

Abstract
Objective
To characterize natural history of Laminin‐α2 related muscular dystrophies (LAMA2‐RD) to help anticipating complications and identifying reliable outcome measures for clinical trial design and powering.
Methods
We conducted a retrospective, single‐center, cross‐sectional and longitudinal study on 46 LAMA2‐RD pediatric patients (37 families). Patients were seen at the Dubowitz Neuromuscular Centre, London between 1985 and 2019. Data were collected by case note reviews. Time‐to‐event analysis was performed to estimate median age at complications occurrence.
Results
Forty two patients had complete deficiency of Laminin‐α2 (CD) and four had partial deficiency (PD). Median age at first and last assessment was 2 years and 12.1 years, respectively. Median follow‐up length was 7.8 years (range 0‐18 years). Seven CD patients died at median age 12 years. One CD and two PD subjects achieved independent ambulation. We observed a linear increase in elbow flexor contractures in CD subjects. Thirty‐two CD and one PD patient developed scoliosis, nine underwent spinal surgery. Twenty‐two CD required nocturnal noninvasive ventilation (median age 11.7 years). CD subjects showed a 2.9% linear annual decline in forced vital capacity % predicted. Nineteen CD and one PD patient required gastrostomy insertion for failure to thrive and/or unsafe swallow (median age 10.9 years). Four CD patients had partial seizures. Mild left cardiac ventricular dysfunction and rhythm disturbances were identified in seven CD patients.
Interpretation
This retrospective longitudinal study provides long‐term natural history of LAMA2‐RD. This will help management and identification of key milestones of disease progression that could be considered for future therapeutic intervention.
Introduction
Laminin α2‐related muscular dystrophies (LAMA2‐RD), previously known as merosin‐deficient congenital muscular dystrophy type 1A (MDC1A), are autosomal recessive disorders caused by pathogenic variants in the LAMA2 gene (6q22–q23; OMIM*156225). 1 , 2 LAMA2 gene encodes for the alpha 2 subunit of the heterotrimeric Laminin‐2 protein (Lm‐211, also called merosin), a tissue‐specific component of the extracellular matrix expressed in skeletal muscles, Schwann cells, placental trophoblast, dermis‐epidermis junction and myocardium, 3 , 4 with a central role in the protein scaffold connecting extracellular matrix to the cell surface. 5 , 6 The clinical manifestations of LAMA2‐RD range from severe, early‐onset congenital muscular dystrophy (CMD), to a milder, later childhood‐onset limb‐girdle muscular dystrophy (LGMD). 7 , 8 , 9 , 10 This phenotypic difference is largely, but not exclusively, explained by the type of mutations and the corresponding amount of residual Lm‐211 protein in muscle. 11 , 12 With an estimated prevalence of 0.6‐0.7/100 000, 13 , 14 LAMA2‐RD is one of the most common type of CMDs. 11 , 15 , 16 , 17 , 18
While few cross‐sectional studies explored the genotype/pathology phenotype correlation, 12 , 19 , 20 , 21 long‐term retrospective or prospective natural history data are limited. As novel therapeutic approaches are getting closer to clinical application, 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 it is crucial to fully clarify the natural history, rate of progression of muscle weakness, 30 , 31 occurrence of complications and time at intervention for disease‐related complications, to help identify the most valid and reliable outcome measure for LAMA2‐RD.
In this study we provide detailed retrospective cross‐sectional and longitudinal natural history data on a large pediatric cohort of LAMA2‐RD patients addressing outstanding needs in the translational path for this rare condition.
Methods
This is a retrospective cross‐sectional and longitudinal study conducted on pediatric patients affected by LAMA2‐RD. All patients included in the study had a diagnosis of LAMA2‐RD confirmed by the presence of biallelic pathogenic variant/s in the LAMA2 gene and/or complete deficiency of laminin alpha 2 in muscle biopsies on immunohistochemistry. Patients with residual (partial) laminin alpha 2 expression (PD) and biallelic pathogenic LAMA2 variants were also included in the study. All patients were under clinical follow‐up at the Dubowitz Neuromuscular Centre (DNC) in the period between 1985 and 2019. The median year at last visit was 2011 (1st quartile, 2004; 3rd quartile 2018).
Clinical, physiotherapy, instrumental, pathological and genetic data were retrospectively collected from medical records of each individual. Collected data were recorded in an anonymized excel database. The following longitudinally collected variables were considered: weight, current motor ability, goniometry measurements of right hip flexion, knee flexion and elbow flexion, scoliosis (defined as Cobb Angle >20°), respiratory complications, spirometry measurements (Forced Vital Capacity percentage‐FVC%‐predicted and cough peak of flow –PEF‐ when available), swallowing and feeding abilities, cardiac function, epilepsy and antiepileptic treatment. For instrumental investigations, we considered observations that were consistent in subsequent visits.
Standard protocol approvals, registrations, and patient consents
The study was registered as clinical audit in agreement with local hospital trust guidelines.
Statistical analysis
Descriptive statistics were generated for all measures, mean and standard deviation were used for normally distributed data and median, IQR for skewed data. Categorical data were summarized as frequency and percentage. Pearson correlation test was used to determine linear correlation between parametric variables. Mann‐Whitney U test was used to compare median for skewed variables, Fisher Exact Test was used to compare frequencies of categorical variables.
For the longitudinal data, very few patients had been followed‐up beyond age 17 years, therefore we included only data prior to this during the pediatric phase. Kaplan‐Meier analyses were used to assess the median time to event for scoliosis, noninvasive ventilation (NIV) initiation and percutaneous endoscopic gastrostomy (PEG) positioning (with IQR range). Mixed effects regression analysis was used to assess the longitudinal trend for right elbow flexion, knee flexion, hip flexion, Cobb Angle and FVC% and estimate the annual change. A P value < 0.05 was considered significant. We used Stata and SPSS software for statistical analysis.
Results
Cohort description
The study cohort included 46 patients (25 (54%) female) from 37 unrelated families. Muscle biopsy was available for 33/46 patients. Result of genetic analysis was available for 40/46 patients. Patients were classified in two categories based on either biopsy results or combination of genotype, presentation and family history (in the absence of muscle biopsy): 1) patients with complete laminin alpha 2 deficiency (CD) and 2) patients with partial laminin alpha 2 deficiency (PD) on immune analysis. Forty‐two of 46 (91%) individuals were classified as CD patients and four as PD (9%) (Sup Table S1). Median clinical follow‐up period was 7.8 years (range 0‐18 years), ranging from one to 22 visits per patient (median seven visits). The median age at first and last assessment was 2 years and 12.1 years, respectively (range 0.1‐14.8 and 12.1‐22.7 years). Demographics, onset, investigations and comorbidities are summarized in Table 1.
Table 1.
Patients’ characteristics and total cohort description
| CD patients (n = 42) | PD patients (n = 4) | |
|---|---|---|
| Onset | ||
| Onset <1 month | 17/31* | 1/4 |
| Onset 1‐6 months | 9/31 | 2/4 |
| Onset >6 months | 5/31 | 1/4 |
| Investigations | ||
| Brain MRI Isolated WM alteration | 11/15 | 1/3 |
| Brain MRI WM + structural abnormalities | 3/15 | 1/3 |
| Normal Brain MRI | 1/15 *at 6 months | 1/3 |
| Abnormal nerve conduction | 6/13 | 0/2 |
| CK levels, IU/L, median (range) | 2665 (394‐36000) | 2318(563‐3375) |
| Prenatal features | ||
| Decreased fetal movement | 5/22 | 1/4 |
| Talipes/contractures | 5/26 | 0/4 |
| Polyhydramnios | 2/22 | 0/4 |
| Breech presentation | 1/24 | 0/4 |
| Speech delay | 2/24 | 0/4 |
| Motor delay | 42/42 | 4/4 |
| Motor attainments | ||
| Independent sitting, median in months (range) | 12 (6‐40) | 10.1 (8.1‐13.1) |
| Best independent walking | 1 | 2 |
| Best walking with support ± KAFO | 5 | 2 |
| Best standing with support | 3 | ‐ |
| Disease‐related complications | ||
| Scoliosis (median age of onset, years; range) | 36 (6.3; 1.9‐14.8) | 1 |
| Scoliosis surgery (median age, years; range) | 9 (11.6; 7.7‐13.5) | 0 |
| Hospital admission for chest infection <2 years | 15/27 | 0/4 |
| Nocturnal NIV (median age, years; range) | 22 (9.5; 0.5‐16.5) | 0 |
| Tracheostomy (age) | 1 (1.8y) | 0 |
| Feeding | ||
| PEG positioning (median age, years; range) | 19 (5, 1.7‐15.5) | 1 (5.3) |
| PEG due to failure to thrive | 10/19 | 1/1 |
| PEG due to unsafe swallow | 1/19 | ‐ |
| PEG due to unsafe swallow/failure to thrive | 5/19 | ‐ |
| PEG+ oral feeds at last follow‐up | 11/19 | 1/1 |
| Cardiac involvement | ||
| Mild LVD | 5 | 0 |
| Rhythm abnormalities | 3 | 0 |
| Severe cardiac involvement# | 0 | 0 |
| Epilepsy (median age of onset, years; range) | 4 (7.7, 5.3‐11) | 0 |
| Learning difficulties | 2 | 0 |
FU, follow‐up; KAFO, Knee ankle foot orthosis; LFD, left ventricular dysfunction; n, number; NIV, non‐invasive ventilation; PEG, Percutaneous endoscopic gastrostomy; WM, white matter; y, years. *values indicate number of patients with this finding in numerator and total of patients for which this information was available in the denominator; # defined as requiring medication.
Pregnancy and birth history
Reduced fetal movements were the most common prenatal finding in CD subjects, reported in 5/22 (23%) patients. Six of 24 CD patients (25%) were born preterm. Eight of 23 CD (35%) required C‐section and one the use of forceps. Three patients with PD had uneventful delivery and one was born by C‐section.
Onset of symptoms
Seventeen of 31 CD subjects (57%) had disease onset between birth and first month of life, 9/31 (30%) between 1 and 6 sixth months and 5/31 (13%) between 6 months and one year (Table 1). The most common symptoms at onset were hypotonia (28/28, 100%), axial weakness (in particular head flexor weakness; 15/15, 100%) and paucity of limb movements (21/23, 91% patients). At onset, feeding difficulties (i.e. poor sucking, failure to thrive) were present in 17/30 (56.7%), limb contractures or talipes were reported in 7/26 (28%), weak cry in 7/31 (22.6%), facial weakness in 4/20 (21%), and torticollis in 3/32 (10%). Respiratory difficulties were reported in 3/29 (10%). One PD patient (25%) presented clinical features at delivery. All four patients with PD had hypotonia, axial and limb weakness at onset, and one had feeding difficulties.
Gross motor function
Information on achievement of developmental milestones and gross motor function was collected retrospectively from longitudinal observations. Where early observations were not available, information was collected from historical recalls of milestones as reported in medical notes. All patients had motor delay.
Information regarding age at independent sitting was available for 20 CD and all four PD subjects. Sitting was achieved by CD patients at a mean age of 13.6 months (median 12 months; range 6‐40 months) and 10.3 months in PD patients (median 10.1 months; range 8.1‐13.1 months) (Fig. 1). Observational data indicate that additional 14 CD patients were able to sit without support when seen in clinic, and at least 11 of them lost this ability during follow‐up (Fig. 2). Eleven CD patients achieved ability to bottom shuffle and 13 ability to roll, for variable duration. Three CD patients acquired the ability to stand with support (age at sitting not available). Five CD patients acquired ability to perform steps with support (provided the use of orthoses) at median age 4.8 years (range 3.4‐8.5 years), four lost such ability after a median time of 4.6 years (range 0.7‐6.5). Age at sitting was available for two of these five CD patients, showing they were able to perform assisted steps 40 and 64 months after sitting. One CD patient (Pt 33) sat at 12 months and acquired independent ambulation at 2.7 years. At age 13.3 years, following scoliosis surgery, support for walking became necessary.
Figure 1.
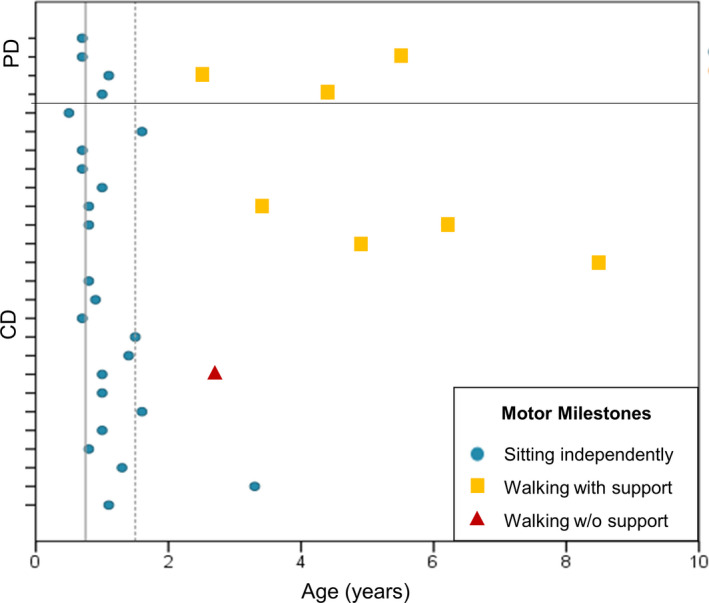
Age at attaining sitting without support and walking with or without support in 24 patients with LAMA2‐RD. Tick marks on vertical axis indicate single patients. Patients with more severe phenotype/complete merosin deficiency (CD) are included in bottom section; patients with milder phenotype/partial deficiency (PD) are in the top section. Dots: age at independent sitting. Squares: age at walking with support. Triangle: age at independent walking. The continuous vertical line at 9 months and dashed line at 18 months indicates WHO thresholds for attaining independent sitting and walking alone in 99% of children, respectively (reference: WHO Multicentre Growth Reference Study Group. WHO Motor Development Study: Windows of achievement for six gross motor development milestones. Acta Paediatrica Supplement 2006;450:86‐95). Observational information on sitting and walking are not included in this figure.
Figure 2.
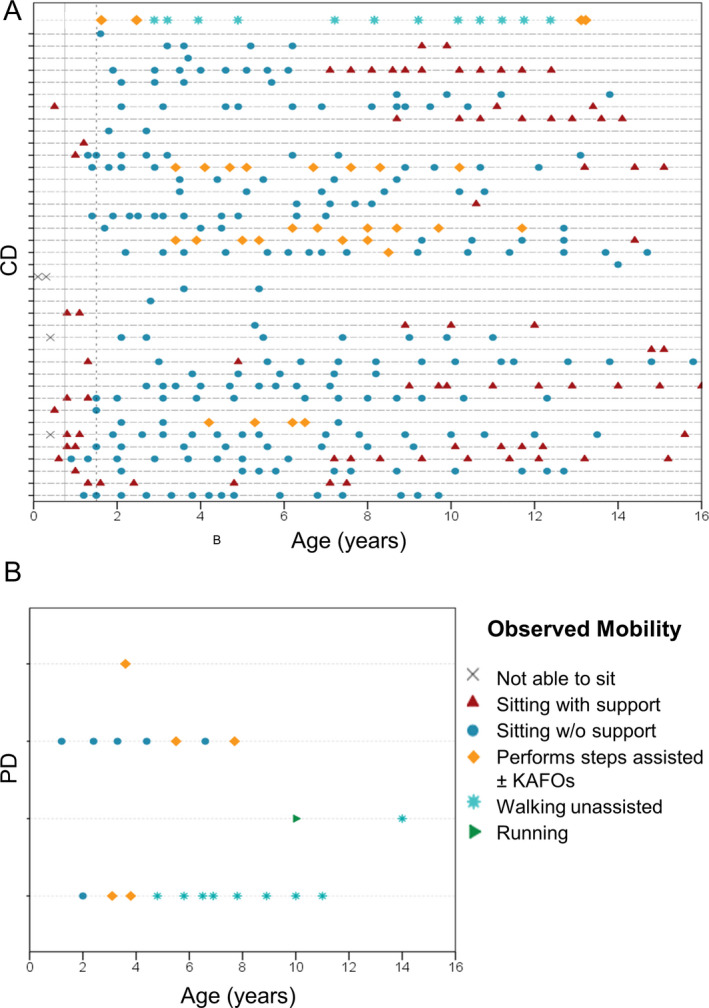
Longitudinal observational data on gross motor abilities in LAMA2‐RD patients. Each horizontal line indicates single patients. Symbol on lines indicates different assessments in each patient. Panel A: patients with severe phenotype/complete deficiency (CD). Panel B: patients with milder phenotype/partial deficiency (PD). Cross: not able to sit with support; triangle: sitting with support, circle: sitting without support; diamond: walking with support; asterisk: walking alone; arrowhead: running. The continuous vertical line at 9 months and dashed line at 18 months indicates WHO thresholds for attaining independent sitting and walking alone in 99% of children.
Two PD subjects, who sat at around age 1 year, achieved independent ambulation at age 2.5 and 4.4 years, respectively, and were ambulant at last follow‐up at age 12 and 17 years. The two remaining PD patients, last seen at age 9 and 3.6 years, were only able to walk with orthoses (data on start of ambulation not available) (Sup Table S1).
Distribution of weakness
The distribution of limb weakness followed a proximodistal gradient in both CD and PD patients. All CD patients had below gravity strength in head flexor muscles. Muscle involvement was predominant in upper limbs compared to lower limbs. In particular, 14 CD patients had lack of full antigravity movements in shoulder abduction from the first two years of life, but full antigravity strength in elbow flexion. At the time of first observation of shoulder abduction weakness, six patients had antigravity movements in lower limbs and eight had antigravity movements in knee extensors and feet dorsiflexors, but not hip flexors. Facial weakness and ophtalmoparesis were observed in 21 and 18 CD patients, respectively, but in no PD.
Orthopedic complications
Joint contractures
Goniometric measurements of right elbow flexion were collected in 34 CD and three PD subjects (174 observations), of right hip flexion in 36 CD and three PD patients (210 observations), and of right knee flexion for 37 CD and three PD patients (214 observations; Fig. 3A‐B). Longitudinal analysis of joint range progression was performed in CD subjects only. Right elbow flexion contractures >30˚ were observed in 26/34 CD (76%) patients at a median age of 6.7 years and >60˚ in 18/34 (5%) CD patients, at a median age of 8.7 years. A single ambulant CD patient (Pt 33) developed elbow flexion contractures >30˚ at 9 years and >60˚ at 12 years. No PD patient had right elbow contractures >30˚. Long finger flexor contractures were reported in 30/42 (71%) CD patients as well as 2/4 PD patients. Analysis of longitudinal data demonstrated a linear yearly increase rate of 6.6˚ for right elbow flexion (95% CI 5.5‐7.8; P < 0.001) and of 3.1˚ for knee flexion contractures (95%CI 2.3‐3.9; P < 0.001). No linear progression was identified in hip flexion.
Figure 3.
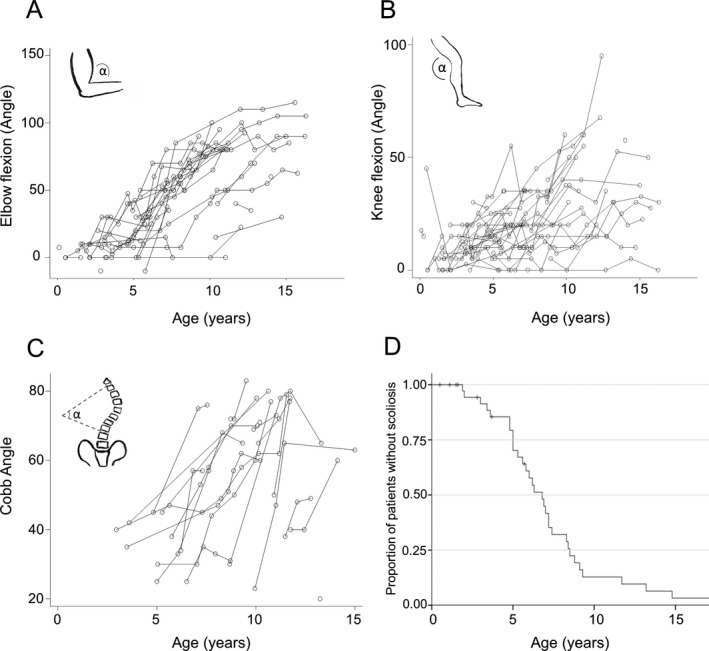
Orthopedic complications. (A‐B) Line charts representing contracture progression by age of right elbow flexion and right knee flexion, as measured by goniometric angle. (C) Line chart representing Cobb Angle° changes by age and (D) Kaplan–Meier estimates for scoliosis onset from birth. Right‐censored data (subject alive without event occurrence at last follow‐up) were indicated with vertical tick‐marks. Only CD patients were considered for analysis.
Spine deformity
Thirty‐three out of 42 CD patients and 1/4 PD patients developed scoliosis. Nine of 33 CD patients required spinal surgery at a median age of 11.6 years (range 7.7‐13.5 years). Spine X‐ray results were available for 19 CD patients (total 67 films). If we assume linearity, Cobb angle increased by 5.3˚/year (95% CI 4.0‐6.6; P < 0.001) (Fig. 3C). Time‐to‐event analysis for CD subjects gave an estimate median age at scoliosis of 6.8 years (95% CI 5.7‐7.9) (Fig. 3D).
Neck rigidity was reported in 21 patients (20 CD and one PD), spine rigidity in 15 (13 CD and two PD), and lordosis in 12 (10 CD and two PD).
Hip dislocation was reported in 12 CD and two PD patients, pectus excavatum in two CD and pectus carinatum in two CD patient.
Respiratory function
Data on hospitalizations were available for 31 patients (27 CD and four PD). Fifteen of 27 CD patients (57.7%) required a hospital admission for respiratory complications during the first two years of life. No PD patient had severe chest infections or hospitalization before the second birthday (P 0.043).
Spirometry was performed in 25 CD patients and three PD, from a minimum age of 4.8 years to a maximum of 18.8 years, for a total of 104 observations. At first spirometry evaluation (median age 8.7 years, range 4.9‐17.3), 23/25 CD patients had FVC%<60%, 14/25 had FVC% < 40%. No statistical difference in median age was observed between patients with FVC%> or <40% at first evaluation. No PD subject had FVC% <60% at baseline (age 3.3, 4.8 and 10 years, respectively) or during follow‐up. We observed a linear annual decline in FVC% predicted of 2.9% (95% CI 2.2‐3.7; P < 0.001) in CD patients (Fig. 4A).
Figure 4.
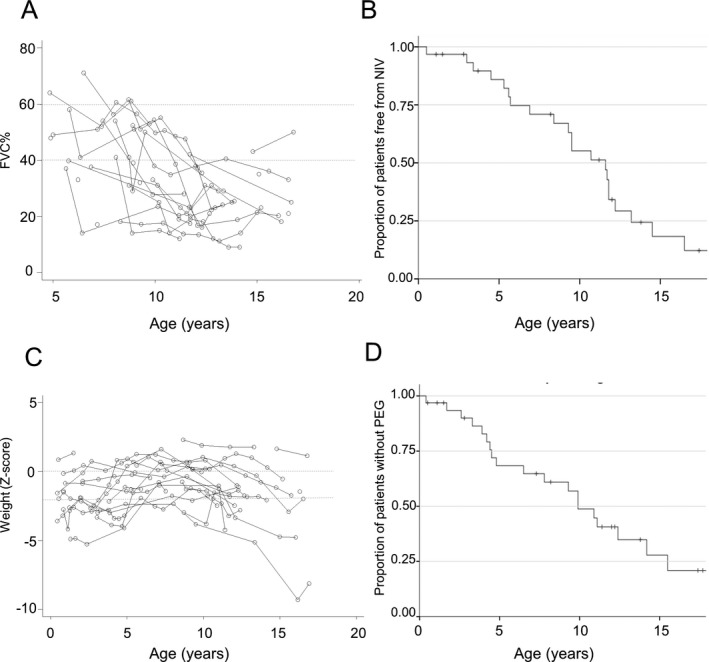
Respiratory function and feeding. (A) Spirometry results displaying progression of FVC% predicted by age in CD patients. Dotted lines indicate FVC% 60 and 40%, respectively. (B) Kaplan–Meier estimates for NIV start from birth in CD subjects. (C) Line charts representing progression of weight values represented by Z‐Scores. Z‐score is derived from normative UK population. Z‐Score 0 equals to 50th centile, Z‐score ‐1 equals to 26th centile, Z‐Score ‐2 equals to 2.5th centile, Z‐score ‐3 equals to 0.3rd centile. Dotted lines represent the 50th and 2.5 centile, respectively. (D) Kaplan–Meier estimates for gastrostomy positioning from birth in CD subjects. Abbreviations: NIV, noninvasive ventilation; PEG, percutaneous endoscopic gastrostomy.
As spirometry trajectory could represent an endpoint for future clinical trials, we calculated the sample size required to detect a 12‐month change in FVC% decline with a level of power of 80% and 5% significance. Forty‐five patients are required to detect a 50% change, 22 for a 75% change and 13 for full restoration.
A linear correlation between Cobb angle and FVC% predicted at baseline (Pearson coefficient −0.66, P = 0.003) was also observed. Cough PEF was recorded in 11 patients (eight CD, three PD), from age 5.5 years to 17.8 years, with seven patients (six CD and one PD) scoring <160 L/min, which is the absolute threshold usually considered as the indication for cough assist device in adolescents and adults with neuromuscular disorders. 32
Twenty‐two CD patients required nocturnal NIV. No patient required NIV for >16 h/d. One patient received tracheostomy at 1.8 years following respiratory failure during chest infection. Time‐to‐event analysis on NIV initiation estimates median age at start of NIV at 11.7 years (95%CI 9.6‐13.8) (Fig. 4B). No PD patients required NIV.
Feeding and nutrition
Weight measurements were available for 29 patients. Twenty‐five had weight <50th centiles and 17 <2.5th centile during follow‐up (Fig. 4C), chronic constipation was reported in 16 individuals (15 CD and one PD), significant gastroesophageal reflux in eight CD, recurrent pancreatitis in one CD and recurrent hypoglycemia in one further CD patient. Nineteen CD and one PD patient had gastrostomy insertion at a median age of 5 years (range 1.7‐15.5 years). Main indications for gastrostomy were failure to thrive (11/20 patients), unsafe swallow (1/20) or combination of the two (5/20). Indication was not clear for three patients. Twelve of 19 patients with gastrostomy (63%) received oral meals at last assessment, regardless of presence of gastrostomy. Gastrostomy was removed in one CD patient two years after positioning as oral feeds were considered safe. Time‐to‐event analysis on 33/42 CD subjects estimates median age at gastrostomy at 10.9 years (95%CI 8.7‐13) (Fig. 4D).
Cardiac involvement
Seven CD patients and no PD subjects had developed subclinical cardiomyopathy and/or rhythm disturbances during follow‐up. Mild persistent left ventricular dysfunction (atrial septum or inferior wall) was observed in three patients at age 9, 14 and 19 years, respectively. The lowest fractional shortening was 24%, observed at 14 years of age. Mild pericardial effusion associated with localized inferior wall hypokinesis and normal fractional shortening was transiently detected in one patient at the age of 12 years. Two patients presented sinus tachycardia and one patient required treatment with amiodarone at the age of 2 years and 2 months because of an episode of supraventricular tachycardia.
Epilepsy
Four CD patients developed seizures at a median age of 7.7 years (range 5.3‐11 years). Epileptic manifestations were focal seizures, absences or complex partial seizures. One patient received multi‐therapy (valproate and vigabatrin) to control seizures, with benefits.
Cognition
Mild learning difficulties were reported in two CD patients. Brain MRI results or formal neuropsychological assessment was not available at the time of this study.
Additional features
Squint was reported in two CD patients and limitation of jaw opening in further two CD.
Survival
Seven CD patients died during follow‐up period at a median age of 12 years (range 7‐17 years; Fig. 5). Causes of death included respiratory and multi‐organ failure following viral chest infection (at 12.8 years), sudden respiratory failure (at 7 years) and cardiopulmonary arrest following sustained vomiting (at 12.1 years). A detailed report of cause of death was not available for the remaining four subjects. Survival data after age 17 years were extremely limited and available only for 9/21 patients. One patient died at the age of 21.2 years, while the oldest patient known to be alive at time of this study is Pt 20 (CD), currently aged 33.5 years.
Figure 5.
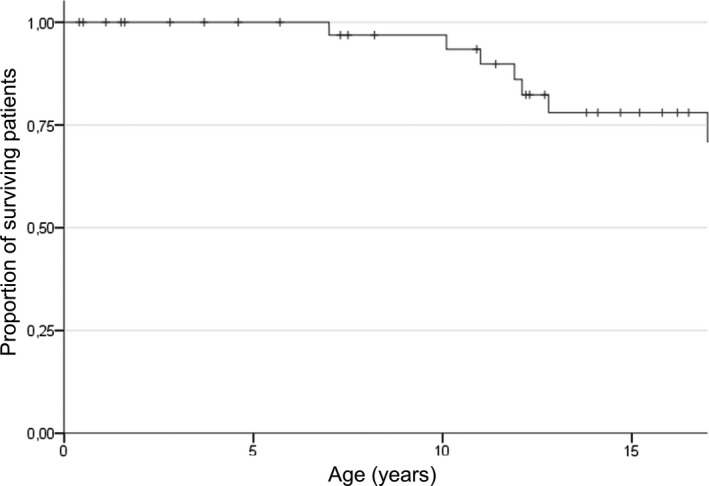
Survival analysis. Kaplan–Meier estimates for survival from birth in CD subjects.
Investigations
Eighteen patients had a brain MRI at the time of diagnosis (15 CD and three PD). Isolated white matter (WM) alteration was the most common finding (11/15 patients) while three patients presented T‐2 signal changes associated with polymicrogyria, pontine hypoplasia or mild cerebellar hypoplasia. One CD patient had normal MRI at age six months of age. One PD patient presented isolated WM abnormality, and one had additional gray matter sub ependymal heterotopia. Pt 44 (PD), currently aged 14 years, had a normal MRI at 7 years 6 months.
Results of nerve conduction studies (NCS) were available for 15 patients (13 CD and two PD). Six out of 13 CD patients showed mildly reduced conduction velocities for age (in ulnar or peroneal motor nerves). Pt 14 (CD) showed sensorimotor axonal loss in addition to mildly reduced nerve conduction velocity. Pt 44 (PD), had a normal NCS performed at 17 days of life, Pt 46 had normal NCS at 6 months of age.
Baseline measurement of creatine kinase (CK) was available for 22 patients. Median CK levels of CD patients were 2665 UI/L (range 394‐36000 UI/L). Three PD subjects had levels of 563, 2318 and 3375 UI/L, respectively.
Genetics
A total of 43 pathogenic variants were identified in this cohort of LAMA2‐RD patients (Sup Table S1, Fig. 6). Seventeen variants were nonsense (39%), 11 frameshift (26%), nine splice‐site (21%), three copy number variations (7%) and three missense (7%). Twenty patients (13 families) were homozygous (consanguinity in nine families), and 19 compound heterozygous for two LAMA2 pathogenic variants. In two patients, genetic analysis was only able to identify a single LAMA2 variant in heterozygosity, while genetic testing was not available at time of diagnosis for 6 patients. Ten variants were novel.
Figure 6.
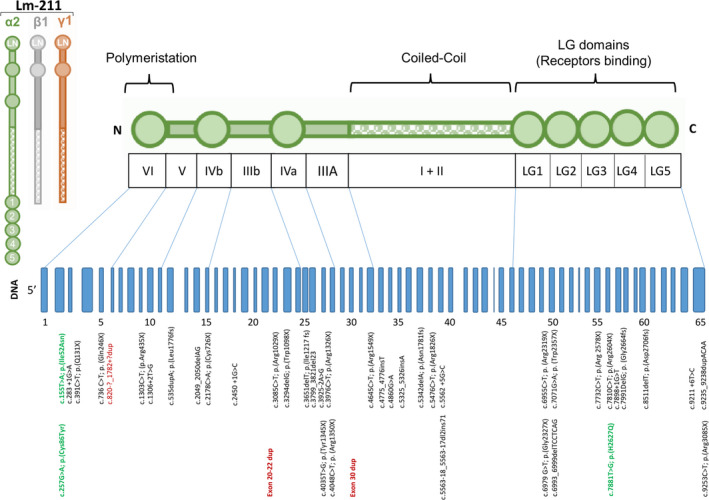
Schematic representation of the Laminin α2 chain and the LAMA2 gene. The 65 exons are represented in blue. All 43 described variants are presented according to cDNA localization. Missense mutations are displayed in green, copy number variations in red.
Discussion
With growing evidence on new therapeutic approaches for LAMA2‐RD, including preclinical studies on use of linker proteins, 29 exogenous administration of Lm‐111 24 or upregulation of the modifier Lama1, 25 there is increasing need for prospective, longitudinal studies to report on natural history and disease progression. Such knowledge is essential to refine standard of care and identify most reliable parameters to function as outcome measures for future therapeutic trials. To date, several cross‐sectional studies provided information on LAMA2‐RD phenotype and genotype. 12 , 19 , 20 , 21 Recently, a prospective natural history study was completed on 24 LAMA2‐RD patients over an average of 3 years (NIH, Bethesda, USA). 33 This study assessed a range of outcome measures and evaluated their rate of change/year. While this work provides useful information towards trial readiness, detailed knowledge on long‐term progression of skeletal and respiratory muscle weakness, occurrence and time of intervention for disease‐related complications are still lacking. Also, no published study addressed early motor function or respiratory outcome measures in the younger LAMA2‐RD population.
Here we present a comprehensive cross‐sectional and retrospective longitudinal natural history analysis on a large pediatric cohort of LAMA2‐RD patients attending a single tertiary neuromuscular center, with the longest follow‐up presented thus far (median 7.8 years, up to 18). We confirm the wide phenotypic spectrum of the disease, from severe, prenatal/neonatal onset in >50% of patients with CD, to milder forms presenting later in life. We provide detailed information on symptoms at onset and respiratory, feeding and orthopedic function during follow‐up. Although not specific for LAMA2‐RD, we also confirm the high prevalence of pre‐/neonatal features, with talipes or mild contractures present in ~20% of CD patients at birth. Feeding difficulties at onset were more common in CD than in PD patients (57% vs 25%, respectively).
As previously reported, 12 we confirm major differences in ambulation for CD and PD patients, with only one CD patient attaining independent walking (2.7 years). However, our data also confirm that achievement of supported ambulation is possible in a small subset of CD up until a median age of 4.8 years, ability that can be retained for up to a median of 4.6 years. The mixed (anamnestic and observational) nature of our data limits ability to draw strong conclusions from these findings.
The combination of severe head/trunk flexor weakness with prevalent upper girdle involvement from onset 11 , 31 in association with raised CK levels, represents valid diagnostic handles for LAMA2‐RD and warrants for use of functional scales specific for axial strength and upper limb function. The D2 domain of the Motor Function Measure (MFM) 32 scale 33 covers axial and proximal motor capacity and is sensitive to yearly changes in patients >5 years, but further validation in younger populations is required.
Besides muscle weakness, the main contributors to disease burden in LAMA2‐ RD are respiratory insufficiency, feeding difficulties, joint contractures and scoliosis, with early restrictive pulmonary insufficiency being the most common cause of morbidity and mortality. 34 , 35 , 36 Here we first provide detailed analysis on progression of lung function in severe LAMA2‐RD patients, over up to 12.8 years (Fig. 4A). Our data indicate severe compromise of respiratory function at first evaluation in CD patients (FVC%<40 in over half of the cohort), and subsequent linear annual decline of 2.9% of FVC%. This finding differs from that seen in PD subjects, with no patients having FVC% <60% at any time during their follow‐up period. These data first provide information for powering future therapeutic trials where FVC could represent one endpoint. Despite a high prevalence of hospital admission due to chest infections in the first 2 years, only one CD patient received tracheostomy, and none required NIV >16 hours. Survival analysis evidenced a significant number of deaths during childhood, mostly due to respiratory impairment. With the limitation of the information available for this study, our data highlight the need for proactive respiratory management from diagnosis onwards. Feeding difficulties with impairment of all stages of swallowing are well documented in LAMA2‐RD, 37 with silent aspiration (and subsequent risk of chest infections) being particular common in early phases of the disease. Our data confirm early feeding impairment and the need for gastrostomy 37 in particular in CD patients (Fig. 4C). Nonetheless, the observation of safe oral feeds in later stages of the disease can help counseling on the benefits and downsides of gastrostomy. Contrary to NIV management, review of age at interventions indicates that gastrostomy insertion has been increasingly anticipated over the last 15 years, suggesting a more proactive approach and increased awareness of aspiration‐derived risks and effect of malnutrition on performance status in these patients (data not shown). Similar to recent reports, we also anecdotally observed repeated episodes of hypoglycemia in one CD patient, highlighting the importance of adequate feeding in this patient population. 38
This study first provides novel longitudinal data on scoliosis and Cobb Angle’s progression for LAMA2‐RD. Our results indicate rapidly progressive scoliosis with an annual progression of 5.3˚ in Cobb angle from as early as 6 years of age, with >80% of patients having scoliosis by 10 years (Fig. 3D). We found a negative linear correlation between Cobb angle and FVC % progression. LAMA2‐RD patients have reduced chest wall movement and scoliosis might further affect lung function capacity, although such correlation could be merely a consequence of disease progression (Yun at al, WMS 2018). The yearly 6.6˚ progression in elbow extension is similar to what reported by Jain et al (4.11˚/year). 33 While such variables might not represent reliable outcome measures for future clinical trials, with the exception of elbow flexion, joint contractures are relevant when assessing the ability to perform activities of daily living and selecting appropriate functional scales. 39
We confirm that severe cardiomyopathy is not common during childhood in LAMA2‐RD. However, as previously indicated, regular cardiac monitoring is recommended from diagnosis and in particular after the third decade, when more severe cardiac abnormalities and sudden death have been more often described. 9 , 40 , 41 , 42 , 43 , 44 , 45
While seizures have been reported in up to 30% of LAMA2‐RD patients older than six years regardless of LAMA2 expression on muscle biopsy, 46 , 47 seizures were not common in this cohort of CD patients and absent in PD subjects. Also, no correlation between structural brain changes, epilepsy or learning difficulties were noted in our cohort. Notably, we observed normal brain MRI in a 7 year old PD subject (Pt 44), an uncommon finding in LAMA2‐RD. 19 , 48 , 49 Finally, we also observed significant axonal loss in one CD patient, expanding previous knowledge on neuropathy in LAMA2‐RD.
Despite considerable differences between CD and PD patients, we did not identify genotype‐phenotype correlation in this cohort (Sup Table S1). 16 , 19 , 20 As previously described, we also observed exceptional intrafamilial variability. 50 In particular, in one family with seven patients (siblings or cousins) harboring a homozygous missense variant (c.7881T>G; p.(H2627Q)) and complete laminin alpha 2 deficiency in all, we observed a more typical CMD like course in six and a significantly milder disease in the 7th patient. This milder patient (Pt 32) presented better‐preserved motor function, maintaining independent ambulation for 11 years, but developed severe joint contractures, scoliosis and respiratory insufficiency, necessitating nocturnal NIV and scoliosis surgery at age 13 years. We also observed in a CD patient a homozygous nonsense variant (c.4645C>T; p(Arg1549Ter)) previously described in a milder patient. 7 , 12 These findings suggest that factors other than the LAMA2 gene variant and the amount of residual laminin alpha 2 in the tissue can modify disease severity and progression.
This study has intrinsic limitations, such as sample size, the retrospective nature of data collection and inconsistencies in the use of functional scales throughout the years (despite many having been longitudinally recorded in standardized assessment forms). Time‐to‐intervention for disease‐related complications was also influenced by local criteria and standard of care at that time point.
In conclusion, this large cross‐sectional and retrospective longitudinal natural history study provides a detailed and comprehensive description of disease progression and management of LAMA2‐RD patients. These results will be valuable to guide anticipation of complications and help prioritize outcome variables for future therapeutic trials. While FVC% progression and dependence on nocturnal NIV could possibly represent meaningful outcome measure in older children, prospective natural history studies on larger and younger populations are warranted to validate these results and identify further measures for younger patients.
Author Contributions
Conception and design of the study: A.A.Z, A.S., F.M. Acquisition and analysis of data: A.A.Z, D.R., M.M., R.M., R.P., A.S., drafting of the manuscript and figures: A.A.Z., D.R. F.M., A.S.
Conflicts of Interest
Nothing to report.
Supporting information
Supplementary Table S1. Legend: New variants are indicated in bold, missense changes in italics. #, age at last assessment; CD, complete merosin deficiency; KAFO, knee ankle foot orthosis; PD, partial merosin deficiency; NA, not available; *, members of the same family (* family 1, ** family 2, *** family 3).
Acknowledgments
Special thanks and appreciation to the neuromuscular team at the DNC, and in particular to Dr Adnan Manzur, Dr Pinki Munot, Dr Stephanie Robb, Dr Ros Quinlivan, Dr Caroline Sewry, Dr Mariacristina Scoto, Dr Giovanni Baranello and the Great Ormond street Hospital Neuromuscular Physiotherapy team for their contribution over the years in the assessment and management of the patients at the DNC. We are grateful to the Great Ormond Street Lung Physiology team for providing the spirometry data. We would like to acknowledge the contribution of the Highly Specialised Service for congenital muscular dystrophies and congenital myopathies at the Dubowitz Neuromuscular Centre and in particular Dr Lucy Feng for her assistance. A special thanks goes to Dr Veronica Pini for her critical review of the work and contribution to figures. This work was supported in part by The Muscular Dystrophy UK Central Grant to the DNC. This research was also supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors, upon request.
References
- 1. Helbling‐Leclerc A, Zhang X, Topaloglu H, et al. Mutations in the laminin α2–chain gene (LAMA2) cause merosin–deficient congenital muscular dystrophy. Nat Genet 1995;11(2):216–218. 10.1038/ng1095-21 [DOI] [PubMed] [Google Scholar]
- 2. Zhang X, Vuolteenaho R, Tryggvason K. Structure of the human laminin alpha2‐chain gene (LAMA2), which is affected in congenital muscular dystrophy. J Biol Chem 1996;271(44):27664–27669. [DOI] [PubMed] [Google Scholar]
- 3. Vainzof M, Richard P, Herrmann R, et al. Prenatal diagnosis in laminin alpha2 chain (merosin)‐deficient congenital muscular dystrophy: a collective experience of five international centers. Neuromuscul Disord. 2005;15(9–10):588–594. [DOI] [PubMed] [Google Scholar]
- 4. Taratuto AL, Lubieniecki F, Díaz D, et al. Merosin‐deficient congenital muscular dystrophy associated with abnormal cerebral cortical gyration: an autopsy study. Neuromuscul Disord 1999;9(2):86–94. [DOI] [PubMed] [Google Scholar]
- 5. McKee KK, Harrison D, Capizzi S, Yurchenco PD. Role of laminin terminal globular domains in basement membrane assembly. J Biol Chem 2007;282(29):21437–21447. [DOI] [PubMed] [Google Scholar]
- 6. Yurchenco PD, McKee KK, Reinhard JR, Ruegg MA. Laminin‐deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Matrix Biol 2018;71–72:174–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Di Blasi C, Mora M, Pareyson D, et al. Partial laminin α2 chain deficiency in a patient with myopathy resembling inclusion body myositis. Ann Neurol 2000;47(6):811–816. [PubMed] [Google Scholar]
- 8. Gavassini BF, Carboni N, Nielsen JE, et al. Clinical and molecular characterization of limb‐girdle muscular dystrophy due to LAMA2 mutations. Muscle Nerve 2011;44(5):703–709. [DOI] [PubMed] [Google Scholar]
- 9. Harris E, McEntagart M, Topf A, et al. Clinical and neuroimaging findings in two brothers with limb girdle muscular dystrophy due to LAMA2 mutations. Neuromuscul Disord 2017;27(2):170–174. [DOI] [PubMed] [Google Scholar]
- 10. Lokken N, Born AP, Duno M, Vissing J. LAMA2‐related myopathy: frequency among congenital and limb‐girdle muscular dystrophies. Muscle Nerve 2015;52(4):547–553. [DOI] [PubMed] [Google Scholar]
- 11. Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14(10):635–649. [DOI] [PubMed] [Google Scholar]
- 12. Geranmayeh F, Clement E, Feng LH, et al. Genotype‐phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010;20(4):241–250. [DOI] [PubMed] [Google Scholar]
- 13. Norwood FLM, Harling C, Chinnery PF, et al. Prevalence of genetic muscle disease in Northern England: in‐depth analysis of a muscle clinic population. Brain 2009;132(11):3175–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mostacciuolo ML, Miorin M, Martinello F, et al. Genetic epidemiology of congenital muscular dystrophy in a sample from north‐east Italy. Hum Genet 1996;97(3):277–279. [DOI] [PubMed] [Google Scholar]
- 15. Sframeli M, Sarkozy A, Bertoli M, et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12‐year period. Neuromuscul Disord 2017;27(9):793–803. [DOI] [PubMed] [Google Scholar]
- 16. Allamand V, Guicheney P. Merosin‐deficient congenital muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for α2 chain of laminin). Eur J Hum Genet 2002;10(2):91–94. [DOI] [PubMed] [Google Scholar]
- 17. Peat RA, Smith JM, Compton AG, et al. Diagnosis and etiology of congenital muscular dystrophy. Neurology 2008;71(5):312–321. [DOI] [PubMed] [Google Scholar]
- 18. Clement EM, Feng L, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord 2012;22(6):522–527. [DOI] [PubMed] [Google Scholar]
- 19. Oliveira J, Santos R, Soares‐Silva I, et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet 2008;74(6):502–512. [DOI] [PubMed] [Google Scholar]
- 20. Xiong H, Tan D, Wang S, et al. Genotype/phenotype analysis in Chinese laminin‐alpha2 deficient congenital muscular dystrophy patients. Clin Genet 2015;87(3):233–243. [DOI] [PubMed] [Google Scholar]
- 21. Pegoraro E, Marks H, Garcia CA, et al. Laminin alpha2 muscular dystrophy: genotype/phenotype studies of 22 patients. Neurology 1998;51(1):101–110. [DOI] [PubMed] [Google Scholar]
- 22. Erb M, Meinen S, Barzaghi P, et al. Omigapil ameliorates the pathology of muscle dystrophy caused by laminin‐alpha2 deficiency. J Pharmacol Exp Ther 2009;331(3):787–795. [DOI] [PubMed] [Google Scholar]
- 23. Yu Q, Sali A, Van der Meulen J, et al. Omigapil treatment decreases fibrosis and improves respiratory rate in dy(2J) mouse model of congenital muscular dystrophy. PLoS One 2013;8(6):e65468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rooney JE, Knapp JR, Hodges BL, et al. Laminin‐111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin‐deficient congenital muscular dystrophy. Am J Pathol 2012;180(4):1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kemaladewi DU, Bassi PS, Erwood S, et al. A mutation‐independent approach for muscular dystrophy via upregulation of a modifier gene. Nature 2019;572(7767):125–130. [DOI] [PubMed] [Google Scholar]
- 26. Moll J, Barzaghi P, Lin S, et al. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature 2001;413(6853):302–307. [DOI] [PubMed] [Google Scholar]
- 27. Qiao C, Li J, Zhu T, et al. Amelioration of laminin‐α2‐deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci USA 2005;102(34):11999–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKee KK, Crosson SC, Meinen S, et al. Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype. J Clin Investig 2017;127(3):1075–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reinhard JR, Lin S, McKee KK, et al. Linker proteins restore basement membrane and correct LAMA2‐related muscular dystrophy in mice. Sci Transl Med. 2017;9(396). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meilleur KG, Jain MS, Hynan LS, et al. Results of a two‐year pilot study of clinical outcome measures in collagen VI‐ and laminin alpha2‐related congenital muscular dystrophies. Neuromuscul Disord 2015;25(1):43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bendixen RM, Butrum J, Jain MS, et al. Upper extremity outcome measures for collagen VI‐related myopathy and LAMA2‐related muscular dystrophy. Neuromuscul Disord 2017;27(3):278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Toussaint M, Chatwin M, Gonzales J, Berlowitz DJ. 228th ENMC International Workshop: Airway clearance techniques in neuromuscular disorders Naarden, The Netherlands, 3–5 March, 2017. Neuromuscul Disord 2018;28(3):289–298. [DOI] [PubMed] [Google Scholar]
- 33. Jain MS, Meilleur K, Kim E, et al. Longitudinal changes in clinical outcome measures in COL6‐related dystrophies and LAMA2‐related dystrophies. Neurology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quijano‐Roy S, Sparks SE, Rutkowski A. LAMA2‐Related Muscular Dystrophy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., Seattle, WA: GeneReviews(®), 1993. [Google Scholar]
- 35. Foley AR, Quijano‐Roy S, Collins J, et al. Natural history of pulmonary function in collagen VI‐related myopathies. Brain 2013;136(Pt 12):3625–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meilleur KG, Linton MM, Fontana J, et al. Comparison of sitting and supine forced vital capacity in collagen VI‐related dystrophy and laminin α2‐related dystrophy. Pediatr Pulmonol 2017;52(4):524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Philpot J, Bagnall A, King C, et al. Feeding problems in merosin deficient congenital muscular dystrophy. Arch Dis Child 1999;80(6):542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hayes LH, Yun P, Mohassel P, et al. Hypoglycemia in patients with congenital muscle disease. BMC Pediatr 2020;20(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oosterwijk AM, Nieuwenhuis MK, van der Schans CP, Mouton LJ. Shoulder and elbow range of motion for the performance of activities of daily living: a systematic review. Physiother Theory Pract 2018;34(7):505–528. [DOI] [PubMed] [Google Scholar]
- 40. Finsterer J, Ramaciotti C, Wang CH, et al. Cardiac findings in congenital muscular dystrophies. Pediatrics 2010;126(3):538–545. [DOI] [PubMed] [Google Scholar]
- 41. Spyrou N, Philpot J, Foale R, et al. Evidence of left ventricular dysfunction in children with merosin‐deficient congenital muscular dystrophy. Am Heart J 1998;136(3):474–476. [DOI] [PubMed] [Google Scholar]
- 42. Gilhuis HJ, ten Donkelaar HJ, Tanke RB, et al. Nonmuscular involvement in merosin‐negative congenital muscular dystrophy. Pediatr Neurol 2002;26(1):30–36. [DOI] [PubMed] [Google Scholar]
- 43. Carboni N, Marrosu G, Porcu M, et al. Dilated cardiomyopathy with conduction defects in a patient with partial merosin deficiency due to mutations in the laminin‐alpha2‐chain gene: a chance association or a novel phenotype? Muscle Nerve 2011;44(5):826–828. [DOI] [PubMed] [Google Scholar]
- 44. Nelson I, Stojkovic T, Allamand V, et al. Laminin alpha2 deficiency‐related muscular dystrophy mimicking emery‐dreifuss and collagen VI related diseases. J Neuromuscul Dis 2015;2(3):229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marques J, Duarte ST, Costa S, et al. Atypical phenotype in two patients with LAMA2 mutations. Neuromuscul Disord 2014;24(5):419–424. [DOI] [PubMed] [Google Scholar]
- 46. Bönnemann CG, Wang CH, Quijano‐Roy S, et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord 2014;24(4):289–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Natera‐de Benito D, Muchart J, Itzep D, et al. Epilepsy in LAMA2‐related muscular dystrophy: an electro‐clinico‐radiological characterization. Epilepsia 2020;61(5):971–983. [DOI] [PubMed] [Google Scholar]
- 48. Leite CC, Lucato LT, Martin MG, et al. Merosin‐deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol 2005;35(6):572–579. [DOI] [PubMed] [Google Scholar]
- 49. Mercuri E, Gruter‐Andrew J, Philpot J, et al. Cognitive abilities in children with congenital muscular dystrophy: correlation with brain MRI and merosin status. Neuromuscul Disord 1999;9(6–7):383–387. [DOI] [PubMed] [Google Scholar]
- 50. Prandini P, Berardinelli A, Fanin M, et al. LAMA2 loss‐of‐function mutation in a girl with a mild congenital muscular dystrophy. Neurology 2004;63(6):1118–1121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Legend: New variants are indicated in bold, missense changes in italics. #, age at last assessment; CD, complete merosin deficiency; KAFO, knee ankle foot orthosis; PD, partial merosin deficiency; NA, not available; *, members of the same family (* family 1, ** family 2, *** family 3).
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors, upon request.