

Summary
Metformin is the first-line therapy for type 2 diabetes, but there are large inter-individual variations in responses to this drug. Its mechanism of action is not fully understood, but activation of AMP-activated protein kinase (AMPK) and changes in the gut microbiota appear to be important. The inhibitory role of microbial metabolites on metformin action has not previously been investigated. Here, we show that concentrations of the microbial metabolite imidazole propionate are higher in subjects with type 2 diabetes taking metformin who have high blood glucose. We also show that metformin-induced glucose lowering is not observed in mice pretreated with imidazole propionate. Furthermore, we demonstrate that imidazole propionate inhibits AMPK activity by inducing inhibitory AMPK phosphorylation, which is dependent on imidazole propionate-induced basal Akt activation. Finally, we identify imidazole propionate-activated p38γ as a novel kinase for Akt and demonstrate that p38γ kinase activity mediates the inhibitory action of imidazole propionate on metformin.
Keywords: microbiota, microbial metabolites, imidazole propionate, diabetes, metformin, individual variations, AMPK, p38γ
Graphical Abstract
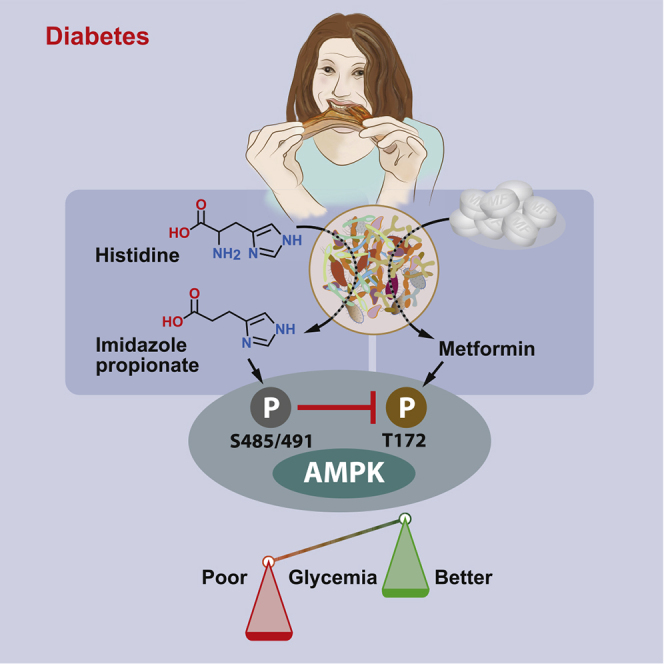
Highlights
-
•
Imidazole propionate is higher in T2D subjects on metformin with high blood glucose
-
•
Imidazole propionate impairs the glucose-lowering effect of metformin in mice
-
•
Imidazole propionate inhibits metformin-induced AMPK activation via p38γ and Akt
-
•
Inhibition of p38γ blocks the inhibitory action of imidazole propionate on metformin
Koh et al. show that imidazole propionate, a microbial metabolite, impairs the glucose-lowering effect of the anti-diabetic drug metformin and inhibits metformin-induced AMPK activation by activating p38γ/Akt/inhibitory AMPK serine phosphorylation. They further show that metformin action is restored by blocking imidazole propionate-activated p38γ.
Context and Significance
Interactions between the gut microbiota and drugs are known to affect the efficacy or adverse effects of commonly used drugs, potentially contributing to individual responses to drugs. The gut microbiota may alter drug responses by producing bioactive microbial metabolites that can be absorbed into the circulation and peripheral tissues and modify drug-induced signaling pathways. Here, Koh et al. show that imidazole propionate, a histidine-derived microbial metabolite, is elevated in subjects with type 2 diabetes with high blood glucose despite treatment with the anti-diabetic drug metformin. They also show that imidazole propionate interacts with metformin-induced signaling pathways, thereby potentially reducing the glucose-lowering response to metformin. Understanding the interaction between microbial metabolites and metformin will help us improve individual responses to metformin.
Introduction
Metformin is currently recommended as the first-line oral drug to manage glucose levels in subjects with type 2 diabetes (Marshall, 2017). However, there are large inter-individual variations in responses to this pharmacotherapy (Brown et al., 2010; Cook et al., 2005). Although metformin has been used in clinical practice for over 60 years, its mechanism of action is still not fully understood. Multiple mechanisms have been proposed, but 5′-AMP-activated protein kinase (AMPK)-dependent pathways are thought to play a key role (Musi et al., 2002; Owen et al., 2000; Shaw et al., 2005; Zhou et al., 2001).
Bidirectional interactions between the gut microbiota and drugs have gained attention in recent years by their potential to explain individual variations in responses to drugs beyond genetic polymorphisms (Haiser et al., 2013; Zimmermann et al., 2019). The gut microbiota can directly metabolize drugs, which affects their efficacy and/or bioavailability (Bachrach, 1988; Colebrook et al., 1936; García-González et al., 2017; Haiser et al., 2013; Saitta et al., 2014; Scott et al., 2017; Spanogiannopoulos et al., 2016; Svartz, 1988). In addition, drugs can modify the microbiota composition and metabolic profile and thereby affect the beneficial effects of the drug; such a phenomenon has been observed for chemotherapy (Lin et al., 2012; Panebianco et al., 2018; van Vliet et al., 2009) and also for metformin (Shin et al., 2014; Sun et al., 2018; Wu et al., 2017; Zhang et al., 2015). The gut microbiota also has the potential to interact with drugs in the host by generating bioactive compounds (microbial metabolites) that can, in some cases, enter the circulation and reach host tissues beyond the gut (Donia and Fischbach, 2015; Uchimura et al., 2018). A recent study showed that metformin acts directly on gut bacteria to promote agmatine production and thereby increases lipid metabolism and lifespan in model organisms (Pryor et al., 2019). However, the existence of microbial metabolites that impair metformin action has not been shown.
We previously reported that the microbial metabolite imidazole propionate, which is elevated in subjects with type 2 diabetes, can impair glucose tolerance (Koh et al., 2018). In this study, we sought to determine whether imidazole propionate could also contribute to the inter-individual variations in response to metformin and to identify potential interactions between imidazole propionate and known metformin-signaling pathways.
Results and Discussion
Imidazole Propionate Levels Are Higher in Metformin-Treated Subjects with Type 2 Diabetes and High Blood Glucose
To investigate whether imidazole propionate is potentially associated with the clinical outcome of metformin treatment, we first asked if circulating imidazole propionate levels differ according to glycemic level in subjects with type 2 diabetes who were taking metformin (Table S1). We found that mean levels of imidazole propionate were higher in metformin-treated subjects with high blood glucose (≥7.8 mM; approximately equivalent to 6.5% HbA1c, a stringent HbA1c goal; American Diabetes Association, 2019) compared to those with blood glucose <7.8 mM (Figure 1A). By contrast, mean levels of urocanate, the imidazole propionate precursor, were similar in both groups (Figure 1A). The mean time from diagnosis with type 2 diabetes was also similar in both groups (Table S1). These results are consistent with a potential negative contribution of imidazole propionate to metformin action in humans, but further prospective studies in larger cohorts are required to confirm this hypothesis.
Figure 1.

Imidazole Propionate Is Associated with Poor Glucose Control on Metformin
(A) Plasma levels of imidazole propionate (ImP) or urocanate in metformin (Met)-treated subjects with type 2 diabetes divided according to glucose control. Poor glucose control is defined as glucose levels ≥7.8 mM, approximately 6.5% HbA1c.
(B–G) Effects of ImP in metformin-treated mice models. Mice were injected intraperitoneally with ImP (100 μg) or vehicle (Veh, 1% DMSO) followed 1 h later by oral administration of Met (200 mg/kg) or water.
(B) Percent change in fasting blood glucose levels in chow-fed mice 45 min after Met (or water) versus start of experiment. Veh+Water (n = 7), ImP+Water (n = 8), Veh+Met (n = 7), and ImP+Met (n = 7).
(C) Intraperitoneal glucose tolerance tests in chow-fed mice treated with Veh+Water (n = 9) or ImP+Water (n = 10) (left) or Veh+Met (n = 9) or ImP+Met (n = 9) (right).
(D) Serum insulin levels during intraperitoneal glucose tolerance tests shown in (C).
(E) Percent changes in fasting blood glucose levels in western diet-fed mice 45 min after Met (or water) versus start of experiment. Veh+Water (n = 4), ImP+Water (n = 6), Veh+Met (n = 7), or ImP+Met (n = 6).
(F) Intraperitoneal glucose tolerance tests in western diet-fed mice treated with Veh+Water (n = 4) or ImP+Water (n = 6) (left) or Veh+Met (n = 7) or ImP+Met (n = 6) (right).
(G) Intraperitoneal glucose tolerance tests in diabetic (db/db) mice treated with Veh+Met (n = 7) or ImP+Met (n = 7).
Data in (A) are presented as box plots showing minimum, 25% quartile, median, 75% quartile, maximum, and mean (marked as +). Other data are mean ± SEM.∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant. p values were determined by Wilcoxon rank-sum test (A), unpaired two-tailed Student’s t tests (C, F, and G), one-way ANOVA with Tukey’s multiple comparisons test (B and E), and two-way ANOVA with Tukey’s multiple comparisons test (D).
Imidazole Propionate Impairs Metformin Action in Mice
To determine whether metformin action is reduced in the presence of imidazole propionate, we tested the effect of imidazole propionate on glucose metabolism in metformin-treated mice. A single injection of imidazole propionate in chow-fed mice increased fasting glucose levels (p = 0.06) and reversed the expected metformin-induced reduction in fasting glucose levels (Figure 1B). We showed that the acute treatment with imidazole propionate was sufficient to impair glucose tolerance in wild-type mice on a chow diet both in the absence of metformin, albeit to a lesser extent than we previously observed after 3-day imidazole propionate treatment (Koh et al., 2018), and in the presence of metformin (Figure 1C). We also showed that metformin promoted glucose-induced increases in insulin levels in mice on a chow diet (Figure 1D), supporting a recently reported role of metformin in insulin secretion (Zhang et al., 2019); however, this metformin response was not affected by imidazole propionate (Figure 1D). Thus, the effects of imidazole propionate on the metformin response are not likely mediated through an effect on insulin secretion. In our previous study, we showed that 3 or 14 days of imidazole propionate treatment did not affect insulin levels, but that imidazole propionate impairs insulin signaling by inhibiting IRS (Koh et al., 2018).
We also investigated if imidazole propionate prevented the glucose-lowering effects of metformin in diabetes models. In mice fed a western diet (i.e., high in fat and sucrose) to induce insulin resistance, we showed that acute treatment with imidazole propionate did not significantly affect fasting glucose levels but reversed the metformin-induced reduction in fasting glucose levels (Figure 1E). Imidazole propionate also reduced glucose tolerance in metformin-treated mice on a western diet (Figure 1F) and in metformin-treated diabetic (db/db) mice (Figure 1G). Taken together, these results indicate that the effects of metformin on glucose control in insulin-sensitive, insulin-resistant, and diabetic mouse models are not observed in the presence of imidazole propionate. It should be noted that here we analyzed acute effects (i.e., we administered one intraperitoneal injection of imidazole propionate 1 h before oral administration of metformin) to avoid the impact of metformin-induced microbial compositional changes (Wu et al., 2017) and to minimize the effects of imidazole propionate per se on blood glucose levels. Further studies in mouse models are required to determine the chronic effects of imidazole propionate on metformin action.
Imidazole Propionate-Induced Inhibitory AMPK Serine Phosphorylation Inhibits Metformin-Induced AMPK Active Site Phosphorylation
In an attempt to elucidate the underlying molecular mechanism through which imidazole propionate affects the metformin response, we tested whether imidazole propionate alters the AMPK signaling pathway. In agreement with our hypothesis, we observed that imidazole propionate inhibited metformin-induced phosphorylation of the AMPK active site T172 in liver tissue 1 h after mice received a single injection of imidazole propionate (Figure 2A).
Figure 2.

Imidazole Propionate Induces Inhibitory AMPK S485 Phosphorylation
(A) Effects of imidazole propionate (ImP) on metformin (Met)-induced AMPK T172 phosphorylation in the liver. Mice were injected intraperitoneally with vehicle (1% DMSO in water) or ImP (100 μg) and after 1 h Met (200 mg/kg) was orally administered; liver was collected 45 min after Met administration (n = 3 mice per group).
(B) Immunoblot (and quantification) of liver lysates taken from mice 105 min after one intraperitoneal injection of ImP. Vehicle (n = 3) and ImP (n = 4).
(C) Immunoblot (and quantification) of liver lysates from mice (taken after an intraperitoneal glucose tolerance test) showing effects of oral administration of Met preceded by an intraperitoneal injection of vehicle or ImP. Vehicle (n = 3), Met (n = 4), and ImP+Met (n = 3).
(D) Time-dependent effects of ImP (100 μM) on AMPK S485 phosphorylation in serum-starved HEK293 cells (n = 3).
(E) Concentration-dependent effects of ImP on AMPK S485 phosphorylation in serum- and amino acid-deprived HEK293 cells (representative of n = 2).
(F) Comparison of effects of ImP (100 μM) or amino acids (a.a) on AMPK S485 and S6K1 phosphorylation in serum- and amino acid-deprived HEK293 cells (representative of n = 2).
(G) Role of AMPK S485 phosphorylation on inhibitory action of ImP on Met-induced AMPK T172 phosphorylation. HEK293 cells were transfected with HA AMPK wild-type (WT), HA AMPK S485A, or HA AMPK T479A mutant and incubated with 0.5 mM Met with or without ImP (100 μM) for 6 h (n = 3).
Data are mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant. One-way ANOVA with Dunnett’s multiple comparisons test (A, D, and G), unpaired two-tailed Student’s t tests (B and C).
We then focused on inhibitory AMPK S485 (α1)/S491 (α2) phosphorylation, which has been shown to act hierarchically upstream to inhibit AMPK T172 phosphorylation and subsequent AMPK activity (Dagon et al., 2012; Hawley et al., 2014; Horman et al., 2006; Ning et al., 2011; Valentine et al., 2014) (Figure S1A). Moreover, AMPK S485 phosphorylation is inversely correlated with insulin sensitivity in human muscle (Heathcote et al., 2016). The single injection of imidazole propionate was sufficient to induce hepatic AMPK S485 phosphorylation without affecting basal AMPK T172 phosphorylation (Figure 2B). In contrast to our earlier results showing that 3-day treatment with imidazole propionate promotes phosphorylation of S6K1, a marker of mTORC1 activation (Koh et al., 2018) (Figure S1B), one injection of imidazole propionate in mice was not sufficient to induce S6K1 phosphorylation (Figure 2B); thus, imidazole propionate appears to affect glucose levels independently of mTORC1 activation in the short term. As expected, imidazole propionate inhibited metformin-induced AMPK T172 phosphorylation and concomitantly increased AMPK S485 phosphorylation (Figure S1C) in the liver (Figures 2C and S2A), muscle (Figure S2B), and white adipose tissue (WAT) (Figure S2C). Imidazole propionate also induced phosphorylation of AMPK S485 (corresponds to S487 in humans) in the human embryonic kidney cell line HEK293 both in the presence and the absence of amino acids (Figures 2D and 2E). Although imidazole propionate shares signaling pathways with amino acids to activate mTORC1 (Koh et al., 2018; Linares et al., 2015), inhibitory AMPK serine phosphorylation was specific to imidazole propionate stimulation as amino acids did not affect S485 phosphorylation (Figures 2F, S1B, and S1C).
Recent reports have questioned the clinical relevance of the high concentrations of metformin used in most cell studies (2–10 mM); indeed, oral administration of metformin in mice (125–200 mg/kg), corresponding to around 20 mg/kg metformin in humans, results in plasma concentrations of 10–126 μM (Chandel et al., 2016; Dowling et al., 2016). Here we showed that 0.5 mM metformin was sufficient to induce AMPK T172 phosphorylation without affecting AMPK S485 and S6K1 phosphorylation in primary hepatocytes and HEK293 cells; however, 2 mM metformin induced inhibitory AMPK phosphorylation in primary hepatocytes and did not induce AMPK T172 phosphorylation in HEK293 cells (Figures S2D and S2E). We therefore used 0.5 mM metformin to determine if AMPK S485 phosphorylation mediates imidazole propionate-induced inhibition of metformin action on AMPK T172 phosphorylation. Under these conditions, metformin-induced AMPK T172 phosphorylation was reduced by imidazole propionate in HEK293 cells (Figures 2G and S2F) and primary hepatocytes (Figure S2G), consistent with the effects of imidazole propionate on metformin in mice (Figure 2A). More importantly, overexpression of the AMPK S485 phosphorylation-deficient mutant (S485A) blocked the inhibitory effect of imidazole propionate on metformin-induced AMPK T172 phosphorylation (Figure 2G). AMPK can also be inhibited by glycogen synthase kinase 3 through phosphorylation of AMPK T479 (Suzuki et al., 2013). However, overexpression of the AMPK T479 phosphorylation-deficient mutant (T479A) did not reduce the inhibitory effect of imidazole propionate (Figure 2G). Together, these data support the role of AMPK S485 in mediating the inhibitory action of imidazole propionate on metformin-induced AMPK T172 phosphorylation.
Imidazole Propionate-Induced Akt Activation Induces Inhibitory AMPK Serine Phosphorylation
Earlier studies have shown that Akt is an AMPK S485 kinase and that basal Akt phosphorylation increases in parallel with AMPK S485 phosphorylation in brains and aortae from diabetic mice (Hawley et al., 2014; Horman et al., 2006; Kim et al., 2015; Ning et al., 2011; Valentine et al., 2014) (Figure S1A). Under insulin-resistant conditions, insulin-induced Akt phosphorylation is often reduced; however, basal Akt activation has been shown to increase and has therefore been suggested as a contributing factor for the development of insulin resistance (Khamzina et al., 2005; Liu et al., 2009; Sajan et al., 2015). We have previously shown in primary hepatocytes that imidazole propionate inhibits insulin-stimulated Akt phosphorylation when IRS levels are reduced (Koh et al., 2018). Here, we confirmed this result in HEK293 cells, which have a rapid turnover of IRS proteins (Figure S3A). However, we also showed that imidazole propionate induced basal Akt phosphorylation and impaired insulin signaling (shown by inhibition of insulin-stimulated IRS tyrosine and Akt S473 phosphorylation) in primary hepatocytes before IRS reduction occurred (Figure S3B). Basal Akt phosphorylation in HEK293 cells was increased by imidazole propionate treatment both in the absence and presence of amino acids (Figures 3A and 3B) but decreased 8 h after imidazole propionate stimulation (Figure 3B). We also observed increased basal Akt phosphorylation in tissues from mice treated with imidazole propionate (either for 3 days or by a single injection) (Figures S3C and S3D).
Figure 3.

Imidazole Propionate-Induced Basal Akt Activation Is Responsible for Inhibitory AMPK S485 Phosphorylation
(A) Effect of 2 h treatment with imidazole propionate (ImP) (at the indicated concentrations) on Akt S473 phosphorylation in amino acid-deprived HEK293 cells (n = 3).
(B) Time-dependent effects of ImP (100 μM) on Akt phosphorylation in serum-starved HEK293 cells (n = 3).
(C) Effects of mTORC1 inhibition (by 200 nM Rapamycin, Rap) or Akt inhibition (by 200 nM MK2206, MK) on ImP-induced Akt or mTORC1 activation. HEK293 cells preincubated with Rap or MK for 30 min were stimulated with 100 μM ImP for 1 h in the absence of amino acids (n = 4).
(D and E) Effects of mTORC1 inhibition by rapamycin (20 nM Rap) or the Akt inhibitor MK (200 nM) on ImP-induced Akt or mTORC1 activation. Serum-starved HEK293 cells were co-incubated with 100 μM ImP and Rap or MK for 6 h (D) (n = 3) and for 8 h (E) (representative of n = 2).
(F) mTORC1- and Akt-dependent inhibitory AMPK S485 phosphorylation by ImP. Serum-starved HEK293 cells were co-incubated with ImP and Rap or MK for 6 h (n = 3).
Data are mean ± SEM. ∗p < 0.05, ∗∗∗p < 0.001. One-way ANOVA with Dunnett’s multiple comparisons test (A–D), one-way ANOVA with Tukey’s multiple comparisons test compared to ImP-treated groups (F).
Although Akt activation is upstream of growth factor-induced mTORC1 activation (Inoki et al., 2002), imidazole propionate-induced S6K1 phosphorylation was not inhibited by treatment with the Akt-specific inhibitor MK2206 (but was efficiently blocked by the mTORC1 inhibitor rapamycin as expected) (Figures 3C–3E). Thus, Akt is not an upstream regulator of mTORC1 in the imidazole propionate signaling pathway. Similarly, phorbol 12,13-myristate acetate (PMA), which has been shown to induce both Akt and mTORC1 activation, can promote S6K1 phosphorylation independently of the PI3K/Akt pathway (Aeder et al., 2004; Yang et al., 2015).
We further showed that imidazole propionate-induced inhibitory AMPK serine phosphorylation was inhibited both by inhibition of Akt and by inhibition of mTORC1 (Figure 3F), consistent with a previous report identifying S6K1 as an AMPKα2 S491 kinase (Dagon et al., 2012) (Figure S1A). Taken together, these results might indicate that imidazole propionate could impair insulin sensitivity through three potential axes: the p62/mTORC1/IRS axis (Koh et al., 2018), the Akt/AMPK axis, or the S6K1/AMPK axis. However, 4 h after a single imidazole propionate injection in mice, we could detect AMPK serine phosphorylation and phosphorylation of the activation loop of Akt (T308) without an increase in phosphorylation of the hydrophobic motif of Akt (S473) or correlation with mTORC1 activation (Figure S3E). Although phosphorylation on both T308 and S473 is required for full activation of Akt, T308 phosphorylation alone can induce one-third maximum activity of Akt (no activity is observed when only Akt S473 is phosphorylated) (Alessi et al., 1996). Therefore, our results suggest that imidazole propionate-induced inhibitory AMPK phosphorylation is mediated through the Akt/AMPK axis in vivo (Figure S1D).
Imidazole Propionate-Activated p38γ Is a Direct Kinase for Akt
We tested whether imidazole propionate could activate mTORC2 given that (1) mTORC2 is known as a major kinase for Akt S473 (Sarbassov et al., 2005), and (2) we have previously shown that imidazole propionate induces the activation of mTORC1 (Koh et al., 2018). Unexpectedly, we showed that imidazole propionate did not promote mTORC2 kinase activity in vitro (Figure S4A). However, there are several reports showing that mTORC2 is not essential for Akt S473 phosphorylation (Bentzinger et al., 2008; Risson et al., 2009; Zhang et al., 2010), and other kinases have been proposed such as the atypical IκB kinase epsilon (IKKϵ) and TANK-binding kinase 1 (TBK1) (Xie et al., 2011). The open source PhosphoNET database (http://www.phosphonet.ca) predicted known kinases such as mTOR, IKKϵ, and TBK1 for S473 phosphorylation of Akt, but also p38δ for all three Akt isoforms (Figure S4B). p38 MAPK can be divided into two subsets based on substrate specificity, sequence homology, and sensitivity to inhibitors: p38α/β and p38γ/δ, also known as alternative p38 (Escós et al., 2016). We have previously shown that both p38δ and p38γ can mediate imidazole propionate-induced S6K1 phosphorylation, although p38γ appears to have a larger impact on imidazole propionate-induced mTORC1 signaling (Koh et al., 2018). We therefore tested whether p38δ and/or p38γ could mediate imidazole propionate-induced activation of Akt. Interestingly, we showed that knockdown of p38γ, but not p38δ, blocked imidazole propionate-induced Akt and AMPK S485 phosphorylation (Figures S4C and S4D), suggesting that the imidazole propionate/Akt/inhibitory AMPK phosphorylation axis is specific to p38γ.
To investigate if p38γ can act as a novel Akt kinase, we next performed an in vitro kinase assay. In the presence of ATP, recombinant p38γ did not induce phosphorylation of inactive recombinant Akt1 in vitro; however, in the presence of imidazole propionate and ATP, p38γ directly phosphorylated both Akt S473 and T308 residues (Figures 4A and S1E). Imidazole propionate-induced p38γ activation was supported by increased p38γ autophosphorylation (Figure 4A), consistent with our previous report (Koh et al., 2018). IKKϵ and TBK1 are the only direct kinases for Akt that have previously been reported to induce phosphorylation at both the activation loop (T308) and the hydrophobic motif (S473) (Xie et al., 2011). Our results indicate that imidazole propionate-activated p38γ is also a kinase for Akt at both of these phosphorylation sites. In line with our in vitro results, p38γ depletion blocked imidazole propionate-induced Akt T308 and S473 phosphorylation in HEK293 cells (Figures 4B and S1E).
Figure 4.

Imidazole Propionate-Activated p38γ Is a Direct Kinase for Akt, Responsible for Mediating Inhibitory AMPK S485 Phosphorylation
(A) In vitro kinase assay (n = 4). p38γ and inactive Akt1 were preincubated and the kinase reaction was started by adding ATP in the absence or presence of ImP at the indicated concentrations.
(B) Effects of p38γ depletion on ImP (100 μM)-induced AMPK and Akt phosphorylation in serum-starved HEK293 cells (n = 3).
(C) Effects of the p38γ inhibitor pirfenidone (Pirf) on ImP-induced inhibitory action on metformin (Met). Serum-starved HEK293 cells were co-incubated with 0.5 mM Met, 100 μM ImP, and Pirf at the indicated concentrations for 6 h (n = 3).
(D and E) Effects of Pirf on ImP-induced inhibition of response to Met. Mice were injected intraperitoneally with vehicle (Veh, 1% DMSO), ImP (100 μg), Pirf (700 μg), or ImP with Pirf followed 1 h later by oral administration of Met (200 mg/kg) or water.
(D) Immunoblot (and quantification) of liver lysates taken from chow-fed mice 45 min after Met (n = 4 mice per group).
(E) Percent change in fasting blood glucose levels in chow-fed mice 45 min after Met (or water) versus start of experiment: Veh+Water (n = 7), Pirf+Water (n = 6), and Pirf+Met (n = 7) (left); Veh+Met (n = 4), ImP+Met (n = 4), and ImP+Met+Pirf (n = 4) (right).
(F) Schematic depiction of imidazole propionate signaling. Interaction between Met and the ImP/p38γ/Akt/AMPK axis investigated in this study was shown together with previously reported ImP/alternative p38/p62/mTORC1 axis (Koh et al., 2018).
Data are mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA followed by Dunnett’s multiple comparisons test (A and B), one-way ANOVA followed by Tukey’s multiple comparisons test (C–E).
Blocking Imidazole Propionate-Activated p38γ Restores Metformin Action on AMPK
We next investigated if imidazole propionate-induced p38γ kinase activity per se is important to mediate the inhibitory effects of imidazole propionate on metformin action. Pirfenidone (5-methyl-1-phenyl-2-(1H)-pyridone) has been shown to specifically inhibit p38γ kinase activity in vivo and in vitro without affecting p38α or CDK2 activity (Ozes and Seiwert, 2008; Tomás-Loba et al., 2019; Yin et al., 2016). Pirfenidone in the absence of imidazole propionate did not affect the phosphorylation of AMPK, Akt, or S6K1 (Figure S4E). However, pirfenidone efficiently blocked imidazole propionate-induced p38γ phosphorylation (activation) and subsequent Akt T308 and inhibitory AMPK serine phosphorylation (and S6K1 phosphorylation as expected from our earlier results; Koh et al., 2018) (Figures 4C and S4F). The inhibitory effects of imidazole propionate on metformin-induced AMPK T172 phosphorylation were reversed by pirfenidone treatment (Figure 4C). These results suggest that imidazole propionate-activated p38γ mediates imidazole propionate-induced inhibition on metformin action.
Pirfenidone has been recently shown to be located in the ATP binding pocket of p38γ (Tomás-Loba et al., 2019), and we performed blind docking of imidazole propionate to p38γ by SwissDock (Grosdidier et al., 2011). Superimposing the structure of p38γ-ATP (PDB: 1CM8) on our predicted docking model revealed that imidazole propionate can also possibly anchor to the ATP binding pocket (Figure S4G), suggesting a competitive mode of action between pirfenidone and imidazole propionate toward the p38γ ATP binding pocket.
Finally, we investigated whether pirfenidone could also reverse the inhibitory action of imidazole propionate on metformin action in vivo. We injected chow-fed mice intraperitoneally with vehicle or imidazole propionate in the absence or presence of pirfenidone before orally administering metformin. Consistent with the results observed in the cell studies (Figure 4C), pirfenidone inhibited the imidazole propionate-induced increase in inhibitory AMPK serine phosphorylation and reversed the inhibitory effects of imidazole propionate on metformin-induced AMPK T172 phosphorylation in the liver (Figure 4D). We further observed that pirfenidone treatment did not lower fasting blood glucose levels when administered alone but blocked the imidazole propionate-induced increase in glucose levels in the presence of metformin (Figure 4E). These results suggest that pirfenidone can efficiently block the inhibitory action of imidazole propionate on metformin at the cellular level and in vivo.
Concluding Remarks
In this study, we showed that the microbial metabolite imidazole propionate can reduce the efficacy of metformin through interaction with AMPK, a known metformin signaling pathway. We have previously shown that imidazole propionate can activate p62/mTORC1 via p38γ, leading to the inhibition of IRS-mediated insulin signaling; this response requires long-term exposure to imidazole propionate (Koh et al., 2018). Here we identified imidazole propionate-activated p38γ as a kinase for Akt in vitro; we showed that imidazole propionate induces inhibitory AMPK serine phosphorylation via Akt and subsequently inhibits metformin-induced AMPK T172 phosphorylation. Thus, imidazole propionate not only promotes insulin resistance through the p62/mTORC1/S6K1/IRS pathway but may also reduce the glucose-lowering response to metformin by promoting basal Akt activation (summarized in Figure 4F).
We also showed that the p38γ inhibitor pirfenidone can reduce imidazole propionate-induced (but not basal) inhibitory AMPK phosphorylation (Figure 4F). Pirfenidone is a relatively non-toxic drug that is currently being used in phase III clinical trials to treat idiopathic pulmonary fibrosis (Moran, 2011). Although additional studies are required to determine whether there is a causal association between imidazole propionate and impaired metformin action in humans and to test the combination of pirfenidone and imidazole propionate in a diabetes animal model, our findings suggest that it would be of potential interest in diabetes research to investigate the combinatorial effects of pirfenidone in subjects with type 2 diabetes who do not achieve adequate glycemic control when on metformin. Our findings also provide mechanistic understanding for how the microbiota or its metabolic products may affect drug responses and thus provide opportunities to modulate personalized drug responses.
Limitations of Study
In this study, we divided metformin-treated subjects with type 2 diabetes into two groups according to their blood glucose level and observed that imidazole propionate levels were higher in those with high blood glucose. However, we used a cross-sectional cohort and thus it is unclear whether the metformin-treated subjects with high blood glucose did not respond to metformin or had more severe diabetes before metformin treatment. Participants in the current study were older than in Koh et al. (2018) (68.9 ± 0.7 years versus 58.4 ± 0.2 years), which may explain the higher imidazole propionate values observed in the current study. Furthermore, we cannot exclude that microbiome changes induced by metformin increase imidazole propionate levels; indeed, we previously showed that metformin treatment increased the abundance of Streptococcus parasanguinis (Wu et al., 2017), a bacteria with the potential to produce imidazole propionate (Koh et al., 2018). Thus, it will be important to clarify in a longitudinal cohort study including different age groups whether imidazole propionate has a direct negative effect on metformin action and whether metformin increases the abundance of imidazole propionate-producing bacteria.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-phospho-Akt (Ser473) (1:1000) | Cell Signaling | Cat# 4058; RRID: AB_331168 |
| Rabbit anti-phospho-Akt (Thr308) (1:500) | Cell Signaling | Cat# 9275; RRID: AB_329828 |
| Rabbit anti-phospho-p70 S6 kinase (Thr389) (1:500) | Cell Signaling | Cat# 9205; RRID: AB_3309 |
| Rabbit p70 S6 kinase (1:500) | Cell Signaling | Cat# 9202; RRID: AB_331676 |
| Rabbit anti-p38γ MAPK (1:500) | Cell Signaling | Cat# 2307; RRID: AB_10860779 |
| Rabbit anti-phospho-p38 MAPK (Thr180/Tyr182) (1:500) | Cell Signaling | Cat# 9211; RRID: AB_331641 |
| Rabbit anti-Akt (pan) (11E7) (1:500) | Cell Signaling | Cat# 4685; RRID: AB_2225340 |
| Rabbit anti-GAPDH (14C10) (1:500) | Cell Signaling | Cat# 2118; RRID: AB_561053 |
| Rabbit anti-β-Actin (13E5) (1:500) | Cell Signaling | Cat# 4970; RRID: AB_2223172 |
| Rabbit anti-IRS1 (1:500) | EMD Millipore | Cat# 06-248; RRID: AB_2127890 |
| Goat anti-SAPK4 (p38δ) (1:500) | Santa Cruz | Cat# sc-7585; RRID: AB_656012 |
| Mouse anti-HA-probe (F-7) (1:1000) | Santa Cruz | Cat# sc-7392; RRID: AB_627809 |
| Rabbit anti-phospho-IRS1 (Tyr612) (1:500) | EMD Millipore | Cat# 09-432; RRID: AB_1163457 |
| Rabbit anti-phospho-AMPK (T172) (1:500) | Cell Signaling | Cat# 2535; RRID: AB_331250 |
| Rabbit anti-phospho-AMPK (S485) (1:500) | Cell Signaling | Cat# 4185, RRID: AB_2169402 |
| Enhanced chemiluminescence | Thermo Fisher | WBKLS0500 |
| HRP-linked anti-rabbit IgG (1:2500) | GE Healthcare | Cat# NA934-1ML; RRID: AB_2750578 |
| HRP-linked anti-mouse IgG (1:2500) | GE Healthcare | Cat# NA9310-1ML; RRID: AB_772193 |
| Biological Samples | ||
| Human peripheral plasma | Malmö Preventive Project | Leosdottir et al., 2010 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Imidazole propionate (deamino histidine) | Santa Cruz | sc294276; CAS 1074-59-5 |
| Metformin (1,1-dimethylbiguanide hydrochloride) | Sigma-Aldrich | D150959 |
| Pirfenidone | Sigma-Aldrich | P2116 |
| Rapamycin | Merk Millipore | 553210 |
| MK2206 | Santa Cruz | Sc-364537 |
| Recombinant p38γ | Abcam | ab125651 |
| Recombinant Akt1 | Abcam | ab116412 |
| Collagenase type IV | Sigma-Aldrich | C5138 |
| Dulbecco’s modified Eagle’s medium | Lonza | BE12614F |
| Ham’s F-12 Nutrient Mix | Hyclone | 10235122 |
| Fetal bovine serum | Hyclone | SV30160.03 |
| Earle’s Balanced Salt Solution | GIBCO | 24010-043 |
| Penicillin/Streptomycin | Hyclone | SV30010 |
| RPMI-HEPES 1640 | Lonza | BE12115F |
| Dexamethasone | Sigma-Aldrich | D8893 |
| Protein A-Sepharose beads | RepliGen | CA-PRI-0005 |
| Poly-L-lysine | Sigma-Aldrich | P6282 |
| Lipofectamine 2000 | Thermo Fisher | 11668019 |
| Recombinant p38γ | Abcam | ab125651 |
| Tris (Trizma Base) | Sigma-Aldrich | T6066 |
| NaCl | Sigma-Aldrich | S3014 |
| EDTA | Sigma-Aldrich | EDS |
| Sodium orthovanadate | Sigma-Aldrich | S6508 |
| Sodium fluoride | Sigma-Aldrich | 201154 |
| PMSF | Sigma-Aldrich | P7626 |
| glycerophosphate | Sigma-Aldrich | G5422 |
| glycerol | Sigma-Aldrich | G7893 |
| CHAPS | Thermo Fisher | 28300 |
| Complete mini, EDTA free protease inhibitor cocktail | Sigma-Aldrich | 11836170001 |
| ATP | Sigma-Aldrich | A2383 |
| MOPS | Sigma-Aldrich | M1254 |
| Beta-glycerol-phosphate | Sigma-Aldrich | 50020 |
| DTT | Sigma-Aldrich | D9163 |
| Glucose | Fresenius Kabi | B05BA03 |
| Western diet | ENVIGO | TD.96132 |
| Critical Commercial Assays | ||
| NuPAGE Novex 4-12% Bis-Tris Gels | Invitrogen | NP0336BOX |
| Novex 4-20% Tris-Glycine Mini gels | Invitrogen | XP04205BOX |
| Ultra-sensitive mouse insulin ELISA | Crystal Chem | 90080 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | CRL-1573 |
| Primary hepatocytes | Male C57BL/6J | N/A |
| Experimental Models: Organisms/Strains | ||
| Male C57BL/6J | N/A | N/A |
| db/db mice | Jackson Laboratory | 000697 |
| Oligonucleotides | ||
| AllStars negative control siRNA | QIAGEN | 1027280 |
| p38δ siRNA | QIAGEN_ Hs_MAPK13_6 | CGGGATGAGCCTCATC CGGAA |
| p38γ siRNA 1 | QIAGEN_ Hs_MAPK12_5 | CTGGACGTATTCACTC CTGAT |
| p38γ siRNA 2 | QIAGEN_ Hs_MAPK12_6 | TGGAAGCGTGTTACTT ACAAA |
| Recombinant DNA | ||
| HA AMPK WT | Dr. Ken Inoki | Suzuki et al., 2013 |
| HA AMPK S485A | Dr. Ken Inoki | Suzuki et al., 2013 |
| HA AMPK T479A | Dr. Ken Inoki | Suzuki et al., 2013 |
| Flag mTOR | Dr. Sung Ho Ryu | Pohang University of Science and Technology, Republic of Korea |
| HA rictor | Dr. Sung Ho Ryu | Pohang University of Science and Technology, Republic of Korea |
| Software and Algorithms | ||
| Prism (version 7) | GraphPad | N/A |
| Other | ||
| TissueLyser II | QIAGEN | 85300 |
| Micro tube 1.3ml K3E | SARSTEDT | 41.1504.105 |
| Microvette CB 300 Z | SARSTEDT | 16.440.100 |
| Glucose meters | Bayer Contour XT | ASCENSIA Diabetes Care |
| Nitrocellulose membrane (0.45 μm pore) | Invitrogen | LC2001 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Fredrik Bäckhed (fredrik@wlab.gu.se).
Materials Availability
Plasmid DNA constructs used in this study were from Dr. Sung Ho Ryu (Pohang University of Science and Technology, Republic of Korea) and from Dr. Ken Inoki (University of Michigan, USA).
Data and Code Availability
This study does not use any custom code, software, or algorithm.
Experimental Model and Subject Details
Human Subjects
To investigate the potential relationship between imidazole propionate levels and metformin response, we analyzed subjects with type 2 diabetes aged over 65 who were treated only with metformin (Table S1). Subjects with prevalent diabetes and using metformin were identified from a re-examination conducted in 2002-2006 of the population-based study Malmö Preventive Project, which includes subjects born 1921-1949 and living in the city of Malmö in southern Sweden, as described previously (Leosdottir et al., 2010). This study was approved by the Ethics review board at Lund University and all participants gave written informed consent. In brief, according to a stringent HbA1c guideline (6.5% HbA1c, approximately equivalent to 7.8 mM blood glucose) by the American Diabetes Association, we divided type 2 diabetic subjects only on metformin with blood glucose more than 7.8 mM (n = 40) and less than 7.8 mM (n = 29). Both men and women were included in the study cohorts and due to the relative few individuals in each group, we did not perform separate analyses of the influence of sex or gender on the imidazole propionate levels.
Mice
8-14-week-old male C57BL/6J mice were used in the animal experiments (unless otherwise indicated). Mice were maintained under a strict 12 h light cycle and had unlimited access to water and food. All mice were fed an autoclaved chow diet (5021 LabDiet) ad libitum unless indicated. To induce glucose intolerance, 8-week-old C57BL/6J mice were fed western diet (ENVIGO, TD.96132) for 6 weeks. Mice with genetic predisposition for diabetes (db/db mice) were purchased from the Jackson Laboratory (Stock number: 000697) and studied at 6 weeks of age. All mice experiments were performed in our animal facility and were approved by the Ethics Committee on Animal Care and Use in Gothenburg, Sweden.
HEK293 Cells
HEK293 cells (ATCC CRL-1573) grown in RPMI-HEPES with 10% (v/v) fetal bovine serum (FBS) were serum-starved for 14 h and stimulated with 100 μM imidazole propionate or otherwise indicated. For imidazole propionate stimulation in the absence of amino acids, HEK293 cells in 4.5 g/l glucose DMEM with 10% (v/v) FBS were serum-starved for 18 h and then amino acid-deprived with EBSS for 1 h. For transfection, HEK293 cells cultured in poly-L-lysine-coated 60 mm dish for 18 h were transfected (with plasmids or siRNA as indicated) for 4 h; after replacing the medium with 10% (v/v) FBS, the cells were cultured for another 24 h and then serum-starved for 14-18 h. For insulin stimulation, serum-starved HEK293 cells were preincubated with imidazole propionate for 8 h and treated with 5 nM insulin. For metformin stimulation, serum-starved HEK293 cells were incubated with imidazole propionate in the presence or absence of metformin for 6 h.
Primary Hepatocytes
Primary hepatocytes were isolated from male C57BL/6J mice as described previously (Zhang et al., 2012). After perfusion, 16 × 105 cells were plated on collagen-coated 60 mm dishes in DMEM/F12 supplemented with FBS, penicillin/streptomycin, and 100 nM dexamethasone. After 4 h, the medium was changed to DMEM/F12 containing penicillin/streptomycin. For insulin stimulation, primary hepatocytes were cultured in DMEM/F12 with or without 100 μM imidazole propionate for 8 h and treated with 5 nM insulin for the indicated times. For metformin stimulation, serum-starved primary hepatocytes were incubated with imidazole propionate in the presence or absence of metformin for 8 h.
Method Details
Metabolite Analysis
For targeted measurement of imidazole propionate and urocanate from subjects with type 2 diabetes, plasma samples were extracted with 3 volumes of ice-cold acetonitrile in 1.5 mL polypropylene tube as previously described (Koh et al., 2018).
Imidazole Propionate Injection
To test the short-term effect of imidazole propionate on metformin-induced glucose lowering, mice were fasted for 4 h and 100 μg imidazole propionate in 200 μL water containing 1% DMSO or vehicle (1% DMSO in water) was injected intraperitoneally; after 1 h 200 mg/kg metformin or water was orally administered. Blood glucose levels were measured using glucose meters (Bayer Contour XT) in tail blood taken before treatment and 45 min after metformin or water administration. Fresh tissues were taken at the end of the experiment, snap-frozen in liquid nitrogen and stored at −80°C until protein extraction.
For the intraperitoneal glucose tolerance test, imidazole propionate and metformin were administered to fasted mice as described above. 30 min or 45 min after metformin or water administration, 1 g/kg glucose (Fresenius Kabi) was injected intraperitoneally. Blood glucose levels were measured from tail blood using glucose meters at 0, 15, 30, 60, 90, and 120 min after glucose injection. Blood samples for analysis of insulin were taken in serum clot activator tubes (Microvette CB 300 Z) before and 15 and 30 min after glucose injection. Serum insulin was measured with Ultra-Sensitive mouse Insulin ELISA kit. Mice were under isoflurane anesthesia and fresh tissues were taken at the end of the intraperitoneal glucose tolerance test, snap-frozen in liquid nitrogen and stored at −80°C until protein extraction.
Protein Analysis
Snap-frozen tissues and harvested cells were lysed in buffer A containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 20 mM NaF, 10 mM glycerophosphate, 1 mM PMSF, 10% glycerol, 1% Triton X-100 and protease inhibitor cocktail. For western blotting, the cell lysates were sonicated and centrifuged at 20,000g for 15 min at 4°C, and the supernatant was mixed with 5X Laemmli buffer [0.156 M Tris-HCl (pH 6.8), 25% glycerol, 12.5% β-mercaptoethanol, 12.5% SDS, 0.1% bromophenol blue], followed by heating at 95°C for 5-10 min. Samples treated with Laemmli buffer were separated in Bis-Tris gels or Tris-glycine gels, transferred to nitrocellulose membrane, blocked with 5% skim milk, and then probed with antibodies indicated at 4°C overnight. Blots were quantified with ImageJ; when independent experimental sets were run on different gels, control sets were set to 1 and relative fold-change was calculated.
Plasmids and siRNAs
HA-Rictor and Flag-mTOR were gifts from Dr. Sung Ho Ryu (Pohang University of Science and Technology, Republic of Korea). HA-AMPK WT, S485A, and T479A constructs were kindly provided by Dr. Sung Ki Hong and Dr. Ken Inoki (University of Michigan, USA) (Suzuki et al., 2013). siRNAs are listed in Key Resources Table.
mTORC2 Kinase Activity Assay
HEK293 cells were transfected with 1 μg of HA Rictor and 0.5 μg of Flag mTOR for mTORC2 activity assay, lysed with 0.3% CHAPS-containing lysis buffer, sonicated and centrifuged at 20,000 g for 15 min at 4°C, and the supernatant was incubated with 2 μg of anti-HA antibody at 4°C for overnight under gentle agitation. Immunocomplexes were then collected with protein A-Sepharose beads at 4°C for 2 h and washed three times with 0.3% CHAPS-containing lysis buffer. Immunoprecipitates were preincubated with 450 ng of inactive Akt1 in kinase assay buffer [12.5 mM MOPS (pH 7.2), 10 mM beta-glycerol-phosphate, 12.5 mM MgCl2, 2.5 mM EGTA, and 1 mM EDTA] for 10 min on ice. Kinase reaction was started by adding 100 μM ATP for 15 min at 37°C and stopped by adding 5x Laemmli buffer.
In Vitro Kinase Assay
200 ng of recombinant p38γ and 450 ng of recombinant inactive Akt1 were incubated in a kinase assay buffer described above for 15 min on ice. In a separate tube, 1 mM ATP was incubated with DMSO or imidazole propionate (final concentration of ATP was 50 or 200 μM and that of imidazole propionate was 10 or 100 nM). The kinase reaction was started by adding the content in the second tube to the p38γ and Akt1 mixture for 8 min at 30°C, and stopped by adding 5x Laemmli buffer.
Quantification and Statistical Analysis
Wilcoxon rank-sum test (Mann-Whitney U test) was used to test if imidazole propionate, urocanate, fasting blood glucose levels, or years with diabetes were different in the human cohorts with type 2 diabetes. One-way ANOVA with Tukey’s test or with Dunnett’s tests was performed when comparing three or more groups. Otherwise, unpaired two-tailed Student’s t tests were used, as indicated.
Acknowledgments
We thank Oskar Persson, Louise Helldén, and Manuela Krämer for technical assistance. We also thank Anna Hallen for producing the graphical abstract. This study was supported by the Swedish Research Council (2013-07800), the Novo Nordisk Foundation (NNF19OC0057271 and NNF15OC0016798), the Leducq Foundation (17CVD01), and grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF agreement (ALFGBG-718101). J.G.S. is supported by grants from the Swedish Heart-Lung Foundation (2016-0134 and 2016-0315), the Swedish Research Council (2017-02554), the ERC (ERC-STG-2015-679242), Skåne University Hospital, governmental funding of clinical research within the Swedish National Health Service, a donation from the Knut and Alice Wallenberg Foundation to the Wallenberg Center for Molecular Medicine in Lund, and funding from the Swedish Research Council (Linnaeus grant Dnr 349-2006-237, Strategic Research Area Exodiab Dnr 2009-1039) and Swedish Foundation for Strategic Research (Dnr IRC15-0067) to the Lund University Diabetes Center. This work is also supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2020R1C1C1003241 to A.K.). F.B. is a recipient of a European Research Council (ERC) Consolidator Grant (615362; METABASE) and is a Wallenberg Scholar and Torsten Söderberg Professor in Medicine.
Author Contributions
A.K. designed and performed the experiments. L.M.-H. performed glucose tolerance tests. P.M.N. and J.G.S. contributed to collection and analysis of human samples. N.-O.Y., S.H.R., and A.M. performed the experiments. A.K., R.P., and F.B. wrote the manuscript, and all authors approved and commented on the manuscript.
Declaration of Interests
A.K. and F.B. are shareholders in Implexion Pharma AB.
Published: August 11, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.07.012.
Contributor Information
Ara Koh, Email: arakoh@skku.edu.
Fredrik Bäckhed, Email: fredrik@wlab.gu.se.
Supplemental Information
References
- Aeder S.E., Martin P.M., Soh J.W., Hussaini I.M. PKC-eta mediates glioblastoma cell proliferation through the Akt and mTOR signaling pathways. Oncogene. 2004;23:9062–9069. doi: 10.1038/sj.onc.1208093. [DOI] [PubMed] [Google Scholar]
- Alessi D.R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association 6. Glycemic targets: standards of medical care in diabetes-2019. Diabetes Care. 2019;42(Suppl 1):S61–S70. doi: 10.2337/dc19-S006. [DOI] [PubMed] [Google Scholar]
- Bachrach W.H. Sulfasalazine: I. An historical perspective. Am. J. Gastroenterol. 1988;83:487–496. [PubMed] [Google Scholar]
- Bentzinger C.F., Romanino K., Cloëtta D., Lin S., Mascarenhas J.B., Oliveri F., Xia J., Casanova E., Costa C.F., Brink M. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008;8:411–424. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Brown J.B., Conner C., Nichols G.A. Secondary failure of metformin monotherapy in clinical practice. Diabetes Care. 2010;33:501–506. doi: 10.2337/dc09-1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel N.S., Avizonis D., Reczek C.R., Weinberg S.E., Menz S., Neuhaus R., Christian S., Haegebarth A., Algire C., Pollak M. Are metformin doses used in murine cancer models clinically relevant? Cell Metab. 2016;23:569–570. doi: 10.1016/j.cmet.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Colebrook L., Buttle G.A.H., O’Meara R.A.Q. The mode of action of p-aminobenzenesulphonamide and prontosil in haemolytic streptococcal infections. Lancet. 1936;228:1323–1326. [Google Scholar]
- Cook M.N., Girman C.J., Stein P.P., Alexander C.M., Holman R.R. Glycemic control continues to deteriorate after sulfonylureas are added to metformin among patients with type 2 diabetes. Diabetes Care. 2005;28:995–1000. doi: 10.2337/diacare.28.5.995. [DOI] [PubMed] [Google Scholar]
- Dagon Y., Hur E., Zheng B., Wellenstein K., Cantley L.C., Kahn B.B. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 2012;16:104–112. doi: 10.1016/j.cmet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia M.S., Fischbach M.A. Human microbiota. Small molecules from the human microbiota. Science. 2015;349:1254766. doi: 10.1126/science.1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling R.J.O., Lam S., Bassi C., Mouaaz S., Aman A., Kiyota T., Al-Awar R., Goodwin P.J., Stambolic V. Metformin pharmacokinetics in mouse tumors: implications for human therapy. Cell Metab. 2016;23:567–568. doi: 10.1016/j.cmet.2016.03.006. [DOI] [PubMed] [Google Scholar]
- Escós A., Risco A., Alsina-Beauchamp D., Cuenda A. p38γ and p38δ mitogen activated protein kinases (MAPKs), new stars in the MAPK galaxy. Front. Cell Dev. Biol. 2016;4:31. doi: 10.3389/fcell.2016.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-González A.P., Ritter A.D., Shrestha S., Andersen E.C., Yilmaz L.S., Walhout A.J.M. Bacterial metabolism affects the C. elegans response to cancer chemotherapeutics. Cell. 2017;169:431–441.e8. doi: 10.1016/j.cell.2017.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosdidier A., Zoete V., Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270-7. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser H.J., Gootenberg D.B., Chatman K., Sirasani G., Balskus E.P., Turnbaugh P.J. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341:295–298. doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley S.A., Ross F.A., Gowans G.J., Tibarewal P., Leslie N.R., Hardie D.G. Phosphorylation by Akt within the ST loop of AMPK-α1 down-regulates its activation in tumour cells. Biochem. J. 2014;459:275–287. doi: 10.1042/BJ20131344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heathcote H.R., Mancini S.J., Strembitska A., Jamal K., Reihill J.A., Palmer T.M., Gould G.W., Salt I.P. Protein kinase C phosphorylates AMP-activated protein kinase α1 Ser487. Biochem. J. 2016;473:4681–4697. doi: 10.1042/BCJ20160211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horman S., Vertommen D., Heath R., Neumann D., Mouton V., Woods A., Schlattner U., Wallimann T., Carling D., Hue L., Rider M.H. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J. Biol. Chem. 2006;281:5335–5340. doi: 10.1074/jbc.M506850200. [DOI] [PubMed] [Google Scholar]
- Inoki K., Li Y., Zhu T., Wu J., Guan K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Khamzina L., Veilleux A., Bergeron S., Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146:1473–1481. doi: 10.1210/en.2004-0921. [DOI] [PubMed] [Google Scholar]
- Kim B., Figueroa-Romero C., Pacut C., Backus C., Feldman E.L. Insulin resistance prevents AMPK-induced Tau dephosphorylation through Akt-mediated increase in AMPKSer-485 phosphorylation. J. Biol. Chem. 2015;290:19146–19157. doi: 10.1074/jbc.M115.636852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A., Molinaro A., Ståhlman M., Khan M.T., Schmidt C., Mannerås-Holm L., Wu H., Carreras A., Jeong H., Olofsson L.E. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175:947–961.e17. doi: 10.1016/j.cell.2018.09.055. [DOI] [PubMed] [Google Scholar]
- Leosdottir M., Willenheimer R., Plehn J., Borgquist R., Gudmundsson P., Harris T.B., Launer L.J., Bjornsdottir H., Nilsson P.M., Gudnason V. Myocardial structure and function by echocardiography in relation to glucometabolic status in elderly subjects from 2 population-based cohorts: a cross-sectional study. Am. Heart J. 2010;159:414–420.e4. doi: 10.1016/j.ahj.2009.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.B., Dieleman L.A., Ketabi A., Bibova I., Sawyer M.B., Xue H., Field C.J., Baracos V.E., Gänzle M.G. Irinotecan (CPT-11) chemotherapy alters intestinal microbiota in tumour bearing rats. PLoS One. 2012;7:e39764. doi: 10.1371/journal.pone.0039764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares J.F., Duran A., Reina-Campos M., Aza-Blanc P., Campos A., Moscat J., Diaz-Meco M.T. Amino acid activation of mTORC1 by a PB1-domain-driven kinase complex cascade. Cell Rep. 2015;12:1339–1352. doi: 10.1016/j.celrep.2015.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.Y., Hong T., Wen G.B., Han J., Zuo D., Liu Z., Cao W. Increased basal level of Akt-dependent insulin signaling may be responsible for the development of insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009;297:E898–E906. doi: 10.1152/ajpendo.00374.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall S.M. 60 years of metformin use: a glance at the past and a look to the future. Diabetologia. 2017;60:1561–1565. doi: 10.1007/s00125-017-4343-y. [DOI] [PubMed] [Google Scholar]
- Moran N. p38 kinase inhibitor approved for idiopathic pulmonary fibrosis. Nat. Biotechnol. 2011;29:301. doi: 10.1038/nbt0411-301. [DOI] [PubMed] [Google Scholar]
- Musi N., Hirshman M.F., Nygren J., Svanfeldt M., Bavenholm P., Rooyackers O., Zhou G., Williamson J.M., Ljunqvist O., Efendic S. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- Ning J., Xi G., Clemmons D.R. Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology. 2011;152:3143–3154. doi: 10.1210/en.2011-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen M.R., Doran E., Halestrap A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- Ozes, O.B.L., and Seiwert, S.D. (2008). Use of pirfenidone in therapeutic regimens. US patent WO2005040758A2, filed October 21, 2004, and granted May 6, 2005.
- Panebianco C., Adamberg K., Jaagura M., Copetti M., Fontana A., Adamberg S., Kolk K., Vilu R., Andriulli A., Pazienza V. Influence of gemcitabine chemotherapy on the microbiota of pancreatic cancer xenografted mice. Cancer Chemother. Pharmacol. 2018;81:773–782. doi: 10.1007/s00280-018-3549-0. [DOI] [PubMed] [Google Scholar]
- Pryor R., Norvaisas P., Marinos G., Best L., Thingholm L.B., Quintaneiro L.M., De Haes W., Esser D., Waschina S., Lujan C. Host-microbe-drug-nutrient screen identifies bacterial effectors of metformin therapy. Cell. 2019;178:1299–1312.e29. doi: 10.1016/j.cell.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risson V., Mazelin L., Roceri M., Sanchez H., Moncollin V., Corneloup C., Richard-Bulteau H., Vignaud A., Baas D., Defour A. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009;187:859–874. doi: 10.1083/jcb.200903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitta K.S., Zhang C., Lee K.K., Fujimoto K., Redinbo M.R., Boelsterli U.A. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica. 2014;44:28–35. doi: 10.3109/00498254.2013.811314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan M.P., Ivey R.A., Lee M.C., Farese R.V. Hepatic insulin resistance in ob/ob mice involves increases in ceramide, aPKC activity, and selective impairment of Akt-dependent FoxO1 phosphorylation. J. Lipid Res. 2015;56:70–80. doi: 10.1194/jlr.M052977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Scott T.A., Quintaneiro L.M., Norvaisas P., Lui P.P., Wilson M.P., Leung K.Y., Herrera-Dominguez L., Sudiwala S., Pessia A., Clayton P.T. Host-microbe co-metabolism dictates cancer drug efficacy in C. elegans. Cell. 2017;169:442–456.e18. doi: 10.1016/j.cell.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R.J., Lamia K.A., Vasquez D., Koo S.H., Bardeesy N., Depinho R.A., Montminy M., Cantley L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin N.R., Lee J.C., Lee H.Y., Kim M.S., Whon T.W., Lee M.S., Bae J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63:727–735. doi: 10.1136/gutjnl-2012-303839. [DOI] [PubMed] [Google Scholar]
- Spanogiannopoulos P., Bess E.N., Carmody R.N., Turnbaugh P.J. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016;14:273–287. doi: 10.1038/nrmicro.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Xie C., Wang G., Wu Y., Wu Q., Wang X., Liu J., Deng Y., Xia J., Chen B. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018;24:1919–1929. doi: 10.1038/s41591-018-0222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Bridges D., Nakada D., Skiniotis G., Morrison S.J., Lin J.D.D., Saltiel A.R., Inoki K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell. 2013;50:407–419. doi: 10.1016/j.molcel.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svartz N. Sulfasalazine: II. Some notes on the discovery and development of salazopyrin. Am. J. Gastroenterol. 1988;83:497–503. [PubMed] [Google Scholar]
- Tomás-Loba A., Manieri E., González-Terán B., Mora A., Leiva-Vega L., Santamans A.M., Romero-Becerra R., Rodríguez E., Pintor-Chocano A., Feixas F. p38γ is essential for cell cycle progression and liver tumorigenesis. Nature. 2019;568:557–560. doi: 10.1038/s41586-019-1112-8. [DOI] [PubMed] [Google Scholar]
- Uchimura Y., Fuhrer T., Li H., Lawson M.A., Zimmermann M., Yilmaz B., Zindel J., Ronchi F., Sorribas M., Hapfelmeier S. Antibodies set boundaries limiting microbial metabolite penetration and the resultant mammalian host response. Immunity. 2018;49:545–559.e5. doi: 10.1016/j.immuni.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine R.J., Coughlan K.A., Ruderman N.B., Saha A.K. Insulin inhibits AMPK activity and phosphorylates AMPK Ser485/491 through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch. Biochem. Biophys. 2014;562:62–69. doi: 10.1016/j.abb.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet M.J., Tissing W.J.E., Dun C.A.J., Meessen N.E.L., Kamps W.A., de Bont E.S.J.M., Harmsen H.J.M. Chemotherapy treatment in pediatric patients with acute myeloid leukemia receiving antimicrobial prophylaxis leads to a relative increase of colonization with potentially pathogenic bacteria in the gut. Clin. Infect. Dis. 2009;49:262–270. doi: 10.1086/599346. [DOI] [PubMed] [Google Scholar]
- Wu H., Esteve E., Tremaroli V., Khan M.T., Caesar R., Mannerås-Holm L., Ståhlman M., Olsson L.M., Serino M., Planas-Fèlix M. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017;23:850–858. doi: 10.1038/nm.4345. [DOI] [PubMed] [Google Scholar]
- Xie X., Zhang D., Zhao B., Lu M.K., You M., Condorelli G., Wang C.Y., Guan K.L. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc. Natl. Acad. Sci. USA. 2011;108:6474–6479. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G., Murashige D.S., Humphrey S.J., James D.E. A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep. 2015;12:937–943. doi: 10.1016/j.celrep.2015.07.016. [DOI] [PubMed] [Google Scholar]
- Yin N., Qi X., Tsai S., Lu Y., Basir Z., Oshima K., Thomas J.P., Myers C.R., Stoner G., Chen G. p38γ MAPK is required for inflammation-associated colon tumorigenesis. Oncogene. 2016;35:1039–1048. doi: 10.1038/onc.2015.158. [DOI] [PubMed] [Google Scholar]
- Zhang D., Contu R., Latronico M.V.G., Zhang J., Rizzi R., Catalucci D., Miyamoto S., Huang K., Ceci M., Gu Y. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Invest. 2010;120:2805–2816. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Sargis R.M., Volden P.A., Carmean C.M., Sun X.J., Brady M.J. PCB 126 and other dioxin-like PCBs specifically suppress hepatic PEPCK expression via the aryl hydrocarbon receptor. PLoS One. 2012;7:e37103. doi: 10.1371/journal.pone.0037103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhao Y., Xu J., Xue Z., Zhang M., Pang X., Zhang X., Zhao L. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci. Rep. 2015;5:14405. doi: 10.1038/srep14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang E., Mohammed Al-Amily I., Mohammed S., Luan C., Asplund O., Ahmed M., Ye Y., Ben-Hail D., Soni A., Vishnu N. Preserving insulin secretion in diabetes by inhibiting VDAC1 overexpression and surface translocation in β cells. Cell Metab. 2019;29:64–77.e6. doi: 10.1016/j.cmet.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M., Zimmermann-Kogadeeva M., Wegmann R., Goodman A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570:462–467. doi: 10.1038/s41586-019-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study does not use any custom code, software, or algorithm.