐value after adjust for age, gender and APOE status (Table 3). Since the lack of significant associations could be due to the sample size, we further compared the frequency of each variant with the East Asian allele frequencies (n = 8,628) in the ExAC database (Table 2). Three missense variants in ABCA7 (P143S and A1507T) and CR1(T239M) were significantly associated with familial AD when compared with the East Asian controls in the ExAC database after correction for multiple testing (P = 0.005437, 0.001383, 0.000549. Bonferroni‐corrected P‐value = 0.0083). A missense mutation in TREM2(S183C) was significantly associated with AD when we compared the total AD carriers with the East Asian controls in the ExAC database after correction for multiple testing (P = 0.000396, Bonferroni‐corrected P value = 0.0083) (Table 3).
Table 2.
Annotation of rare variants in AD risk genes found in at least two unrelated families.
| Gene | Position | ID | Ref | Alt | AA change | Effect | POLYPHEN2 | SIFT | Mutationtaster |
|---|---|---|---|---|---|---|---|---|---|
| TREM2 | 6‐41126454 | rs200820365 | A | T | S183C | nonsynonymous | PD | T | P |
| ABCA1 | 9‐107581120 | rs13306073 | G | A | V1096I | nonsynonymous | B | T | D |
| ABCA7 | 19‐1042325 | / | C | T | P143S | nonsynonymous | B | T | P |
| ABCA7 | 19‐1056431 | rs192694824 | G | A | A1507T | nonsynonymous | B | T | P |
| CR1 | 1‐207758129 | rs374551420 | T | C | I1363T | nonsynonymous | PD | T | P |
| CR1 | 1‐207697184 | rs199598381 | C | T | T239M | nonsynonymous | PD | T | P |
PD, Possible damaging; T, Tolerable; P, Polymorphism; B, Benign; D, Damaging.
Table 3.
Allele frequency and association test in case‐control dataset and East Asian of ExAC database.
| Gene | rsID | AA change | Number and frequency in targeted sequencing families | Number and frequency in cases | Number and frequency in controls | ExAC Frequecny in East Asian | P a, Fisher test | Corrected P a | P b, Fisher test | P c, Fisher test |
|---|---|---|---|---|---|---|---|---|---|---|
| TREM2 | rs200820365 | S183C | 3 (0.04) | 11 (0.022) | 6 (0.012) | 0.007646 | 0.328 | 0.461 | 0.021637 | 0.000396 |
| ABCA1 | rs13306073 | V1096I | 2 (0.027) | 0 | 1 (0.002) | 0.001992 | / | / | 0.01163 | 0.3394 |
| ABCA7 | 19‐1042325‐C‐T | P143S | 2 (0.027) | 1 (0.002) | 0 | 0.001284 | / | / | 0.005437 | 0.05442 |
| ABCA7 | rs192694824 | A1507T | 3 (0.04) | 2 (0.004) | 2 (0.004) | 0.002664 | 1.0 | 0.984 | 0.001383 | 0.02789 |
| CR1 | rs374551420 | I1363T | 2 (0.027) | 0 | 1 (0.002) | 0.000261 | / | / | 0.000549 | 0.02654 |
| CR1 | rs199598381 | T239M | 2 (0.027) | 0 | 1 (0.002) | 0.001861 | / | / | 0.010268 | 0.3118 |
P a, Fisher test between SAD cases and controls.
Corrected P a, P a after the adjustment of age, gender, and APOE ε4 status.
Pb, Fisher test between targeted sequencing affected carriers and East Asian in ExAC.
Pc, Fisher test between both FAD and SAD carriers and East Asian in ExAC.
(Nominally significant variants are highlighted in bold).
Variants of other neurodegenerative disease‐related genes and functionally related genes
A total of 211 rare variants were found in other neurodegenerative disease‐related genes and functional related genes (Table S3). The variants were extremely rare in the general population (max MAF = 5%). A majority of them were predicted to be deleterious. The variant G223del in FUS was shared by three FAD families (2 LOAD and 1 EOAD) and was absent in the Exome Sequencing Project, 1000 Genomes Project, and ExAC database. We further tested its association with risk of AD in our case‐control datasets and found that G223del is frequent in AD (0.04 in family cases and 0.006 in sporadic AD cases) compared with controls (0.002). After Fisher exact test, the variant was significantly associated with FAD (P = 0.008). We also detected a missense variant A104T of PARK7/DJ‐1 in 2 EOAD families. The A104T was previously detected in sporadic PD patients and reported as a risk factor for EOPD.
Discussion
Mutations of APP, PSEN1, and PSEN2 account for only a small portion of FAD, leaving most of the genetic factors for the other AD families unexplained. Recent studies identified that rare variants in several GWAS loci of AD were segregated in LOAD families and may confer risk of AD. 10 , 11 , 12 Pathogenic mutations of FTD and PD related genes were presented in dementia patients or families that formerly diagnosed of AD, 19 , 20 suggesting that rare, pathogenic variants of neurodegenerative genes other than APP, PSEN1 and PSEN2 may account for etiology of the remaining AD families. In this study, we analyzed the association of rare variants in neurodegenerative diseases genes as well as APOE in a Chinese FAD cohort. As a result, the frequency of APOE ε4 allele was higher in LOAD families, we also detected causal and risk variants that segregated within AD families, and showed strong association with AD when combined with ExAC East Asian control data and our case‐control cohort. To our knowledge, there lacked studies on thoroughly analysis of rare variants in neurodegenerative genes towards AD families in Chinese cohort. Our result indicated a potential role for rare variants from AD risk genes and other neurodegenerative diseases genes in FAD etiology.
In this study, a novel missense variant, P410S, of PLD3 was identified in an EOAD families. PLD3 was identified as a new risk gene by a next‐generation sequencing study on a large LOFAD cohort of European descent. V232M of PLD3 was found to segregate with disease status in two large late‐onset families and showed strong association with disease status in a large LOAD cohort (4,998 cases and 6,356 controls). 22 By now, multiple replication studies in different ethnicities (Belgian, Germany, French, Chinese, etc) have been performed on rare variants of PLD3, but they showed controversial association consequences with AD risk. 25 , 26 , 27 In a northern Han Chinese population study of 960 LOAD cases and 2,290 controls, the authors found the rare variants p.I163M and c.1020‐8G > A that conferred considerable risk of LOAD in their cohort; they detected a p.V232M carrier but found no association of p.V232M with the LOAD risk in their cohort, 28 which indicated that the role of PLD3 to AD risk was still unclear and that it showed strong ethnic diversity. The P410S identified in our study was detected in a dementia patient and a SCD individual in an EOAD family and absent in unaffected siblings of the family and our case‐control cohort. Due to lack of functional and population studies, P410S of PLD3 gene was divided as variant of uncertain significance according to the ACMG guideline, however, multiple lines of computational evidence indicated that P410S might be a pathogenic variant. Our result demonstrated that rare variants of PLD3 existed in EOAD family that may confer risk of EOAD as well as LOAD. The pathogenicity of P410S and the role of PLD3 in EOAD family need functional studies and large family sample studies to further investigate.
Mutations of LRRK2 is a main genetic cause for both familial and sporadic PD firstly identified by Zimprich et al. in 2004. 29 The I2012T is a causative mutation that mostly detected in Asian PD patients. 30 , 31 I2012T lies in the activation loop of kinase domain and could decrease LRRK2 kinase activity in several functional studies. 32 , 33 The clinical spectrum of I2012T mutation carriers varies widely, ranges from typical late‐onset levodopa‐responsive PD to FTD with parkinsonism. 34 To date, dementia without parkinsonism phenotype has not been reported in I2012T mutation carriers. In the study, we detected I2012T mutation of LRRK2 in an EOAD family. The proband showed a dementia clinical course with progressive episodic memory loss and psychiatry symptoms with apathy and fewer words. No typical FTD symptoms or parkinsonism were found in the proband and his relatives. Brain MRI showed mild to moderate atrophy mainly in the hippocampus, temporal and parietal areas. According to NIA‐AA criteria, the I2012T carrier was currently diagnosed as probable AD since his symptom of episodic memory loss was initial and predominant. However, because lack of pathological evidence of AD, the diagnosis of atypical FTD cannot be excluded by now. Alternatively, due to the incomplete penetrance of LRRK2 mutations, it could be that the I2012T mutation presented with low penetrance and there was a possibility of other unknown causative mutation that responsible for the EOAD family. The penetrance of I2012T has not been widely studied. However, in previous studies on other LRRK2 mutations, incomplete penetrance was presented in G2019S, 30 G2385R, R1628P, 35 and R1441C 36 which was modified by age 30 and multiple PD‐associated genetic factors. 35 Nevertheless, our study amplified the pleomorphic range of LRRK2 I2012T variants. The association of LRRK2 I2012T with dementia or AD need functional and large sample studies to confirm. Before that, we propose that screening of LRRK2 is needed in the genetic cause unknown FAD patients.
In the targeted sequencing, we detected 51 rare variants of the known AD risk gene 36 probands of EOAD families and 22 probands of LOAD families. Although not statistically significant, the prevalence of rare variants in AD risk genes were more frequent in EOAD families. We also detected several families had co‐occurrence of 2 or more variants, but we didn’t find their association with AAO and Goldman score since the pathogenesis of the rare variants was uncertain. To evaluate the pathogenesis of the rare variants of AD risk genes, we further tested their association with AD in a case‐control cohort. In the follow‐up genotyping, P143S, A1507T in ABCA7 and T239M in CR1 were significantly associated with FAD risk, and S183C in TREM2 was significantly associated with AD risk when they were compared with the allele frequency of the Asian data in ExAC. No previously reported risk variants were found in this study, suggesting region specificity and race specificity of the rare variants in AD risk. ABCA7 and CR1 are LOAD susceptibility loci identified in SNP‐based GWAS study. Common SNPs of ABCA7 and CR1 were identified as risk factors of LOAD, 8 , 37 , 38 but their effect size was much smaller than for APOE. 7 , 37 Recently, a sequencing study found that GWAS loci could also harbor rare damaging variants that were independently associated with LOAD in Caucasian and Caribbean Hispanic population. 39 In an European population study, targeted resequencing identified an intronic variant in ABCA7, 12 whereas among African‐Americans, the most strongly associated SNP is rs115550680. 40 No rare variants in CR1 have been reported to be associated with AD by now. In this study, P143S, A1507T in ABCA7 and T239M in CR1 that showed strong association were identified in Chinese FAD patients. Our study confirmed that rare coding variants of GWAS loci were existed and may confer risk of AD in Chinese population. The variants differed between people of different ethnic origins. The rare variant R47H in TREM2 was first detected with strong association with LOAD with an effect size similar to that of APOE ε4 from two NGS‐based studies. 9 The genetic association between TREM2 R47H and AD has been widely replicated. R47H was absent in our sequencing data; instead, we found that S183C in TREM2 showed a strong association with AD in this study. S183C was overrepresented in EOAD families and LOAD cases, supporting its causative role in both EOAD and LOAD.
A total of 211 rare coding variants in FTD‐, VD‐, PD‐, and ALS‐related and functionally related genes were identified in this study. Their association with AD needs association study in large cohort to elucidate. The G223del in FUS was frequent in AD cases. Through follow‐up genotyping in our case‐control cohort, it was found in three FAD families and three sporadic AD cases. By Fisher’s exact test, G223del in FUS was significantly associated with FAD. Variants in FUS have been identified as causal and risk for ALS, essential tremor, and FTD. 41 , 42 , 43 , 44 The G223del was located in the G‐rich region of FUS that is not strongly conserved in closely related organisms, suggesting it may be a benign variant. We also detected two families carrying A104T in PARK7/DJ‐1. The A104T mutation was first reported in a sporadic PD patient in 2003. 45 Previous studies on PARK7/DJ‐1 and PD supposed that mutations in the heterozygous state of parkin gene may represent risk factors. 46 As the role of A104T in PD remains unclear, the association of A104T mutation with FAD needs further large cohort and functional studies for confirmation.
In summary, of the 75 FAD families that non‐carrying mutation of causal dementia genes, we identified 2 EOAD families that carrying likely pathogenic variant of PLD3 and causative mutation of LRRK2, respectively, 15 AD families that carrying risk variants of TREM2, ABCA7, CR1, FUS, and PARK7/DJ‐1. The results here implied that multiple rare coding variants of causal and risk neurodegenerative genes were present in familial AD that may confer risk of AD. Further confirmation and characterization of these rare variants will be important for understanding the biology of AD and developing therapeutic strategies. Our data supported that the clinical, pathological, and genetic architectures of AD, PD, and FTD/ALS may be overlapping. Thus, we propose that targeted sequencing on neurodegenerative disease genes is necessary for genetic unclear AD families.
This study had several limitations. First, the AD families involved in this study were probable AD according to the NIA‐AA criteria, which were diagnosed mainly based on their clinical symptoms and brain imaging. We cannot rule out the presence of misdiagnose and comorbidities of AD and other neurodegenerative diseases due to lack of AD pathological evidence. This may limit the power of this study to demonstrate pathogenicity and risk of detected rare variants in AD. Second, in spite of 39 probands of AD families got a Goldman score of 1 that followed the autosomal dominant inheritance, the sample size of available family members was still not enough to perform linkage analysis within families. As a result, it was uncertain whether the likely pathogenic variants detected in families were responsible for family aggregation of AD phenotype. Functional study and genotype‐phenotype correlation analysis in large pedigrees with AD are needed to confirm the role of these variants. At last, the comparison with East Asian data of ExAC also limits the statistical power to establish the risk of rare variants in AD risk genes. We will amplify the sample size of local case‐control cohort to validate the potential variants in the follow‐up study.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Table S1. List of 277 neurodegenerative genes.
Table S2. Rare variants of AD risk genes.
Table S3. Rare variants of other neurodegenerative‐related genes and functionally‐related genes.
Acknowledgment
The authors thank all the subjects for participation in this study. This study was supported by the National Natural Science Foundation of China (No.81671075 to Lu Shen, No.81971029 to Lu Shen, No.81701134 to Bin Jiao), the National Key R&D Program of China (No.2017YFC0840100 and 2017YFC0840104 to Lu Shen), the Provincial Key Plan for Research and Development of Hunan (No.2017SK2031 to Lu Shen), Provincial Technology Innovation Guidance Plan Project of Hunan (No.2018SK52601 to Bin Jiao), and the Youth Program of Science Foundation of Xiangya Hospital (No.2018Q017 to Weiwei Zhang).
Funding Information
This study was supported by the National Natural Science Foundation of China (No. 81671075 to Lu Shen, No. 81971029 to Lu Shen, No. 81701134 to Bin Jiao), the National Key R&D Program of China (No. 2017YFC0840100 and 2017YFC0840104 to Lu Shen), the Provincial Key Plan for Research and Development of Hunan (No. 2017SK2031 to Lu Shen), Provincial Technology Innovation Guidance Plan Project of Hunan (No. 2108SK52601 to Bin Jiao), and the Youth Program of Science Foundation of Xiangya Hospital (No. 2018Q017 to Weiwei Zhang).
Funding Statement
This work was funded by National Natural Science Foundation of China grants 81671075, 81971029, and 81701134; National Key R&D Program of China grants 2017YFC0840100 and 2017YFC0840104; the Provincial Key Plan for Research and Development of Hunan grant 2017SK2031; Provincial Technology Innovation Guidance Plan Project of Hunan grant 2018SK52601; Youth Program of Science Foundation of Xiangya Hospital grant 2018Q017.
References
- 1. Prince M, Bryce R, Albanese E, et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 2013;9:63–75.e2. [DOI] [PubMed] [Google Scholar]
- 2. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: a collaborative study of population‐based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54:S4–S9. [PubMed] [Google Scholar]
- 3. Chan KY, Wang W, Wu JJ, et al. Epidemiology of Alzheimer's disease and other forms of dementia in China, 1990–2010: a systematic review and analysis. Lancet 2013;381:2016–2023. [DOI] [PubMed] [Google Scholar]
- 4. Alzheimer’s Disease International Consortium . World Alzheimer Report 2018.
- 5. Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature 1995;375:754–760. [DOI] [PubMed] [Google Scholar]
- 6. Goate A. Segregation of a missense mutation in the amyloid beta‐protein precursor gene with familial Alzheimer's disease. J Alzheimers Dis 2006;9:341–347. [DOI] [PubMed] [Google Scholar]
- 7. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997;278:1349–1356. [PubMed] [Google Scholar]
- 8. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol 2015;77:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pottier C, Hannequin D, Coutant S, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early‐onset Alzheimer disease. Mol Psychiatry 2012;17:875–879. [DOI] [PubMed] [Google Scholar]
- 12. Cuyvers E, De Roeck A, Van den Bossche T, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. Lancet Neurol 2015;14:814–822. [DOI] [PubMed] [Google Scholar]
- 13. Hagenaars SP, Radakovic R, Crockford C, et al. Genetic risk for neurodegenerative disorders, and its overlap with cognitive ability and physical function. PLoS One 2018;13:e0198187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tofaris GK, Buckley NJ. Convergent molecular defects underpin diverse neurodegenerative diseases. J Neurol Neurosurg Psychiatry 2018;89:962–969. [DOI] [PubMed] [Google Scholar]
- 15. Jendroska K, Lees AJ, Poewe W, Daniel SE. Amyloid beta‐peptide and the dementia of Parkinson's disease. Mov Disord 1996;11:647–653. [DOI] [PubMed] [Google Scholar]
- 16. Jellinger KA, Seppi K, Wenning GK, Poewe W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson's disease. J Neural Transm 2002;109:329–339. [DOI] [PubMed] [Google Scholar]
- 17. Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology 2008;70:144–152. [DOI] [PubMed] [Google Scholar]
- 18. Talbot K, Ansorge O. Recent advances in the genetics of amyotrophic lateral sclerosis and frontotemporal dementia: common pathways in neurodegenerative disease. Hum Mol Genet 2006;15(suppl_2):R182–R187. [DOI] [PubMed] [Google Scholar]
- 19. Fernandez MV, Kim JH, Budde JP, et al. Analysis of neurodegenerative Mendelian genes in clinically diagnosed Alzheimer Disease. PLoS Genet 2017;13:e1007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giau VV, Senanarong V, Bagyinszky E, et al. Analysis of 50 neurodegenerative genes in clinically diagnosed early‐onset Alzheimer's disease. Int J Mol Sci 2019;20:1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005;65:1817–1819. [DOI] [PubMed] [Google Scholar]
- 22. Cruchaga C, Karch CM, Jin SC, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature 2014;505:550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jessen F, Amariglio RE, van Boxtel M, et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer's disease. Alzheimers Dement 2014;10:844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schulte EC, Kurz A, Alexopoulos P, et al. Excess of rare coding variants in PLD3 in late‐ but not early‐onset Alzheimer's disease. Hum Genome Var 2015;2:14028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van der Lee SJ, Holstege H, Wong TH, et al. PLD3 variants in population studies. Nature 2015;520:E2–E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiao B, Liu X, Tang B, et al. Investigation of TREM2, PLD3, and UNC5C variants in patients with Alzheimer's disease from mainland China. Neurobiol Aging 2014;35:2422.e9–2422.e11. [DOI] [PubMed] [Google Scholar]
- 28. Tan MS, Zhu JX, Cao XP, et al. Rare variants in PLD3 increase risk for Alzheimer's disease in Han Chinese. J Alzheimers Dis 2018;64:55–59. [DOI] [PubMed] [Google Scholar]
- 29. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601–607. [DOI] [PubMed] [Google Scholar]
- 30. Healy DG, Falchi M, O'Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2‐associated Parkinson's disease: a case‐control study. Lancet Neurol 2008;7:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomiyama H, Li Y, Funayama M, et al. Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson's disease patients from 18 countries. Mov Disord 2006;21:1102–1108. [DOI] [PubMed] [Google Scholar]
- 32. Luzon‐Toro B, Rubio de la Torre E, Delgado A, et al. Mechanistic insight into the dominant mode of the Parkinson's disease‐associated G2019S LRRK2 mutation. Hum Mol Genet 2007;16:2031–2039. [DOI] [PubMed] [Google Scholar]
- 33. Cardona F, Tormos‐Perez M, Perez‐Tur J. Structural and functional in silico analysis of LRRK2 missense substitutions. Mol Biol Rep 2014;41:2529–2542. [DOI] [PubMed] [Google Scholar]
- 34. Fan TS, Wu RM, Chen PL, et al. Clinical heterogeneity of LRRK2 p. I2012T mutation. Parkinsonism Relat Disord 2016;33:36–43. [DOI] [PubMed] [Google Scholar]
- 35. Wang C, Cai Y, Zheng Z, et al. Penetrance of LRRK2 G2385R and R1628P is modified by common PD‐associated genetic variants. Parkinsonism Relat Disord 2012;18:958–963. [DOI] [PubMed] [Google Scholar]
- 36. Gosal D, Lynch T, Ross OA, et al. Global distribution and reduced penetrance: Lrrk2 R1441C in an Irish Parkinson's disease kindred. Mov Disord 2007;22:291–292. [DOI] [PubMed] [Google Scholar]
- 37. Lambert JC, Heath S, Even G, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 2009;41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 38. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011;43:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome‐wide association studies loci. Ann Neurol 2015;78:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cukier HN, Kunkle BW, Vardarajan BN, et al. ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol Genet 2016;2:e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 42. Broustal O, Camuzat A, Guillot‐Noel L, et al. FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J Alzheimers Dis 2010;22:765–769. [PubMed] [Google Scholar]
- 43. Van Langenhove T, van der Zee J, Sleegers K, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 2010;74:366–371. [DOI] [PubMed] [Google Scholar]
- 44. Merner ND, Girard SL, Catoire H, et al. Exome sequencing identifies FUS mutations as a cause of essential tremor. Am J Hum Genet 2012;91:313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hague S, Rogaeva E, Hernandez D, et al. Early‐onset Parkinson's disease caused by a compound heterozygous DJ‐1 mutation. Ann Neurol 2003;54:271–274. [DOI] [PubMed] [Google Scholar]
- 46. West A, Periquet M, Lincoln S, et al. Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet 2002;114:584–591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of 277 neurodegenerative genes.
Table S2. Rare variants of AD risk genes.
Table S3. Rare variants of other neurodegenerative‐related genes and functionally‐related genes.