

Abstract
Background
Chlorine gas (Cl2) exposure remains a public health concern in household, occupational, and transportation accidents around the world. The death rate associated with acute respiratory distress syndrome (ARDS) caused by high concentrations of Cl2 is very high, mainly because the pathogenesis of ARDS remains unclear. Histone H4 has been identified as an important endogenous pro-inflammatory molecule. The present study aimed to examine the pathogenic role of histone H4 in Cl2-induced ARDS.
Methods
ARDS was induced by Cl2 exposure in male C57BL/6 mice. Circulating histone H4, blood gas, pulmonary edema, endothelial activation, and neutrophil infiltration were measured during acute lung injury (ALI). Histone H4 or anti-H4 antibody was administered through the tail vein 1 h prior to Cl2 exposure to study the pathogenic role of histone H4. Toll-like receptor 2 knock-out (Tlr2-KO) and Tlr4-KO mice were used in conjunction with blocking antibody against TLR1, TLR2, TLR4, or TLR6 to explore the mechanism involved in histone H4-mediated injury.
Results
Cl2 exposure induced a concentration-dependent ALI. The levels of circulating histone H4 were positively correlated with Cl2 concentrations. Pretreatment with intravenous histone H4 further aggravated lethality rate, blood gas, endothelial activation, and neutrophil infiltration, while anti-H4 antibody showed protective effects. Tlr4 deficiency improved lethality rate, blood gas, and pulmonary edema, and prevented endothelial and neutrophil activation caused by Cl2 exposure. More importantly, Tlr4 gene deletion greatly diminished the effect of histone H4 or anti-H4 antibody observed in wild-type (WT) mice. The impact of Tlr2 on inflammatory injury was not significant. The role of TLRs was also validated by endothelial activation mediated by histone H4 in vitro.
Conclusions
Circulating histone H4 played a pro-inflammatory role in ARDS caused by Cl2. TLR4 was closely involved in histone H4-mediated inflammatory injury. Therefore, intervention targeting histone H4 is potentially protective.
Keywords: Chlorine gas, Acute respiratory distress syndrome, Histone H4, Toll-like receptor, Pulmonary inflammation
Background
Chlorine gas (Cl2) exposure remains a public health concern in household, occupational, and transportation accidents around the world. It has also been used as a chemical warfare agent and weapon of mass destruction by terrorists [1, 2]. High concentrations of Cl2 can result in acute respiratory distress syndrome (ARDS), which manifests as pulmonary edema, hypoxemia, and uncontrolled overwhelming inflammation [3, 4].
The death rate associated with ARDS is very high, primarily because the interventions are mostly supportive and symptom-oriented. This is because the pathogenesis and underlying causes of ARDS remains unclear [5–7]. Respiratory system damage inflicted by Cl2 can be divided into two phases. The first phase is direct chemical toxicity to the airway, and the second is the more severe ensuing inflammatory response, which can result in ARDS. Few studies have assessed how direct injury triggers uncontrolled, overwhelming inflammation [8, 9].
Extracellular histones increase during acute inflammatory diseases such as trauma, aspiration, and sepsis, which can mediate distant organ damage [10, 11]. The cytotoxicity of extracellular histones is primarily due to histone H4 [12, 13].
This work aimed to study the pathogenic role of circulating histone H4 in Cl2-induced ARDS.
Materials and methods
Chemicals and reagents
Histone H4 was purchased from Millipore (Billerica, MA, USA). Blocking antibodies against TLR1 (GD2.F4), TLR2 (TL2.1), and TLR4 (HTA125) were purchased from eBioscience (San Diego, CA, USA). Antibodies for TNF-α, IL-1β, P-selectin, Ly6G, H4, Ac-H4(acetyl K5 + K8 + K12 + K16) and blocking antibody against TLR6 (TLR6.127) were purchased from Abcam (Cambridge, UK). An ELISA kit for von Willebrand factor (vWF) and a myeloperoxidase (MPO) detection kit were purchased from Jiancheng Biotech (Nanjing, China). The blocking antibody against histone H4 (anti-H4) was prepared following the previously described protocol involving autoimmune mice [14].
Animals
Eight-week-old wild-type (WT) C57BL/6 mice, Toll-like receptor 2 knock-out (Tlr2-KO) mice and Tlr4-KO mice were provided by Peking University Animal Center (Beijing, China). The Tlr4 gene (NCBI Reference Sequence: NM_021297) is located on mouse chromosome 4. Exon 2 was selected as the target site among the three exons identified. The Tlr2 gene (NCBI Reference Sequence: NM_011905) is located on mouse chromosome 3. Exon 3 was selected as the target site among the three exons identified. Cas9 and gRNA were co-injected into fertilized eggs for KO mouse production. The genotype was confirmed by polymerase chain reaction (PCR) and sequencing analysis [15]. All the genetically deficient mice were backcrossed at least six generations onto the B6 background before the experiment. The protocols for the animal experiment were reviewed and permitted by the Peking University Animal Care and Use Committee (No. LA201783). Mice were housed in an air-conditioned room at 25 °C with a 12 h dark-light cycle. To minimize animal suffering, mice were administered anesthesia before surgery and euthanized humanely by intravenous injection of xylazine (6 mg/kg) and ketamine (90 mg/kg) followed by cervical dislocation.
Chlorine gas exposure
Cl2 exposure was performed in a special chamber (no more than six mice at a time) where Cl2 released from a cylinder was mixed with air as previously described [16]. The concentration of Cl2 was monitored by a chlorine detector. When the Cl2 concentration decreased, the Cl2-air mixture was immediately replaced. The exposure conditions were 10, 20, 50, 100, 200, 400, 600, and 800 ppm respectively for 30 min. The control mice stayed in the same chamber for 30 min without Cl2. Immediately after Cl2 exposure, mice were returned to room air. They were monitored hourly for 12 h, and every 4 h thereafter for 72 h. The mouse model of Cl2-induced ARDS was validated by blood gas analysis (PaO2/FiO2 ≤ 300 mmHg).
Measurement of circulating histone H4 and blood gas analysis
To obtain blood samples, a catheter with 11 mM sodium citrate was inserted into the abdominal aorta after the mice were anesthetized. To measure arterial partial oxygen pressure (PaO2), whole blood (0.1–0.2 ml) was measured with a blood gas analyzer (Ciba Corning, Canada). To measure histone H4, the plasma was collected from the whole blood after centrifugation and then analyzed with a histone H4 detection kit (USCN, Wuhan, China).
Cell culture
Human pulmonary vascular endothelial cells were purchased from the Peking Union Medical College (Beijing, China). They were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) at 37 °C. To determine which TLR was involved, a blocking antibody (10 mg/L) against TLR1, TLR2, TLR4, or TLR6 was concurrently administered when the endothelial cells were stimulated by histone H4 (10 mg/L) for 12 h. Each group was tested in triplicate at each time point.
Real-time PCR
Quantitative PCR was performed with TNFA (MP200370) and IL1B (MP200131) primers (Sino Biological, Radnor, PA, USA). The reverse transcription reactions were conducted with a Cells-to-CT kit (Invitrogen, Carlsbad, CA, USA) in 7300 Real-Time PCR System (AB Biosciences, Concord, MA, USA). PCR conditions included initial denaturation at 95°Cfor 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, and annealing at 60 °C for 1 min. GAPDH was used as the housekeeping gene. The relative abundance of mRNA expression in each sample was calculated as 2-∆∆Ct.
Western blot
Protein concentration was measured by the Bio-Rad Protein Assay Kit. For all groups, proteins (40 μg) were mixed with loading buffer. After electrophoresis, the separated proteins were transferred onto PVDF membranes. The membranes were probed with anti-TNF-α antibody (1:2000), anti-IL-1β antibody (1: 2000), anti-H4 antibody (1: 1000), and anti-GAPDH antibody (1: 5000) overnight at 4 °C.
Statistical analysis
The data are shown as mean ± standard deviation (SD) and analyzed with Prism (GraphPad Software). Pearson correlation analysis was used to analyze correlation and a log-rank test was used to analyze survival rate. An analysis of variance (ANOVA) was used to analyze the statistical differences among groups and the Student-Newman-Keuls test was used to analyze the differences between groups. A p-value < 0.05 was viewed as statistically significant.
Results
Circulating histone H4 increased in Cl2-induced ARDS in mice
Different concentrations of Cl2 (10, 20, 50, 100, 200, 400, 600, 800 ppm) were used to induce ARDS in mice (n = 12, exposure time 30 min). The effect of Cl2 on the severity of ALI was concentration dependent. Low Cl2 concentrations (≤ 100 ppm) merely caused brief tachypnea, and the mice (12/12) all survived. When the concentration increased to 200 ppm, Cl2 caused dyspnea and eight mice (8/12) survived for 72 h. A concentration of 400 ppm caused evident dyspnea, and five mice (5/12) survived for 72 h. A concentration of 600 ppm caused serious dyspnea and only three mice (3/12) survived for 72 h. Almost, all mice (11/12) died within 72 h after exposure to a Cl2 concentration of 800 ppm. Pulmonary interstitial edema, inflammatory cell infiltration, hemorrhage, atelectasis, microthrombus formation, and epithelium necrosis were evident. The pathological changes in lung tissue 24 h after Cl2 exposure were shown in Figure S1 (supplemental data).
Circulating histone H4 was very low in the normal state. After Cl2 exposure, circulating histone H4 increased, particularly when the concentration of Cl2 reached 400 ppm. As shown in Fig. 1a, there was a significant positive correlation between the concentrations of Cl2 (from 10 to 800 ppm) and histone H4 in plasma (r = 0.8017, p = 0.0289). As shown in Fig. 1b, western blot was used to further prove the histone H4 changes in the plasma.
Fig. 1.
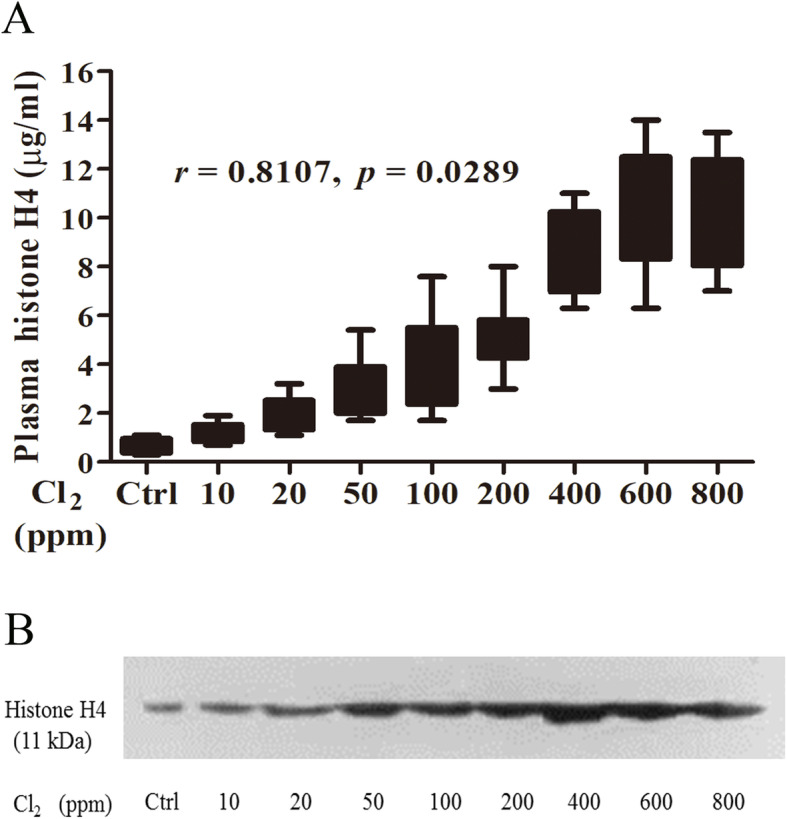
Change of circulating histone H4 in Cl2-induced ARDS. Twenty four hours after mice were treated (30 min) with different concentrations of Cl2, circulating histone H4 was measured with an ELISA kit (a) and western blot (b). Data were presented as mean ± SD (n = 6). Western blot was performed in triplicate
Effects of histone H4 and TLRs on survival rate
In order to study the role of histone H4, mice were injected via tail vein with histone H4 or anti-H4 antibody 30 min prior to Cl2 exposure. As shown in Fig. 2a, nine mice (9/12) died within 72 h after exposure to lethal Cl2 concentration (600 ppm, 30 min). When pretreated with intravenous histone H4 (10 mg/kg), nearly all mice (11/12) died within 72 h after Cl2 exposure (p = 0.1574, compared with the mice solely exposed to Cl2). Only three mice (3/12) died when pretreated with intravenous anti-H4 antibody (20 mg/kg) (p = 0.0223, compared with the mice solely exposed to Cl2). The difference in lethality rate between the mice pretreated with histone H4 and those pretreated with anti-H4 antibody was statistically significant (p = 0.0010).
Fig. 2.
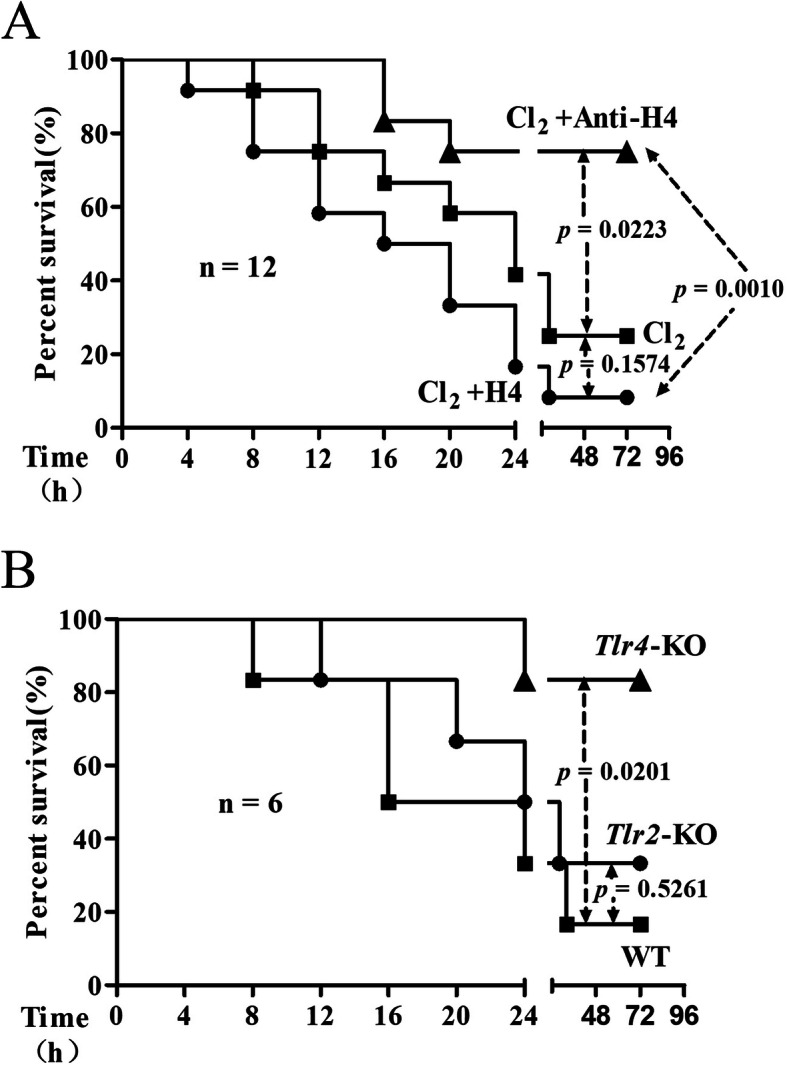
Effects of histone H4 and TLRs on survival rate. After exposure to lethal concentration of Cl2 (600 ppm, 30 min), the survival rate was recorded at predetermined time points (every 4 h for 96 h). Histone H4 (10 mg/kg) or anti-H4 antibody (20 mg/kg) was delivered through the tail vein 1 h prior to Cl2 exposure in the WT mice (n = 12, 2A). Tlr2-KO and Tlr4-KO mice were used to explore the role of TLRs (n = 6, 2B). The p values are compared with the WT mice treated solely with Cl2
Tlr2-KO and Tlr4-KO mice were used to investigate the role TLRs in ARDS. As shown in Fig. 2b, five WT mice (5/6) died within 72 h after exposure to lethal Cl2 concentration (600 ppm, 30 min). Compared with the WT mice, four Tlr2-KO mice (4/6) and only one Tlr4-KO mouse (1/6) died within 72 h after Cl2 exposure. The difference in lethality rate between the WT mice and the Tlr4-KO mice was statistically significant (p = 0.0201). Thus, the Tlr2-KO and Tlr4-KO mice were resistant to Cl2 exposure of lethal concentrations to some extent.
Roles of histone H4 and TLRs in Cl2-induced ALI
As shown in Fig. 3a, Cl2 exposure caused obvious hypoxemia (48.73 ± 14.23 mmHg) in the WT mice (p = 0.0083, compared with the control group). In mice pretreated with intravenous histone H4 (10 mg/kg), PaO2 decreased much more seriously to 36.35 ± 15.17 mmHg (p = 0.0067, compared with the control group). On the contrary, PaO2 increased to 70.35 ± 11.51 mmHg (p = 0.0474, compared with the control group) when mice were pretreated with intravenous anti-H4 antibody (20 mg/kg). Compared with the WT mice treated in the same manner, PaO2 in the Tlr4-KO mice only decreased slightly to 67.31 ± 10.66 mmHg 24 h after Cl2 exposure (p = 0.0443). More importantly, the damaging effect of histone H4 was diminished greatly in the Tlr4-KO mice compared with the WT mice treated in the same manner (p = 0.0268). Furthermore, the effect of the anti-H4 antibody was also decreased in the Tlr4-KO mice. In contrast to the Tlr4-KO mice, the PaO2 changes and the effects of histone H4 or anti-H4 antibody in the Tlr2-KO mice were similar to those in the WT mice.
Fig. 3.

Roles of histone H4 and TLRs in Cl2 induced inflammatory lung injury. Twenty four hours after Cl2 exposure (400 ppm, 30 min) in WT, Tlr2-KO, and Tlr4-KO mice, PaO2 (3A) and lung wet/dry mass ratio (3B) were measured. Pulmonary endothelial activation was represented by P-selectin expression (3C) (Scale bars: 100 μm) and circulating vWF (3D). Pulmonary neutrophil infiltration was shown by Ly6G marker staining (3E) (Scale bars: 50 μm), and neutrophilic activation was indicated by MPO activity (3F). The protein levels of inflammatory cytokines TNF-α (3G) and IL-1β (3H) were measured by western blot. Histone H4 (10 mg/kg) or anti-H4 antibody (20 mg/kg) was delivered through the tail vein 1 h prior to Cl2 exposure. N = 6 independent replicates for all groups. *p < 0.05, **p < 0.01 compared with the control group; #p < 0.05, ##p < 0.01 compared with the WT mice treated solely with Cl2; $p < 0.05, $$p < 0.01 compared with the WT mice treated in the same manner (Cl2 + H4); &p < 0.05, &&p < 0.01 compared with the WT mice treated in the same manner (Cl2 + Anti-H4).
As shown in Fig. 3b, pulmonary edema was serious after Cl2 exposure in the WT mice (p = 0.0092, compared with the control group). Pretreatment with histone H4 (10 mg/kg) further increased the lung wet/dry mass ratio to 4.81 ± 0.97 (p = 0.0073, compared with the control group). However, pretreatment with anti-H4 antibody (20 mg/kg) decreased the lung wet/dry mass ratio to 3.16 ± 0.68 (p = 0.0319, compared with the control group). Compared with the WT mice treated in the same manner, the lung wet/dry mass ratio in the Tlr4-KO mice merely increased to 2.96 ± 0.71 (p = 0.0457) after Cl2 exposure. Additionally, the damaging effect of histone H4 was diminished in the Tlr4-KO mice compared with the WT mice treated in the same manner (p = 0.0338). The effect of anti-H4 antibody was also decreased. In contrast to the Tlr4-KO mice, the Tlr2-KO mice did not show obvious differences in pulmonary edema when compared with the WT mice.
Cl2 exposure caused evident endothelial activation that manifested as elevated P-selectin expression (Fig. 3c) and vWF release from endothelial Weibel-Palade bodies (WPBs) (Fig. 3d) (p = 0.0038, compared with the control group). Pretreatment with intravenous histone H4 (10 mg/kg) aggravated P-selectin expression and caused further release of vWF (p = 0.0017, compared with the control group). However, pretreatment with the anti-H4 antibody (20 mg/kg) produced some protective effects on P-selectin overexpression and vWF release (p = 0.0089, compared with the control group). Compared with the WT mice treated in the same manner, P-selectin overexpression and vWF release in the Tlr4-KO mice were decreased (p = 0.0093) after Cl2 exposure. The effects of histone H4 (p = 0.0075) and anti-H4 antibody (p = 0.0377) were diminished greatly in the Tlr4-KO mice compared with the WT mice treated in the same manner. However, changes in endothelial activation and the effect of histone H4 or anti-H4 antibody in the Tlr2-KO mice were similar to those in the WT mice.
Pulmonary neutrophil infiltration and activation were prominent after Cl2 exposure, and were measured by using neutrophil specific marker Ly6G (Fig. 3e) and MPO activity (Fig. 3f) (p = 0.0031, compared with the control group). Pretreatment with intravenous histone H4 (10 mg/kg) increased neutrophil infiltration and MPO activity further (p = 0.0017, compared with the control group) while pretreatment with anti-H4 antibody (20 mg/kg) showed some antagonistic effects (p = 0.0165, compared with the control group). Compared with the WT mice treated in the same manner, neutrophil infiltration and activation in the Tlr4-KO mice were inhibited after Cl2 exposure (p = 0.0234). The effects of histone H4 (p = 0.0341) and anti-H4 antibody (p = 0.0972) were also diminished in the Tlr4-KO mice compared with the WT mice treated in the same manner. In contrast to the Tlr4-KO mice, the Tlr2-KO mice did not show evident differences in neutrophil infiltration and activation compared with the WT mice.
As shown in Fig. 3g and h, Cl2 exposure triggered the expression of inflammatory cytokines, such as TNF-α and IL-1β, in WT mice compared with the control group. Pretreatment with intravenous histone H4 (10 mg/kg) further increased the protein level of TNF-α and IL-1β. However, pretreatment with anti-H4 antibody (20 mg/kg) inhibited the protein expression of TNF-α and IL-1β caused by Cl2 exposure. In the Tlr4-KO mice, the protein level of TNF-α and IL-1β was much lower than that in the WT mice. In contrast to the Tlr4-KO mice, the Tlr2-KO mice did not show obvious differences in TNF-α and IL-1β protein expression compared with the WT mice.
TLRs involved in histone H4-mediated endothelial inflammation in vitro
Blocking antibodies against TLR1, TLR2, TLR4, and TLR6 were used to investigate the role of TLRs in histone H4-mediated endothelial inflammation. Histone H4 (10 mg/L) treatment triggered the expression of inflammatory cytokines in the pulmonary vascular endothelial cells, including TNF-α and IL-1β. As shown in Fig. 4a, a blocking antibody against TLR4 distinctly reduced the transcription of TNFA (38% decrease versus H4 group, p = 0.0373). Additionally, blocking antibody against TLR2 slightly reduced the transcription of TNFA (p > 0.05). However, blocking antibodies against TLR1 and TLR6 showed little effect. The effects of the blocking antibodies against TLR1, TLR2, TLR4, and TLR6 on TNF-α protein expression were similar to their effects on transcription (Fig. 4c).
Fig. 4.

TLRs involved in histone H4 mediated expression of inflammatory cytokines. The blocking antibody (10 mg/L) against TLR1, TLR2, TLR4, or TLR6 was concurrently administered when pulmonary vascular endothelial cells were challenged by histone H4 (10 mg/L) for 12 h. The mRNA of TNFA and IL1B was measured by real-time qPCR (a, b), and protein was measured by western blot (c, d). N = 3 independent replicates for all groups. *p < 0.05, **p < 0.01 compared with the control group; #p < 0.05, ##p < 0.01 compared with the histone H4 group
As shown in Fig. 4b, the blocking antibody against TLR4 markedly reduced the transcription of IL1B (41% decrease versus H4 group, p = 0.0211), while the effect of the blocking antibody against TLR2 on IL1B transcription was much weaker (p > 0.05). The blocking antibodies against TLR1 and TLR6 showed little effect on IL1B transcription. The effect of the blocking antibodies on IL-1β was also validated by protein expression (Fig. 4d).
Discussion
Exposure to high levels of Cl2 remains a documented public threat. The severity of Cl2-induced injury depends primarily on the concentration and duration of exposure [3]. ARDS is the most severe consequence, and the overwhelming inflammatory response triggered by the initial injury is considered as the ARDS keystone [17, 18].
In addition to the classic pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs) are another significant pathway of uncontrolled inflammation. Extracellular histones were recognized as DAMP molecules in 2009 [12], and are essential inflammatory mediators in a variety of diseases such as sepsis, trauma, and multiple organ dysfunction syndrome [19, 20].
In this study, it was shown that Cl2 induced concentration-dependent lung injury. The levels of circulating histone H4 were closely correlated with Cl2 concentrations (ranging from 10 to 800 ppm). The concentration of Cl2 was a major determinant for the level of circulating histone H4. The pathogenic role of histone H4 in Cl2-induced ARDS was proven by histone H4 or specific blocking anti-H4 antibody intervention. Pretreatment with histone H4 further worsened the lethality rate and PaO2, while anti-H4 antibody showed some protective effects. Compared with the control mice, both endothelial and neutrophil activation were much more distinct in mice with Cl2-induced ARDS, which was indicated by P-selectin expression, vWF release, pulmonary neutrophil infiltration, and elevated MPO activity. Pretreatment with intravenous histone H4 aggravated endothelial and neutrophil activation, while anti-H4 antibody played an antagonistic role. Thus, it can be deduced that circulating histone H4 was a critical inflammatory mediator in Cl2-induced ARDS.
Additionally, TLRs were closely involved with histone H4-mediated inflammatory injury during Cl2-induced ARDS. The Tlr4-KO mice were resistant to exposure to lethal Cl2 concentrations. Tlr4 deficiency improved lethality rate, PaO2, and pulmonary edema, and prevented the endothelial and neutrophil activation caused by Cl2 exposure. More importantly, Tlr4 gene deletion greatly diminished the effect of histone H4 or anti-H4 antibody observed in WT mice. In contrast to Tlr4 deficiency, the impact of Tlr2 on inflammatory injury was not evident. In addition to TLR4 and TLR2, the roles of TLR1 and TLR6 were also screened. The blocking antibody against TLR4 or TLR2 decreased the histone H4-mediated expression of TNFA and IL1B in human pulmonary vascular endothelial cells, while the blocking antibody against TLR1 or TLR6 showed little effect.
Histones are structural elements of nuclear chromatin that mainly regulate gene expression. When histones are released passively from necrotic cells or actively by cell death such as NETosis, extracellular histones produce toxic effects on adjacent and circulating cells [21, 22]. The predominant source of histones may be neutrophils that have been activated by C5a to form neutrophil extracellular traps (NETs) in ALI [23, 24]. In contrast, nucleosomes, composed of histones and DNA, appear to be less toxic [25, 26]. Extracellular histones are DAMP molecules that can cause systemic inflammation involved in a wide range of inflammatory conditions, such as acute lung injury, liver injury, kidney injury, myocardial injury, cerebral stroke, coagulopathy, systemic lupus erythematosus, and even hair follicle death [27–34]. Extracellular histones can induce the release of chemokines, and activate the vascular endothelium, so as to facilitate leukocyte adhesion and transmigration [35, 36]. Both endothelial and neutrophilic activation are key events during the ARDS inflammatory response [37, 38]. Elevated P-selectin expression is an important marker for endothelial activation. Additionally, P-selectin translocation is a prerequisite for neutrophil adhesion to the pulmonary vascular endothelium during inflammation [39, 40]. Along with P-selectin translocation, abundant vWF is released from the endothelia, which excessively activates the coagulation cascade and aggravates lung injury [41, 42]. In addition to the endothelia and neutrophils, Westman et al. showed that extracellular histones induced monocytes to produce chemokines such as CXCL9 and CXCL10, which triggered neutrophil recruitment [36]. Fuchs et al. showed that extracellular histones bound to platelets, inducing calcium influx, recruiting plasma adhesion proteins such as fibrinogen, and triggering microaggregation [43].
Extracellular histones serve as DAMP molecules to promote inflammation. ARDS is an overwhelming inflammatory response that is triggered by various damaging factors. The initial detrimental stimuli are sensed by pattern recognition receptors (PRRs) [44]. TLRs are responsible for sensing invading pathogens and injury stimuli outside of the cell, as well as in intracellular endosomes and lysosomes. TLR1, TLR2, TLR4, TLR5, and TLR6 are present in the plasma membrane, while TLR3, TLR7, and TLR9 are mainly present in the membrane of the endoplasmic reticulum [45]. Thus, TLR1, TLR2, TLR4, TLR5, and TLR6 may mediate the inflammation caused by extracellular histones. In accordance with the results of this study, both TLR2 and TLR4 were previously found to be involved with extracellular histones-induced liver and kidney injury [39, 46]. In addition to TLR2 and TLR4, other PRRs are also involved in extracellular histones-induced inflammation. Huang et al. demonstrated that Tlr9-KO mice were protected from histone-mediated ischemia/reperfusion-induced liver injury [47]. TLR9 is generally viewed as a receptor mediating signaling brought about by endogenous circulating DNA released from dying cells. The exogenous histones may act as a cofactor of DNA, and thus, they can amplify TLR9-mediated signaling.
Histones are unique cytotoxic DAMP molecules that elicit both PRR-dependent pro-inflammatory signaling and PRR-independent direct cytotoxicity. Rationally, the affinity of histones for phosphodiester bonds may ensure their avid binding to both DNA in the nucleus and phospholipids in the plasma membrane [48, 49]. Abrams et al. demonstrated that FITC-labeled histones directly bind to the surface of cultured EA. hy926 endothelial cells, subsequently inducing an influx of calcium, ultimately resulting in cell lysis [11]. Silvestre-Roig et al. demonstrated that when smooth muscle cells (SMCs) were exposed to histone H4, alterations in the membrane were characterized by dynamic bending and pore formation. The activated SMCs attracted neutrophils and triggered the ejection of NETs. NETs, or histone H4 could induce SMC swelling and release of ATP. Finally, extracellular histone H4 triggered inflammation and arterial tissue damage by mediating SMC membrane lysis [50].
It is well-known that histone-acetylation (open chromatin) promotes gene expression, whereas histone deacetylation (closed chromatin) represses gene expression in living cells [51, 52]. To clarify whether extracellular histones underwent acetylation modification, this study detected histone H4 in plasma samples by western blot using an anti-acetylated histone H4-antibody (K5, K8, K12, K16). As shown in Figure S2, the acetylation status of plasma histone H4 in Cl2 exposure groups (10, 50 ppm) was similar to its status in the control group. An increase in the acetylation level of plasma histone H4 in the Cl2 exposure groups (200, 800 ppm) was not evident compared with the control group. Extracellular histones are released by NETs and apoptotic/necrotic cells. The main role of histones released from NETs is to kill bacteria instead of regulating gene transcription. Thus, acetylation modification was not applicable to them [53]. For the histones released from apoptotic/necrotic cells, they might be acetylated before release and retained the acetylation modification after release. It can be deduced that the pro-inflammatory effect of extracellular histones is mainly concentration dependent.
Because extracellular histones are cytotoxic DAMP molecules, it is rational that the damaging effects can be ameliorated through the application of histone-targeted interventions [54]. Administration of specific blocking antibodies or peptides targeted to these extracellular histones could inhibit inflammatory damage and improve outcomes in several types of animal models, such as sepsis, acute lung injury, liver injury, acute pancreatitis, and multiple organ injury [10, 55–58]. The direct toxicity of extracellular histones is dependent on the electrostatic membrane’s interaction with target cells. Accordingly, histone neutralizing agents have been identified as therapeutic options in treating extracellular histone-mediated tissue injury. Heparin is a highly sulfated negatively charged proteoglycan. Heparin and its derivatives can bind histones through electrostatic interactions, and they have demonstrated the ability to inhibit ALI, vascular endothelial injury, and thrombocytopenia caused by histones released [59, 60]. In addition to heparin and its derivatives, many innate and synthetic substances have been proven to prevent histone-related toxicity, including plasma albumin, C-reactive protein, bacterial O-antigen, polyglutamic acid and polysialic acid [25, 61–64].
Conclusions
This study showed that circulating histone H4 played a pro-inflammatory role primarily through TLR4 in ARDS caused by Cl2. This may help to explain the pathogenesis of ARDS and promote potentially effective intervention. Surely, the mechanisms for Cl2-induced inflammatory injury are very complex, so more thorough investigation is required to further understand the pathogenesis. Since the data originate from animal experiments, caution must be exercised in extrapolating these findings to more complicated clinical situations.
Supplementary information
Additional file 1: Figure S1. Pathological changes in lung tissue after Cl2 exposure. Twenty four hours after exposure (30 min) to different concentrations (10 to 800 ppm) of Cl2, pulmonary histopathology was analyzed, including alveolar hemorrhage, interstitial edema, infiltration of inflammatory cells, and disruption of the alveolar wall. Lung sections were stained with hematoxylin and eosin (HE). N = 6 for all groups. Scale bars: 100 μm. Figure S2 Acetylation analysis of circulating histone H4 after Cl2 exposure. Twenty four hours after mice were treated (30 min) with different concentrations (10, 50, 200, and 800 ppm) of Cl2, the acetylation status of circulating histone H4 was analyzed by western blot. The concentration of histone H4 was measured with a histone H4 detection kit. Equal amounts of histone H4 were mixed with loading buffer and subjected to electrophoresis. Western blot was performed in triplicate.
Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Abbreviations
- ALI
Acute lung injury
- ARDS
Acute respiratory distress syndrome
- Cl2
Chlorine gas
- PaO2
Arterial partial oxygen pressure
- MPO
Myeloperoxidase
- vWF
von Willebrand factor
- WPBs
WEIBEL-Palade bodies
- PAMPs
Pathogen-associated molecular patterns
- DAMPs
Damage-associated molecular patterns
- PRRs
Pattern recognition receptors
- TLR
TOLL-like receptor
- WT
Wild-type
- KO
Knock-out
Authors’ contributions
YLZ and SQL designed the experiments. JZ, LG, and LJM performed the animal and cellular experiments. YLZ, JZ, and JYZ analyzed and interpreted the data. YLZ and JYZ were major contributors in writing the manuscript. YLZ and SQL contributed the reagents and materials. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant 81773374) and the Natural Science Foundation of Beijing Municipality (Grant 7182179).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The protocols for the animal experiment were reviewed and permitted by the Peking University Animal Care and Use Committee (No. LA201783).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yanlin Zhang and Jian Zhao contributed equally to this work.
Contributor Information
Yanlin Zhang, Email: zhangyanlin@bjmu.edu.cn.
Shuqiang Li, Email: shuqiangli@263.net.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12995-020-00282-z.
References
- 1.Zellner T, Eyer F. Choking agents and chlorine gas-history, pathophysiology, clinical effects and treatment. Toxicol Lett. 2020;320:73–79. doi: 10.1016/j.toxlet.2019.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Winder C. The toxicology of chlorine. Environ Res. 2001;85(2):105–114. doi: 10.1006/enrs.2000.4110. [DOI] [PubMed] [Google Scholar]
- 3.Van Sickle D, Wenck MA, Belflower A, Drociuk D, Ferdinands J, Holguin F, et al. Acute health effects after exposure to chlorine gas released after a train derailment. Am J Emerg Med. 2009;27(1):1–7. doi: 10.1016/j.ajem.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5(1):18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Definition Task Force ARDS, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 6.Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome: advances in diagnosis and treatment. JAMA. 2018;319(7):698–710. doi: 10.1001/jama.2017.21907. [DOI] [PubMed] [Google Scholar]
- 7.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White CW, Martin JG. Chlorine gas inhalation: human clinical evidence of toxicity and experience in animal models. Proc Am Thorac Soc. 2010;7(4):257–263. doi: 10.1513/pats.201001-008SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Govier P, Coulson JM. Civilian exposure to chlorine gas: a systematic review. Toxicol Lett. 2018;293:249–252. doi: 10.1016/j.toxlet.2018.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Kawai C, Kotani H, Miyao M, Ishida T, Jemail L, Abiru H, et al. Circulating extracellular histones are clinically relevant mediators of multiple organ injury. Am J Pathol. 2016;186(4):829–843. doi: 10.1016/j.ajpath.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma associated lung injury. Am J Respir Crit Care Med. 2013;187(2):160–169. doi: 10.1164/rccm.201206-1037OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15(11):1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via TlR2 and TlR4. J Am Soc Nephrol. 2012;23(8):1375–1388. doi: 10.1681/ASN.2011111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monestier M, Fasy TM, Losman MJ, Novick KE, Muller S. Structure and binding properties of monoclonal antibodies to core histones from autoimmune mice. Mol Immunol. 1993;30(12):1069–1075. doi: 10.1016/0161-5890(93)90153-3. [DOI] [PubMed] [Google Scholar]
- 15.Kiani S, Chavez A, Tuttle M, Hall RN, Chari R, Ter-Ovanesyan D, et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat Methods. 2015;12(11):1051–1054. doi: 10.1038/nmeth.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zarogiannis SG, Jurkuvenaite A, Fernandez S, Doran SF, Yadav AK, Squadrito GL, et al. Ascorbate and deferoxamine administration after chlorine exposure decrease mortality and lung injury in mice. Am J Respir Cell Mol Biol. 2011;45(2):386–392. doi: 10.1165/rcmb.2010-0432OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nanchal RS, Truwit JD. Recent advances in understanding and treating acute respiratory distress syndrome. F1000Res. 2018;7:F1000 Faculty Rev-1322. doi: 10.12688/f1000research.15493.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33(4):319–327. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Z, Abrams ST, Alhamdi Y, Toh J, Yu W, Wang G, et al. Circulating histones are major mediators of multiple organ dysfunction syndrome in acute critical illnesses. Crit Care Med. 2019;47(8):e677–e684. doi: 10.1097/CCM.0000000000003839. [DOI] [PubMed] [Google Scholar]
- 20.Patel S. Danger-associated molecular patterns (DAMPs): the derivatives and triggers of inflammation. Curr Allergy Asthma Rep. 2018;18(11):63. doi: 10.1007/s11882-018-0817-3. [DOI] [PubMed] [Google Scholar]
- 21.Magna M, Pisetsky DS. The alarmin properties of DNA and DNA-associated nuclear proteins. Clin Ther. 2016;38(5):1029–1041. doi: 10.1016/j.clinthera.2016.02.029. [DOI] [PubMed] [Google Scholar]
- 22.Sørensen OE, Borregaard N. Neutrophil extracellular traps - the dark side of neutrophils. J Clin Invest. 2016;126(5):1612–1620. doi: 10.1172/JCI84538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grailer JJ, Ward PA. Lung inflammation and damage induced by extracellular histones. Inflamm Cell Signal. 2014;1(4):e131. doi: 10.14800/ics.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosmann M, Grailer JJ, Ruemmler R, Russkamp NF, Zetoune FS, Sarma JV, et al. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J. 2013;27(12):5010–5021. doi: 10.1096/fj.13-236380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abrams ST, Zhang N, Dart C, Wang SS, Thachil J, Guan Y, et al. Human CRP defends against the toxicity of circulating histones. J Immunol. 2013;191(5):2495–2502. doi: 10.4049/jimmunol.1203181. [DOI] [PubMed] [Google Scholar]
- 26.Gauthier VJ, Tyler LN, Mannik M. Blood clearance kinetics and liver uptake of mononucleosomes in mice. J Immunol. 1996;156(3):1151–1156. [PubMed] [Google Scholar]
- 27.Zhang Y, Wen Z, Guan L, Jiang P, Gu T, Zhao J, et al. Extracellular histones play an inflammatory role in acid aspiration-induced acute respiratory distress syndrome. Anesthesiology. 2015;122(1):127–139. doi: 10.1097/ALN.0000000000000429. [DOI] [PubMed] [Google Scholar]
- 28.Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol. 2013;191(5):2665–2679. doi: 10.4049/jimmunol.1202733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar SV, Kulkarni OP, Mulay SR, Darisipudi MN, Romoli S, Thomasova D, et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J Am Soc Nephrol. 2015;26(10):2399–2413. doi: 10.1681/ASN.2014070673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah M, Yellon DM, Davidson SM. The role of extracellular DNA and histones in ischaemia-reperfusion injury of the myocardium. Cardiovasc Drugs Ther. 2020;34(1):123–131. doi: 10.1007/s10557-020-06946-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Meyer SF, Suidan GL, Fuchs TA, Monestier M, Wagner DD. Extracellular chromatin is an important mediator of ischemic stroke in mice. Arterioscler Thromb Vasc Biol. 2012;32(8):1884–1891. doi: 10.1161/ATVBAHA.112.250993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakahara M, Ito T, Kawahara K, Yamamoto M, Nagasato T, Shrestha B, et al. Recombinant thrombomodulin protects mice against histone-induced lethal thromboembolism. PLoS One. 2013;8(9):e75961. doi: 10.1371/journal.pone.0075961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amoura Z, Piette JC, Chabre H, Cacoub P, Papo T, Wechsler B, et al. Circulating plasma levels of nucleosomes in patients with systemic lupus erythematosus: correlation with serum antinucleosome antibody titers and absence of clear association with disease activity. Arthritis Rheum. 1997;40(12):2217–2225. doi: 10.1002/art.1780401217. [DOI] [PubMed] [Google Scholar]
- 34.Shin SH, Joo HW, Kim MK, Kim JC, Sung YK. Extracellular histones inhibit hair shaft elongation in cultured human hair follicles and promote regression of hair follicles in mice. Exp Dermatol. 2012;21(12):956–958. doi: 10.1111/exd.12033. [DOI] [PubMed] [Google Scholar]
- 35.Chen R, Xie Y, Zhong X, Fu Y, Huang Y, Zhen Y, et al. Novel chemokine-like activities of histones in tumor metastasis. Oncotarget. 2016;7(38):61728–61740. doi: 10.18632/oncotarget.11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Westman J, Papareddy P, Dahlgren MW, Chakrakodi B, Norrby-Teglund A, Smeds E, et al. Extracellular histones induce chemokine production in whole blood ex vivo and leukocyte recruitment in vivo. PLoS Pathog. 2015;11(12):e1005319. doi: 10.1371/journal.ppat.1005319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Looney MR, Matthay MA. Neutrophil sandwiches injure the microcirculation. Nat Med. 2009;15(4):364–366. doi: 10.1038/nm0409-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Hinsbergh VW. Endothelium-role in regulation of coagulation and inflammation. Semin Immunopathol. 2012;34(1):93–106. doi: 10.1007/s00281-011-0285-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang HB, Wang JT, Zhang L, Geng ZH, Xu WL, Xu T, et al. P-selectin primes leukocyte integrin activation during inflammation. Nat Immunol. 2007;8(8):882–892. doi: 10.1038/ni1491. [DOI] [PubMed] [Google Scholar]
- 40.Jin H, Gebska MA, Blokhin IO, Wilson KM, Ketsawatsomkron P, Chauhan AK, et al. Endothelial PPAR-γ protects against vascular thrombosis by downregulating P-selectin expression. Arterioscler Thromb Vasc Biol. 2015;35(4):838–844. doi: 10.1161/ATVBAHA.115.305378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ochoa CD, Wu S, Stevens T. New developments in lung endothelial heterogeneity: Von Willebrand factor, P-selectin, and the Weibel-Palade body. Semin Thromb Hemost. 2010;36(3):301–308. doi: 10.1055/s-0030-1253452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ware LB, Matthay MA, Parsons PE, Thompson BT, Januzzi JL, Eisner MD. Pathogenetic and prognostic significance of altered coagulation and fibrinolysis in acute lung injury/acute respiratory distress syndrome. Crit Care Med. 2007;35(8):1821–1828. doi: 10.1097/01.CCM.0000221922.08878.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118(13):3708–3714. doi: 10.1182/blood-2011-01-332676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 45.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 46.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187(5):2626–2631. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through toll-like receptor 9 in mice. Hepatology. 2011;54(3):999–1008. doi: 10.1002/hep.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furnrohr BG, Groer GJ, Sehnert B, Herrmann M, Voll RE. Interaction of histones with phospholipids–implications for the exposure of histones on apoptotic cells. Autoimmunity. 2007;40(4):322–326. doi: 10.1080/08916930701356457. [DOI] [PubMed] [Google Scholar]
- 49.Kleine TJ, Lewis PN, Lewis SA. Histone-induced damage of a mammalian epithelium: the role of protein and membrane structure. Am J Phys. 1997;273(6):C1925–C1936. doi: 10.1152/ajpcell.1997.273.6.C1925. [DOI] [PubMed] [Google Scholar]
- 50.Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. 2019;569(7755):236–240. doi: 10.1038/s41586-019-1167-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389(6649):349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 52.Koprinarova M, Schnekenburger M, Diederich M. Role of histone acetylation in cell cycle regulation. Curr Top Med Chem. 2016;16(7):732–744. doi: 10.2174/1568026615666150825140822. [DOI] [PubMed] [Google Scholar]
- 53.Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. 2019;133(20):2178–2185. doi: 10.1182/blood-2018-11-844530. [DOI] [PubMed] [Google Scholar]
- 54.Szatmary P, Huang W, Criddle D, Tepikin A, Sutton R. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J Cell Mol Med. 2018;22(10):4617–4629. doi: 10.1111/jcmm.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alhamdi Y, Abrams ST, Cheng Z, Jing S, Su D, Liu Z, et al. Circulating histones are major mediators of cardiac injury in patients with sepsis. Crit Care Med. 2015;43(10):2094–2103. doi: 10.1097/CCM.0000000000001162. [DOI] [PubMed] [Google Scholar]
- 56.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122(7):2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–614. doi: 10.1002/hep.27841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Merza M, Rahman M, Zhang S, Hwaiz R, Regner S, Schmidtchen A, et al. Human thrombin-derived host defense peptides inhibit neutrophil recruitment and tissue injury in severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2014;307(9):G914–G921. doi: 10.1152/ajpgi.00237.2014. [DOI] [PubMed] [Google Scholar]
- 59.Alcantara FF, Iglehart DJ, Ochs RL. Heparin in plasma samples causes nonspecific binding to histones on Western blots. J Immunol Methods. 1999;226(1–2):11–18. doi: 10.1016/S0022-1759(99)00043-5. [DOI] [PubMed] [Google Scholar]
- 60.Wang F, Zhang N, Li B, Liu L, Ding L, Wang Y, et al. Heparin defends against the toxicity of circulating histones in sepsis. Front Biosci (Landmark Ed) 2015;20:1259–1270. doi: 10.2741/4370. [DOI] [PubMed] [Google Scholar]
- 61.Lam FW, Cruz MA, Leung HC, Parikh KS, Smith CW, Rumbaut RE. Histone induced platelet aggregation is inhibited by normal albumin. Thromb Res. 2013;132(1):69–76. doi: 10.1016/j.thromres.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 62.Gamberucci A, Fulceri R, Marcolongo P, Pralong WF, Benedetti A. Histones and basic polypeptides activate Ca2+/cation influx in various cell types. Biochem J. 1998;331(Pt2):623–630. doi: 10.1042/bj3310623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. 2015;149(7):1920–1931. doi: 10.1053/j.gastro.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 64.Chaput C, Spindler E, Gill RT, Zychlinsky A. O-antigen protects gram-negative bacteria from histone killing. PLoS One. 2013;8(8):e71097. doi: 10.1371/journal.pone.0071097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Pathological changes in lung tissue after Cl2 exposure. Twenty four hours after exposure (30 min) to different concentrations (10 to 800 ppm) of Cl2, pulmonary histopathology was analyzed, including alveolar hemorrhage, interstitial edema, infiltration of inflammatory cells, and disruption of the alveolar wall. Lung sections were stained with hematoxylin and eosin (HE). N = 6 for all groups. Scale bars: 100 μm. Figure S2 Acetylation analysis of circulating histone H4 after Cl2 exposure. Twenty four hours after mice were treated (30 min) with different concentrations (10, 50, 200, and 800 ppm) of Cl2, the acetylation status of circulating histone H4 was analyzed by western blot. The concentration of histone H4 was measured with a histone H4 detection kit. Equal amounts of histone H4 were mixed with loading buffer and subjected to electrophoresis. Western blot was performed in triplicate.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.