

Abstract
Objective
To examine whether apolipoprotein B (ApoB), apolipoprotein A‐1 (ApoA1), or their ratio (ApoB/A1) were associated with early changes in cerebrospinal fluid (CSF) biomarkers of Alzheimer’s disease (AD) pathology in elderly adults with subjective cognitive decline (SCD).
Methods
This study included 507 objective cognitive normal participants from the Chinese Alzheimer’s Biomarker and LifestylE (CABLE) database including 288 cognitive normal participants (CN) and 219 SCD. Multiple linear regression models were used to examine the associations of apolipoproteins with CSF AD biomarkers.
Results
Compared with control group, SCD participants with significant AD biological characteristics had lower ApoB levels (P = 0.0461). In total participants, lower level of serum ApoB was associated with decreases in CSF Aβ42 (P = 0.0015) and Aβ42/40 (P = 0.0081) as well as increases in CSF p‐tau/Aβ42 (P < 0.0001) and t‐tau/Aβ42 (P = 0.0013), independent of APOEɛ4 status. In further subgroup analysis, these associations were more significant in SCD participants (ApoB × Diagnose: P < 0.05). In addition, lower levels of ApoB were also found associated with increases in p‐tau in the SCD subgroup (P = 0.0263). Furthermore, these protective associations were more significant in the overweight participants (ApoB × weight: P < 0.05). Results showed no association between ApoA1 and CSF biomarkers.
Interpretation
This study is the first to find protective associations of serum ApoB with CSF AD core biomarkers, especially in SCD individuals. It indicated that ApoB may be a potential biomarker for preclinical AD and may play different roles in different stages of AD.
Introduction
It has been widely accepted that neuropathological processes associated with Alzheimer’s disease (AD) begin a decade or more before the emergence of obvious objective cognitive impairment. 1 , 2 , 3 Biomarker research proved that levels of cerebrospinal fluid (CSF) β‐amyloid 42 (Aβ42), phosphorylated tau (p‐tau), and total tau (t‐tau) have been significantly abnormal in the preclinical stage of AD. 3 , 4 , 5 The 2018 National Institute on Aging and Alzheimer’s Association (NIAA) research framework further included the three CSF biomarkers in the ATN framework, reflecting brain amyloidosis (A: Aβ42), tau pathology (T: p‐tau), and neurodegeneration (N: t‐tau). 6 All these theories shifted the definition of AD from a syndromic to a biological construct and leaded us to focus on disease’s earlier stage such as preclinical stage.
Subjective cognitive decline (SCD) in individuals without objective cognitive impairment was recognized as a transitional stage between a fully asymptomatic stage and a mild cognitively impaired (MCI) stage of the disease. 7 , 8 It has been suggested to be a possible first symptomatic expression of preclinical AD and a target stage for future AD prevention trials. Therefore, finding some available blood biomarkers associated with early pathological changes in AD biomarkers in SCD individuals may contribute to creating more practical early diagnostic methods or finding some new factors involved in AD early pathogenesis.
Apolipoproteins are key proteins in the lipid metabolism of peripheral blood and central nervous system and also play an important role in the transport of some key proteins. 9 , 10 Previous studies have indicated that Apolipoprotein B (ApoB) and Apolipoprotein A‐1 (ApoA1) may be involved in the pathogenesis of AD. Some studies have shown that AD patients had higher levels of circulating ApoB and lower levels of circulating ApoA1 compared with healthy controls. 11 , 12 ApoB may promote the formation of amyloid plaques and vascular pathological changes. 13 , 14 ApoA1 may have a neuroprotective effect by forming a complex with the Aβ peptide. 12 However, some studies showed different results. 15 Overall, the number of studies on these factors is still limited. Moreover to the best of our knowledge, no studies have been reported on the associations of serum ApoB, ApoA1, and the ratio of ApoB and ApoA1 (ApoB/A1) with CSF AD biomarkers, especially in the preclinical stage.
The purpose of our study was to examine whether ApoB, ApoA1, and ApoB/A1 can reflect the early pathological changes in CSF AD biomarkers in elderly adults with subjective cognitive decline and to find some potential candidate early biomarkers in blood for AD.
Methods
CABLE database
Since 2017, the Chinese Alzheimer’s Biomarker and LifestylE (CABLE) database has been an ongoing large‐scale study majorly focused on AD’s risk factors and biomarkers in Chinese Northern Han population. CABLE aimed to determine the genetic and environmental modifiers of AD biomarkers and to identify lifestyle factors that may affect the risk of AD in the non‐demented northern Chinese Han population, thus providing a basis for disease prevention and early diagnosis. In conjunction with the investigation, oral informed consent for future use of their CSF and blood samples for research purposes was obtained. All patients were later instructed to withdraw their permission if they changed their minds. The CABLE database was conducted in accordance with the Helsinki declaration, and the research program was approved by the Institutional Ethics Committee of Qingdao Municipal Hospital.
Participants
All enrolled participants in the CABLE were Han Chinese aged between 40 and 90 years old. The exclusion criteria include: (1) central nervous system infection, head trauma, neurodegenerative diseases other than AD (e.g., epilepsy, Parkinson's Disease), or other major neurological disorders; (2) major psychological disorders; (3) severe systemic diseases (e.g., malignant tumors) that may affect CSF or blood levels of AD biomarkers including Aβ and tau; (4) family history 141 of genetic diseases. All participants received clinical and neuropsychological assessments, biochemical testing, as well as blood and CSF sample collection. Comprehensive questionnaire, electronic medical record system, and a laboratory inspection management system were used to collect demographic information, AD risk factor profile and medical history.
The diagnosis of preclinical AD was defined according to the criteria of National Institute on Aging‐Alzheimer's Association (NIA‐AA) workgroups criteria 16 that is, normal cognition but abnormal AD biomarkers. We first screened cognitive state of participants using the China Modified Mini‐Mental State Examination (CM‐MMSE) and Montreal Cognitive Assessment (MoCA). The cut‐off values after correcting the years of education were used in the MMSE (≤24 for 6 + y of education, ≤20 for 6‐ y of education, ≤17 for 0 y of education) and MOCA (<26 for 12 + y of education, <25 for 12‐ y of education). Basic living ability was assessed by basic Activities of Daily Living score (ADL). Behavioral or psychological symptoms were assessed by Geriatric Depression Scale (GDS), Hamilton Rating Scale for Depression (HAMD) and Hamilton Rating Scale for Anxiety (HAMA). Vascular factors were assessed by Hachinski Inchemic Score (HIS). All diagnoses were evaluated by two medical doctors with extensive experience in cognitive disorders through intact performance on neuropsychological tests combined with CSF biomarkers and MRI examinations. Subjective cognitive decline was assessed by a subjective cognitive decline scale (SCDS) which was designed based on SCD‐I recommendations. 17 , 18 We distinguished participants with SCD accompanied by particular concerns (worries) from those without objective cognitive decline by the first section of SCDS which included a dichotomous question. Then we chose SCD participants who met the SCD‐plus criteria as much as possible. 18
Finally, we included 507 participants without objective cognitive impairment from CABLE database, including 288 CN and 219 SCD participants. None of the participants had a history of diabetes or high blood pressure. None of the participants were taking lipid‐lowering drugs or on antihypertensive and antidiabetic treatment. All the participants provided both blood and CSF samples.
CSF sample collection and measurements
Collection of fasting lumbar CSF samples was performed at Qingdao Municipal Hospital. CSF was drawn and processed within 2 hours after collection. Each tube was centrifuged at 2000× g for 10 minutes, and CSF was separated and stored in enzyme‐free EP (Eppendorf) tube (AXYGEN; PCR‐02‐C) at −80 centigrade until further use in this study. The thaw/freezing cycle was limited and did not surpass two times. Baseline CSF Aβ42, Aβ40, t‐tau, and p‐tau181 were determined with the ELISA kit (Innotest β‐AMYLOID (1‐42), β‐AMYLOID (1‐40), hTAU‐Ag, and PHOSPHO‐TAU (181p); Fujirebio, Ghent, Belgium) on the microplate reader (Thermo Scientific Multiskan MK3). The within‐batch coefficient of variation (CV) was <5% (4.5% for Aβ42, 3.5% for Aβ40, 4.4% for t‐tau, and 2.5% for p‐tau). The inter‐batch CV was <15% (9.6% for Aβ42, 8.1% for Aβ40, 12.2% for t‐tau, and 11.0% for p‐tau). Furthermore, Additional File S1 showed that CSF Aβ42 was reduced with APOE ε4 when stratifying the whole cohort for APOE genotype and that it decreased with age. These results were consistent with measurements in previous studies, 19 which indicated that raw data were sound with no technical problems.
Amyloid imaging 19 , 20 and neuropathological studies 21 , 22 , 23 , 24 showed that approximately one‐third of older adults without objective cognitive impairment had AD pathology in their brains. Therefore, in our study, CSF biomarker positive participants were defined as having CSF Aβ42 levels in the lower one‐third of the distribution of participants (A+: ≤113.84 pg/mL) or having p‐tau (T+: ≥39.11 pg/mL) or t‐tau (N+: ≥182.49 pg/mL) levels in the upper one‐third of the distribution. This cut‐off setting method was also applied by a study in JAMA Neurology and received reasonable results. 25 Then, according to the 2018 NIAA research framework, we resulted in three different biomarker group combinations including stage 0, stage 1, stage 2. Individuals with normal measures of Aβ42, p‐tau, and t‐tau (A‐T‐N‐) were classified as stage 0. Individuals with abnormal Aβ1‐42 but no abnormal p‐tau or t‐tau (A+T‐N‐) were classified as stage 1. Individuals with abnormal Aβ1‐42, and abnormal p‐tau or t‐tau (A+T+N‐, A+T‐N+, A+T+N+) were classified as stage 2.
Blood sample collection and measurements
Blood samples of all the participants were drawn after an overnight fast. Serum levels of ApoB and ApoA1 were assayed by immunonephelometry using a BN II analyzer (Siemens Healthcare, Marburg, Germany) within two hours after collection. The within‐batch CV was <2%. The inter‐batch CV was <5%. The lowest detectable concentrations for ApoB and ApoA1 were 0.021 mg/mL and 0.005 mg/mL, respectively.
QIAamp® DNA Blood Mini Kit (250) was used to extract DNA from blood samples. And the extracted DNA was separated and stored in enzyme free EP tube at −80 centigrade until the apolipoprotein E (APOE) 4 genotyping was completed in this study. Specific loci were selected for genotyping with restriction fragment length polymorphism (RFLP) technology, including the two loci related to APOE4 status (rs7412 and rs429358).
Standard protocol approvals, registrations, and patient consents
The CABLE database was conducted in accordance with the Helsinki declaration, and the research program was approved by the Institutional Ethics Committee of Qingdao Municipal Hospital. All study participants or their caregivers provided written informed consent directly.
Statistical analysis
Kruskall–Wallis test and Chi‐square test were applied in the comparison among groups. All serum and CSF variables were log‐transformed to normalize the distributions (see Additional File S2). Multiple linear regression models were used to examine the associations adjusting for age, sex, education, and APOEɛ4 status. What’s more, body mass index (BMI) was also further added to multiple linear regression models as a possible confounder. Bonferroni correction was used for multiple comparisons. Statistical analyses were conducted using R, version 3.5.1. A two‐tailed P < 0.05 was considered significant.
Results
Characteristics of participants
For the current study, we analyzed data from 507 participants of the CABLE cohort, including 288 CN and 219 SCD (Table 1). The average age of participants was 63 years; 312 (61.54%) participants were male; and 79 (15.58%) participants were APOEɛ4 positive. We found no significant difference in sex distribution or level of education between the two subgroups. The age of SCD subgroup was older than CN subgroup. Frequency of positive APOEε4 status showed an increasing trend in SCD subgroup (CN: 13.14%, SCD: 18.72%), although these results were not statistically significant.
Table 1.
Characteristics of study participants from CABLE database.
| Variable | CN | SCD | Total | P |
|---|---|---|---|---|
| N | 288 | 219 | 507 | ‐ |
| AGE (year) mean (SD) | 61.16 (11.00) | 65.64 (9.86) | 63.09 (10.75) | <0.0001 # |
| SEX (Female/Male) | 111/177 | 84/135 | 195/312 | 1.0000* |
| Education (year) mean (SD) | 9.75 (4.36) | 9.87 (4.26) | 9.80 (4.31) | 0.7365 # |
| APOEε4 N (%) | 38 (13.14) | 41 (18.72) | 79 (15.58) | 0.1150* |
| CSF Aβ42 (pg/mL) mean (SD) | 172.31 (102.97) | 157.24 (106.06) | 165.80 (104.48) | 0.0203 # |
| CSF p‐tau (pg/mL) mean (SD) | 37.42 (9.82) | 38.18 (10.10) | 37.75 (9.94) | 0.3348 # |
| CSF t‐tau (pg/mL) mean (SD) | 173.05 (77.34) | 180.90 (90.56) | 176.44 (83.31) | 0.4108 # |
| CSF Aβ40 (pg/mL) mean (SD) | 6195.56 (2958.88) | 6244.04 (2573.09) | 6216.50 (2796.21) | 0.5972 # |
| CSF Aβ42/40 mean (SD) | 0.03 (0.03) | 0.027 (0.01) | 0.03 (0.02) | 0.0125 # |
| CSF p‐tau/Aβ42 mean (SD) | 0.25 (0.09) | 0.29 (0.12) | 0.27 (0.10) | 0.0058 # |
| CSF t‐tau/Aβ42 mean (SD) | 1.15 (0.60) | 1.34 (0.87) | 1.23 (0.73) | 0.0072 # |
| CM‐MMSE mean (SD) | 27.91 (2.16) | 27.64 (2.30) | 27.79 (2.22) | 0.1160 # |
| MOCA mean (SD) | 26.80 (0.98) | 26.62 (0.99) | 26.74 (0.99) | 0.0810 # |
| ApoB (mg/mL) mean (SD) | 1.03 (0.29) | 1.06 (0.25) | 1.04 (0.27) | 0.0665 # |
| ApoA1 (mg/mL) mean (SD) | 1.43 (0.32) | 1.47 (0.28) | 1.45 (0.31) | 0.0634 # |
| ApoB/A1 mean (SD) | 0.74 (0.22) | 0.74 (0.19) | 0.74 (0.21) | 0.7304 # |
| BMI mean (SD) | 26.00 (5.92) | 25.35 (3.74) | 25.72 (5.10) | 0.1969 # |
Abbreviations: CN, cognitively normal participants; SCD, participants with subjective cognitive decline; APOE, apolipoprotein E gene; CSF, cerebrospinal fluid; Aβ, amyloid‐β; p‐tau, phosphorylated tau protein; t‐tau, total tau protein; CM‐MMSE, China‐Modified Mini‐Mental State Examination; MOCA, Montreal Cognitive Assessment; ApoB, Apolipoprotein B; ApoA1, Apolipoprotein A‐1; BMI, Body Mass Index; SD, standard deviation.
Bold indicated that the results were statistically significant.
Intergroup comparisons were tested by Kruskall‐Wallis test.
Intergroup comparisons were tested by Chisquare test.
As for cognitive levels, we did not find a significant cognitive difference between CN subgroup and SCD subgroup (CM‐MMSE: P = 0.1160; MOCA: P = 0.0810). As for CSF biomarkers, compared with CN subgroup, SCD subgroup had lower levels of Aβ42 (P = 0.0203) and Aβ42/40 (P = 0.0125) and higher levels of p‐tau/Aβ42 (P = 0.0058) and t‐tau/Aβ42 (P = 0.0072).
Levels of serum ApoB and ApoA1 in different diagnostic groups
As for levels of serum ApoB, in a clinical diagnostic construct, we did not find difference between CN and SCD subgroups (Fig. 1A). In an ATN biological construct, A+ subgroups had lower levels of serum ApoB compared with A‐ subgroups (Fig. 1D–F). Then, according to the 2018 NIAA research framework, we resulted in three different biomarker group combinations including stage 0, stage 1, stage 2. Results showed that stage 2 subgroup showed significantly lower levels of serum ApoB than stage 0 (P = 0.0004) (Fig. 1B). Furthermore, in a diagnostic structure combining clinical diagnosis and biomarkers, CN and SCD participants with A+ had significantly lower levels of serum ApoB than participants without pathologic changes in CSF biomarkers (Fig. 1C). As for ApoA1, we did not find any difference in both clinical construct and biological construct (results were not shown).
Figure 1.
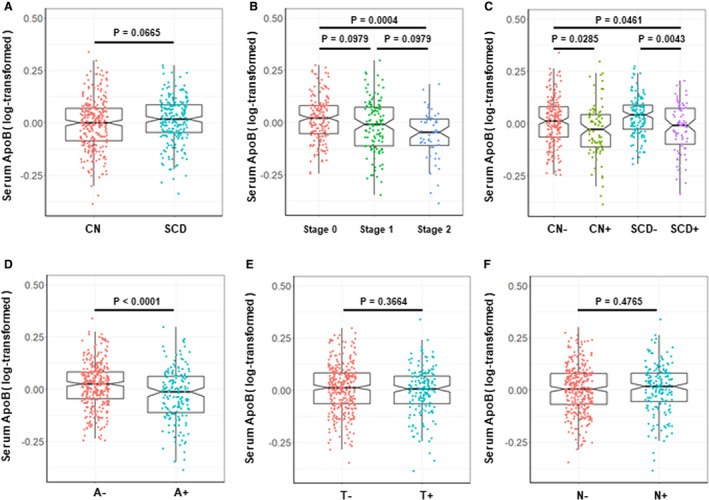
Levels of serum ApoB in different groups. (A) Levels of serum ApoB in a clinical diagnostic construct. CN (N = 288): cognitively normal individuals without subjective and objective cognitive decline; SCD (N = 219): individuals with subjective cognitive decline; (B) Levels of serum ApoB in an ATN biological construct. Stage 0: A‐T‐N‐ (N = 182), Stage 1: A + T‐N‐ (N = 117), Stage 2: A + T+N‐ (N = 8), A + T‐N+ (N = 6), A + T+N+ (N = 38); (C) Levels of serum ApoB in a diagnostic structure combining clinical diagnosis and biomarkers. CN‐ (N = 203): cognitively normal individuals without subjective and objective cognitive decline and with negative CSF Aβ42 [A‐]; CN+ (N = 85): cognitively normal individuals without subjective and objective cognitive decline and with positive CSF Aβ42 [A+]; SCD‐ (N = 135): individuals with subjective cognitive decline and negative CSF Aβ42 [A‐]; SCD+ (N = 84): individuals with subjective cognitive decline and positive CSF Aβ42 [A+]. (D–F) Levels of serum ApoB in A or T or N construct respectively. All intergroup comparisons were tested by Kruskall–Wallis test and Wilcoxon test.
Associations of ApoB, ApoA1, and ApoB/A1 with CSF AD core biomarkers in total participants without objective cognitive impairment
The results on associations of ApoB, ApoA1, and ApoB/A1 with CSF biomarkers in total participants without objective cognitive impairment were shown in Table 2. Results showed that lower levels of serum ApoB were significantly associated with decreased levels of CSF Aβ42 (β = 0.23, P = 0.0015) and Aβ42/40 (β = 0.22, P = 0.0081) but not with Aβ40. Although no association of ApoB was found with t‐tau and p‐tau, lower levels of serum ApoB were significantly associated with increased levels of CSF p‐tau/Aβ42 (β = −0.29, P < 0.0001) and t‐tau/Aβ42 (β = −0.26, P = 0.0013). Similar associations were found of ApoB/A1 with Aβ42 and p‐tau/Aβ42, but these associations seemed less significant than those of ApoB. We did not find any association between ApoA1 and any of the CSF biomarkers (Aβ42, p‐tau, t‐tau, Aβ40, Aβ42/40, p‐tau/Aβ42, and t‐tau/Aβ42).
Table 2.
Results of associations in total participants..
| Variable | ApoB | ApoA1 | ApoB/A1 | |||
|---|---|---|---|---|---|---|
| β | P | β | P | β | P | |
| CSF Aβ42 | 0.23 | 0.0015 | 0.11 | 0.2335 | 0.14 | 0.0363 |
| CSF p‐tau | −0.06 | 0.1122 | −0.06 | 0.2031 | −0.02 | 0.5867 |
| CSF t‐tau | −0.03 | 0.6450 | −0.09 | 0.2920 | 0.02 | 0.7230 |
| CSF Aβ40 | 0.01 | 0.9050 | −0.04 | 0.7140 | 0.03 | 0.6990 |
| CSF Aβ42/40 | 0.22 | 0.0081 | 0.15 | 0.1615 | 0.11 | 0.1496 |
| CSF p‐tau/Aβ42 | −0.29 | <0.0001 | −0.17 | 0.0515 | −0.16 | 0.0140 |
| CSF t‐tau/Aβ42 | −0.26 | 0.0013 | −0.20 | 0.0528 | −0.12 | 0.1148 |
Abbreviations: CSF, cerebrospinal fluid; Aβ, amyloid‐β; p‐tau, phosphorylated tau protein; t‐tau, total tau protein; ApoB, Apolipoprotein B; ApoA1, Apolipoprotein A‐1.
Bold indicated that the results were statistically significant.
Multiple linear regression models were used to examine the associations of serum ApoB, ApoA1, and ApoB/A1 levels with CSF biomarkers, adjusting for age, sex, education, and APOEɛ4 status.
Furthermore, we added the interaction between APOEɛ4 status and ApoB into multiple linear regression models. Results showed that these associations between ApoB and Aβ‐related biomarkers (Aβ42, Aβ42/40, p‐tau/Aβ42, and t‐tau/Aβ42) were independent of APOEε4 status. In these models, both ApoB and APOEɛ4 status were associated with Aβ‐related biomarkers, but their interactions (ApoB × APOEε4) were not (see Additional File S3). Moreover subgroup analyses of APOEɛ4 status also showed that associations between ApoB and Aβ‐related biomarkers were similar for different APOEɛ4 statuses (Fig. 2).
Figure 2.
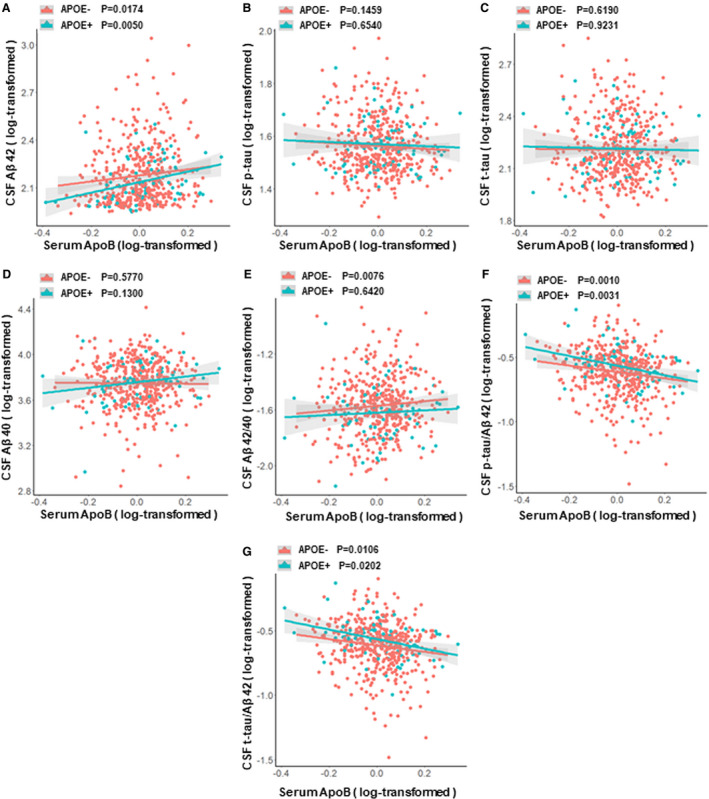
Associations between ApoB and CSF AD biomarkers among participants with different APOEε4 status. (A) Associations of ApoB with CSF Aβ42 in different APOEε4 status. (B) Associations of ApoB with CSF p‐tau in different APOEε4 status. (C) Associations of ApoB with CSF t‐tau in different APOEε4 status. (D) Associations of ApoB with CSF Aβ40 in different APOEε4 status. (E) Associations of ApoB with CSF Aβ42/40 in different APOEε4 status. (F) Associations of ApoB with CSF p‐tau/Aβ42 in different APOEε4 status. (G) Associations of ApoB with CSF t‐tau/Aβ42 in different APOEε4 status. Abbreviations: CSF, cerebrospinal fluid; Aβ, amyloid‐β; p‐tau, phosphorylated tau protein; t‐tau, total tau protein; ApoB, Apolipoprotein B; APOE, apolipoprotein E gene. Multiple linear regression models were used to examine the associations between serum ApoB levels and CSF biomarkers, adjusting for age, sex and education.
Associations of ApoB, ApoA1, and ApoB/A1 with CSF AD core biomarkers in different diagnostic subgroups
The results from interaction analyses and subgroup analyses of different diagnostic groups were shown in Additional File S4, Figure 3 and Additional File S5. As for ApoB, the interaction of ApoB and diagnose had obvious influences on the levels of CSF AD biomarkers (ApoB × diagnose: Aβ42, P = 0.0416; Aβ42/40, P = 0.0137; p‐tau/Aβ42, P = 0.0029; t‐tau/Aβ42, P = 0.0021) (see Additional File S4). In subgroup analyses, these associations between ApoB and Aβ‐related biomarkers (Aβ42, Aβ42/40, p‐tau/Aβ42, and t‐tau/Aβ42) still remained significant in SCD subgroup but not in CN subgroup. In addition, we found that lower levels of ApoB were also associated with increased CSF p‐tau (β = −0.15, P = 0.0263) in SCD subgroup, while these associations were not found in the CN subgroup. As for ApoB/A1, interaction between ApoB/A1 and diagnose was obviously associated with the CSF Aβ42/40 (P = 0.0179), p‐tau/Aβ42 (P = 0.0472), and t‐tau/Aβ42 (P = 0.0086) (see Additional File S4). Subgroup analyses showed that significant associations between ApoB/A1 and these CSF biomarkers (Aβ42, Aβ42/40, p‐tau/Aβ42, and t‐tau/Aβ42) were found only in SCD subgroup but not in CN subgroup (see Additional File S5). We did not find any association between ApoA1 and any of the CSF biomarkers (Aβ42, p‐tau, t‐tau, Aβ40, Aβ42/40, p‐tau/Aβ42, and t‐tau/Aβ42).
Figure 3.
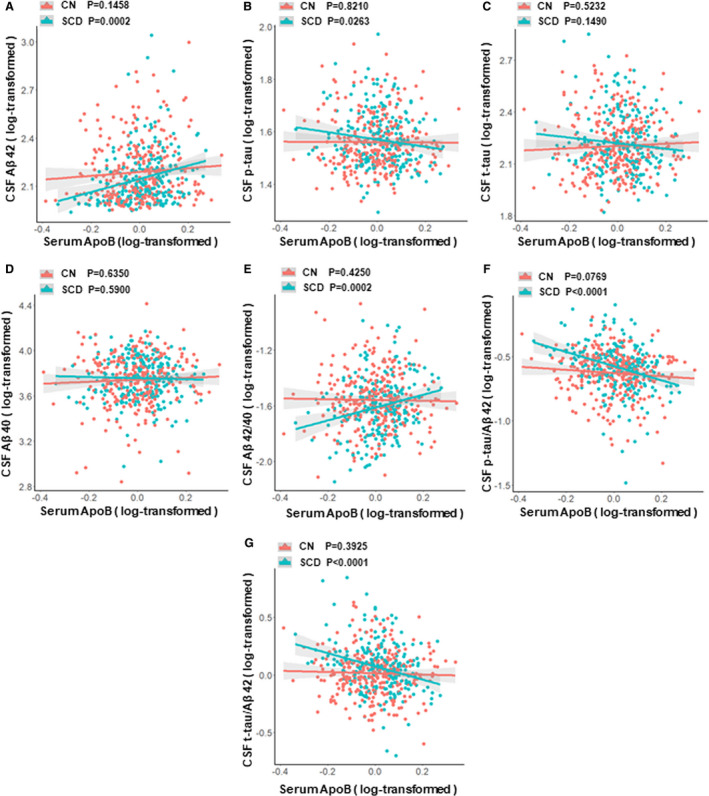
Associations between ApoB and CSF AD biomarkers in different diagnostic subgroups. (A) Associations of ApoB with CSF Aβ42 in CN and SCD subgroups. (B) Associations of ApoB with CSF p‐tau in CN and SCD subgroups. (C) Associations of ApoB with CSF t‐tau in CN and SCD subgroups. (D) Associations of ApoB with CSF Aβ40 in CN and SCD subgroups. (E) Associations of ApoB with CSF Aβ42/40 in CN and SCD subgroups. (F) Associations of ApoB with CSF p‐tau/Aβ42 in CN and SCD subgroups. (G) Associations of ApoB with CSF t‐tau/Aβ42 in CN and SCD subgroups. Abbreviations: CN, cognitively normal participants; SCD, participants with subjective cognitive decline; CSF, cerebrospinal fluid; Aβ, amyloid‐β; p‐tau, phosphorylated tau protein; t‐tau, total tau protein; ApoB, Apolipoprotein B. Multiple linear regression models were used to examine the associations between serum ApoB levels and CSF biomarkers, adjusting for age, sex, education, and APOEɛ4 status.
Then we tested the associations of BMI with serum ApoB and ApoA1. As expected, higher BMI was associated with higher levels of serum ApoB (P = 0.0416) and lower levels of serum ApoA1 (P = 0.0183). After adding BMI as an additional covariable to multiple linear regression models, above associations in total and different diagnostic subgroups still remained (see Additional File S6). Given that there may be different metabolic states of apolipoproteins between overweight and normal weight populations, we further tested our results found above in different weight participants that were classified according to BMI (normal weight participants: BMI < 25, overweight participants: BMI ≥ 25). Interestingly, the interactions between apolipoproteins and weight had obvious influences on the levels of CSF AD biomarkers in total participants and in both diagnostic subgroups (ApoB × weight: P < 0.05) (see Additional File S7). Moreover subgroup analysis showed that, in both CN subgroup and SCD subgroup, almost all of associations found above were more significant in the overweight participants compared with normal weight participants (Tables 3 and 4, Fig. 4).
Table 3.
Results of associations among different weight participants in CN subgroup.
| Variable | Normal‐weight (BMI < 25) N = 124 | Overweight (BMI ≥ 25) N = 164 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ApoB | ApoA1 | ApoB/A1 | ApoB | ApoA1 | ApoB/A1 | |||||||
| β | P | β | P | β | P | β | P | β | P | β | P | |
| CSF Aβ42 | −0.18 | 0.1780 | 0.10 | 0.5250 | −0.21 | 0.1040 | 0.42 | 0.0021 | 0.06 | 0.7382 | 0.37 | 0.0055 |
| CSF p‐tau | −0.10 | 0.1860 | 0.07 | 0.4600 | −0.12 | 0.0746 | 0.05 | 0.4633 | −0.11 | 0.2166 | 0.12 | 0.0889 |
| CSF t‐tau | −0.05 | 0.7070 | −0.11 | 0.4790 | 0.02 | 0.8600 | 0.13 | 0.2766 | −0.11 | 0.4611 | 0.19 | 0.0965 |
| CSF Aβ40 | −0.17 | 0.2670 | 0.00 | 0.9840 | −0.15 | 0.2960 | 0.24 | 0.1340 | −0.21 | 0.3000 | 0.37 | 0.0209 |
| CSF Aβ42/40 | −0.01 | 0.9495 | 0.10 | 0.5373 | −0.06 | 0.6054 | 0.17 | 0.2923 | 0.27 | 0.1920 | 0.00 | 0.9820 |
| CSF p‐tau/Aβ42 | 0.08 | 0.4510 | −0.04 | 0.7810 | 0.09 | 0.3650 | −0.36 | 0.0038 | −0.17 | 0.2811 | −0.18 | 0.2085 |
| CSF t‐tau/Aβ42 | 0.13 | 0.3150 | −0.22 | 0.1790 | 0.21 | 0.1025 | −0.29 | 0.0453 | −0.17 | 0.3588 | −0.25 | 0.0449 |
Abbreviations: CN, cognitively normal participants; BMI, Body Mass Index; N, number of samples; CSF, cerebrospinal fluid; Aβ, amyloid‐β; p‐tau, phosphorylated tau protein; t‐tau, total tau protein; ApoB, Apolipoprotein B; ApoA1, Apolipoprotein A‐1.
Bold indicated that the results were statistically significant.
Multiple linear regression models were used to examine the associations of serum ApoB, ApoA1, and ApoB/A1 levels with CSF biomarkers, adjusting for age, sex, education, and APOEɛ4 status.
Table 4.
Results of associations among different weight participants in SCD subgroup.
| Variable | Normal‐weight (BMI < 25) N = 107 | Overweight (BMI ≥ 25) N = 112 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ApoB | ApoA1 | ApoB/A1 | ApoB | ApoA1 | ApoB/A1 | |||||||
| β | P | β | P | β | P | β | P | β | P | β | P | |
| CSF Aβ42 | 0.40 | 0.0102 | 0.33 | 0.1014 | 0.17 | 0.2272 | 0.40 | 0.0138 | 0.09 | 0.6850 | 0.33 | 0.0364 |
| CSF p‐tau | −0.03 | 0.7480 | −0.08 | 0.5370 | 0.01 | 0.8860 | −0.27 | 0.0021 | −0.29 | 0.0160 | −0.11 | 0.2220 |
| CSF t‐tau | 0.14 | 0.3530 | 0.10 | 0.6003 | 0.07 | 0.6301 | −0.48 | 0.0016 | −0.40 | 0.0555 | −0.25 | 0.1008 |
| CSF Aβ40 | 0.12 | 0.4910 | 0.23 | 0.3060 | −0.01 | 0.9260 | −0.29 | 0.1037 | −0.18 | 0.4591 | −0.18 | 0.2967 |
| CSF Aβ42/40 | 0.28 | 0.1270 | 0.10 | 0.6810 | 0.19 | 0.2680 | 0.69 | 0.0004 | 0.27 | 0.3190 | 0.51 | 0.0076 |
| CSF p‐tau/Aβ42 | −0.43 | 0.0087 | −0.40 | 0.0585 | −0.16 | 0.2960 | −0.67 | 0.0002 | −0.38 | 0.1229 | −0.44 | 0.0131 |
| CSF t‐tau/Aβ42 | −0.26 | 0.1663 | −0.22 | 0.3534 | −0.11 | 0.5414 | −0.88 | <0.0001 | −0.49 | 0.0847 | −0.58 | 0.0042 |
Abbreviations: SCD, participants with subjective cognitive decline; BMI, Body Mass Index; N, number of samples; CSF, cerebrospinal fluid; Aβ, amyloid‐β; p‐tau, phosphorylated tau protein; t‐tau, total tau protein; ApoB, Apolipoprotein B; ApoA1, Apolipoprotein A‐1.
Bold indicated that the results were statistically significant.
Multiple linear regression models were used to examine the associations of serum ApoB, ApoA1, and ApoB/A1 levels with CSF biomarkers, adjusting for age, sex, education, and APOEɛ4 status.
Figure 4.
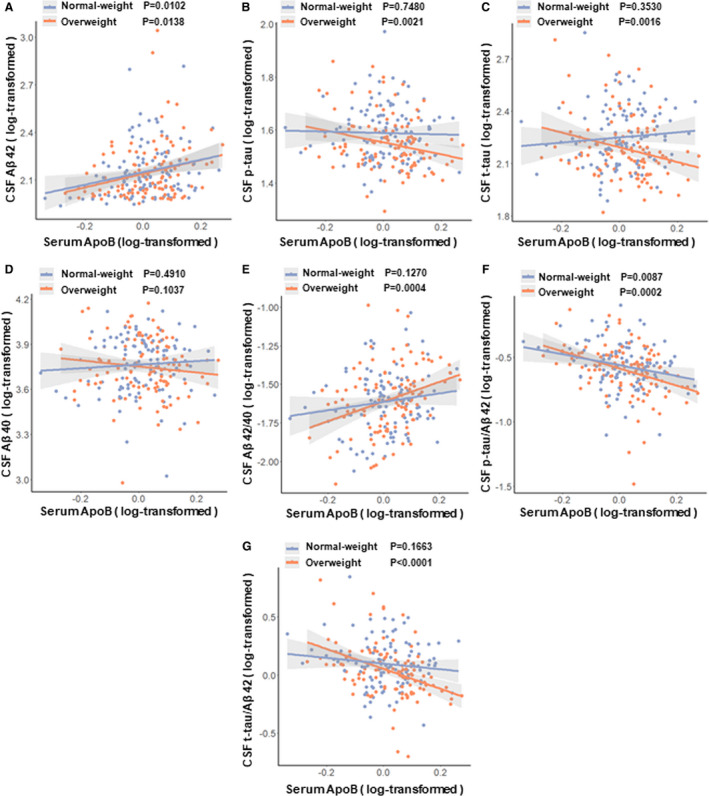
Associations between ApoB and CSF AD biomarkers among different weight participants in SCD subgroup. (A) Associations of ApoB with CSF Aβ42 in normal‐weight and overweight participants. (B) Associations of ApoB with CSF p‐tau in normal‐weight and overweight participants. (C) Associations of ApoB with CSF t‐tau in normal‐weight and overweight participants. (D) Associations of ApoB with CSF Aβ40 in normal‐weight and overweight participants. (E) Associations of ApoB with CSF Aβ42/40 in normal‐weight and overweight participants. (F) Associations of ApoB with CSF p‐tau/Aβ42 in normal‐weight and overweight participants. (G) Associations of ApoB with CSF t‐tau/Aβ42 in normal‐weight and overweight participants. Abbreviations: CSF, cerebrospinal fluid; Aβ, amyloid‐β; p‐tau, phosphorylated tau protein; t‐tau, total tau protein; ApoB, Apolipoprotein B. Multiple linear regression models were used to examine the associations between serum ApoB levels and CSF biomarkers, adjusting for age, sex, education and APOEɛ4 status.
Discussion
This study is the first to systematically explore associations of CSF AD core biomarkers with ApoB, ApoA1, and ApoB/A1 in elderly adults without objective cognitive impairment. The main finding of this study was that the levels of ApoB were associated with CSF AD core biomarkers in elderly adults without objective cognitive impairment independently of APOEε4 genotype, especially in the SCD population. This finding was novel and potentially important, because it suggested that levels of serum ApoB added information beyond APOEε4 status on the level of CSF AD core biomarkers in the preclinical stage, which may contribute to the discovery of new pathogenic mechanisms and therapeutic targets.
First of all, we analyzed the characteristics of SCD participants included in our study. Results showed that, compared with the CN subgroup, although there was no significant cognitive decline, there were obvious changes in CSF AD core biomarkers (especially Aβ‐related biomarkers) in the SCD subgroup, which were consistent with previous studies. 8 , 26 In fact, accumulating cross‐sectional or longitudinal evidence has supported that SCD occurred at the preclinical stage of AD and might serve as a symptomatic indicator of preclinical AD. 7 , 8 , 26 , 27 , 28 , 29 , 30 , 31 , 32 Our analysis of population characteristics showed that, in our study, SCD participants had preclinical AD characteristics.
Then we explored the distribution of ApoB and ApoA in the diagnostic and biological constructs. Results showed that participants with significant AD biological characteristics had a lower ApoB level suggesting that the level of ApoB may decrease in the preclinical stage of AD.
Finally, we detailed the associations of ApoB, ApoA1, and ApoB/A1 with CSF AD core biomarkers in total participants or different diagnostic subgroups and analyzed the factors that might affect these associations. We firstly found that serum ApoB and ApoB/A1 were associated with CSF AD core biomarkers, especially with the Aβ‐related biomarkers (Aβ42, Aβ42/40, p‐tau/Aβ42, and t‐tau/Aβ42). It is worth noting that these associations were much more significant in SCD participants. Surprisingly, inconsistent with previous studies, the results found above showed protective associations. To be precise, lower ApoB and ApoB/A1 were associated with decreased CSF Aβ42 and Aβ42/40, as well as with increased CSF p‐tau, p‐tau/Aβ42, and t‐tau/Aβ42. No previous studies have focused on the associations of serum ApoB and ApoB/A1 with CSF AD core biomarkers in cognitively normal or SCD participant. However, two autopsy studies in AD patients indicated that ApoB may be a risk factor for the late stage of AD. One showed that serum ApoB were positive associated with the amount of Aβ42 in AD brains. 14 Another showed that ApoB immunoreactivity was positively associated with cerebral amyloids. 13 The exact causes of these seemingly contradictory results were still unclear, but we thought that there were several possible reasons as follows.
First, different stages of the disease might contribute to this contradiction. Previous studies have found that changes in the blood–brain barrier contributed differently to the dynamic changes in CSF AD core biomarkers in different stages of disease. CSF AD core biomarker (especially Aβ42) may be temporarily elevated and its significant deposition in brain may occur when the blood–brain barrier was significantly impaired. 33 As a possible transporter of CSF biomarkers, ApoB may facilitate the processes of early clearance and late deposition of biomarkers in brain. In addition, in the late stage of the disease, atherosclerosis and other vascular damages caused by ApoB‐LDL may also aggravate the pathological changes, which may not be apparent in the early stage of the disease.
Second, genetic factors may also contribute to this contradiction. In this study, we found that these protective associations were independent of the APOEε4 status, based on the following evidences. a) Both ApoB and APOEε4 status were associated with CSF AD core biomarkers, but their interaction was not. b) The results of the subgroups analysis were similar among population with different APOEε4 status. This independence on APOEε4 was also seen in a previous study on the association between serum ApoB and amount of Aβ42 in AD brains. 14 Collectively, these data provided evidences that ApoB and APOEε4 status may contribute to the changes in CSF AD core biomarkers, independently of each other. However, a recent study found that rare genetic coding variants of APOB gene were strongly associated with familial AD, which indicated that APOB gene might also harbor protective and deleterious variants in sporadic AD similar to the APOEε2 and APOEε4 alleles. 34 Identifying such a protective coding variant of APOB gene would greatly strengthen the link between ApoB and AD pathogenesis.
In addition, some other factors may also contribute to this contradiction, such as changes involved low density lipoprotein cholesterol metabolism (the low density lipoprotein receptor and proprotein convertase subtilisin/kexin type 9). So given the concept that normal weight populations and overweight populations often have different lipid metabolism status, we further tested our results found above in different weight participants. It is worth noting that these protective associations of serum ApoB with CSF AD core biomarkers were particularly stronger in overweight participants compared with the normal weight participants. These results indicated that different lipid metabolism status may also influence above associations, which may contribute to the contradiction. Moreover growing body of studies suggested that overweight in older age may be a protective factor for AD. Overweight older adults, especially metabolic health individuals that were similar to the participants in our study, had a lower incidence or a later onset of AD than those of normal weight or low weight. 35 , 36 , 37 Our results indicated that the protective effect of serum ApoB on CSF AD core biomarkers may be the potential mechanism. In other words, the serum ApoB's potential ability to alleviate early pathological changes may contribute to slow the progression of the disease in overweight older adults, but not in normal weight older adults.
Overall, we found protective associations of serum ApoB with CSF AD core biomarkers, these associations may be affected by disease’s stages and lipid metabolism status or some other indefinite factors such as genetic factors. In this study we did not find any associations between ApoA1 and CSF AD core biomarkers. However, a recent liquid chromatography‐tandem mass spectrometry based proteomics study highlighted plasma ApoA1 as a biomarker in Alzheimer's disease and Aβ burden, even in cognitively unimpaired individuals. 38 , 39 So the effects of ApoA1 on early pathological changes in population without objective cognitively impairment still need to be tested in more studies.
There were some potential limitations in our study. For example, this was a cross‐sectional study and this protective association found in this study did not confirm any causative effects. However, it did give us new perspectives to explore the potential causes or new mechanisms. All these results still needed to be tested in larger longitudinal cohort. In addition, the associations of CSF apolipoproteins with CSF AD‐related biomarkers and serum apolipoproteins in preclinical stage of AD were still worth to explore in further studies, which will help to further elucidate the relationship between apolipoprotein and AD.
In conclusion, this study was the first to find some protective associations of serum ApoB, but not ApoA1, with CSF AD core biomarkers in the preclinical stage of AD. This finding indicated that ApoB may play different roles in different stages of AD. Further studies to identify the underlying mechanisms would greatly strengthen the link between ApoB and AD pathogenesis, which may contribute to the discovery of new pathogenic mechanisms and therapeutic targets.
Conflict of Interest
The authors declare that they have no competing interests.
Supporting information
Additional File S1. Associations of age and APOE ε4 status for CSF measures of Aβ42.
Additional File S2. The frequency distribution histogram of serum ApoB and ApoA1.
Additional File S3. Associations between ApoB and CSF AD biomarkers after adjusting for interaction between ApoB and APOEε4 status.
Additional File S4. Influences of the interactions between apolipoproteins and diagnose on CSF AD biomarkers.
Additional File S5. Results of associations in different diagnostic subgroups.
Additional File S6. Results of associations after adjusting BMI in different diagnostic groups.
Additional File S7. Influences of the interactions between ApoB and weight on CSF AD biomarkers.
Acknowledgments
The authors thank all the participants of the present study as well as all colleagues who have made contributions to build the CABLE cohort. This study was supported by grants from the National Natural Science Foundation of China (91849126), the National Key R&D Program of China (2018YFC1314700), Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01) and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University. The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Funding information
This study was supported by grants from the National Natural Science Foundation of China (91849126), the National Key R&D Program of China (2018YFC1314700), Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01).
Funding Statement
This work was funded by Frontiers Center for Brain Science of Ministry of Education, Fudan University grant ; ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute grant ; National Natural Science Foundation of China grant 91849126; Shanghai Municipal Science and Technology Major Project grant 2018SHZDZX01; National Key R&D Program of China grant 2018YFC1314700.
References
- 1. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 2016;8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lane CA, Hardy J, Schott JM. Alzheimer's disease. Eur J Neurol 2018;25:59–70. [DOI] [PubMed] [Google Scholar]
- 3. Weiner MW, Aisen PS, Jack CR Jr, et al. The Alzheimer's disease neuroimaging initiative: progress report and future plans. Alzheimers Dement 2010;6(202–211):e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta‐analysis. Lancet Neurol 2016;15:673–684. [DOI] [PubMed] [Google Scholar]
- 5. Molinuevo JL, Ayton S, Batrla R, et al. Current state of Alzheimer's fluid biomarkers. Acta Neuropathol 2018;136:821–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glodzik‐Sobanska L, Reisberg B, De Santi S, et al. Subjective memory complaints: presence, severity and future outcome in normal older subjects. Dement Geriatr Cogn Disord 2007;24:177–184. [DOI] [PubMed] [Google Scholar]
- 8. Wolfsgruber S, Molinuevo JL, Wagner M, et al. Prevalence of abnormal Alzheimer's disease biomarkers in patients with subjective cognitive decline: cross‐sectional comparison of three European memory clinic samples. Alzheimer's Res Ther 2019;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown MS, Goldstein JL. How LDL receptors influence cholesterol and atherosclerosis. Sci Am 1984;251:58–66. [DOI] [PubMed] [Google Scholar]
- 10. Emerging Risk Factors C , Di Angelantonio E, Gao P, et al. Lipid‐related markers and cardiovascular disease prediction. JAMA 2012;307:2499–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raygani AV, Rahimi Z, Kharazi H, et al. Association between apolipoprotein E polymorphism and serum lipid and apolipoprotein levels with Alzheimer's disease. Neurosci Lett 2006;408:68–72. [DOI] [PubMed] [Google Scholar]
- 12. Merched A, Xia Y, Visvikis S, et al. Decreased high‐density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer's disease. Neurobiol Aging 2000;21:27–30. [DOI] [PubMed] [Google Scholar]
- 13. Namba Y, Tsuchiya H, Ikeda K. Apolipoprotein B immunoreactivity in senile plaque and vascular amyloids and neurofibrillary tangles in the brains of patients with Alzheimer's disease. Neurosci Lett 1992;134:264–266. [DOI] [PubMed] [Google Scholar]
- 14. Kuo YM, Emmerling MR, Bisgaier CL, et al. Elevated low‐density lipoprotein in Alzheimer's disease correlates with brain abeta 1–42 levels. Biochem Biophys Res Comm 1998;252:711–715. [DOI] [PubMed] [Google Scholar]
- 15. Tynkkynen J, Hernesniemi JA, Laatikainen T, et al. Apolipoproteins and HDL cholesterol do not associate with the risk of future dementia and Alzheimer's disease: the National Finnish population study (FINRISK). Age 2016;38:465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jessen F, Amariglio RE, van Boxtel M, et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer's disease. Alzheimers Dement 2014;10:844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Molinuevo JL, Rabin LA, Amariglio R, et al. Implementation of subjective cognitive decline criteria in research studies. Alzheimers Dement 2017;13:296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid‐beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010;67:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275–1283. [DOI] [PubMed] [Google Scholar]
- 21. Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid‐beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci USA 2009;106:6820–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community‐based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 23. Hulette CM, Welsh‐Bohmer KA, Murray MG, et al. Neuropathological and neuropsychological changes in "normal" aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol 1998;57:1168–1174. [DOI] [PubMed] [Google Scholar]
- 24. Knopman DS, Parisi JE, Salviati A, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 2003;62:1087–1095. [DOI] [PubMed] [Google Scholar]
- 25. Soldan A, Pettigrew C, Cai Q, et al. Hypothetical preclinical Alzheimer disease groups and longitudinal cognitive change. JAMA Neurol 2016;73:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miebach L, Wolfsgruber S, Polcher A, et al. Which features of subjective cognitive decline are related to amyloid pathology? Findings from the DELCODE study. Alzheimer's Res Ther 2019;11:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jessen F, Wiese B, Bachmann C, et al. Prediction of dementia by subjective memory impairment: effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry 2010;67:414–422. [DOI] [PubMed] [Google Scholar]
- 28. Reisberg B, Shulman MB, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement 2010;6:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y, Risacher SL, West JD, et al. Altered default mode network connectivity in older adults with cognitive complaints and amnestic mild cognitive impairment. J Alzheimers Dis 2013;35:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perrotin A, Mormino EC, Madison CM, et al. Subjective cognition and amyloid deposition imaging: a Pittsburgh Compound B positron emission tomography study in normal elderly individuals. Arch Neurol 2012;69:223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prichep LS, John ER, Ferris SH, et al. Prediction of longitudinal cognitive decline in normal elderly with subjective complaints using electrophysiological imaging. Neurobiol Aging 2006;27:471–481. [DOI] [PubMed] [Google Scholar]
- 32. van Harten AC, Visser PJ, Pijnenburg YA, et al. Cerebrospinal fluid Abeta42 is the best predictor of clinical progression in patients with subjective complaints. Alzheimers Dement 2013;9:481–487. [DOI] [PubMed] [Google Scholar]
- 33. Potter R, Patterson BW, Elbert DL, et al. Increased in vivo amyloid‐beta42 production, exchange, and loss in presenilin mutation carriers. Sci Transl Med 2013;5:189ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wingo TS, Cutler DJ, Wingo AP, et al. Association of early‐onset Alzheimer disease with elevated low‐density lipoprotein cholesterol levels and rare genetic coding variants of APOB. JAMA Neurol 2019;76:809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hughes TF, Borenstein AR, Schofield E, et al. Association between late‐life body mass index and dementia: The Kame Project. Neurology 2009;72:1741–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee JY, Han K, Han E, et al. Risk of incident dementia according to metabolic health and obesity status in late life: a population‐based cohort study. J Clin Endocrinol Metab 2019;104:2942–2952. [DOI] [PubMed] [Google Scholar]
- 37. Tolppanen AM, Ngandu T, Kareholt I, et al. Midlife and late‐life body mass index and late‐life dementia: results from a prospective population‐based cohort. J Alzheimer's Dis 2014;38:201–209. [DOI] [PubMed] [Google Scholar]
- 38. Westwood S, Leoni E, Hye A, et al. Blood‐based biomarker candidates of cerebral amyloid using PiB PET in non‐demented elderly. J Alzheimers Dis 2016;52:561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Slot RE, Van Harten AC, Kester MI, et al. Apolipoprotein A1 in cerebrospinal fluid and plasma and progression to Alzheimer's disease in non‐demented elderly. J Alzheimer's Dis 2017;56:687–697. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional File S1. Associations of age and APOE ε4 status for CSF measures of Aβ42.
Additional File S2. The frequency distribution histogram of serum ApoB and ApoA1.
Additional File S3. Associations between ApoB and CSF AD biomarkers after adjusting for interaction between ApoB and APOEε4 status.
Additional File S4. Influences of the interactions between apolipoproteins and diagnose on CSF AD biomarkers.
Additional File S5. Results of associations in different diagnostic subgroups.
Additional File S6. Results of associations after adjusting BMI in different diagnostic groups.
Additional File S7. Influences of the interactions between ApoB and weight on CSF AD biomarkers.