

Abstract
Identifying disease-causing pathways and drugs that target them in Parkinson’s disease (PD) has remained challenging. We uncovered a PD-relevant pathway in which the stress-regulated heterodimeric transcription complex CHOP/ATF4 induces the neuron prodeath protein Trib3 that in turn depletes the neuronal survival protein Parkin. Here we sought to determine whether the drug adaptaquin, which inhibits ATF4-dependent transcription, could suppress Trib3 induction and neuronal death in cellular and animal models of PD. Neuronal PC12 cells and ventral midbrain dopaminergic neurons were assessed in vitro for survival, transcription factor levels and Trib3 or Parkin expression after exposure to 6-hydroxydopamine or 1-methyl-4-phenylpyridinium with or without adaptaquin co-treatment. 6-hydroxydopamine injection into the medial forebrain bundle was used to examine the effects of systemic adaptaquin on signaling, substantia nigra dopaminergic neuron survival and striatal projections as well as motor behavior. In both culture and animal models, adaptaquin suppressed elevation of ATF4 and/or CHOP and induction of Trib3 in response to 1-methyl-4-phenylpyridinium and/or 6-hydroxydopamine. In culture, adaptaquin preserved Parkin levels, provided neuroprotection and preserved morphology. In the mouse model, adaptaquin treatment enhanced survival of dopaminergic neurons and substantially protected their striatal projections. It also significantly enhanced retention of nigrostriatal function. These findings define a novel pharmacological approach involving the drug adaptaquin, a selective modulator of hypoxic adaptation, for suppressing Parkin loss and neurodegeneration in toxin models of PD. As adaptaquin possesses an oxyquinoline backbone with known safety in humans, these findings provide a firm rationale for advancing it towards clinical evaluation in PD.
Keywords: Parkinson’s disease, Trib3, ATF4, CHOP, Parkin, Adaptaquin, Neuroprotection, Oxyquinoline
1. Introduction
Current treatments for Parkinson’s disease (PD) temporarily relieve associated motor symptoms, but do not stop or slow loss of affected neuronal populations (Olanow et al., 2009; Schapira et al., 2014). There is therefore a critical need in PD to uncover disease-related pathways and to identify suitable therapeutics that target such pathways to suppress neurodegeneration and disease progression.
Tribbles pseudokinase 3 (Trib3) is a protein mediating cell death and neurodegeneration in multiple PD models and is highly elevated with a dramatically altered distribution in dopaminergic substantia nigral (SN) neurons of PD patients (Aimé et al., 2015). Trib3 is induced by a range of PD-relevant stresses (Kiss-Toth, 2015) including robust transcriptional activation in 6-hydroxydopamine (6-OHDA) models of PD both in vitro (Ryu et al., 2005; Aimé et al., 2015) and in vivo (Kanaan et al., 2015). Such induction is observed before measurable cell death in cellular PD models, including neuronal PC12 cells and rat ventral midbrain dopaminergic (VM DA) neurons treated with 6-OHDA, 1-methyl-4-phenylpyridinium (MPP+), or α-synuclein (αSYN) fibrils (Ryu et al., 2005; Aimé et al., 2015; Aimé et al., 2018). Trib3 over-expression is sufficient to induce neuron apoptosis and Trib3 knockdown protects from 6-OHDA, MPP+ and αSYN-induced death (Aimé et al., 2015). Regarding mechanism, Trib3 physically interacts with, and interferes with expression of Parkin (Aimé et al., 2015), a pro-survival protein whose loss-of-function is linked to both familial and sporadic forms of PD (Dawson and Dawson, 2010; Dawson and Dawson, 2014). Such findings therefore identify Trib3 as a promising therapeutic target for PD.
Several transcriptional regulators mediate Trib3 induction, including activating transcription factor 4 (ATF4) (Ohoka et al., 2005; Ord and Ord, 2005; Han et al., 2012; Aimé et al., 2015). ATF4 is highly expressed in the SN of PD patients (Sun et al., 2013) and in cellular PD toxin models, ATF4, along with its binding partner CHOP (product of the Ddit3 gene), mediates Trib3 induction (Aimé et al., 2015). Because Trib3 induction occurs before and promotes cell death in PD cellular models, we reasoned that impeding its transcriptional activation by ATF4 and/or CHOP would be an attractive strategy to suppress neuronal degeneration in PD.
As a potential inhibitor of the ATF4/CHOP-Trib3 prodeath pathway in PD, we considered the small molecule adaptaquin (AQ). AQ is an oxyquinoline inhibitor of hypoxia inducible factor prolyl hydroxylases (HIF PHDs), metalloenzymes that hydroxylate prolines and destabilize HIF1α under normoxia (Smirnova et al., 2010; Karuppagounder and Ratan, 2012; Lee et al., 2014). Although HIF1α is a canonical HIF PHD substrate, PHDs also hydroxylate and regulate other substrates (Gorres and Raines, 2010) including ATF4 (Koditz et al., 2007). siRNA-mediated HIF PHD3 silencing or mutation of proline residues stabilize ATF4 under conditions of anoxia (Koditz et al., 2007). It was recently reported that HIF PHD inhibition by AQ reduces ATF4 proline hydroxylation, represses ATF4 dependent pro-death genes and improves functional outcomes in rodent models of intra-cerebral hemorrhage (Karuppagounder et al., 2016). Trib3 was among the most responsive ATF4 targets in this model and AQ reduced ATF4 occupancy and activation of the Trib3 promoter and suppressed Trib3 expression (Karuppagounder et al., 2016). These findings thus identify AQ as a promising drug to prevent ATF4-dependent Trib3 induction. Given the apparent role of Trib3 in PD and its regulation by ATF4 in PD models, we were prompted to evaluate AQ’s capacity to suppress Trib3 induction and to provide neuroprotection in in vitro and in vivo PD models.
2. Material and methods
2.1. Cell culture
PC12 cells were cultured as described previously (Greene and Tischler, 1976; Aimé et al., 2015; Aimé et al., 2018) on plastic cell culture dishes coated with rat tail collagen (Roche). Non-differentiated PC12 cells were grown in RPMI 1640 cell culture medium supplemented with 10% heat inactivated horse serum (Sigma), 5% fetal bovine serum (FBS) and penicillin/streptomycin. For neuronal differentiation, cells were grown in RPMI 1640 cell culture medium supplemented with 1% horse serum, penicillin/streptomycin, and a 100 ng/ml final concentration of human recombinant nerve growth factor (Gemini Bioproducts). Cell culture medium was changed every other day.
HEK293T/17 cells were grown in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin.
Ventral midbrain dopaminergic (VM DA) neurons from P0–P3 rats were dissected, dissociated, and plated on a confluent glial monolayer following the protocol kindly provided by Dr. David Sulzer, Columbia University and as described previously (Rayport et al., 1992).
2.2. Parkinson’s disease toxins
For PC12 cells, 10 mM stock solutions of 6-hydroxydopamine (6-OHDA) (Tocris) or 1-methyl-4-phenylpyridinium (MPP+) (Sigma) diluted in water were freshly prepared just before each experiment. 6-OHDA was used at a final concentration of 150 μM and MPP+ was used at a final concentration of 1 mM. For VM DA neurons, the 10 mM stock solution of 6-OHDA was prepared in MEM supplemented with ascorbic acid (Sigma) to prevent 6-OHDA oxidation and degradation (Ding et al., 2004). 6-OHDA was used at a final concentration of 40 μM in 0.015% ascorbic acid. MPP+ was diluted in water and used at a final concentration of 40 μM.
2.3. In vitro adaptaquin treatment
For PC12 cells and VM DA neurons, 10 mM stock solution of adaptaquin diluted in anhydrous DMSO was prepared and stored at −20 °C. On the day of treatment, stock solution was diluted 1:100 in cell culture medium and added to the cell culture wells at final concentrations ranging from 0.1 μM to 5 μM.
2.4. Lentiviral preparation and transduction
The following sequence was used for shRNA-mediated down-regulation of ATF4: shATF4, 5′-GCCTGACTCTGCTGCTTATAT-3′ and Parkin: sh-Parkin 5’-GGACACATCAGTAGCTTTG-3′. A mutated version of shATF4 (mutated bases are underlined; shControl 5′-GCCAGATTCAGCGGCCTACAT-3′ was used as a control shRNA. The transfer plasmid used for ATF4 and Parkin shRNAs and shControl expression was pLVTHM (Addgene), expressing shRNAs from the H1 promoter along with GFP from the EF1-α promoter. Lentiviruses were prepared in HEK293T/17 cells by co-transfecting pLVTHM expression plasmids along with second-generation lentiviral packaging plasmids (AddGene) using the calcium phosphate transfection method. Lentiviral particles were collected twice (48 h and 72 h after transfection) and concentrated using Lenti-X concentrator (Clontech #631231) following the manufacturer’s protocol, resuspended in PBS and stored at −80 °C. For lentiviral transduction, 0.1 up to 5 × 107 viral particles were added per cm2 of culture area, directly into the cell medium. The transduced neurons were analyzed after 5–14 days.
2.5. Quantitative real-time PCR
PC12 cells were lysed and total RNA was extracted using TRI reagent (Molecular Research Center) following the manufacturer’s protocol. RNA concentration and purity were assessed by measuring the optical density at 260 and 280 nm with a NanoDrop (Thermo Scientific). cDNA was synthesized using first-strand cDNA synthesis kit (Origene) with 1 μg of total RNA, following the manufacturer’s instructions. Quantitative real-time PCR was performed using FastStart SYBR Green Master Mix (Roche) and an Eppendorf Realplex Mastercyler with the following settings: 1 cycle at 95 °C for 10 min and 40 cycles of amplification: 95 °C for 15 s, 58–60 °C for 30–60 s, 72 °C for 30–60 s. The amounts of Trib3, CHOP and ATF4 mRNAs were quantified and normalized to 18S rRNA or GAPDH mRNA using the following primer pairs: Trib3 Fwd 5′-GTTGCGTCGATTTGTCTTCA-3′ and Rvrs 5′-CGGGAGCTGAGTATCTCTGG-3′; ATF4 Fwd 5′- CCTTCGACCAGTCG GGTTTG-3′ and Rvrs 5′-CTGTCCCGGAAAAGGCATCC-3′; CHOP Fwd 5′-CTGGAAGCCTGGTATGAGGA-3′ and Rvrs 5′-AGGTGCTTGTGACCTCTGCT-3′; 18S Fwd 5′- TTGATTAAGTCCCTGCCCTTTGT-3′ and reverse 5′- CGATCCGAGGGCCTCACTA-3′; GAPDH Fwd 5′-GACATCAAGAAGGTGGTGAA-3′ and Rvrs 5′-TGTCATACCAGGAAATGAGC-3′. The threshold cycles were determined for each gene of interest and normalized to the threshold cycles of a housekeeping gene. Relative mRNA levels for our genes of interest were expressed as a fold induction in an experimental condition compared to a control condition.
2.6. Western immunoblotting
Cells were homogenized in 1× cell lysis buffer (Cell Signaling) supplemented with complete mini protease inhibitor cocktail (Roche). Samples were sonicated and protein concentrations were determined by BCA assay according to the manufacturer’s protocol (Thermo Scientific #23225). Protein samples were prepared for loading with LDS-sample buffer (Life Technologies) supplemented with 50 mM dithiothreitol. Twenty microgram of proteins were loaded per well of 10% Bis-Tris polyacrylamide gels (Life technologies) and separated by electrophoresis for 1 h at 110 V. Proteins were then transferred onto a PVDF membrane (Bio Rad) for 1 h 30 min at 40 V. Membranes were blocked with 5% powdered milk in TBS containing 0.1% of Tween-20 (TBST) and incubated overnight at 4 °C with primary antibodies. The following primary antibodies were used for Western blotting: rabbit anti-ATF4 (diluted a 1:1000, Cell Signaling #11815), mouse anti-CHOP/GADD153 (diluted a 1:500, Santa Cruz Biotechnology #sc-7351), rabbit anti-Trib3 (diluted at 1:2000, Calbiochem #ST1032), mouse anti-Parkin (diluted at 1:500, Santa Cruz Biotechnology #sc-32282), rabbit anti-Erk1/2 (diluted a 1:5000, Santa Cruz Biotechnology #sc-93). Membranes were washed 3 times with TBST and incubated with HRP-conjugated secondary antibodies diluted at 1:2500. After 3 final washes with TBST, blots were incubated with ECL reagents (GE Amersham Biosciences) and chemiluminescent signals were detected by exposure to autoradiography film. Films were scanned using a desktop scanner and band intensities were quantified using ImageJ software.
2.7. Immunofluorescence
Cells were fixed for 12–15 min in 4% PFA and washed 3 times with PBS. Cells were blocked with Superblock (Thermo Scientific) supplemented with 0.3% Triton X-100 for 1 h at room temperature and incubated overnight at 4 °C with primary antibodies. The following primary antibodies were used for immunofluorescence: rabbit anti-Phospho-Histone H2A.X (diluted at 1:500, Cell Signaling #9718), mouse anti-tyrosine hydroxylase (diluted at 1:500, Millipore #MAB318). Cells were washed 3 times with PBS and incubated with fluorescent secondary antibodies for 2 h at room temperature: Alexa Fluor 568 anti-Mouse or -Rabbit, Alexa Fluor 488 anti-Mouse or -rabbit (diluted at 1:2500, Life Technologies). For PC12 cells grown in multi-well dishes, Hoescht 33328 was added to the secondary antibody solution, cells were washed in PBS and observed with an Olympus inverted fluorescence microscope. For ventral midbrain dopaminergic neurons grown on glass coverslips, after 3 final washes with PBS, coverslips were mounted on slides with Vectashield mounting medium containing DAPI for nuclear staining (Vector Laboratories). Images were acquired using Olympus inverted and upright fluorescent microscopes equipped with digital camera and cellSens software.
2.8. Survival assays
For PC12 cells treated with Parkinson’s disease toxins and/or adaptaquin, cell survival was assessed on the total cell population by incubating the cell cultures with a detergent solution that lyses the plasma membrane and leaves the nuclei intact (10× counting lysis buffer: 5 g of cetyldimethyl-ethanolammonium bromide, 0.165 g of NaCl, 2.8 ml of glacial acetic acid, 50 ml of 10% TritonX-100, 2 ml of 1 M MgCl2, 10 ml of 10 × PBS, 35.2 ml of H2O). Two hundred and fifty microliters of 1 X counting lysis buffer was added per cm2 of culture dish area and the suspended nuclei were counted into a hemacytometer. For ventral midbrain dopaminergic neurons, cell survival was assessed by performing immunofluorescence and counting TH+ cells, or TH/GFP+ cells in the case of transduced neurons.
2.9. Animals and study approval
Adult male C57BL/6 mice (10–12 weeks) were purchased from Charles River Laboratories (Wilmington, MA, USA) and housed in a temperature, humidity controlled and pathogen-free environment under 12-h light/dark cycle conditions with access to food and water ad libitum. All procedures were performed under NIH guidelines for care and use of animals and approved by the Institutional Animal Care and Use Committee of the Weill Cornell Medical College, NY, USA.
2.10. In vivo 6-OHDA lesion model
6-OHDA hydrobromide was purchased from Sigma (St. Louis, MO) and prepared at a concentration of 6 μg/μl in sterile normal saline (0.9%)/ascorbate (0.2%) solution. To denervate nigrostriatal pathway in mice, 6-OHDA was stereotaxically injected in the right median forebrain bundle (MFB) at the following coordinates: anterior posterior, −1.2 mm; medio-lateral, 1.2 mm; dorsoventral, 5.0 mm (Paxinos and Watson, 2007). Each mouse received 1 μl of 6-OHDA (6 μg/μl) at a flow rate of 0.2 μl/min using a Nano-mite pump (Harvard Apparatus) and a Hamilton syringe. Sham mice received an injection of one μl of vehicle in the right MFB. The needle was left in place for 5 min after the injection was complete and was slowly withdrawn. Proper post-operative care was taken until the animals recovered completely.
2.11. In vivo adaptaquin treatment
Adaptaquin was prepared in a 5% DMSO and 95% sesame oil emulsion. A 30 mg/kg dose of adaptaquin was injected intraperitoneally (IP) starting 2 h after the 6-OHDA lesion. The 6-OHDA-only and sham groups received IP injections of 5% DMSO and 95% sesame oil (100 μl) only. Animals were randomized for sham or 6-OHDA groups. The surgeon who performed the 6-OHDA lesions was blinded for the treatments that were revealed after the data was collected.
2.12. RNAscope multiplex fluorescent in situ hybridization
Two hours after the 6-OHDA lesion, mice received a single injection of 30 mg/kg of adaptaquin IP or a single IP injection of vehicle. Eight hour after the 6-OHDA lesion, mice were anesthetized and transcardially perfused with 0.9% NaCl followed by 4% paraformaldehyde in 0.1 M phosphate buffer for 10 min. Brains were dissected and postfixed in 4% PFA for 24 h at 4 °C. The brains were cryoprotected in a sucrose gradient by immersion in 10% sucrose at 4 °C overnight, then by repeating this step in 20% followed by 30% sucrose solutions. The brains were then rapidly frozen by immersion in isopentane on dry ice. Fourteen micrometer sections of the substantia nigra were cut and collected in PBS. Sections were immediately mounted on superfrost plus slides and air-dried. Chosen sections underwent the multiplex fluorescent in situ hybridization procedure using ACD’s RNAscope multiplex fluorescent reagent kit v2 (ACD #323100) and following the manufacturer’s instructions. Briefly, sections were treated with hydrogen peroxide for 10 min at room temperature and washed twice in distilled water. Target retrieval was performed by incubating the sections in a Black & Decker steamer containing target retrieval reagent at > 99 °C for 5 min. Slides were rinsed in distilled water, incubated in 100% ethanol for 3 min and air-dried. A hydrophobic barrier was created around each section and the sections were treated with RNAscope protease plus for 30 min at 40 °C in ACD’s HybEZ incubation oven. After a wash in distilled water, sections were incubated with a mix of mouse-Trib3-C1 (ACD #506301), mouse-Slc6a3-C2 (ACD# 315441-C2) and mouse-Ddit3-C3 (ACD #317661-C3) RNAscope Probes for 2 h at 40 °C. Control sections were incubated with a set of negative control probes (targeting the bacterial gene DapB) or a set of positive control probes targeting the ubiquitous genes POLR2A, PPIB and UBC, that should display low, moderate and high levels of expression, respectively. All sections then underwent three amplification steps by sequentially applying Amp1 (30 min), Amp2 (30 min) and Amp3 (15 min) reagents at 40 °C after 2 washes in wash buffer. Trib3 mRNA signal was revealed by incubating the sections with HRP-C1 for 15 min followed by TSA plus Cyanine 5 (Perkin Elmer # NEL760001KT) for 30 min, and HRP blocker for 15 min at 40 °C. Slc6a3 (coding for the dopamine transporter, DAT) mRNA signal was revealed by incubating the sections with HRP-C2 for 15 min, followed by TSA plus Fluorescein for 30 min and HRP blocker for 15 min at 40 °C. Ddit3 (coding for CHOP) mRNA signal was revealed by incubating the sections with HRP-C3 for 15 min, followed by TSA plus Cyanine 3 for 30 min and HRP blocker for 15 min at 40 °C. All steps were preceded by two washes in wash buffer. After a final wash, sections were incubated with DAPI for 30 s and mounted with Prolong Gold Antifade mounting medium (Life Technologies). Images were acquired using a Zeiss epifluorescence microscope equipped with a digital camera and Neurolucida software.
2.13. Behavioral analysis
Starting 2 h after the 6-OHDA lesion, mice received a daily IP injection of 30 mg/kg of adaptaquin or an IP injection of vehicle for 7 consecutive days. In order to test for behavioral defects associated with lesions of the dopaminergic nigrostriatal system, 7, 14, 21 and 28 days after the 6-OHDA lesion, the mice were challenged with a subcutaneous dose of 0.5 mg/kg of apomorphine (Sigma, St. Louis, MO) and the contralateral rotational behavior was measured. Mice were placed in a hemispherical bowl (Schwarting and Huston, 1996) and apomorphine-induced rotational behavior was quantified by measuring the contralateral turns made by each animal over a 40 min period.
2.14. TH Immunohistochemistry and unbiased stereology
Starting 2 h after the 6-OHDA lesion, mice received a daily injection of 30 mg/kg of adaptaquin IP or an IP injection of vehicle for 7 consecutive days. After 28 days, mice were anesthetized and transcardially perfused with 30 ml of 0.9% NaCl followed by 50 ml of 4% paraformaldehyde in 0.1 M phosphate buffer for 10 min. Brains were dissected and postfixed in 4% PFA for 48 h at 4 °C before being cut at the level of the optic chiasm. The forebrain portion of the brain containing the striatum was rapidly frozen by immersion in isopentane on dry ice and cut in 30 μm serial sections collected in PBS. The midbrain portion of the brain containing the substantia nigra (SN) was postfixed for another 5 days in 4% PFA, cryoprotected in 20% sucrose for 48 h at 4 °C and then rapidly frozen by immersion in isopentane on dry ice. The brains were cut into 30 μm serial sections, were then cut through the
SN, collected in PBS and every 4th section underwent TH immunostaining. Striatal (ST) sections showing alignment of the lateral ventricles and the anterior commissures were chosen for TH immunostaining. Floating SN and ST sections were washed twice in PBS, blocked in PBS supplemented with 0.5% BSA and then permeabilized in PBS with 0.5% BSA and 0.1% Triton X-100 for 30 min. After 2 washes with PBS, the sections were incubated with rabbit anti-TH primary antibody (Calbiochem, #657012) diluted at 1:750 in PBS with 0.5% BSA for 48 h at 4 °C. Sections were washed twice in PBS with 0.5% BSA and incubated with biotinylated protein A for 1 h at room temperature. After 2 washes in PBS with 0.5% BSA, sections were incubated with avidin-biotinylated horseradish peroxidase complexes (ABC, Vector Labs) diluted at 1:600 in PBS for 1 h at room temperature. Sections were washed twice with PBS and immunoperoxidase staining was developed with diaminobenzidine. Sections were washed in Tris buffer and mounted on slides. ST sections were dehydrated, coverslipped and analyzed for TH-DAB+ optical density. The relative optical density of striatal TH immunostaining was determined with an Analytical Image Station (Imaging Research, St. Catherines, ON, Canada). All immunocytochemical analyses of ST and SN sections for all animals were performed at the same time using the same material/reagents/conditions to allow for unbiased comparison.
SN sections were counterstained with Thionin, coverslipped and analyzed for TH-DAB+ neuronal counts using unbiased stereological analysis (Chen et al., 2012). In order to count the TH-positive neurons in the control and experimental sides of the SN for each mouse, an area corresponding to the SN was manually delimited on each section and a fractionator probe was created with the StereoInvestigator program (MicroBrightField, Williston, VT). The number of TH-positive neurons in each counting frame was determined by focusing down through the section, using an 100× objective with immersion oil. Our criterion for counting an individual TH-positive neuron was the presence of its nucleus either within the counting frame or crossing the right or top limits of the counting frame. The total number of TH-positive neurons for each side of the SN was then estimated using the StereoInvestigator program.
2.15. Experimental design and statistical analysis
For in vitro experiments, Comparisons of two experimental groups were performed with a t-test and multiple comparisons of more than two treatment groups were performed using one-way analysis of variance (ANOVA) and Tukey’s multiple comparisons post-hoc test
For in vivo experiments, multiple comparisons were performed on raw TH+ numbers using two-way ANOVA with the treatment groups (sham, 6-OHDA and 6-OHDA + AQ) and the brain side (control and experimental) defined as factors. Post-hoc analyses were performed using Sidak’s multiple comparisons test. Multiple comparisons of the percentages of TH+ neurons and level of striatal TH immunoreactivity in the experimental side compared to the control side across the three animal groups (sham, 6-OHDA and 6-OHDA + AQ) were performed using one-way analysis of variance (ANOVA) and Tukey’s multiple comparisons post-hoc test. Multiple comparisons of the numbers of net contralateral rotations across the three animal groups (sham, 6-OHDA and 6-OHDA + AQ) under apomorphine challenge were performed using one-way analysis of variance (ANOVA) and Tukey’s multiple comparisons post-hoc test. The strength of experimental correlations between the percentages of TH+ neurons, the relative level of striatal TH immunoreactivity and the numbers of net contralateral rotations in the 6-OHDA + AQ group at 28 days was measured by the Pearson correlation coefficient. All statistical analyses were performed using GraphPad Prism software. The threshold of significance was set at α = 0.05 for all experiments.
3. Results
3.1. Adaptaquin blocks 6-OHDA and MPP+ induced death in vitro
To evaluate the neuroprotective potential of AQ, we first assessed survival and cellular morphology in a well-characterized PD cellular model of PC12 cells differentiated into neuron-like cells with NGF (Greene and Tischler, 1976) and treated with 6-OHDA (Ryu et al., 2002; Sun et al., 2013; Aimé et al., 2015; Aimé et al., 2018). Cultures were treated with diluent (DMSO) or 0.1, 0.5, 1 and 5 μM AQ ± 150 μM 6-OHDA (Fig. 1A,B). Twenty-four hour after 6-OHDA treatment alone, less than half the cells survived and those that remained presented an abnormal morphology, with scant neurites (Fig. 1A,B). The range of AQ concentrations tested alone showed no effect on either cell numbers or morphology. Although 0.1 μM AQ provided no significant protection from 6-OHDA, neurites were visible in some surviving cells. By contrast, cultures treated with 0.5, 1 and 5 μM AQ displayed robust protection from 6-OHDA, both in cell numbers and morphology, with extensive neurite preservation (Fig. 1A,B). We further confirmed protection by immunostaining phosphorylated (Ser139) histone H2A.X (PH2AX), an apoptotic marker (Lu et al., 2006). In cultures treated with 6-OHDA ± AQ (Fig. 1C), 6-OHDA alone induced a robust increase in nuclear PH2AX staining while co-treatment with 0.5 μM AQ blocked this response. Altogether, these results demonstrate that AQ prevents apoptosis of 6-OHDA-treated neuronal PC12 cells.
Fig. 1.
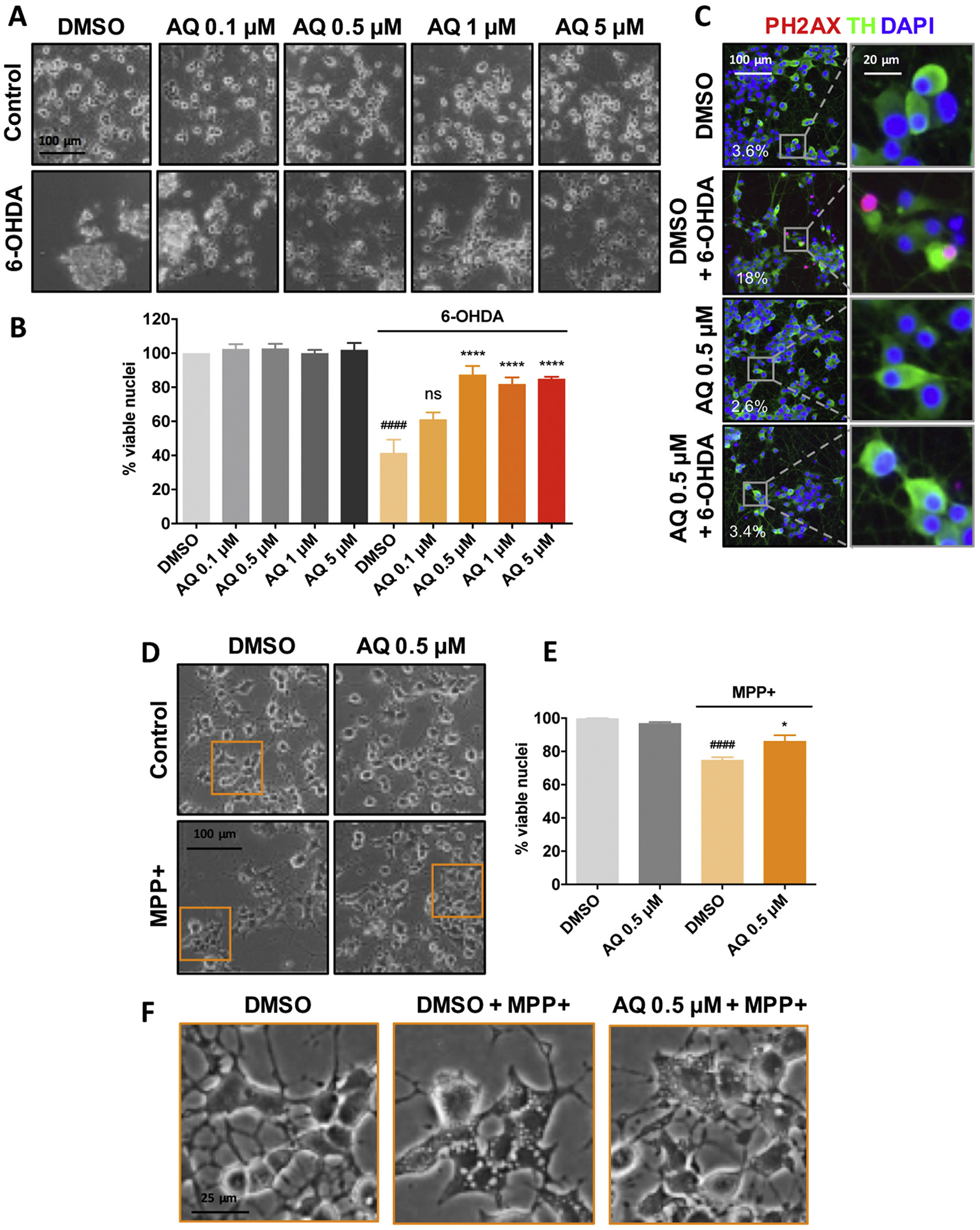
Adaptaquin protects neuronal PC12 cells against 6-OHDA and MPP+ − induced cell death. Phase contrast images (A) and corresponding quantifications (B) of survival assay showing the remaining viable nuclei in neuronal PC12 cell cultures either untreated (control, DMSO) or treated with 150 μM 6-OHDA and indicated AQ concentrations for 24 h. 6-OHDA induces significant cell death. ANOVA with Tukey’s post-hoc test ####p < .0001; ****p < .0001; ns = not significant. Values are mean +/− SEM of 3 independent experiments. Representative immunofluorescence images (C) showing that AQ decreases the phosphorylation of histone 2A.X (PH2AX), a marker of apoptosis. Neuronal PC12 cells were treated with DMSO or 0.5 μM AQ and 150 μM 6-OHDA for 24 h and stained for the catecholaminergic marker tyrosine hydroxylase (TH, green), nuclei (DAPI, blue) and PH2AX (red). The percentage of PH2AX+ cells for each condition is annotated on the images. Phase contrast images (D) and corresponding quantifications (E) of survival assay showing the remaining viable nuclei in neuronal PC12 cell cultures either untreated (control, DMSO) or treated with 1 mM MPP+ and 0.5 μM AQ for 48 h. ANOVA with Tukey’s post-hoc test: ####p < .0001) and *p < .05). Values are mean +/− SEM of 3 independent experiments. Higher magnification images (F) showing that MPP+ elicits the formation of large intracellular vacuoles and that treatment with 0.5 μM AQ reduces their size.
Next, we examined the protective potential of AQ in a second PD-relevant cellular model, exposure to MPP+. Cultures were treated with DMSO or 0.5 AQ ± 1 mM MPP+ for 48 h (Fig. 1D–F). MPP+ alone significantly affected viability with a loss of about 25% (Fig. 1D,E). Many surviving cells displayed large intracellular vacuoles (Fig. 1F) similar to those previously identified as autophagic vacuoles in MPP+-treated dopaminergic neurons (Zhu et al., 2007). Co-treatment with 0.5 μM AQ significantly increased the survival and appeared to substantially reduce vacuolar size (Fig. 1F).
To extend these findings, we evaluated the protective potential of AQ in cultured postnatally-derived rat ventral midbrain dopaminergic (VM DA) neurons that have been used for PD-related studies (Gearan et al., 2001; Ding et al., 2004; Mosharov et al., 2009; Aimé et al., 2015). We measured viability of tyrosine hydroxylase (TH) immunopositive neurons in cultures treated ± AQ and either 40 μM 6-OHDA or MPP+ for 24 h (Fig. 2). Consistent with previous literature (Gearan et al., 2001; Ding et al., 2004; Aimé et al., 2015), either toxin alone caused extensive VM DA neuron death. Strikingly, 0.5 μM AQ robustly protected VM DA neurons from 6-OHDA and MPP+ (Fig. 2A–D). In addition, in MPP+ treated cultures, the complexity of VM DA neuron arborization appeared reduced compared to controls. AQ co-treatment appeared to at least partially restore the morphology and complexity of TH+ processes (Fig. 2C). To test whether the protective 0.5 μM AQ dose might be toxic to neurons after prolonged exposure, we measured viability in cultures treated for up to 14 days. AQ was well tolerated and the viability of VM DA neurons treated for 1, 4, 10 or 14 days was slightly better or comparable to that of DMSO-treated controls (Fig. 2E).
Fig. 2.
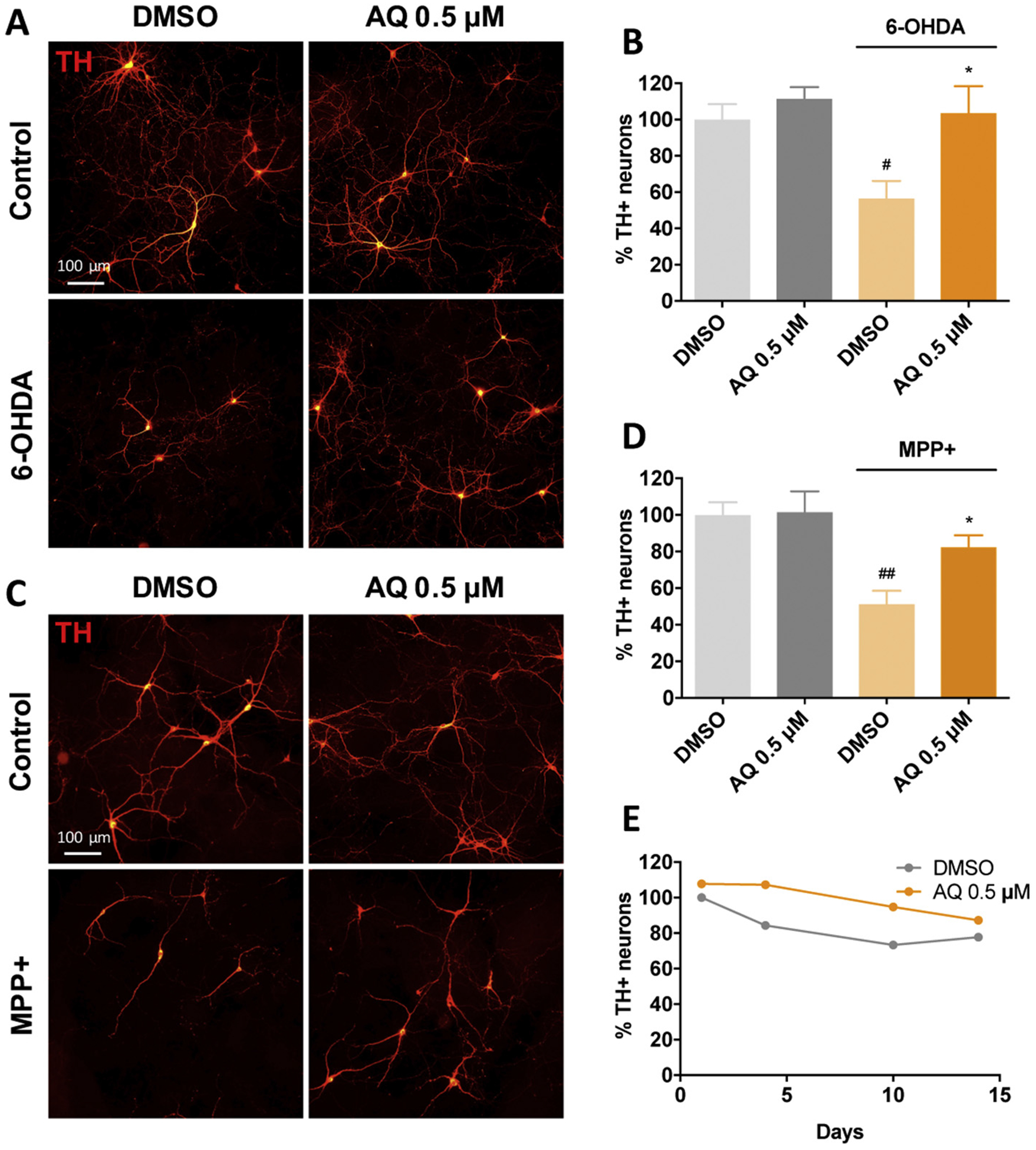
Adaptaquin protects ventral midbrain dopaminergic neurons against 6-OHDA and MPP+-induced cell death. Representative immunofluorescence images (A, C) of untreated ventral midbrain primary cultures (Control, DMSO) or treated with 0.5 μM AQ and 40 μM 6-OHDA (A) or 40 μM MPP+ (C) for 24 h and immunostained for tyrosine hydroxylase (TH, red). Corresponding quantifications (B, D) of 3 independent experiments showing the percentage of remaining viable TH+ neurons in control cultures (DMSO) or ventral midbrain cultures treated with 0.5 μM AQ and 40 μM 6-OHDA (B) or 0.5 μM AQ and 40 μM MPP+ (D) for 24 h. Both 6-OHDA and MPP+ induce significant death of dopaminergic neurons (ANOVA with Tukey’s post-hoc tests, #p < .05, ##p < .005) while AQ restores cell viability of both 6-OHDA and MPP+ treated dopaminergic neurons (ANOVA with Tukey’s post-hoc tests *p < .05). Time-course (E) showing the percentage of remaining viable dopaminergic neurons in ventral midbrain primary cultures treated with DMSO or 0.5 μM AQ for up to 14 days and immunostained for TH.
3.2. Adaptaquin blocks the Trib3 pro-death pathway in cellular PD models
Past work established that Trib3 is induced and plays a necessary role in neuronal death in multiple cellular PD models and that such induction is partly dependent on the transcription factors ATF4 and CHOP (Aimé et al., 2015). Studies in non-PD models indicate that AQ provides neuroprotection at least in part by preventing ATF4-dependent Trib3 transcriptional induction (Karuppagounder et al., 2016). To test the idea that AQ protects in PD models by interfering with Trib3 induction, we measured Trib3 mRNA in PC12 cultures treated with 150 μM 6-OHDA ± AQ for 8 h (Fig. 3A), a time before detectable cell death, but at which Trib3 is transcriptionally up-regulated (Aimé et al., 2015). Consistent with previous findings (Aimé et al., 2015), Trib3 mRNA was induced nearly 4-fold by 6-OHDA. Co-treatment with the non-protective 0.1 μM AQ dose had no significant effect on Trib3 induction while co-treatment with a protective 0.5 μM AQ dose significantly reduced Trib3 mRNA induction (Fig. 3A). Exposure of PC12 cells to 1 mM MPP+ for 16 h (Fig. 3D) also significantly elevated Trib3 mRNA levels and this underwent significant reduction by co-treatment with 0.5 μM AQ. These results demonstrate that protective levels of AQ interfere with Trib3 transcriptional up-regulation in PD models.
Fig. 3.
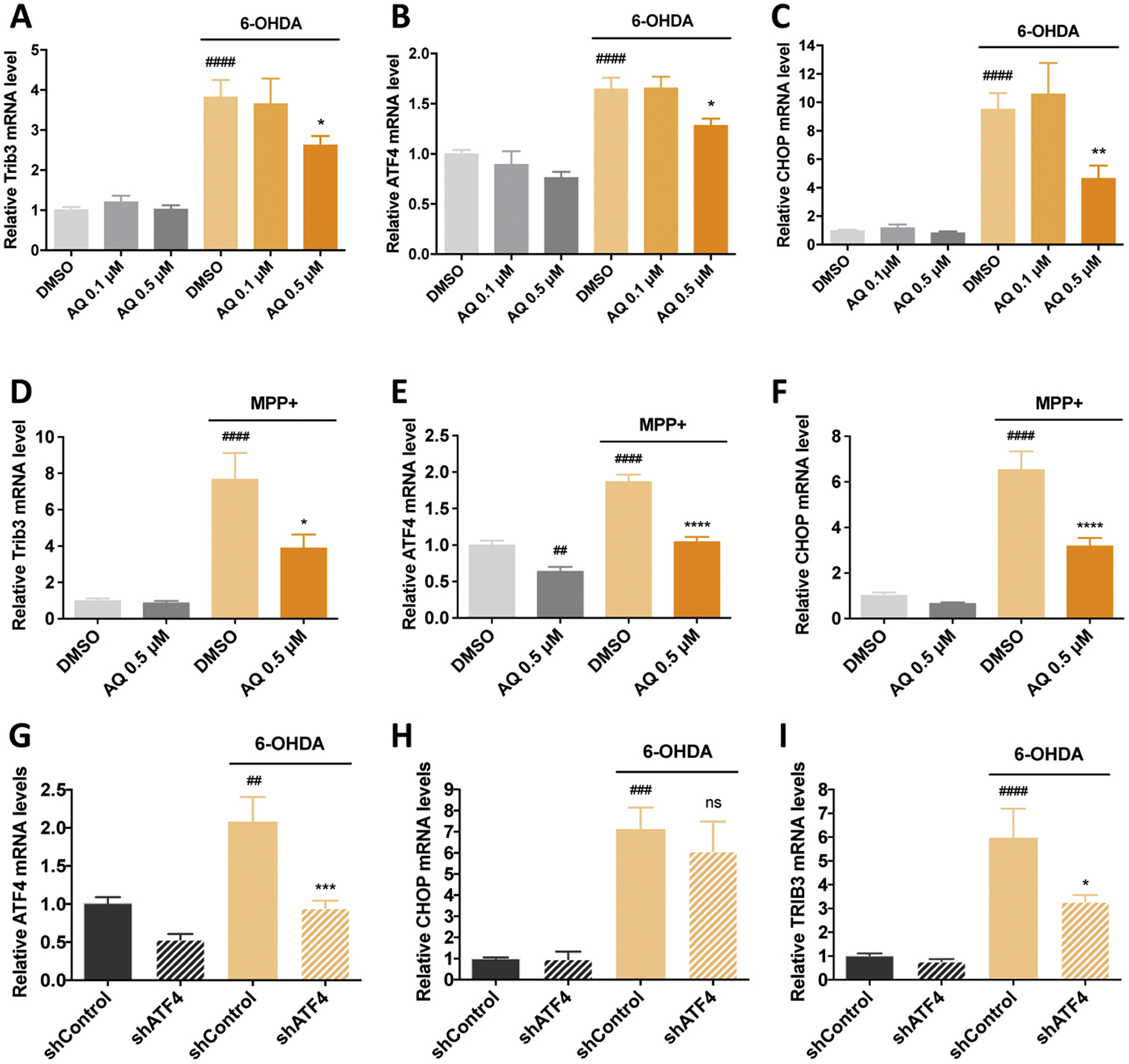
A protective dose of adaptaquin blocks the induction of ATF4, CHOP and Trib3 mRNAs and ATF4 knockdown reduces Trib3 mRNA but not CHOP mRNA induction in response to 6-OHDA in neuronal PC12 cells. qPCR analysis of Trib3 (A, D), ATF4 (B, E) and CHOP (C, F) mRNA levels in neuronal PC12 cells treated with DMSO, 0.1 μM or 0.5 μM AQ and 150 μM 6-OHDA for 8 h (A–C); DMSO, 0.5 μM AQ and 1 mM MPP+ for 16 h (D–F). In DMSO-treated cultures, 6-OHDA induces an increase in Trib3, ATF4 and CHOP mRNA (ANOVA with Tukey’s post-hoc tests, ####p < .0001). For cultures treated with 6-OHDA and a 0.5 μM AQ significant decreased Trib3, ATF4 and CHOP mRNA levels (ANOVA with Tukey’s post-hoc tests, *p < .05, **p < .005). In DMSO-treated cultures, MPP+ increases Trib3, ATF4 and CHOP mRNA (ANOVA with Tukey’s post-hoc tests, ####p < .0001). In MPP+ treated cultures, 0.5 μM AQ significantly decreases Trib3, ATF4 and CHOP mRNA levels (ANOVA with Tukey’s post-hoc tests, *p < .05, ****p < .0001). In absence of MPP+, 0.5 μM AQ decreases ATF4 mRNA levels at 16 h (ANOVA with Tukey’s post-hoc tests, ##p < .005). All mRNA levels are expressed as mean +/− SEM of 3 independent experiments. qPCR analysis shows the effects of lentivirally-delivered ATF4 shRNA (shATF4) on levels of ATF4 (G), CHOP (H) and Trib3 (I) mRNA after treatment with 150 μM 6-OHDA for 8 h. Control cultures were infected with a control shRNA (shControl). In control cultures, 6-OHDA induces Trib3, ATF4 and CHOP mRNA (ANOVA with Tukey’s post-hoc tests, ##p < .005, ###p < .0005, ####p < .0001). ATF4 knockdown reduces the induction of ATF4 and Trib3 mRNA in response to 6-OHDA (ANOVA with Tukey’s post-hoc tests, *p < .05, ***p < .001). By contrast ATF4 knockdown does not reduce the induction of CHOP mRNA in response to 6-OHDA (ns). All mRNA levels are expressed as mean +/− SEM of 3 independent experiments.
Because ATF4 and CHOP are potential upstream regulators of Trib3 (Ohoka et al., 2005; Ord and Ord, 2005; Han et al., 2012) and contribute to Trib3 regulation in PD models (Aimé et al., 2015), we also examined ATF4 and CHOP/Ddit3 mRNA levels. Consistent with previous reports (Ryu et al., 2002; Holtz and O’Malley, 2003; Ryu et al., 2005; Sun et al., 2013; Aimé et al., 2018), both 6-OHDA and MPP+ significantly upregulated ATF4 (Fig. 3B,E) and CHOP/Ddit3 (Fig. 3C,F) transcripts. This induction was unaffected by co-treatment with non-protective 0.1 μM AQ, while in contrast, the protective 0.5 μM AQ dose significantly reduced ATF4 (Fig. 3B,E) and CHOP/Ddit3 (Fig. 3C,F) mRNA induction by 6-OHDA and MPP+. These observations indicate that a protective dose of AQ that reduces Trib3 induction also reduces ATF4 and CHOP/Ddit3 mRNA induction.
CHOP has been described as an ATF4 target in several cellular stress contexts (Fawcett et al., 1999; Oyadomari and Mori, 2004) and we therefore assessed whether the effects of AQ on CHOP reflect its actions on ATF4. We accordingly used lentiviral-delivery of a previously validated shATF4 construct (Sun et al., 2013; Aimé et al., 2015) to knock down ATF4 in 6-OHDA-treated PC12 cells and assessed induction of ATF4, Ddit3/CHOP and Trib3 mRNAs (Fig. 3G–I). We confirmed that 6-OHDA induced all three transcripts in cells expressing control shRNA (Fig. 3G–I) and that ATF4 knockdown significantly decreased the 6-OHDA-promoted inductions of ATF4 and Trib3 mRNAs (Fig. 3G–I). However, in contrast, ATF4 knockdown did not significantly reduce 6-OHDA promoted Ddit3/CHOP mRNA induction (Fig. 3H). Together, these findings suggest that the effects of AQ on CHOP are not mediated via ATF4 regulation.
We next assessed how the transcriptional effects of AQ reflect expression of proteins in the Trib3 pro-death pathway. PC12 cells were exposed to carrier or 150 μM 6-OHDA with 0, 0.1 or 5 μM AQ and harvested 8 h later for quantification of ATF4, CHOP and Trib3 proteins by Western immunoblotting (Fig. 4A–D). AQ alone did not significantly change levels of ATF4, CHOP or Trib3 protein (Fig. 4A–D). Consistent with previous findings (Aimé et al., 2015; Aimé et al., 2018), 6-OHDA alone elevated ATF4, CHOP and Trib3 proteins (Fig. 4A–D). Moreover, while non-protective 0.1 μM AQ had no significant effect on induction of the 3 proteins (Fig. 4A–D), the protective 0.5 μM AQ dose significantly decreased 6-OHDA-dependent induction of ATF4, CHOP and Trib3 proteins (Fig. 4A–D). Thus a protective dose of AQ suppresses the Trib3 apoptotic pathway at the protein as well as transcriptional levels.
Fig. 4.

A protective dose of adaptaquin block the induction of ATF4, CHOP and Trib3 proteins in neuronal PC12 cells treated with 6-OHDA and maintains Parkin protein levels. Western blot images (A) and corresponding quantifications (B–E) of 5 independent experiments showing the protein levels of ATF4 (B), CHOP (C), Trib3 (D) and Parkin (E) in neuronal PC12 cells treated with DMSO, 0.1 μM or 0.5 μM AQ and 150 μM 6-OHDA for 8 h. In DMSO-treated cultures, 6-OHDA induces ATF4, CHOP and Trib3 proteins and decreases Parkin protein (ANOVA with Tukey’s post-hoc tests, #p ≤ .05). By contrast, cultures treated with 6-OHDA and 0.5 μM displayed a significant decrease in Trib3, ATF4 and CHOP protein levels (ANOVA with Tukey’s post-hoc tests, *p < .05, **p < .001). In addition, a 0.5 μM dose of AQ significantly increased Parkin levels (ANOVA with Tukey’s post-hoc tests, *p = .05). Data are normalized to ERK. Non specific bands are marked with asterisks. Survival assay (F) showing the percentage of remaining dopaminergic neurons in ventral midbrain cultures infected with a lentivirus carrying a control shRNA (shControl) or an shRNA against Parkin (shParkin) for 14 days and immunostained for TH and GFP. Parkin knockdown induces the death of 31% of TH/GFP+ neurons (t-test, ***p < .001).
3.3. Adaptaquin maintains Parkin levels in cellular PD models
We next examined Parkin expression in our models. Parkin loss-of-function causes juvenile parkinsonism (Dawson and Dawson, 2010) and is suggested to contribute to pathogenesis in sporadic PD (Dawson and Dawson, 2014). We and others have reported that Parkin protein (but not mRNA) decreases in in vitro and in vivo models of PD (Kuhn et al., 2003; Sonia Angeline et al., 2012; Sun et al., 2013; Aimé et al., 2015). Of particular relevance, we found that Trib3 interacts with Parkin, and causes its depletion (Aimé et al., 2015). Such observations raise the idea that by diminishing Trib3 induction, AQ may maintain Parkin levels that in turn contribute to AQ’s pro-survival effects in PD models. To explore this, we first determined whether Parkin loss promotes death of dopaminergic neurons. We used lentiviral-delivery of a previously validated shParkin construct co-expressing GFP (Sun et al., 2013; Aimé et al., 2015) to knock down Parkin in cultured VM DA neurons, and assessed survival of the infected TH+ cells after 14 days. Under these conditions, there was an approximate 30% loss of VM DA neurons compared to cells expressing control shRNA (Fig. 4F).
Given its effects on Trib3 expression, we next determined whether AQ prevents loss of Parkin in cellular PD models. Consistent with previous findings (Sun et al., 2013; Aimé et al., 2015), there was significant depletion of Parkin protein in PC12 cells treated with 150 μM 6-OHDA for 8 h (Fig. 4A,E). The non-protective 0.1 μM AQ had no significant effect on Parkin loss, whereas co-treatment with the protective 0.5 μM AQ dose fully maintained Parkin protein (Fig. 4A,E). These findings are consistent with the idea that as a consequence of reducing Trib3 induction, AQ maintains Parkin levels in a cellular PD model.
3.4. Adaptaquin blocks Trib3 mRNA induction in an animal PD model
Gene microarray analysis demonstrated that Trib3 and Ddit3 (encoding CHOP) are upregulated in rat SN following intrastriatal 6-OHDA lesion (Kanaan et al., 2015). It was also reported that 6-OHDA injection into the rat MFB induces markers of neurodegeneration in the SN as early as 6 h post-lesion (Zuch et al., 2000). Taking advantage AQ’s capacity to readily cross the blood brain barrier (Karuppagounder et al., 2016), we next tested whether 6-OHDA administration in the nigrostriatal pathway induces Trib3 and CHOP/Ddit3 mRNAs and whether this is prevented by systemic AQ administration. Adult male mice received a single dose of either vehicle or AQ (30 mg/kg) IP 2 h after unilateral 6-OHDA injection into the MFB (Fig. 5A). This dose was chosen because past work has shown that it reaches at least several regions in brain and inhibits HIF PHD activity (Karuppagounder et al., 2016). Eight hour later, brains were collected, sectioned and Trib3, Ddit3/CHOP and Slc6a3 (encoding the dopamine transporter, DAT) mRNA levels were assessed in the control, unlesioned and 6-OHDA-lesioned sides of the SN by multiplex fluorescent in situ hybridization (FISH) (Fig. 5B,C). As anticipated (Bowenkamp et al., 1996) Slc6a3 mRNA was robustly expressed by dopaminergic neurons in the control side but greatly reduced on the lesioned side (Fig. 5B). Infrequently, small subsets of dopaminergic neurons retained detectable Slc6a3 expression in the lesioned side (Fig. 5B, insets). Although neither Trib3 nor Ddit3/CHOP signals were detectable in the control side of 6-OHDA-injected mice, robust and colocalized Trib3 and Ddit3/CHOP signals were detected on the lesioned side. In striking contrast, in AQ-treated mice, Slc6a3 expression was maintained on the lesioned side and Trib3 and Ddit3/CHOP signals were robustly reduced. FISH performed using negative and positive controls validated the sensitivity and specificity of the procedure (Fig. 5C). Altogether, these results indicate that Trib3 and Ddit3/CHOP are rapidly induced during 6-OHDA-promoted neurodegeneration in vivo and that, consistent with in vitro findings, a single AQ dose prevents Trib3 and Ddit3/CHOP upregulation and preserves a dopaminergic neuron marker in this model.
Fig. 5.
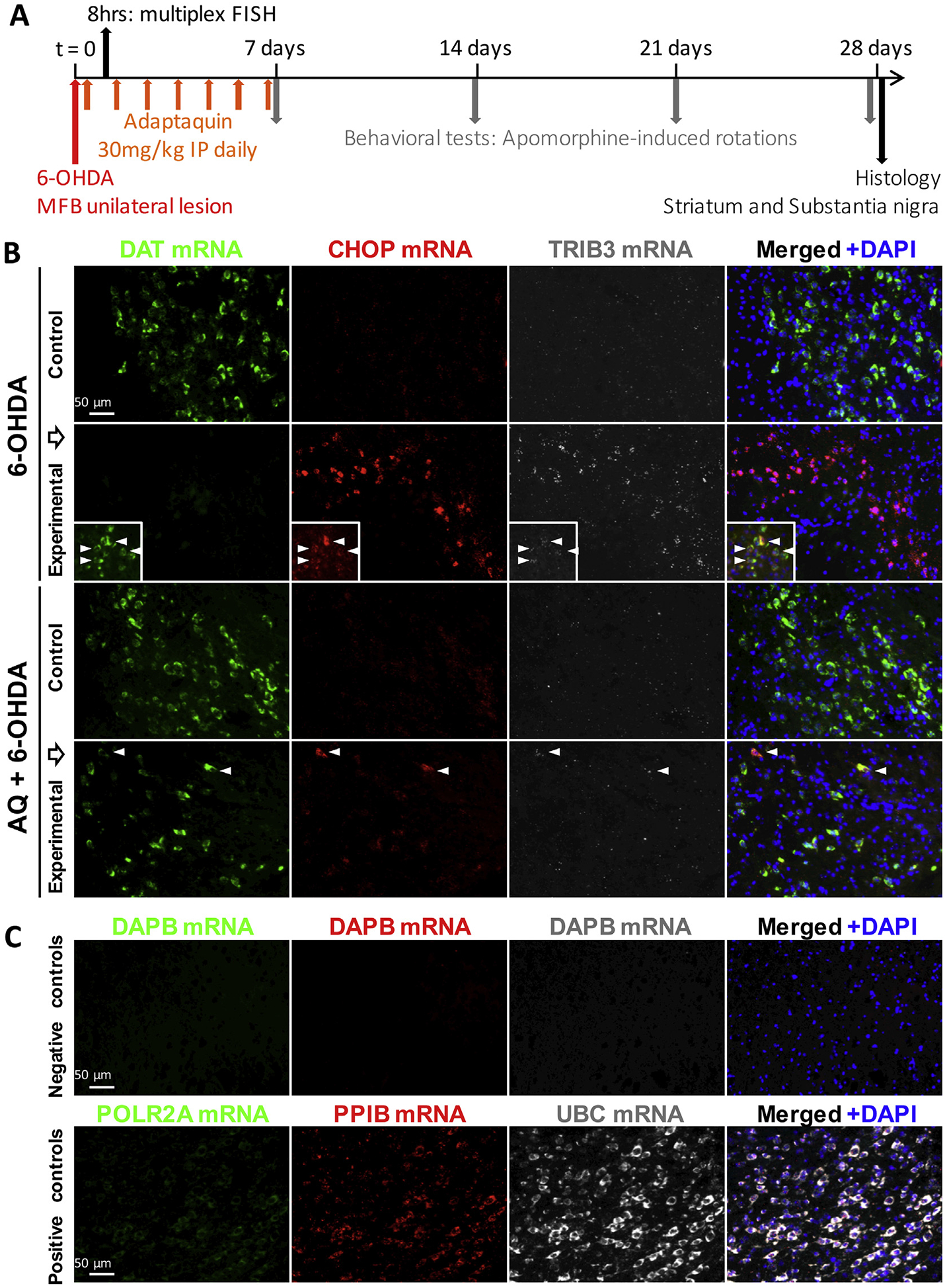
Adaptaquin blocks Trib3 and Ddit3/CHOP mRNA induction in the substantia nigra of 6-OHDA-injected mice. (A) Timeline of in vivo experiments: Stereotaxic surgery was performed and mice received a unilateral injection 6-OHDA in the right median forebrain bundle (MFB) at t = 0. After 2 h, a sub-group of mice received a single intraperitoneal (IP) injection of adaptaquin (AQ). Mice were sacrificed 8 h after the 6-OHDA lesion and their brains were processed for multiplex fluorescent in situ hybridization (FISH) and Trib3, Ddit3/CHOP and Slc6a3/DAT mRNA levels were analyzed. Another sub-group of mice started receiving daily IP AQ injections 2 h after the 6-OHDA lesion and for 7 consecutive days. At 7, 14, 21 and 28 days, mice were challenged by subcutaneous injections of the dopamine agonist apomorphine and contralateral rotations were measured to assess the functional integrity of the nigrostriatal system. Twenty-eight days after the 6-OHDA lesion, mice were sacrificed and their brains were processed for histology and tyrosine hydroxylase immunoreactivity was analyzed in the substantia nigra and the striatum. (B) Representative images of multiplex fluorescent in situ hybridization (FISH) showing Slc6a3/DAT mRNA expressed by midbrain dopaminergic neurons (green); Ddit3/CHOP (red) and Trib3 (white) mRNA signals in the unlesioned (control) or 6-OHDA lesioned (experimental) side of the substantia nigra of mice receiving either a vehicle IP injection 2 h after 6-OHDA treatment (6-OHDA) or a 30 mg/kg IP injection of AQ 2 h after 6-OHDA treatment (AQ + 6-OHDA). Eight hour after the 6-OHDA lesion, Slc6a3/DAT mRNA expression is decreased and only small subsets of dopaminergic neurons retain Slc6a3/DAT mRNA expression (inserts). Strong Ddit3/CHOP and Trib3 mRNA signals are detected in the experimental side of the substantia nigra of vehicle-treated mice. A single IP injection of AQ 2 h after the 6-OHDA lesion maintains Slc6a3/DAT mRNA expression and decreases Ddit3/CHOP and Trib3 mRNA expression in the substantia nigra. Arrow heads indicate neurons showing a colocalization of Slc6a3/DAT, Ddit3/CHOP and Trib3 mRNAs. (C) Representative images of control multiplex FISH experimental procedures using probes targeting the bacterial gene dapb (negative control) and the ubiquitous genes Polr2a, Pp1b and Ubc showing low, moderate and high mRNA expression levels, respectively (positive controls).
3.5. Adaptaquin protects the nigrostriatal system in an animal PD model
Given the above findings, we next examined whether AQ protects the nigrostriatal pathway in the 6-OHDA MFB model of PD. This model is rather stringent and causes massive death of dopaminergic neurons and robust changes in behavior. Adult male mice received a daily dose of vehicle or AQ (30 mg/kg) IP for 7 consecutive days starting 2 h after unilateral injection of 6-OHDA or vehicle (Fig. 5A). This regimen was previously shown to suppress ATF4-dependent gene regulation and improve behavioral outcomes in a model of intracerebral hemorrhage (Karuppagounder et al., 2016). The sham group received a unilateral injection of vehicle in the MFB and vehicle IP injections. Twenty-eight days after the lesion, brains were collected, sectioned and TH immunoreactivity was analyzed in SN (Fig. 6) and striatum (Fig. 7).
Fig. 6.

Adaptaquin partially prevents the loss of dopaminergic neurons in the substantia nigra of 6-OHDA-injected mice. Representative images (A) of TH immunohistochemistry (brown) with thionin counterstain (blue) showing dopaminergic neurons in the unlesioned (control) or 6-OHDA lesioned (experimental) side of the posterior and anterior regions of the substantia nigra of mice who received a sham surgery or a 6-OHDA lesion and either a control IP injection (6-OHDA) or a daily 30 mg/kg IP injection of AQ for 7 consecutive days (AQ + 6-OHDA). Higher magnification images (B) showing the dopaminergic neurons in the experimental side of the anterior substantia nigra of sham, 6-OHDA and AQ + 6-OHDA mice. Unbiased stereology counts (C) and proportions between the experimental and the control side (D) of the total number of dopaminergic TH+ neurons in all three groups showing that 6-OHDA induces a drastic reduction of the counts (ANOVA with Sidak’s multiple comparison test ****p < .0001) and the proportion (ANOVA with Tukey’s post-hoc test ****p < .0001) of dopaminergic neurons in the substantia nigra, compared to sham mice. AQ post-treatment for 7 days significantly increases both the counts (ANOVA with Sidak’s multiple comparison test ***p < .0005) and the proportion (ANOVA with Tukey’s post-hoc test ***p < .0005) of nigral dopaminergic neurons.
Fig. 7.
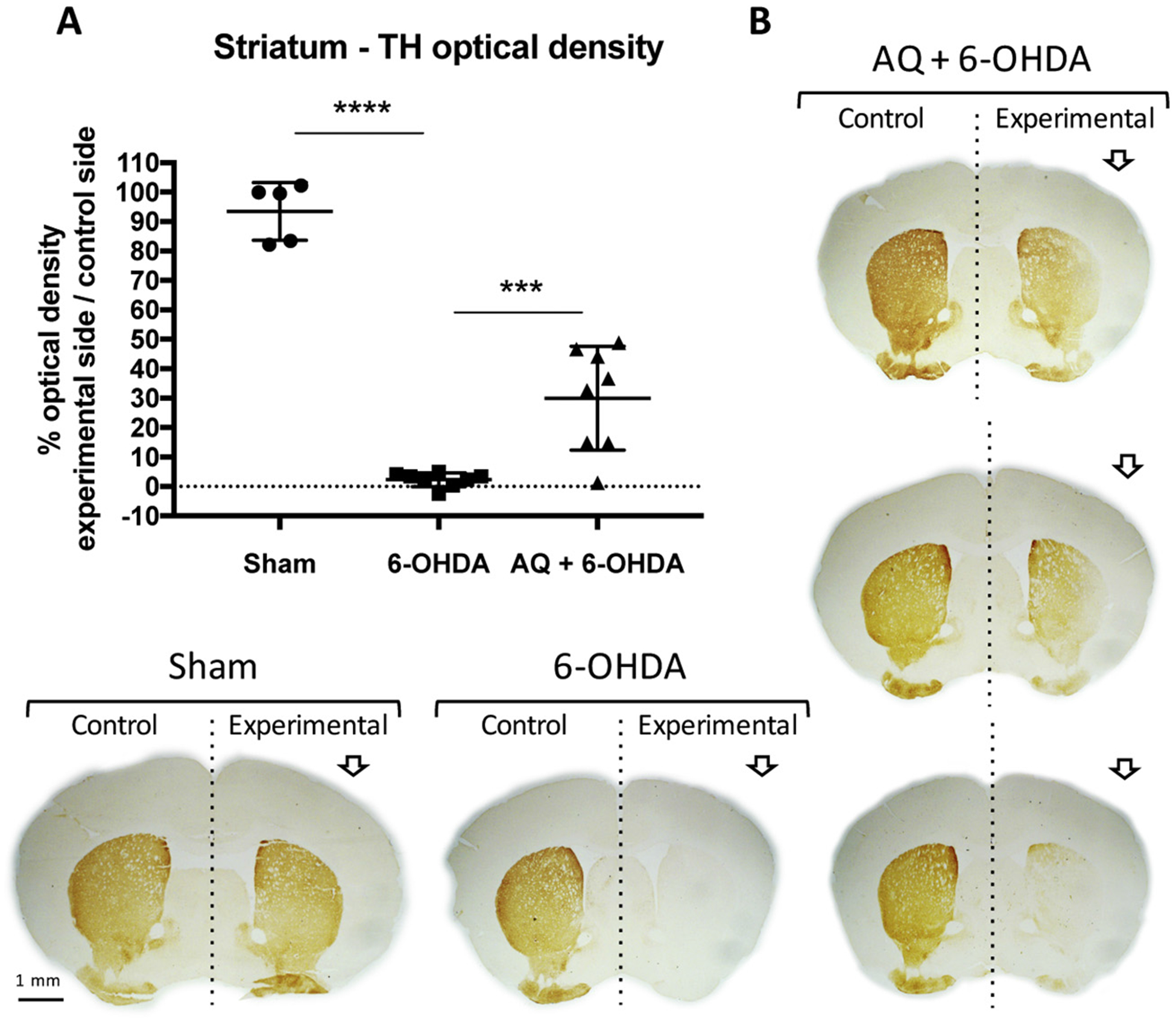
Adaptaquin partially restores the loss of TH immunoreactivity in the striatum of 6-OHDA-injected mice. Quantification (A) and representative images (B) of TH immunohistochemistry (brown) showing the optical density of the TH signal from dopaminergic fibers in the unlesioned (control) or 6-OHDA lesioned (experimental) side of the striatum of mice who received a sham surgery or a 6-OHDA lesion and either a control IP injection (6-OHDA) or a daily 30 mg/kg IP injection of AQ for 7 consecutive days (AQ + 6-OHDA). 6-OHDA induces a drastic reduction of TH immunostaining in experimental side of the striatum, relative to the control side and compared to sham mice (ANOVA with Tukey’s post-hoc test ****p < .0001). AQ post-treatment for 7 days significantly increases the proportion of TH immunoreactivity in the striatum (ANOVA with Tukey’s post-hoc test ***p < .001). Given the variability in the AQ + 6-OHDA group, striatum images of 3 individual mice are shown in B.
In the SN, total TH+ neuron numbers were estimated by unbiased stereology. In the sham group, similar numbers were present on control and experimental sides as anticipated (Fig. 6A,C). By contrast, in the 6-OHDA + vehicle group, there was a > 90% reduction of TH+ neurons (Fig. 6A–D). Consistent with the reduction of Trib3 induction by AQ in this model, AQ significantly increased numbers of surviving TH+ neurons on the lesioned side by an average of nearly 4-fold, although one mouse showed no response (Fig. 6D). Also of note, the prolonged regimen of AQ administration showed no overt dopaminergic toxicity since numbers of TH+ neurons on the control and sham sides of 6-OHDA + AQ groups were not significantly different from those on the control sides of the sham group (Fig. 6C).
Next, we assessed integrity of dopaminergic projections by measuring TH immunoreactivity in the striatum (Fig. 7). In the sham group, TH immunostaining density was comparable between control and experimental sides whereas only about 2% of TH immunostaining remained in the striatum of lesioned mice (Fig. 7A,B). AQ treatment increased TH levels in striata of lesioned animals by about 13-fold to reach, on average, 30% of that in controls (Fig. 7A,B).
Taken together, these results establish that AQ, given after a stringent 6-OHDA lesion, provides long-term protection of both dopaminergic neuron cell bodies in the SN and of TH immunostaining in the striatum.
3.6. Adaptaquin restores function of the nigrostriatal pathway
We also determined whether protection of the nigrostriatal pathway conferred by AQ translated into preservation of function, i.e. movement control. Denervation of the striatum induces supersensitivity of post-synaptic dopamine receptors which causes unilaterally-lesioned mice to respond to dopaminergic agonists by turning away from the ipsilateral side of the lesion (Ungerstedt, 1971). At 7, 14, 21 and 28 days, the sham, 6-OHDA and 6-OHDA + AQ groups described above were given a subcutaneous dose of apomorphine and assessed for rotational behavior (Fig. 8). Consistent with an unaffected nigrostriatal system, sham group mice displayed no rotational behavior. In contrast, mice receiving only 6-OHDA showed a significant increase in contralateral rotations at all tested times (Fig. 8A–D). Strikingly, 6-OHDA-lesioned mice receiving AQ showed a significantly decreased number of contralateral rotations at 14, 21 and 28 days (Fig. 8B–D). These results thus demonstrate that along with protecting dopaminergic cell bodies and projections in a PD model, AQ treatment preserved functional integrity of the nigrostriatal pathway.
Fig. 8.
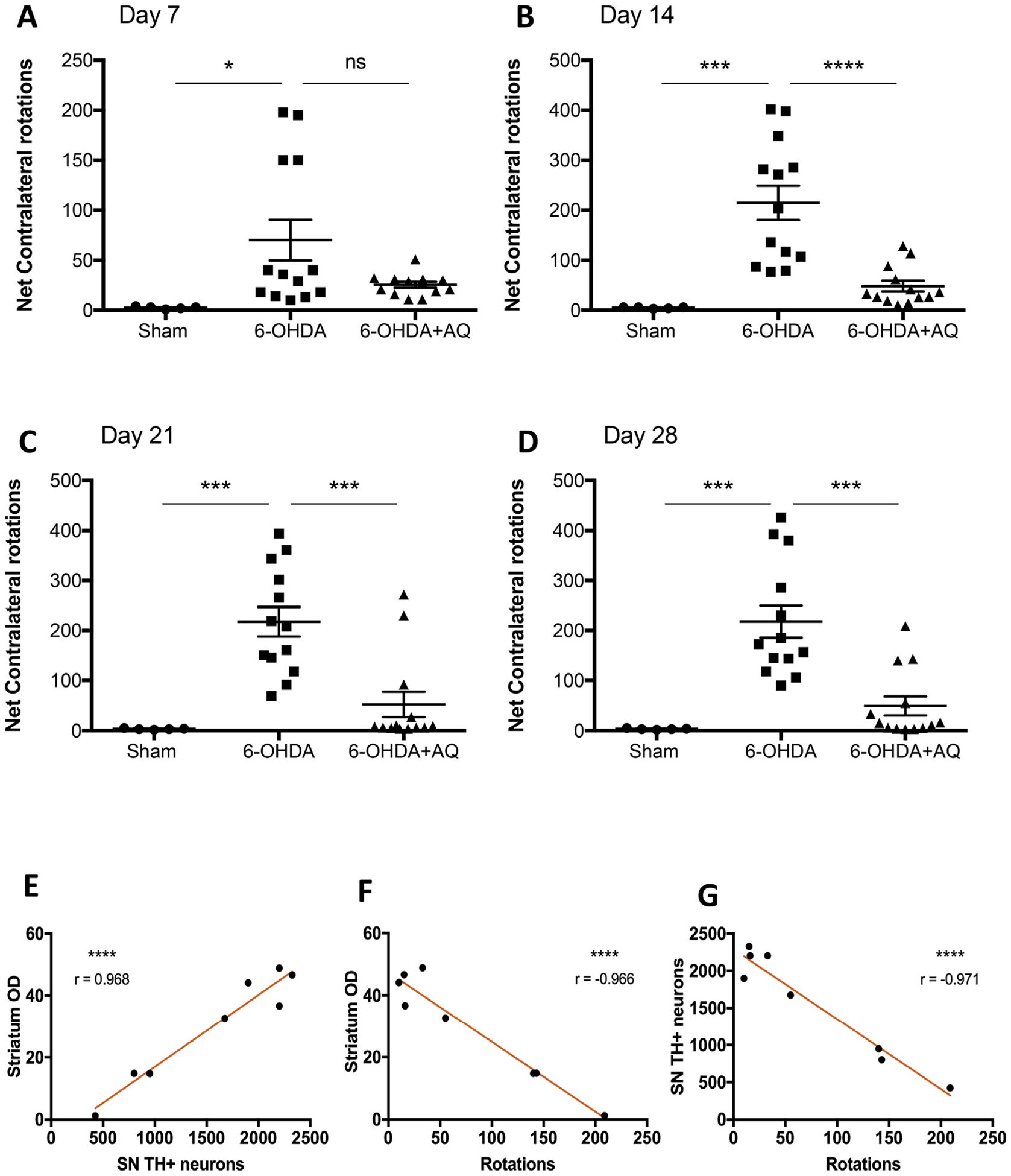
Adaptaquin partially restores movement control in 6-OHDA-injected mice. (A–D) Behavioral analysis by quantification of the net contralateral rotations induced by a subcutaneous injections of apomorphine at 7 days (A), 14 days (B), 21 days (C) and 28 days after mice received a sham surgery or a 6-OHDA lesion and either a control IP injection (6-OHDA) or a daily 30 mg/kg IP injection of AQ for 7 consecutive days (AQ + 6-OHDA). n = 5 for sham, n = 13 for 6-OHDA alone and n = 13 for AQ + 6-OHDA group. 6-OHDA induces a drastic increase in the number of contralateral rotations at all time points measured compared to sham mice (ANOVA with Tukey’s post hoc test; *p < .05, ***p < .0005). AQ post-treatment for 7 days significantly decreases the number of contralateral rotations at 14 days, 21 days and 28 days (ANOVA with Tukey’s post hoc test; ***p < .0005, ****p < .0001). (E–G) Correlation analysis of the results of the AQ-treated group showing that the number of dopaminergic neurons in the substantia nigra is highly correlated to the density of TH immunoreactivity in the striatum (E, Pearson correlation coefficient r = 0.968, p < .0001), the density TH immunoreactivity in the striatum is highly correlated to the number of apomorphine-induced rotations (F, Pearson correlation coefficient r = −0.966, p < .0001) and the number of dopaminergic neurons in the substantia nigra is highly correlated to the number of apomorphine-induced rotations (G, Pearson correlation coefficient r = −0.971, p < .0001).
Lastly, we assessed correlations in the responses of individual mice to AQ treatment. As anticipated, the extents of preservation of striatal TH immunoreactvity and of SN dopaminergic cell body numbers of each individual mouse in the AQ-treated group were highly correlated (Fig. 8E). Similarly, preservation of striatal TH immunoreactivity and of numbers of dopaminergic cell bodies of individual mice in the AQ-treated group were highly correlated with the numbers of apomorphine-induced rotations at 28 days (Fig. 8F,G). Thus, the extents of protection of nigral dopaminergic neurons and of striatal TH immunoreactivity conferred by AQ in individual mice correlates strongly with the improvement of motor function.
4. Discussion
This study identifies AQ as a potential therapeutic to prevent neuronal death and degeneration in PD. AQ was protective in multiple cellular PD models and prevented degeneration and functional impairment of the nigrostriatal pathway in an animal PD model. The model used, 6-OHDA injection in the MFB, is stringent and allowed assessment of the neuroprotective potential of AQ in conditions that lead to the rapid and near complete destruction of the nigrostriatal system (Zuch et al., 2000). In this model markers of neurodegeneration are detectable in the substantia nigra as early as 6 h post-lesion and there is a near-complete (> 90%) loss of nigral dopaminergic neurons and their dopaminergic fibers in the striatum by 4 weeks (Zuch et al., 2000). Strikingly, AQ administered 2 h after 6-OHDA and then daily for 1 week, maintained a mean of about 30% of dopaminergic cell viability in the SN, and a similar proportion of dopaminergic striatal tyrosine hydroxylase immunostaining, as assessed 4 weeks later. In some animals, protection reached 45–50%. These results correlated with functional preservation of the nigrostriatal system.
In this study, we used an AQ treatment regimen previously shown to reduce neuronal death and improve functional recovery in a mouse model of intracerebral hemorrhage (Karuppagounder et al., 2016). It is possible that, with further treatment optimization, AQ may provide even greater protection in the context of PD. AQ is well tolerated in rodents and an analog of the 8-hydroxyquinoine family to which AQ belongs has shown safety and tolerability in phase II human trials (Faux et al., 2010).
Although AQ provided significant protection in our animal model, the effect was not as robust as observed in our culture models. One major difference between the two models is that the initial and ongoing concentrations of AQ in brain were unknown as compared with the culture system in which various AQ concentrations were tested and the dose needed for maximal protection was determined. Also, while in vivo AQ levels likely vary over time after treatment due to pharmacokinetic events, the levels of AQ in culture medium likely show less temporal variation. Another considerable difference between the two systems is that the in vitro models used toxin concentrations that yielded 30–60% cell death whereas the in vivo model resulted in > 90% loss of SN dopaminergic neurons. This raises the possibility that AQ might be more effective in a setting of progressive neuron loss such as occurs in PD. Taken together, these considerations support the idea raised above that further optimization of in vivo AQ dosing remains to be carried out and may yield levels of protection more comparable to that observed in vitro.
Mechanism studies indicate that AQ is a selective HIF PHDs inhibitor (Karuppagounder et al., 2016; Neitemeier et al., 2016) and our results support growing evidence that PHD inhibition is a relevant therapeutic strategy for PD. Structurally diverse PHD inhibitors maintain mitochondrial function, reduce formation of ROS and protect neuronal cells against oxidative cell death (Karuppagounder et al., 2016; Neitemeier et al., 2016). Pharmacological inhibition of PHDs prevents mitochondrial toxin-induced cell death in neuronal cultures (Lee et al., 2009; Niatsetskaya et al., 2010) and prevents nigral dopaminergic cell loss induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in vivo (Lee et al., 2009). Pharmacological inhibitors do not distinguish the 3 PHD isoforms and in silico modeling predicts that AQ fits into the active site of all 3 PHDs (Karuppagounder et al., 2016). All 3 PHD isoforms are expressed in dopaminergic neurons (Rajagopalan et al., 2016) and there is conflicting evidence about which are involved in neuron death (Lee et al., 2005; Siddiq et al., 2009; Myllyharju and Koivunen, 2013; Neitemeier et al., 2016; Rajagopalan et al., 2016). Thus, it remains to be determined whether inhibition of one or a combination of PHDs might contribute to the neuroprotective effects of AQ observed here.
Our results are consistent with the idea that AQ confers neuroprotection in PD models at least partly by suppressing ATF4-CHOP-dependent Trib3 induction. In a mouse model of intracerebral hemorrhage AQ reduced ATF4 protein elevation and abrogated Trib3 protein and mRNA induction (Karuppagounder et al., 2016). Here, a protective dose of AQ suppressed Trib3 as well as ATF4 and CHOP mRNA and protein induction in a cellular PD model, and inhibited Trib3 and Ddit3/CHOP upregulation in the MFB lesion model. There is growing evidence for roles of Trib3 in a variety of neurodegenerative conditions, including a recent association with Alzheimer’s disease (Lorenzi et al., 2018), and it would be of interest to assess AQ’s effect on Trib3 induction and therapeutic efficacy in these cases as well.
The role of CHOP described here is intriguing. CHOP, an ER-stress associated protein along with ATF4, is highly upregulated in both culture (Ryu et al., 2002; Holtz and O’Malley, 2003) and animal (Silva et al., 2005) PD models and there is substantial evidence linking ER-stress to PD (Martinez et al., 2019). For example, dopaminergic neurons in CHOP knockout mice are protected from intrastriatal injected 6-OHDA (Silva et al., 2005). Past findings indicate that CHOP cooperates with ATF4 to induce Trib3 and neuron death (Ohoka et al., 2005; Aimé et al., 2015). The present and past (Sun et al., 2013) data show that CHOP can be induced independently of ATF4 induction, and it thus appears that AQ suppresses CHOP independently of its effects on ATF4. The mechanism by which AQ affects CHOP expression remains to be explored, but represents an additional and significant means by which AQ provides neuroprotection.
Our findings indicate that in consequence of its actions on ATF4 and CHOP, AQ suppresses pro-apoptotic Trib3 induction and maintains expression of prosurvival Parkin, a protein whose loss-of-function causes a familial form of PD and that may be linked to sporadic PD (Dawson and Dawson, 2010; Dawson and Dawson, 2014). Trib3 has scaffold-like properties and interacts with multiple partners (Kiss-Toth, 2015) in some cases to promote their degradation (Hegedus et al., 2007; Hua et al., 2011; Kiss-Toth, 2015). We have reported that Trib3 interacts with Parkin and leads to its cellular depletion (Aimé et al., 2015). We further reported an inverse relationship between Parkin and Trib3 expression in dopaminergic neurons of PD and control patients (Aimé et al., 2015). Here, we provide additional evidence for Parkin’s protective role in dopaminergic neurons by showing that prolonged Parkin knockdown triggers their death. Consistent with the idea that AQ protects dopaminergic neurons at least in part by repressing Trib3 induction, and consequently Parkin depletion, we found that a protective AQ dose that decreases Trib3 induction by 6-OHDA also maintains Parkin expression.
5. Conclusions
In summary, we find that AQ provides substantial neuroprotection in culture and animal models of PD and provide evidence that this includes interference with an ATF4-CHOP-Trib3 pro-death pathway and consequent preservation of Parkin levels. As in a past study (Karuppagounder et al., 2016), AQ showed effective in vivo bioavailability, efficacy and apparent safety. These findings thus support further investigation of AQ as a potential neuroprotective therapeutic in PD.
Acknowledgements
The authors thank Dr. Patricia Tagliaferro and Tinmarlar F. Oo for sharing their expertise in nigrostriatal dopaminergic immunohistochemistry, optical density as well as unbiased stereology. The authors would like to dedicate this manuscript to the memory of our colleague, Dr. Robert E. Burke.
Funding
This work was supported by the National Institutes of Health [grant numbers R01-NS072050 (LAG) and P01 NIA AG014930, Project 1 (RRR)], the Parkinson’s Disease Foundation (LAG and REB), the Sperling Center for Hemorrhagic Stroke Recovery at Burke Neurological Institute (RRR), the Burke Foundation (RRR), and the Dr. Miriam and Sheldon G Adelson Medical Research Foundation (RRR).
Abbreviations:
- AQ
adaptaquin
- ATF4
Activating transcription factor 4
- CHOP
C/EBP homologous protein
References
- Aimé P, et al. , 2015. Trib3 is elevated in Parkinson’s disease and mediates death in Parkinson’s disease models. J. Neurosci 35, 10731–10749. 10.1523/JNEUROSCI.0614-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimé P, Sun X, Greene LA, 2018. Manipulation and study of gene expression in neurotoxin-treated PC12 and SH-SY5Y cells for in vitro studies of Parkinson’s disease gene expression and regulation in mammalian cells. In: Transcription Toward the Establishment of Novel Therapeutics. IntechOpen 2018. 10.5772/intechopen.71811. [DOI] [Google Scholar]
- Bowenkamp KE, et al. , 1996. 6-hydroxydopamine induces the loss of the dopaminergic phenotype in substantia nigra neurons of the rat. A possible mechanism for restoration of the nigrostriatal circuit mediated by glial cell line-derived neurotrophic factor. Exp. Brain Res 111, 1–7. 10.1007/bf00229549. [DOI] [PubMed] [Google Scholar]
- Chen X, et al. , 2012. Neurotrophic effects of serum- and glucocorticoid-inducible kinase on adult murine mesencephalic dopamine neurons. J. Neurosci 32, 11299–11308. 10.1523/JNEUROSCI.5910-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL, 2010. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord 25 (Suppl. 1), S32–S39. 10.1002/mds.22798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL, 2014. Parkin plays a role in sporadic Parkinson’s disease. Neurodegener. Dis 13, 69–71. 10.1159/000354307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding YM, et al. , 2004. Effects of 6-hydroxydopamine on primary cultures of substantia nigra: specific damage to dopamine neurons and the impact of glial cell line-derived neurotrophic factor. J. Neurochem 89, 776–787. 10.1111/j.1471-4159.2004.02415.x. [DOI] [PubMed] [Google Scholar]
- Faux NG, et al. , 2010. PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J. Alzheimers Dis 20, 509–516. 10.3233/JAD-2010-1390. [DOI] [PubMed] [Google Scholar]
- Fawcett TW, et al. , 1999. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J 339, 135–141. [PMC free article] [PubMed] [Google Scholar]
- Gearan T, Castillo OA, Schwarzschild MA, 2001. The parkinsonian neurotoxin, MPP + induces phosphorylated c-Jun in dopaminergric neurons of mesencephalic cultures. Parkinsonism Relat. Disord 8 (1), 19–22. 10.1016/s1353-8020(00)00078-x. [DOI] [PubMed] [Google Scholar]
- Gorres KL, Raines RT, 2010. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol 45, 106–124. 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS, 1976. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. U. S. A 73, 2424–2428. 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, et al. , 2012. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol 15, 481–490. 10.1038/ncb2738. Epub 2013 Apr 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus Z, Czibula A, Kiss-Toth E, 2007. Tribbles: a family of kinase-like proteins with potent signalling regulatory function. Cell. Signal 19, 238–250. 10.1016/j.cellsig.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O’Malley KL, 2003. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J. Biol. Chem 278, 19367–19377. 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Hua F, et al. , 2011. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J. Cell Sci 124, 3235–3246. 10.1242/jcs.082875. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, et al. , 2015. The longitudinal transcriptomic response of the substantia nigra to intrastriatal 6-hydroxydopamine reveals significant upregulation of regeneration-associated genes. PLoS One 10, e0127768. 10.1371/journal.pone.0127768. eCollection 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Ratan RR, 2012. Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J. Cereb. Blood Flow Metab 32, 1347–1361. 10.1038/jcbfm.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, et al. , 2016. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci. Transl. Med 8 10.1126/scitranslmed.aac6008. 328ra329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss-Toth E, 2015. Tribbles: ‘puzzling’ regulators of cell signalling. Biochem. Soc. Trans 39, 684–687. 10.1042/BST0390684. [DOI] [PubMed] [Google Scholar]
- Koditz J, et al. , 2007. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 110, 3610–3617. 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- Kuhn KJ, et al. , 2003. The mouse MPTP model: gene expression changes in dopaminergic neurons. Eur. J. Neurosci 17, 1–12. 10.1046/j.1460-9568.2003.02408.x. [DOI] [PubMed] [Google Scholar]
- Lee S, et al. , 2005. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 8, 155–167. 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Lee DW, et al. , 2009. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: model for the potential involvement of the hypoxia-inducible factor pathway in Parkinson disease. J. Biol. Chem 284, 29065–29076. 10.1074/jbc.M109.000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, et al. , 2014. Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp. Mol. Med 36, 1–12. 10.1038/emm.2004.1. [DOI] [PubMed] [Google Scholar]
- Lorenzi M, et al. , 2018. Susceptibility of brain atrophy to TRIB3 in Alzheimer’s disease, evidence from functional prioritization in imaging genetics. Proc. Natl. Acad. Sci. U. S. A 115, 3162–3167. 10.1073/pnas.1706100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, et al. , 2006. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 23, 121–132. 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, et al. , 2019. Targeting of the unfolded protein response (UPR) as therapy for Parkinson’s disease. Biol. Cell 111, 161–168. 10.1111/boc.201800068. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, et al. , 2009. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229. 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllyharju J, Koivunen P, 2013. Hypoxia-inducible factor prolyl 4-hydroxylases: common and specific roles. J. Biol. Chem 394, 435–448. 10.1515/hsz-2012-0328. [DOI] [PubMed] [Google Scholar]
- Neitemeier S, et al. , 2016. Inhibition of HIF-prolyl-4-hydroxylases prevents mitochondrial impairment and cell death in a model of neuronal oxytosis. Cell Death Dis. 7, e2214. 10.1038/cddis.2016.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niatsetskaya Z, et al. , 2010. HIF prolyl hydroxylase inhibitors prevent neuronal death induced by mitochondrial toxins: therapeutic implications for Huntington’s disease and Alzheimer’s disease. Antioxid. Redox Signal 12, 435–443. 10.1089/ars.2009.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, et al. , 2005. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 24, 1243–1255. 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Stern MB, Sethi K, 2009. The scientific and clinical basis for the treatment of Parkinson disease. Neurology 72, S1–136. 10.1212/WNL.0b013e3181a1d44c. [DOI] [PubMed] [Google Scholar]
- Ord D, Ord T, 2005. Characterization of human NIPK (TRB3, SKIP3) gene activation in stressful conditions. Biochem. Biophys. Res. Commun 330, 210–218. 10.1016/j.bbrc.2005.02.149. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M, 2004. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389. 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C, 2007. The Rat Brain in Stereotaxic Coordinates, 6th ed. Academic Press, Massachusetts. [Google Scholar]
- Rajagopalan S, et al. , 2016. Regulation of ATP13A2 via PHD2-HIF1alpha signaling is critical for cellular iron homeostasis: implications for Parkinson’s disease. J. Neurosci 36, 1086–1095. 10.1523/JNEUROSCI.3117-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayport S, et al. , 1992. Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology. J. Neurosci 12, 4264–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, et al. , 2002. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J. Neurosci 22, 10690–10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Angelastro JM, Greene LA, 2005. Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol. Dis 18, 54–74. 10.1016/j.nbd.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Schapira AH, et al. , 2014. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet 384, 545–555. 10.1016/S0140-6736(14)61010-2. [DOI] [PubMed] [Google Scholar]
- Schwarting RK, Huston JP, 1996. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog. Neurobiol 50, 275–331. 10.1016/s0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- Siddiq A, et al. , 2009. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J. Neurosci 29, 8828–8838. 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva RM, et al. , 2005. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem 95, 974–986. 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova N, et al. , 2010. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem. Biol 17, 380–391. 10.1016/j.chembiol.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonia Angeline M, et al. , 2012. Rotenone-induced parkinsonism elicits behavioral impairments and differential expression of parkin, heat shock proteins and caspases in the rat. Neuroscience 220, 291–301. 10.1016/j.neuroscience.2012.06.021. [DOI] [PubMed] [Google Scholar]
- Sun X, et al. , 2013. ATF4 protects against neuronal death in cellular Parkinson’s disease models by maintaining levels of parkin. J. Neurosci 33, 2398–2407. 10.1523/JNEUROSCI.2292-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U, 1971. Postsynaptic supersensitivity after 6-hydroxy-dopamine induced degeneration of the nigro-striatal dopamine system. Acta Physiol. Scand. Suppl 367, 69–93. 10.1111/j.1365-201x.1971.tb11000.x. [DOI] [PubMed] [Google Scholar]
- Zhu JH, et al. , 2007. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol 170, 75–86. 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuch CL, et al. , 2000. Time course of degenerative alterations in nigral dopaminergic neurons following a 6-hydroxydopamine lesion. J. Comp. Neurol 427, 440–454. . [DOI] [PubMed] [Google Scholar]