

Abstract
Objective
To characterize the progression of brain structural abnormalities in adults with pediatric and adult/late onset DM1, as well as to examine the potential predictive markers of such progression.
Methods
21 DM1 patients (pediatric onset: N = 9; adult/late onset: N = 12) and 18 healthy controls (HC) were assessed longitudinally over 9.17 years through brain MRI. Additionally, patients underwent neuropsychological, genetic, and muscular impairment assessment. Inter‐group comparisons of total and voxel‐level regional brain volume were conducted through Voxel Based Morphometry (VBM); cross‐sectionally and longitudinally, analyzing the associations between brain changes and demographic, clinical, and cognitive outcomes.
Results
The percentage of GM loss did not significantly differ in any of the groups compared with HC and when assessed independently, adult/late DM1 patients and their HC group suffered a significant loss in WM volume. Regional VBM analyses revealed subcortical GM damage in both DM1 groups, evolving to frontal regions in the pediatric onset patients. Muscular impairment and the outcomes of certain neuropsychological tests were significantly associated with follow‐up GM damage, while visuoconstruction, attention, and executive function tests showed sensitivity to WM degeneration over time.
Interpretation
Distinct patterns of brain atrophy and its progression over time in pediatric and adult/late onset DM1 patients are suggested. Results indicate a possible neurodevelopmental origin of the brain abnormalities in DM1, along with the possible existence of an additional neurodegenerative process. Fronto‐subcortical networks appear to be involved in the disease progression at young adulthood in pediatric onset DM1 patients. The involvement of a multimodal integration network in DM1 is discussed.
Introduction
Myotonic Dystrophy type 1 (DM1) is the most common form of adult muscular dystrophy. It is an autosomal dominant disorder affecting multiple systems. The severity of symptoms varies according to age of onset, with earlier onset patients being more severely affected.
Aside from the known progressive nature of the muscular impairment, many of the clinical symptoms have been suggested to be part of an accelerated aging process. CNS studies with a focus on brain pathology have defined DM1 as a combination of tauopathy, spliceopathy, and RNAopathy, all of which contribute to neurodegeneration. 1 Furthermore, from a neuropsychological perspective, recent findings suggest that cognitive functions suffer a decline in several areas, beyond that expected in normal aging. 2 , 3 , 4 , 5 , 6
Although neuroimaging data are thought to support the previously suggested hypothesis of neurodegenerative processes in DM1, to the best of our knowledge, there have been only two previous attempts to longitudinally study a hypothesized progressive impairment in brain structures. While Gliem et al 7 did not find a greater volume loss over time in DM1 for either gray or white matter tissue, Conforti et al. found a progression in white matter lesions along with greater brain atrophy assessed by the ventricular/brain ratio. 8
The aim of this study was to longitudinally assess the structural brain changes of DM1 patients over a period of more than 9 years and to delineate the pathway of disease progression. Additionally, we aimed to examine the potential genetic, muscular, clinical, and cognitive markers of this progression.
Materials and Methods
Participants
The DM1 patients analyzed in this work were selected from those attending the Neurology Department of the Donostia University Hospital (Gipuzkoa, Spain). Healthy controls (HC) included accompanying relatives and non‐relatives of DM1 patients using the service.
Inclusion criteria for DM1 patients included being older than 18 years with molecular confirmation of the clinical diagnosis. Patients were excluded if any of the following criteria were met both at baseline and at follow‐up: congenital form, history of major psychiatric or somatic disorder, acquired brain damage or alcohol or drug abuse, presence of corporal paramagnetic body devices that could impede an MRI study, and the presence of cerebral anomalies that could affect the volumetric analysis. HC participants were required to satisfy the same inclusion criteria, except for the clinical diagnosis. The DM1 participants were classified into two groups according to their age of onset based on the most recently proposed classification (OMMYD‐4): adults with pediatric onset DM1 when age of onset was between 1‐18 years old, and adults with adult and late onset DM1 (from now on adult/late DM1) when patients had either adult onset (18‐40 years old) or late onset (>40 years old).
Figure 1 shows flow chart of the recruitment process. From the healthy volunteers with a valid MRI at baseline, only those whose age, gender and years of education were equal or closely similar to any re‐scanned patient were invited to participate in order to form demographically equivalent groups. The first 20 participants that agreed to be re‐scanned were suitable for forming two groups equivalent in sex, age, and years of education with a valid sample size for purposes of comparison with each of the DM1 groups.
Figure 1.
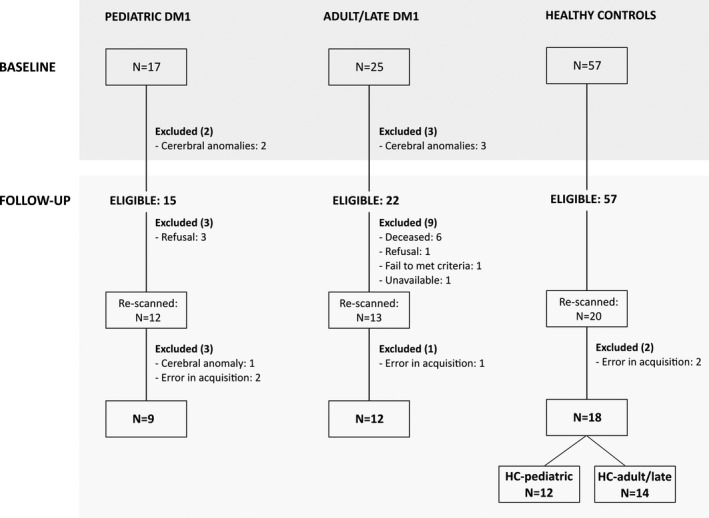
Flow‐chart of sample recruitment from baseline to follow‐up. DM1 = Myotonic Dystrophy Type 1; HC = healthy controls.
A final group of 21 DM1 patients (9 pediatric and 12 adult/late) and one of 18 HC were included for the analysis. The 18 HC controls were then subdivided to form the two comparison groups: one for comparisons with pediatric onset DM1 (HC‐pediatric: N = 12) and one for comparisons with adult/late onset DM1 (HC‐adult/late: N = 14). The healthy volunteers were included to form each control group in a controlled selection process, ensuring gender equivalence, and mean age differences between groups of less than 4 years, which were non‐significant and with small effect sizes. All participants were informed of the objectives and details of the study and signed an informed consent form. The study was approved by the Ethics Committee of the Donostia University Hospital.
Clinical and neuropsychological assessment
Clinical data were extracted from medical records. Additionally, both at baseline and at follow‐up, all patients were clinically examined by a neurologist with the Muscular Impairment Rating Scale (MIRS) 9 and underwent a neuropsychological assessment.
All patients were examined by an experienced neuropsychologist who was blind to the patient’s clinical condition (CTG expansion size, clinical form, maternal or paternal inheritance pattern, muscular impairment, and MRI results). Neuropsychological assessment included the following subtests from the Wechsler Adult Intelligence Scale III (WAIS III) 10 : Block design, Digit span, Object assembly, Arithmetic, Similarities and Vocabulary. An estimated IQ score was calculated from a two subtest short form (Block design and Vocabulary) with high reliability (rxx=.93) and validity (r=.87) based on Sattler and Ryan. 11 Other cognitive tests used were: Stroop test, 12 California Computerized Assessment Package (CALCAP), 13 Rey Auditory Verbal Learning Test (RAVLT), 14 phonemic (P) and semantic (animals) verbal fluency test, 15 , 16 Rey‐Osterrieth Complex Figure test (ROCF), 17 Raven’s progressive matrices, 18 Benton’s Judgement of Line Orientation, 19 and the Wisconsin Card Sorting Test. 20 Raw scores were converted into standardized T values according to Spanish norms for each test.
MRI acquisition and data preprocessing
MR scanning was conducted on a 1.5 Tesla scanner (Achieva Nova, Philips). The current results are based on a high‐resolution volumetric “turbo field echo” (TFE) series (Sagital 3D T1 weighted acquisition, TR = 7.2, TE = 3.3, flip angle = 8, matrix = 256 x 232, slice thickness 1mm, voxel dimensions of 1mm x 1mm x 1mm, NSA = 1, no slices 160, gap = 0, total scan duration 5´34¨). All the scans, both at baseline and at follow‐up, were acquired on the same MR scanner.
To study voxel‐based GM volume loss in DM1 patients and its association with different clinical and neuropsychological outcomes, FSL (version 6.01) Voxel Based Morphometry (VBM) was used, 21 which is an optimized VBM protocol 22 carried out with FSL tools. 23 First, structural images were brain‐extracted and GM‐segmented before being registered to the MNI 152 standard space using non‐linear registration. 24 The resulting images were averaged and flipped along the x‐axis to create a left‐right symmetric, study‐specific GM template. Second, all native GM images were non‐linearly registered to this study‐specific template and "modulated" to correct for local expansion (or contraction) due to the non‐linear component of the spatial transformation. The modulated GM images were then smoothed with an isotropic Gaussian kernel with a sigma of 3.
The same VBM procedure was applied to the WM‐segmented images.
To estimate global GM and WM brain tissue volume, normalized for subject head size, the SIENAX tool was used. 25
Statistical analysis
For all statistical analyses listwise deletion was used to deal with missing values. Demographic (sex, age at baseline, age at follow‐up, years of education) and clinical (CTG repeats, inheritance pattern, and MIRS score) data were analyzed using the SPSS (IBM SPSS Statistics 24) statistical package. Inter‐group comparisons were conducted to compare DM1 patients and HC, as well as the two DM1 groups, using contingency analysis (Chi‐square) for categorical data and parametric (t‐test) or non‐parametric (Mann–Whitney U) for interval data, where appropriate. In order to dismiss the possibility of a higher functioning sample in the re‐tested groups (selective attrition), the same analyses were employed to compare those patients that failed to re‐test at follow‐up and those who were retested. Intra‐group analysis of the longitudinal evolution of clinical and neuropsychological data was conducted using the Wilcoxon signed‐rank test.
In order to address the general objective of this study, three main analyses were carried out (note that no direct statistical comparison was made between pediatric onset and adult/late onset DM1 groups):
Total GM and WM volume analyses: Cross‐sectional and longitudinal: Intra‐group and inter‐group comparisons
Total GM and WM volume comparisons, both inter‐group and intra‐group, were analyzed using the SPSS statistical package. All analyses were conducted by correcting the volume with the head size. Inter‐group cross‐sectional comparisons of total GM and WM volumes were conducted to compare DM1 patients and HC at baseline and at follow‐up, using a parametric t‐test (the data met the assumptions for parametric tests). Longitudinal analyses of GM and WM volumes were separately assessed for DM1 patients and HC using the Wilcoxon signed‐rank test. Finally, the percentage of volume loss from baseline to follow‐up in each group was calculated and comparisons were made between each DM1 group and their HC group, using a univariate ANOVA test corrected for time between the baseline and the follow‐up scan.
Regional GM and WM volume analysis from baseline to follow‐up: VBM inter‐group analysis
A general linear model was used to compute the inter‐group statistical analysis using FSL, controlling for age and head size. All the results were obtained using two‐tailed tests and corrected for multiple comparisons using the Monte Carlo simulation cluster‐wise correction, as implemented in the AFNI software (version 19.3.00) (https://afni.nimh.nih.gov/) with 10,000 iterations to estimate the probability of false positive clusters with a P value < 0.05. Only clusters of more than 50 contiguous voxels were reported.
Association between brain volume and demographic, clinical, and neuropsychological outcomes: VBM intra‐group analysis
To study the potential predictive capacity of demographic, clinical, and neuropsychological variables at baseline, three separate analyses were conducted.
First, in order to assess the predictive capacity of these variables at baseline (CTG, MIRS, years of education, disease inheritance, and neuropsychological scores) for volume loss (volume variations between follow‐up and baseline), partial correlation analyses controlling for age, head size, and time span from baseline to follow‐up scan were conducted. The results were further corrected by multiple comparisons using the false discovery rate (FDR) strategy. These analyses were conducted separately for pediatric and adult/late onset DM1 groups.
Second, in order to assess the predictive capacity of the same variables for the image at follow‐up, partial correlation analyses were conducted, controlling for age, head size and time from baseline assessment to follow‐up scan, between predictive variables and global GM and WM volume at follow‐up. These analyses were conducted separately for pediatric and adult/late onset DM1 groups.
Finally, to assess the association between the previous variables and regional GM volume at follow‐up, a general linear model analysis was applied to evaluate the relationship between GM volume and CTG, MIRS clinical scale, years of education, and inheritance pattern of the disease, as well as the outcomes of neuropsychological tests. The results were controlled for age, head size and time span between the baseline scan and the follow‐up (except for neuropsychological variables, which were adjusted for time between the baseline neuropsychological assessment and the follow‐up scan). The statistical tests were two‐tailed and corrected for multiple comparisons using Monte Carlo simulations with cluster‐wise correction after 10,000 iterations to estimate the probability of false positive clusters with a P value < 0.05. After corrections, a mask was applied using the results of the group difference to look for any overlap with the regions where DM1 patients show a significant GM volume loss compared with controls. Only clusters of more than 50 contiguous voxels were reported. Analyses were conducted separately for pediatric and adult/late onset DM1 groups.
Results
Statistically significant differences were found between the excluded and the included DM1 patients for the following variables: CTG expansion size at baseline (t(34) = 3.276; p= .002, d = 1.1) with more repetitions in the excluded DM1 (mean = 1002.88, SD = 432.88) than the included DM1 (mean = 534.4, SD = 421.21) and MIRS score at baseline (U = 45.5; P = 0.001; r = 0.58), with the excluded DM1 presenting greater muscular impairment (mean = 3.62, SD = 0.87, mean rank = 24.5) than the included DM1 (mean = 2.29, SD = 0.96 mean rank = 13.17). No difference was found between the excluded and the included DM1 patients in terms of sex, inheritance pattern, years of education, age, and IQ estimate.
The demographic and clinical characteristics of the sample are summarized in Tables 1 and 2. None of the DM1 patient groups differed from the corresponding HC groups in terms of sex, age at baseline, age at follow‐up or years of education. Pediatric onset DM1 patients were significantly younger and had a greater CTG expansion at both baseline and follow‐up compared with adult/late onset DM1 patients. Both groups showed a significant increase in MIRS score and CTG repeat size from baseline to follow up. The mean time from baseline to follow‐up was 9.17 (SD = 0.47) years for the complete sample. Neuropsychological outcomes of the DM1 groups are shown in Supplementary Table S1.
Table 1.
Baseline and follow‐up demographic characteristics of the sample divided into 4 subgroups: pediatric DM1, adult/late DM1, HC‐pediatric, HC‐adult/late.
| Pediatric DM1 (N = 9) | HC‐pediatric (N = 12) | Statistic | P | Effect size | |||
|---|---|---|---|---|---|---|---|
| Mean/N(%) | (SD) | Mean/N(%) | (SD) | ||||
| Sex | |||||||
| Male | 4 (44.4%) | 5 (41.7%) | X 2 = .016 | 0.899 | V = .028 | ||
| Female | 5 (55.6%) | 7 (58.3%) | |||||
| Age at baseline | 30 | (6.59) | 33.5 | (8.32) | t = 1.039 | 0.312 | d = .46 |
| Age at follow‐up | 39.67 | (6.61) | 42.5 | (8.06) | t = 0.858 | 0.401 | d = .36 |
| Years of education | 14.22 | (4.26) | 18.89 | (6.49) | U = 22.5 | 0.111 | r = .35 |
| Adult/late DM1 (N = 12) | HC‐adult/late (N = 14) | Statistic | P | Effect size | |||
|---|---|---|---|---|---|---|---|
| Mean/N(%) | (SD) | Mean/N(%) | (SD) | ||||
| Sex | |||||||
| Male | 6 (50%) | 8 (57.1%) | X2 = 0.133 | 0.716 | V = 0.071 | ||
| Female | 6 (50%) | 6 (42.9%) | |||||
| Age at baseline | 45.83 | (9.05) | 43.64 | (8.11) | t = −0.651 | 0.521 | d = 0.26 |
| Age at follow‐up | 55.17 | (8.89) | 52.71 | (8.27) | t = −0.728 | 0.473 | d = 0.29 |
| Years of education | 13.42 | (6.68) | 17.64 | (5.93) | t = 0.976 | 0.340 | d = 0.41 |
| Pediatric DM1 (N = 9) | Adult/late DM1 (N = 12) | Statistic | P | Effect size | |||
|---|---|---|---|---|---|---|---|
| Mean/N(%) | (SD) | Mean/N(%) | (SD) | ||||
| Sex | |||||||
| Male | 4 (44.4%) | 6 (50%) | X2 = 0.064 | 0.801 | V = 0.055 | ||
| Female | 5 (55.6%) | 6 (50%) | |||||
| Age at baseline | 30 | (6.59) | 45.83 | (9.05) | t = −4.427 | 0.000 | d = 1.95 |
| Age at follow‐up | 39.67 | (6.61) | 55.17 | (8.89) | t = −4.387 | 0.000 | d = 1.93 |
| Inheritance | |||||||
| Maternal | 6 (66.7%) | 3 (27.3%) | X2 = 3.104 | 0.078 | V = 0.394 | ||
| Paternal | 3 (33.3%) | 8 (72.7%) | |||||
| CTG at baseline | 851.89 | (444.77) | 362.83 | (325.54) | t = 2.916 | 0.009 | d = 1.29 |
| CTG at follow‐up | 1044.44 | (444.43) | 500.75 | (504.03) | U = 21.500 | 0.021 | r = 0.05 |
| MIRS at baseline | 2,56 | (0.88) | 2.08 | (1) | U = 41.000 | 0.382 | r = 0.22 |
| MIRS at follow‐up | 3.11 | (1.17) | 2.67 | (1.37) | U = 46.500 | 0.602 | r = 0.12 |
DM1: Myotonic Dystrophy Type 1; HC: Healthy controls; SD: Standard Deviation. Descriptive data are shown as mean and SD for age at baseline, age at follow‐up and years of education. Frequency (N) and percentage (%) are shown only for sex.
Table 2.
Longitudinal evolution of clinical features in pediatric DM1 and adult/late DM1 groups.
| N | Baseline | Follow‐up | Statistic | P | Effect size | |||
|---|---|---|---|---|---|---|---|---|
| Mean | (SD) | Mean | (SD) | |||||
| Pediatric DM1 | ||||||||
| CTG | 9 | 851.89 | (444.77) | 1044.44 | (444.43) | t = −3.181 | 0.013 | d = 1.5 |
| MIRS | 9 | 2.56 | (0.88) | 3.11 | (1.17) | t = −3.162 | 0.013 | d = .75 |
| Adult/late DM1 | ||||||||
| CTG | 12 | 362.83 | (325.54) | 500.75 | (504.03) | Z = −2.31 | 0.021 | r = .47 |
| MIRS | 12 | 2.08 | (0.99) | 2.67 | (1.37) | Z = −2.33 | 0.020 | r = .48 |
DM1: Myotonic Dystrophy Type 1; HC: Healthy controls; SD: Standard Deviation.
Total GM and WM volume analyses: cross‐sectional and longitudinal: Intra‐group and inter‐group analyses
With regard to cross‐sectional comparisons of brain volume, patients in the pediatric onset group presented lower volumes of both GM and WM compared with controls, both at baseline and at follow up. Conversely, the adult and late onset group obtained lower volumes only for GM when compared with controls (Table 3). Longitudinal analyses of GM and WM volume loss over time showed that none of the groups suffered a significant decrease in GM volume, and only adult/late DM1 patients and their HC group suffered a significant loss in WM volume. Nonetheless, when comparing the longitudinal variations between groups, the percentage of volume loss did not differ between pediatric onset DM1 and their controls, or between adult/late onset DM1 and their controls. However, the global GM volume decrease in the pediatric group reached 3.86% compared with a 0.63% decrease in HC, and, despite being non‐significant, the effect size was large (ES = 0.94). Similarly, a medium effect size (ES = 0.57) was found for the difference between the percentage of GM volume loss in adult/late onset DM1 (2.71%) and HC‐adult/late (0.85%). Accordingly, moderate effect sizes were found in patients’ GM volume loss when intra‐group analyses were conducted for both pediatric and adult/late onset patients (r = 0.43 and r = 0.4, respectively).
Table 3.
Cross‐sectional (inter‐group) and longitudinal (intra‐group and inter‐group) comparisons for GM and WM volume.
| N | Mean | SD | DM1 versus HC | Mean | SD | DM1 versus HC | Intra‐group Follow‐up ‐ Baseline | Intergroup comparison of percentage of volume loss | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Z | P | Mean | F | P | gHedges | ||||||||
| Baseline | Follow‐up | ||||||||||||
| Pediatric DM1 versus HC‐pediatric | |||||||||||||
| GM volume | t = 4.208 | t = 5.071 | |||||||||||
| DM1 | 9 | 728492.55 | (59435.88) | p = 0.000 | 703854.60 | (53407.41) | P = 0.000 | −1.836 | 0.066 | −3.86 ¶ | 3.497 | 0.078 | 0.94 |
| HC | 12 | 829069.52 | (50050.01) | d = 1.86 | 819800.71 | (50693.01) | d = 2.24 | −1.569 | 0.117 | −0.63 ¶ | |||
| WM volume | |||||||||||||
| DM1 | 9 | 656340.98 | (45830.39) | t = 4.334 | 658010.04 | (28468.26) | t = 4.643 | −0.296 | 0.767 | −0.25 ¶ | 0.097 | 0.759 | 0.16 |
| HC | 12 | 721993.51 | (22597.64) | p = 0.000 | 712342.09 | (25040.87) | p = 0.000 | −1.412 | 0.158 | −0.77 ¶ | |||
| d = 1.91 | d = 2.05 | ||||||||||||
| Adult/late DM1 versus HC‐adult/late | |||||||||||||
| GM volume | t = 2.172 | t = 2.324 | |||||||||||
| DM1 | 12 | 756500.85 | (42058.37) | p = .040 | 737651.22 | (52119.48) | p = 0.029 | −1.961 | 0.050 | −2.71 $ | 1.721 | 0.203 | 0.57 |
| HC | 14 | 787456.64 | (30440.24) | d = .85 | 779936.16 | (40616.75) | d = 0.91 | −1.475 | 0.140 | −0.85 $ | |||
| WM volume | |||||||||||||
| DM1 | 12 | 703532.11 | (40638.08) | t = 1.582 | 685715.24 | (36237.65) | t = 1.256 | −3.059 | 0.002 | −2.63 $ | 0.106 | 0.748 | 0.14 |
| HC | 14 | 727201.95 | (35684.99) | p = 0.127 | 704947.47 | (41037.58) | p = 0.221 | −2.856 | 0.004 | −2.97 $ | |||
| d = 0.62 | d = 0.49 | ||||||||||||
DM1: Myotonic Dystrophy Type 1; HC: Healthy controls; SD: Standard Deviation. Longitudinal inter‐group comparisons of percentage of volume loss are calculated using time to follow‐up as a covariate.
Covariate value: 9.1624.
Covariate value: 9.0973.
Regional GM and WM volume analysis from baseline to follow‐up: VBM inter‐group analysis
Results from the GM VBM analyses are depicted in Figure 2 (and Supplementary Tables S2, S3, and S4 for a detailed report on significant clusters). At baseline, areas where pediatric onset DM1 showed a decrease in GM volumes compared with the HC‐pediatric group were the bilateral thalamus, caudate, putamen, parahippocampal gyrus, right hippocampus, right lingual gyrus, left Rolandic operculum, left middle and superior temporal gyrus, left Heschl’s gyrus, left parietal operculum, left supramarginal gyrus, and left postcentral gyrus. At follow‐up, these areas were still decreased in patients and, additionally, new cortical areas were involved, particularly the left precentral gyrus, bilateral inferior frontal gyrus (triangular part and orbital part), right inferior frontal gyrus (opercular part), bilateral parietal operculum, and left insula. Adult/late onset DM1 patients at baseline showed GM decrease in the bilateral caudate, putamen and thalamus and left insula in comparison with the HC‐adult/late group, and at follow‐up, these areas expanded to adjacent regions such as the right hippocampus and para‐hippocampal gyrus, together with the cerebellum. Additionally, a decrease was found in the HC‐adult/late group compared with adult/late onset DM1 patients, located at baseline in the following areas of the left hemisphere: inferior, middle and superior temporal gyri, middle and superior temporal poles, fusiform gyrus, inferior and middle frontal gyri (orbital part), parahippocampal gyrus, and the cerebellum. At follow‐up, a significant decrease remained in only some of these areas in the HC‐adult/late group compared with adult/late DM1 patients, again in the following regions of the left hemisphere: the middle and superior temporal lobes, fusiform gyrus, and the parahippocampal gyrus.
Figure 2.

Voxel‐Based Morphometry analyses showing significantly decreased regions in patients compared with HC at both baseline (blue) and follow‐up (red). The depicted regions are those that survived multiple comparisons adjusted for age and brain size. Panel 2A) shows the masks where pediatric onset patients obtained lower gray matter values than their corresponding HC‐pediatric group. Panel 2B) shows the masks where adult/late onset patients had lower gray matter values than their corresponding HC‐adult/late group. The mask at baseline is represented with transparency in order to visualize the areas of overlap between baseline and follow‐up (dark blue and dark brown) and the non‐overlapping areas (light blue and light brown).
Results from WM VBM analysis are shown in Supplementary Tables S5 and S6. At baseline, areas where pediatric onset DM1 showed a decrease in WM volumes compared with the HC‐pediatric group were the bilateral posterior limb of internal capsule, left retrolenticular part of internal capsule and bilateral cerebral peduncle. At follow‐up, these areas were still decreased in patients and, additionally, new areas were involved: left anterior corona radiata, left sagittal stratum, left fornix and stria terminalis and right retrolenticular part of internal capsule. Adult/late onset DM1 patients at baseline showed WM decrease in the right anterior corona radiata and the genu of corpus callosum. At follow‐up, these areas were still decreased in patients and, additionally, new areas were involved: left anterior corona radiata, bilateral superior corona radiata, body of corpus callosum and left superior longitudinal fasciculus.
Association between brain volume and demographic, clinical, and neuropsychological outcomes: VBM intra‐group analysis
For pediatric and adult/late onset DM1 patients, none of the clinical (MIRS score, CTG expansion size, inheritance pattern) or demographic (years of education) variables at baseline were significantly associated with the percentage of GM and WM volume loss from baseline to follow‐up. However, among the neuropsychological tests, the ROCF copy and two CALCAP measures (Sequential 1 reaction time (RT) and Sequential 2 RT) were positively correlated with the percentage of WM volume loss only in adult/late onset DM1 patients (Figure 3). Lower scores on neuropsychological tests were associated with a greater percentage of total WM volume loss.
Figure 3.
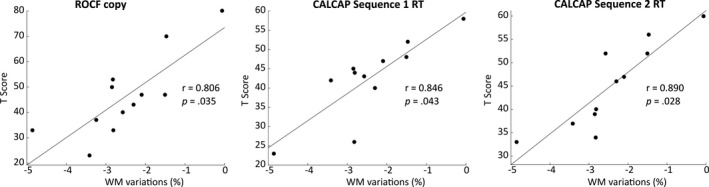
Partial correlation analyses of neuropsychological T scores with percentage of WM volume loss from baseline to follow‐up in adult/late onset DM1 patients. Only the results that survived a false discovery rate (FDR) correction are presented.
No association was found between any clinical, demographic, or neuropsychological variable with the total GM and WM volume at follow‐up in either DM1 group. However, the outcomes of some neuropsychological tests were significantly correlated with specific regions of reduced GM and WM at follow‐up. In the pediatric onset DM1 group the results were inconclusive due to the reduced sample size available in most of the neuropsychological tests, which strongly affected the sensitivity of the statistical procedure. Only the results of the Stroop task (color‐word and interference) were regionally correlated with decreased GM at follow‐up (data not shown). Results in the adult/late onset group for GM are shown in Supplementary Figure S1 (see Supplementary Table S7 for a detailed report on significant clusters) and for WM in Supplementary Table S8. To focus on the association between neuropsychological outcomes and affected brain GM areas, the regions defined by the follow‐up mask of group differences between adult/late DM1 and HC‐adult/late are indicated by black transparent shading.
The MIRS scale was the only clinical variable that significantly correlated with lower GM and WM volume at follow‐up. The specific areas where lower GM volumes at follow‐up were associated with the MIRS score were the bilateral putamen, thalamus, left caudate, right amygdala, hippocampus, left olfactory cortex, right anterior cingulum, left Rolandic operculum, left superior temporal gyrus, left Heschl’s gyrus, left parietal operculum (particularly the OP4 area), left supramarginal gyrus, left postcentral gyrus, right medial and superior frontal orbital gyrus and bilateral gyrus rectus. The specific areas where lower WM volumes at follow‐up were associated with the MIRS score were the left anterior corona radiata, bilateral superior corona radiata, body of corpus callosum and right superior longitudinal fascisulus (see Figure 4 and Supplementary Table S9 for a detailed report on significant clusters on GM and Supplementary Table S10 for WM).
Figure 4.

Gray matter (GM) volume decrease at follow‐up associated with MIRS score of adult/late DM1 patients at baseline. The T‐statistic showing the relationship between GM volume and MIRS score is displayed. Only results surviving multiple comparisons are shown, correcting for age, head size, and time from baseline assessment to follow‐up scan. A black‐transparency mask is displayed to show the damage mask of adult/late onset DM1 compared with HC‐adult/late at follow‐up.
Discussion
This study tests the hypothesis of a neurodegenerative process in DM1, examining for the first time, the different profiles of structural brain involvement in pediatric and adult/late DM1. For this purpose, the same patients and controls were followed across a timespan of almost a decade.
The categorization of DM1 patients is still a matter of debate, and the one employed in this study follows the recently proposed classification system agreed in the latest international workshop (OMMYD‐4) held in Sweden (June 2019). This study sheds light on biological signatures of brain deterioration that support this classification.
Regarding cross‐sectional data, we confirmed that global GM atrophy occurs in DM1 patients, which has been well documented in other studies. 26 , 27 , 28 , 29 A detailed examination of the global volume results in each group yielded some interesting findings. In accord with normal expected aging, when inspecting the raw GM scores, the older HC group (HC‐adult/late) showed a lower total volume score than the younger HC group (HC‐pediatric). However, the raw scores of the pediatric patients showed, in all cases, lower total GM volumes than adult/late patients, even though the former are significantly younger. This reinforces the notion that CNS damage (at least with regard to GM volume atrophy) is greater in pediatric onset forms of the disease. Moreover this result suggests a greater importance of the disease form itself over the mere age of the patients, and thus provides support for the newly‐proposed classification of patients in DM1.
When analyzing the longitudinal data, some striking results emerged. In the pediatric onset group, the loss of volume over time was not significant when compared with that of their HC group. This could be taken to suggest that the brain structure of these patients is developmentally marked as opposed to suffering from an ongoing degenerative process. However, the results do not allow us to completely rule out the possibility of a neurodegenerative process in the pediatric patients, based on the large effect size. Moreover, these patients were not yet in their 40’s at follow‐up. Taken together, these data indicate a tendency toward a greater progressive loss than that expected in normal aging in the pediatric onset DM1 patients. However, the adult/late group did not suffer a significant decrease in their total brain volume over time compared with their HC group, but again, the effect size was moderate. Although the findings point more to the probable occurrence of brain abnormal maturation during neurodevelopment in adult/late DM1 patients, these data do not completely rule out the existence of such progressive degeneration. Indeed, it is important to consider that adult/late patients are in their mid 40s at baseline and in their mid 50s at follow‐up, meaning that, at most, the accelerated brain degeneration in adult/late onset DM1, if it does occur, does not start before the age of 55. Indeed, in most neurogenerative diseases (e.g., Alzheimer’s Disease, Parkinson’s Disease) the prevalence of the affected population significantly increases from the age of 60‐65. 30
The idea of accelerated brain degeneration in DM1 was first described by Conforti et al, 8 who found an increase in brain atrophy, as measured by the ventricular/brain ratio. However, these results contrast with other attempts at longitudinal MRI assessment that found no loss over a period of 5 years. 7 The longer timespan employed in the present work compared with the latter, could explain the potential progression of atrophy found in this study.
The results of this study show that the disease traces a brain signature in patients that develops in a way that is different to what might be expected during the normal aging process; leading to a disease‐specific developmental trajectory regarding the CNS, as observed in other autosomal dominant diseases such as Neurofibromatosis type 1. 31 or Huntington’s Disease. 32 , 33 These different trajectories are confirmed when observing the specific regions of decreased GM in the VBM analyses, and even allow for clearly showing that pediatric and adult/late onset patients display distinctive patterns of damage in a given time‐spot and different trajectories from a longitudinal perspective. Specifically, pediatric DM1 patients showed reduced GM volume, mainly located at subcortical level at baseline, which additionally extended to cortical regions at follow‐up, particularly the frontal lobe. Similar brain involvement is described in other neurodegenerative diseases, often classified as fronto‐subcortical dementias (e.g., Huntington’s disease or Parkinson’s disease). 34 Likewise, fronto‐striatal atrophy has been described as a hallmark of the behavioral variant of frontotemporal dementia. 35
A further observation worth noting is that perisylvian areas such as the parietal operculum are affected in pediatric onset patients. The introduction of new techniques such as stepwise functional connectivity (SFC), 36 , 37 have allowed for identifying the above mentioned areas as key regions in the multimodal integration network, a cortical network where connectivity from somatosensory, auditory, motor, and visual primary cortices converge. 36 , 37 This involvement in pediatric onset DM1 patients could be associated with the observed difficulties in higher‐order cognitive processes, for which these areas establish a link with basic sensorimotor regions. The impairment of such connectivity networks has been suggested in other clinical disorders. This is, for instance, the case for Autism Spectrum Disorder (ASD), 38 in which the disrupted connectivity between primary sensory and sensory integration areas has been hypothesized to be at the basis of, among other features, the weak central coherence proposed in ASD. In fact, a higher than expected prevalence of autistic‐like conditions has been reported in pediatric DM1. 39
Further, adult/late onset patients showed a GM decrease that remained confined to the basal ganglia. Subcortical involvement in DM1 has repeatedly been reported 26 , 28 , 29 , 40 , 41 , 42 and this is confirmed in our study. Moreover only in the adult/late subgroup was the opposite result found, primarily located in the temporal lobe. This result is in accordance with expected normal aging, in which there is a decreased temporal lobe volume, 43 , 44 reinforcing the hypothesis that DM1 natural brain history follows a different neurodegenerative trajectory than that found in healthy aging.
Of the clinical variables assessed in this study, the MIRS score stands out as the only measure that was associated with lower GM volumes at follow‐up in adult/late onset DM1. Furthermore, the high correspondence between the regions implicated in this association and the affected areas (when compared with HC) indicates that this muscular impairment measure could be a potential marker of progressive GM impairment in adult/late onset patients. Indeed, these areas (such as the basal ganglia), have been suggested to play a role in the contribution of the CNS to the motor impairments observed in DM1 patients. 45 , 46 Muscular impairment has been reported to correlate with other brain volumetric measures, as well as WM integrity. 29 , 41 , 47
Neuropsychological correlates with neuroimaging findings are still scarce and inconclusive in the literature. In contrast, the results of this study highlight the potential capacity of certain neuropsychological tools for predicting the progressive loss of WM. Among these neuropsychological tests, ROCF emerges as a powerful measure that needs to be considered not only for characterizing the cognitive profiles of patients, but also as a sensitive prognostic measure. Performance on the ROCF has previously been shown to correlate with brain atrophy measures and WM microstructural damage. 27 , 40 , 48 Regionally, data obtained from a previous study in our laboratory 49 are compatible with the results of this study in the sense that neuropsychological tools appear to be sensitive but still unspecific measures of brain regional correlates. Considering the lack of topological specificity, a network‐wise organization hypothesis must be considered. In this regard, the involvement of areas from the multimodal integration network (such as the supplementary motor area, insula, Rolandic operculum, superior parietal cortex, and anterior cingulated cortex), in association with neuropsychological outcomes, is striking and could form the basis of impaired higher‐order cognitive functions in DM1 patients due to disrupted connectivity between primary sensory regions and higher‐order cortical hubs. Previous studies combining cognitive and imaging outcomes have often failed to find a systematic association between performance on neuropsychological tasks and GM or WM impairment, possibly due to variations in neuropsychological assessment protocols or sample characteristics. It is worth noting, however, that this is the first time that these tests have been employed as potential tools for predicting progressive brain degeneration in DM1. Future research studies should attempt to combine neuropsychological and neuroimaging longitudinal data in order to gain a deeper understanding of the capacity of such variables to serve as potential markers of neurodegeneration.
This study is not without limitations. The main flaw in this study is the small sample size, which forces us to be cautious when generalizing our findings, particularly those derived fromu regression analyses (associations between variables) where the sample size is a strong determinant of statistical power. A major strength, however, is the fact that, for the first time in a longitudinal study of DM1, a sex‐age‐years of education equivalent HC group was included for comparison purposes. Moreover this work constitutes a longitudinal study that applies sophisticated brain volumetric techniques across the largest observation period reported to date. Other shortcomings of this study include the selective attrition bias inherent to any longitudinal study. It should be noted, however, that patients who did not remain in the study until follow‐up had longer CTG expansion sizes, and exhibited greater muscular impairment compared with those who were re‐scanned, which would have, in any case, led to an underestimation of the observed changes in brain structure over time. Finally, whilst a two‐time‐point longitudinal study can shed light on a trajectory, it is insufficient for delineating an established direction in disease progression. Therefore, successive scans (>2 records) of a given cohort should be used in order to clarify whether the trajectories found in this study are maintained over time in a lineal or nonlinear course. Further, future studies are needed that include a broader scope of age‐groups to clarify the trajectory of decline covering the whole life span, particularly older ages.
Conclusion
The findings obtained in this study contribute toward a characterization of the natural progression of a specific CNS feature, that is, brain structure, in DM1. The outcomes show the need to further investigate brain differences over time with a focus on potential neurodevelopmental anomalies preceding potential neurodegeneration, which, in light of the present results, appears to be more pronounced in pediatric onset DM1. Future studies should attempt to recruit larger cohorts of patients with (if feasible) the earliest possible onset of the disease, to allow for a more accurate depiction of the natural history of gray and white matter alterations as neurodevelopmental, neurodegenerative, or both. So far, our results support the recently proposed disease classification by identifying distinct patterns of brain atrophy and its progression over time, and therefore encourage upcoming research to more deeply analyze these differences and to extend the study to other potential variations associated with each phenotype, that is, neuropsychological profiles or biological correlates.
Conflict of Interest
Authors declare that there is no potential conflict of interest.
Author Contributions
Garazi Labayru contributed to conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, visualization, project administration, and funding acquisition. Antonio Jimenez‐Marin contributed to conceptualization, methodology, formal analysis, data curation, writing original draft, and visualization. Esther Fernández, Jorge Villanúa, and Miren Zulaica contributed to investigation, resources, and writing – review & editing. Jesus M. Cortes contributed to conceptualization, methodology, and writing – review & editing. Ibai Díez contributed to methodology, formal analysis, and writing – review & editing. Jorge Sepulcre contributed to writing ‐ review & editing. Adolfo López de Munain contributed to conceptualization, resources, writing – review & editing, supervision, and funding acquisition. Andone Sistiaga contributed to conceptualization, methodology, writing – review & editing, supervision, project administration, and funding acquisition.
Supporting information
Supplementary Figure S1. Gray matter (GM) volume decrease at follow‐up associated with neuropsychological outcome of DM1 patients at baseline. The T‐statistic showing the relationship between GM volume and neuropsychological test is displayed. Only results surviving multiple comparisons are shown, correcting for age, head size and time from baseline assessment to follow‐up scan. A black‐transparency mask is displayed to show the damage mask of adult/late onset DM1 compared with HC‐adult/late at follow‐up. Panel a) shows the GM areas at follow‐up with a significant decrease in relation to lower scores on the WCST (categories completed) at baseline. Panel b) shows the GM areas at follow‐up with a significant decrease in relation to lower scores on Block design at baseline. Panel c) shows the GM areas at follow‐up with a significant decrease in relation to lower scores on Object assembly at baseline. Panel d) shows the GM areas at follow‐up with a significant decrease (red‐yellow) or increase (blue) in relation to lower scores on ROCF at baseline. Panel e) shows the GM areas at follow‐up with a significant increase in relation to lower scores on CALCAP simple RT at baseline. Panel f) shows the GM areas at follow‐up with a significant increase in relation to lower scores on CALCAP Sequential 1 RT at baseline.
Supplementary Table S1. Wilcoxon repeated measures analysis of neuropsychological outcomes at baseline and at follow‐up in pediatric and adult/late onset DM1.
Supplementary Table S2. VBM analyses. Brain areas with reduced gray matter volume in pediatric onset DM1 patients compared with healthy controls, at baseline and at follow‐up.
Supplementary Table S3. VBM analyses. Brain areas with reduced gray matter volume in adult/late onset DM1 patients compared with healthy controls, at baseline and at follow‐up.
Supplementary Table S4. VBM analyses. Brain areas with reduced gray matter volume in healthy controls compared to adult/late onset DM1 patients, at baseline and at follow‐up.
Supplementary Table S5. VBM analyses. Brain areas with reduced white matter volume in pediatric onset DM1 patients compared with healthy controls, at baseline and at follow‐up.
Supplementary Table S6. VBM analyses. Brain areas with reduced white matter volume in adult/late onset DM1 patients compared with healthy controls, at baseline and at follow‐up.
Supplementary Table S7. Gray matter volume at follow‐up associated with baseline neuropsychological outcomes of adult/late DM1 patients.
Supplementary Table S8. White matter volume at follow‐up associated with baseline neuropsychological outcomes of adult/late DM1 patients.
Supplementary Table S9. Gray matter volume at follow‐up associated with baseline MIRS score in adult/late DM1 patients.
Supplementary Table S10. White matter volume at follow‐up associated with baseline MIRS score in adult/late DM1 patients.
Acknowledgments
This study was supported by funding from the Institute of Health Carlos III co‐founded by Fondo Europeo de Desarrollo Regional‐FEDER [grant numbers PI17/01231 and PI17/01841], CIBERNED (grant number: 609) and the Basque Government [SAIO08‐PE08BF01]. G. Labayru was supported by a predoctoral grant from the Basque Government [PRE_2016_1_0187]. A. Jiménez‐Marín was supported by a predoctoral grant from the Basque Government [PRE_2019_1_0070].
Funding Statement
This work was funded by CIBERNED grant 609; Eusko Jaurlaritza grants PRE_2016_1_0187, PRE_2019_1_0070, and SAIO08‐PE08BF01; Institute of Health Carlos III co‐founded by Fondo Europeo de Desarrollo Regional‐FEDER grants PI17/01231 and PI17/01841.
References
- 1. Caillet‐Boudin ML, Fernandez‐Gomez FJ, Tran H, et al. Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci 2014;6(57):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sansone V, Gandossini S, Cotelli M, et al. Cognitive impairment in adult myotonic dystrophies: a longitudinal study. Neurol Sci 2007;28(1):9–15. [DOI] [PubMed] [Google Scholar]
- 3. Modoni A, Silvestri G, Vita MG, et al. Cognitive impairment in myotonic dystrophy type 1 (DM1): a longitudinal follow‐up study. J Neurol 2008;255(11):1737–1742. [DOI] [PubMed] [Google Scholar]
- 4. Winblad S, Samuelsson L, Lindberg C, Meola G. Cognition in myotonic dystrophy type 1: a 5‐year follow‐up study. Eur J Neurol 2016;23(9):1471–1476. [DOI] [PubMed] [Google Scholar]
- 5. Gallais B, Gagnon C, Mathieu J, Richer L. Cognitive decline over time in adults with myotonic dystrophy type 1: a 9‐year longitudinal study. Neuromuscul Disord 2017;27(1):61–72. [DOI] [PubMed] [Google Scholar]
- 6. Labayru G, Aliri J, Zulaica M, et al. Age‐related cognitive decline in myotonic dystrophy type 1: an 11‐year longitudinal follow‐up study. J Neuropsychol 2020;14(1):121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gliem C, Minnerop M, Roeske S, et al. Tracking the brain in myotonic dystrophies: a 5‐year longitudinal follow‐up study. PLoS One 2019;14(3):e0213381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Conforti R, de Cristofaro M, Cristofano A, et al. Brain MRI abnormalities in the adult form of myotonic dystrophy type 1: a longitudinal case series study. Neuroradiol J 2016;29(1):36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mathieu J, Boivin H, Meunier D, et al. Assessment of a disease‐specific muscular impairment rating scale in myotonic dystrophy. Neurology 2001;56(April):336–340. [DOI] [PubMed] [Google Scholar]
- 10. Wechsler D. WAIS‐III: Escala de Inteligencia de Wechsler para Adultos III. Madrid: TEA Ediciones, 1999. [Google Scholar]
- 11. Sattler JM, Ryan JJ. Wechsler Adult Intelligence Scale‐III (WAIS‐III): Description In: Sattler JM, ed. Assessment of Children. San Diego, CA: Cognitive applications. San Diego, 2001. [Google Scholar]
- 12. Golden CJ. STROOP . Test de Colores y Palabras, 3rd ed Madrid: TEA Ediciones, 2001. [Google Scholar]
- 13. Miller EN. CalCAP: California Computerized Assessment Package. 1990. [Google Scholar]
- 14. Lezak M, Howieson DB, Loring D. Neuropsychological Assessment, 4th ed New York: Oxford University Press, 2004. [Google Scholar]
- 15. Casals‐Coll M, Sánchez‐Benavides G, Quintana M, et al. Estudios normativos españoles en población adulta joven (proyecto NEURONORMA jóvenes): normas para los test de fluencia verbal. Neurología 2013;28(1):33–40.22652141 [Google Scholar]
- 16. Peña‐Casanova J, Quiñones‐Úbeda S, Gramunt‐Fombuena N, et al. Spanish Multicenter Normative Studies (NEURONORMA Project): Norms for Verbal Fluency Tests. Arch Clin Neuropsychol 2009;24:395–411. [DOI] [PubMed] [Google Scholar]
- 17. Rey A. Test de copia y de reproducción de memoria de figuras geométricas complejas. Madrid: TEA Ediciones, 2009. [Google Scholar]
- 18. Raven JC, Court JH, Raven J. Raven: Standard Progressive Matrices. Madrid: TEA Ediciones, 2001. [Google Scholar]
- 19. Benton A, Sivan A, Hamsher K, et al. Contributions to Neuropsychological Assessment, 2nd ed New York: Oxford University Press, 1994. [Google Scholar]
- 20. Heaton RK, Chelune GJ, Talley JL, et al. Test de clasificación de tarjetas de Wisconsin. Madrid: 2001. [Google Scholar]
- 21. Douaud G, Smith S, Jenkinson M, et al. Anatomically related grey and white matter abnormalities in adolescent‐onset schizophrenia. Brain 2007;130(9):2375–2386. [DOI] [PubMed] [Google Scholar]
- 22. Good CD, Johnsrude IS, Ashburner J, et al. A voxel‐based morphometric study of ageing in 465 normal adult human brains. NeuroImage 2001;14(1):21–36. [DOI] [PubMed] [Google Scholar]
- 23. Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage 2004;23(SUPPL. 1):208–219. [DOI] [PubMed] [Google Scholar]
- 24. Andersson JLR, Jenkinson M, Smith S. Non‐linear registration, aka spatial normalisation. FMRIB Technial Report TR07JA2. Oxford, UK: Oxford Centre for Functional Magnetic Resonance Imaging of the Brain, Department of Clinical Neurology, Oxford University; 2007. [Google Scholar]
- 25. Smith SM, Zhang Y, Jenkinson M, et al. Accurate, robust, and automated longitudinal and cross‐sectional brain change analysis. NeuroImage 2002;17(1):479–489. [DOI] [PubMed] [Google Scholar]
- 26. Caso F, Agosta F, Peric S, et al. Cognitive impairment in myotonic dystrophy type 1 is associated with white matter damage. PLoS One 2014;9(8):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baldanzi S, Cecchi P, Fabbri S, et al. Relationship between neuropsychological impairment and grey and white matter changes in adult‐onset myotonic dystrophy type 1. Neuroimage Clin 2016;12:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sugiyama A, Sone D, Sato N, et al. Brain gray matter structural network in myotonic dystrophy type 1. PLoS One 2017;12(11):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schneider‐Gold C, Bellenberg B, Prehn C, et al. Cortical and subcortical grey and white matter atrophy in myotonic dystrophies type 1 and 2 is associated with cognitive impairment, depression and daytime sleepiness. PLoS One 2015;10(6):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 2019;15(10):565–581. [DOI] [PubMed] [Google Scholar]
- 31. Moore BD, Slopis JM, Jackson EF, et al. Brain volume in children with neurofibromatosis type 1: Relation to neuropsychological status. Neurology 2000;54(4):914–920. [DOI] [PubMed] [Google Scholar]
- 32. Conforti P, Besusso D, Bocchi VD, et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc Natl Acad Sci USA 2018;115(4):E762–E771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van der Plas E, Langbehn DR, Conrad AL, et al. Abnormal brain development in child and adolescent carriers of mutant huntingtin. Neurology 2019;93(10):e1021–e1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bonelli RM, Cummings JL. Frontal‐subcortical dementias. The Neurologist 2008;14(2):100–107. [DOI] [PubMed] [Google Scholar]
- 35. Bertoux M, O’Callaghan C, Flanagan E, et al. Fronto‐striatal atrophy in behavioral variant frontotemporal dementia and alzheimer’s disease. Front Neurol 2015;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sepulcre J. Integration of visual and motor functional streams in the human brain. Neurosci Lett 2014;567:68–73. [DOI] [PubMed] [Google Scholar]
- 37. Sepulcre J, Sabuncu MR, Yeo TB, et al. Stepwise connectivity of the modal cortex reveals the multimodal organization of the human brain. J Neurosci 2012;32(31):10649–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stevenson RA. Using functional connectivity analyses to investigate the bases of autism spectrum disorders and other clinical populations. J Neurosci 2012;32(50):17933–17934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ekström AB, Hakenäs‐Plate L, Samuelsson L, et al. Autism spectrum conditons in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet 2008;147(6):918–926. [DOI] [PubMed] [Google Scholar]
- 40. Weber YG, Roebling R, Kassubek J, et al. Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology 2010;74(14):1108–1117. [DOI] [PubMed] [Google Scholar]
- 41. Minnerop M, Weber B, Schoene‐Bake JC, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 2011;134(12):3527–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zanigni S, Evangelisti S, Giannoccaro MP, et al. Relationship of white and gray matter abnormalities to clinical and genetic features in myotonic dystrophy type 1. Neuroimage Clin 2016;11:678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vinke EJ, de Groot M, Venkatraghavan V, et al. Trajectories of imaging markers in brain aging: the Rotterdam Study. Neurobiol Aging 2018;71:32–40. [DOI] [PubMed] [Google Scholar]
- 44. Lockhart SN, DeCarli C. Structural imaging measures of brain aging. Neuropsychol Rev 2014;24(3):271–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Serra L, Mancini M, Silvestri G, et al. Brain connectomics’ modification to clarify motor and nonmotor features of myotonic dystrophy type 1. Neural Plast 2016;2016:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Toth A, Lovadi E, Komoly S, et al. Cortical involvement during myotonia in myotonic dystrophy: an fMRI study. Acta Neurol Scand 2015;132(1):65–72. [DOI] [PubMed] [Google Scholar]
- 47. Wozniak JR, Mueller BA, Lim KO, et al. Tractography reveals diffuse white matter abnormalities in myotonic dystrophy type 1. J Neurol Sci 2014;341(1–2):73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Dorst M, Okkersen K, Kessels RPC, et al. Structural white matter networks in myotonic dystrophy type 1. NeuroImage Clin 2019;21:101615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Labayru G, Diez I, Sepulcre J, et al. Regional brain atrophy in gray and white matter is associated with cognitive impairment in myotonic dystrophy type 1. Neuroimage Clin 2019;24:102078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Gray matter (GM) volume decrease at follow‐up associated with neuropsychological outcome of DM1 patients at baseline. The T‐statistic showing the relationship between GM volume and neuropsychological test is displayed. Only results surviving multiple comparisons are shown, correcting for age, head size and time from baseline assessment to follow‐up scan. A black‐transparency mask is displayed to show the damage mask of adult/late onset DM1 compared with HC‐adult/late at follow‐up. Panel a) shows the GM areas at follow‐up with a significant decrease in relation to lower scores on the WCST (categories completed) at baseline. Panel b) shows the GM areas at follow‐up with a significant decrease in relation to lower scores on Block design at baseline. Panel c) shows the GM areas at follow‐up with a significant decrease in relation to lower scores on Object assembly at baseline. Panel d) shows the GM areas at follow‐up with a significant decrease (red‐yellow) or increase (blue) in relation to lower scores on ROCF at baseline. Panel e) shows the GM areas at follow‐up with a significant increase in relation to lower scores on CALCAP simple RT at baseline. Panel f) shows the GM areas at follow‐up with a significant increase in relation to lower scores on CALCAP Sequential 1 RT at baseline.
Supplementary Table S1. Wilcoxon repeated measures analysis of neuropsychological outcomes at baseline and at follow‐up in pediatric and adult/late onset DM1.
Supplementary Table S2. VBM analyses. Brain areas with reduced gray matter volume in pediatric onset DM1 patients compared with healthy controls, at baseline and at follow‐up.
Supplementary Table S3. VBM analyses. Brain areas with reduced gray matter volume in adult/late onset DM1 patients compared with healthy controls, at baseline and at follow‐up.
Supplementary Table S4. VBM analyses. Brain areas with reduced gray matter volume in healthy controls compared to adult/late onset DM1 patients, at baseline and at follow‐up.
Supplementary Table S5. VBM analyses. Brain areas with reduced white matter volume in pediatric onset DM1 patients compared with healthy controls, at baseline and at follow‐up.
Supplementary Table S6. VBM analyses. Brain areas with reduced white matter volume in adult/late onset DM1 patients compared with healthy controls, at baseline and at follow‐up.
Supplementary Table S7. Gray matter volume at follow‐up associated with baseline neuropsychological outcomes of adult/late DM1 patients.
Supplementary Table S8. White matter volume at follow‐up associated with baseline neuropsychological outcomes of adult/late DM1 patients.
Supplementary Table S9. Gray matter volume at follow‐up associated with baseline MIRS score in adult/late DM1 patients.
Supplementary Table S10. White matter volume at follow‐up associated with baseline MIRS score in adult/late DM1 patients.