

Abstract
CARD9 is an immune adaptor protein in myeloid cells that is involved in C-type lectin signaling and antifungal immunity. CARD9 is implicated in autoimmune and inflammatory-related diseases, such as rheumatoid arthritis, IgA nephropathy, ankylosing spondylitis, and inflammatory bowel disease (IBD). Given that Lyn-deficient (Lyn−/−) mice are susceptible to both autoimmunity and IBD, we investigated the immunological role of CARD9 in the development of these diseases using the Lyn−/− mouse model. We found that genetic deletion of CARD9 was sufficient to reduce the development of both spontaneous autoimmune disease as well as DSS- or IL-10-deficiency–associated colitis in Lyn−/− mice. Mechanistically, CARD9 was a vital component of the Lyn-mediated regulation of Toll-like receptor (TLR2 and TLR4) signaling in dendritic cells, but not in macrophages. In the absence of Lyn, signaling through a CD11b-Syk-PKCδ-CARD9 pathway was amplified, leading to increased TLR-induced production of inflammatory cytokines. Dendritic cell–specific deletion of CARD9 reversed the development of autoimmune and experimental colitis observed in dendritic cell–specific, Lyn-deficient mice. These findings suggest that targeting CARD9 may suppress the development of colitis and autoimmunity by reducing dendritic cell–driven inflammation.
One-sentence summary:
Without the kinase Lyn, the adaptor protein CARD9 amplifies cytokine production in autoimmune disease.
Editor’s summary:
Lyn-deficient autoimmunity shows its CARDs
Both the adaptor protein CARD9 and loss of the kinase Lyn are associated with autoimmune disease, notably colitis and inflammatory bowel disease. Ma et al. explored this connection in mice and found that CARD9 amplified Toll-like receptor signaling and subsequent cytokine production in Lyn-deficient bone marrow–derived dendritic cells, but not macrophages. Deleting Card9 or genes encoding Src-family kinases in dendritic cells prevented the development of Lyn deficiency–associated colitis in mice. These findings suggest that targeting CARD9 or its associated kinases may therapeutically relieve inflammation in patients with autoimmune disease and that exploring the biological consequences of CARD9 polymorphisms is warranted.
Introduction
Caspase recruitment domain family member 9 (CARD9) is a myeloid-specific regulatory protein that is critical for signaling through multiple immune pathways. Dendritic cells (DCs) and macrophages lacking CARD9 have signaling defects downstream of C-type lectin receptors (CLRs), nucleotide-binding oligomerization domain (NOD-like) receptors, retinoic acid-inducible gene-I-like (RIG-like) receptors and Toll-like receptors (TLRs) (1). The cellular mechanism connecting CARD9 to CLRs, in particular Dectin-1-mediated signaling, has been established (2-4). Upon Dectin-1 receptor ligation, phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAM) by Src-family kinases (SFKs) leads to recruitment and activation of the kinase Syk. Activated Syk triggers the formation of a CARD9-BCL10 (B-cell lymphoma/leukemia 10)-Malt1 (Mucosa-associated lymphoid tissue lymphoma translocation protein 1) complex, which leads to the activation of the nuclear factor-kappaB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways (5). Cells lacking CARD9 fail to produce cytokines upon Dectin-1 ligation and show defective fungal killing (4). Consequently, mice lacking CARD9 exhibit a defect in their ability to control fungal infections (4).
Besides CLR signaling, CARD9 has also been implicated in TLR-related signaling (6). DCs lacking CARD9 produce lower levels of cytokines upon TLR2 or TLR4 stimulation, and mice lacking CARD9 exhibit defects in controlling bacterial infection by Listeria monocytogenes (6, 7). Unlike CLRs, TLRs do not contain ITAMs and do not recruit Syk, so even though CARD9 clearly plays a role in signaling downstream of TLRs, the cellular mechanism coupling CARD9 to TLR signaling pathways is unclear.
Consistent with its role in C-type lectin signaling, mutations in human CARD9 that result in premature termination of the protein have been associated with immune deficiencies to fungal infections (8, 9). However, a different set of polymorphisms in CARD9 have also been associated with diseases exhibiting symptoms of hyper-inflammation, including ankylosing spondylitis (AS) and inflammatory bowel disease (IBD) (10-12). In particular, the CARD9-S12N variant is strongly associated with both AS and IBD in various cohorts (13, 14). Xu et al. showed that the S12N mutation leads to polarization of avelolar macrophages into interleukin-5 (IL-5)–producing cells which facilitates TH-2 immune response in an aspergillosis lung infection model (15). CARD9-S12N promotes increased activation of the non-canonical NF-κB factor RelB to drive IL-5 production, which was also seen in peripheral blood monocytes (PBMs) from patients with homozygous S12N mutations in CARD9 and chronic bronchopulmonary aspergillosis. PBMs from CARD9-S12N patients have no defect in tumor necrosis factor-alpha (TNF-α), IL-1β, IL-6, IL-8, or interferon-gamma (IFNγ) production in response to exposure to Candida albicans conidia, potentially explaining why these individuals are not susceptible to Candida infections (16). The CARD9Δ11 variant is associated with protection against IBD in humans (17). The CARD9Δ11 variant produces a protein that is truncated at the C-terminus that functions as a dominant-negative mutation resulting in decreased NF-κB activation in mouse DCs (18). A pharmacological molecule mimicking CARD9Δ11 also inhibits NF-κB activity, suggesting that the inhibition of CARD9 can be a potential strategy to reduce IBD severity by blocking NF-κB activity (19). These studies suggest that increased CARD9 signaling drives NF-κB activation to promote inflammation. Consistent with this, Card9−/− mice show reduced inflammatory responses to a high-fat diet (20). Contrary to these studies, Sokol et al. reported that deleting Card9 exacerbates inflammation in the DSS-experimental mouse model of colitis by altering the microbiome metabolism of tryptophan (21). Conflicting results have been reported on tumor formation in the azoxymethane (AOM)-DSS model in Card9−/− mice (22, 23). Clearly, further investigation into the role of CARD9 in IBD development and other inflammatory-related diseases is needed to interpret these results.
Lyn kinase, a member of the Src-family of tyrosine kinases, plays an intricate role in both the activation and inhibition of the immune system. In vitro, deletion of Lyn leads to hyper-activation of TLR-triggered signaling in DCs, which results in spontaneous development of autoimmune disease with characteristics similar to human systemic lupus erythematosus (24). The reproducible development of autoantibody and hyper-inflammation reported by many laboratories has established Lyn−/− (or Lyn KO) mice as a reliable in vivo model for immune-driven autoimmunity (25, 26). In addition to the autoimmune phenotype, the hyper-immune activity in Lyn KO mice leads to intensified intestinal disease in a DSS-colitis mouse model (27, 28). Thus, the Lyn KO mouse is a model of both immune-driven autoimmunity and inflammatory bowel disease.
In this study, we tested the hypothesis that CARD9 is a critical downstream signaling component linking Lyn kinase to the TLR2 and TLR4 pathways. Deletion of CARD9 reversed the increase in TLR-triggered cytokine induction in Lyn-deficient DCs and protected Lyn−/− mice from the development of spontaneous autoimmunity and experimental colitis. Thus, inhibition of CARD9 activity may protect against immune-driven development of autoimmunity and colitis by lowering TLR-related signaling and cytokine production.
Results
CARD9 promotes colitis development in Lyn−/− mice
To elucidate the effects of CARD9 in an immune-driven IBD model, we used a 3.5% DSS model to induce acute colitis in WT or Lyn-deficient mice. We observed that Lyn−/− mice exhibited more severe disease than WT mice as demonstrated by greater weight loss, while the Lyn−/−;Card9−/− mice exhibited an amount of weight loss that was comparable to WT (Fig. 1A). Similarly, Lyn−/−;Card9−/− mice showed significantly less rectal bleeding and prolapse compared to Lyn−/− mice (Fig. 1B), as well as significantly longer colon length after DSS treatment (Fig. 1C). Furthermore, Lyn−/−;Card9−/− mice showed dramatically improved survival compared to the Lyn−/− mice (Fig. 1D). The Card9−/− mice exhibited phenotypes comparable to WT controls but trended towards shorter colon length after 2.5% DSS treatment compared to WT controls.
Fig. 1. CARD9 promotes colitis in Lyn−/− mice.

(A) The indicated mice were given drinking water treated with 3.5% DSS for days 0-5, followed by untreated drinking water for the remainder of the experiment. The weights of the animals were monitored daily and are plotted as a % of starting weight at day 0. (B) Bleeding/prolapse score of the indicated mice after DSS treatment described in (A) was analyzed and reported with the scale of 0, 0.5, 1.0, 1.5, 2.0 for increasing severity. (C) Colon lengths of the indicated mice after the indicated DSS treatment. (D) The survival of mice following DSS treatment described in (A) was assessed daily and plotted as % survival. (E and F) The indicated mice described in (A) were monitored for spontaneous rectal prolapse and the ages of prolapse development. * p<0.05, **p<0.01; by one-way ANOVA with Tukey HSD test for (A, B, C, and F), Z test for proportions (D and E). Each group represents n of 5-10 mice at 8-10 weeks of age and 17-19 grams at day 0. Data are representative of at least 3 independent experiments.
Mice deficient in IL-10 provide a well established model of immune-driven spontaneous colitis (29). By generating Lyn−/−;Il10−/− mice on the C57BL/6 background, we showed that the deletion of Lyn could drive spontaneous rectal prolapse after 10 weeks when IL-10 was also deleted (Fig. 1, E and F). This is consistent with the hyper-inflammatory environment previously described in Lyn−/−;Il10−/− double-KO mice (30). Mice deficient in IL-10 alone did not develop spontaneous colitis because Il10−/− mice on the C57BL/6 background are less susceptible to spontaneous colitis development compared to BALB/c mice (31). Deletion of Card9 completely reversed rectal prolapse in Lyn−/−;Il10−/− mice. There was no rectal prolapse in the Lyn−/−;Il10−/−;Card9−/− triple-KO, WT, or single-KO Il10−/− or Lyn−/− animals during 20 weeks of monitoring. Together, these data demonstrated that Card9 was crucial for the development of immune-driven colitis in Lyn−/− mice.
CARD9 plays a major role in the development of autoimmunity in Lyn−/− mice
CARD9 has been recently implicated in the development of human autoimmune-related diseases by genetic studies (10) and in mice in an auto-antibody-induced autoimmune mouse model (32). Lyn-deficient mice spontaneously develop auto-antibodies and glomerulonephritis (26, 33), making them a suitable model to investigate the role of CARD9 in the development of autoimmunity. By comparing aged WT, Lyn−/−, Lyn−/−;Card9−/−, and Card9−/− mice, we found that IgG and IgM serum autoantibody production against single-strand RNA (ssRNA) (Fig. 2, A and B) and double-strand DNA (dsDNA) (Fig. 2, C and D) were significantly higher in the Lyn−/− mice compared to WT mice, while both Lyn−/−;Card9−/− and Card9−/− animals had significantly lower serum autoantibody levels that were similar to WT mice. Increased serum autoantibody production in Lyn−/− mice is known (24, 34), but data indicating that CARD9 is critical to the development of autoantibody production in the absence of Lyn has not been previously reported. Additionally, hematoxilyn and eosin (H&E) staining of kidneys from 3.5-month-old Lyn−/− mice showed development of glomerulonephritis, whereas age-matched Lyn−/−;Card9−/− and Card9−/− mice did not develop this phenotype (Fig. 2E). Together, these data suggest that CARD9 plays a major role in the development of autoimmunity in Lyn−/− mice.
Fig. 2. CARD9 plays a major role in the development of autoimmunity in Lyn−/− mice.

(A-D) Serum was collected from the indicated mice at the indicated age. The amount of anti-ssRNA or anti-dsDNA antibodies in the serum was analyzed by ELISA. * p<0.05, **p<0.01, one way ANOVA with Tukey HSD test, each bar represents an n of 5-10 mice per group. Results are representative of at least 2 independent experiments. (E) H&E-stained kidney sections from the indicated mice at 3.5 months of age. Scale bars: upper panels = 10 μm, lower panels = 1 μm.
CARD9 mediates cellular activation in Lyn-deficient mice
Because we found CARD9 to be a major component of Lyn-mediated inflammatory and autoimmune disease, we investigated the in vivo immunological mechanism linking CARD9 to disease development. By analyzing the spleens of two month old mice, we found that Lyn-deficient splenic DCs produced significantly more TNF-α (Fig. 3A) and IL-6 (Fig. 3B) during TLR2 or TLR4 stimulation. This was not observed in the double-deficient Lyn−/−;Card9−/− or in Card9−/− splenic DCs. We found that the number of total splenic CD4+ and CD8+ T-cells remained similar across the different groups of mice (Fig. 3C) but Lyn−/− mice exhibited increased numbers of activated CD4+ T cells compared to WT, Lyn−/−;Card9−/− and Card9−/− mice (Fig. 3D). The number of activated CD8+ T cells remained similar across the different groups (Fig. 3D). CARD9 is only expressed in myeloid cells and not in lymphoid cells (Immgen database); therefore, these data suggested that intact CARD9 in myeloid cells leads to increased activation of CD4+ T cells, which likely contributed to the Lyn-deletion-mediated autoimmune development. Besides a difference in activated T cells, we also observed an increase in the total number of plasma cells in the Lyn−/− mice compared to WT, Lyn−/−;Card9−/− and Card9−/− animals (Fig. 3E). This suggested that CARD9 in myeloid cells leads to an increase in the number of plasma cells which was likely responsible for the elevated production of autoantibodies detected in the Lyn−/− mice.
Fig. 3. CARD9 is required for cellular activation in Lyn deficient mice.

(A and B) CD11c+ DCs were isolated from the spleen of the indicated mice by negative selection, plated and stimulated with 100 ng/ml Pam3CSK4 or 100 ng/ml LPS for the indicated times. Amount of TNF-α (A) or IL-6 (B) in the supernatants were measured by ELISA. (C to E) Cells were harvested from spleens of indicated mice at 2 months of age, stained with fluorescently labeled antibodies and analyzed by flow cytometry using the gating strategy shown in fig. S7. * p<0.05, **p<0.01, one way ANOVA with Tukey HSD test, each bar represents an n of 5-10 mice per group or cells pooled from 3 to 5 mice per group. Results were representative of at least 3 independent cohorts of mice.
CARD9 promotes increased TLR-triggered signaling and cytokine production in Lyn-deficient bone-marrow DCs, but not macrophages.
Myd88 in DCs is required for autoimmune development in Lyn-deficient mice, indicating that intact TLR signaling is necessary for autoimmunity (24). Because our in vivo data showed that CARD9 is also vital for autoimmune development in Lyn-deficient mice, we examined the role of CARD9 in TLR2 and TLR4 signaling in both bone marrow-derived dendritic cells (BMDCs) and bone marrow-derived macrophages (BMMs). We observed that Lyn-deficient BMDCs exhibited increased TNF-α and IL-6 production when stimulated by the TLR2 agonist Pam3CSK4 or the TLR4 agonist LPS (Fig. 4, A and B), consistent with previous reports (24, 35). This difference in cytokine production was not observed in bone marrow-derived macrophages (Fig. 4C). Additional deletion of Card9 in the Lyn−/− BMDCs resulted in TNF-α and IL-6 levels similar to those seen in WT BMDCs (Fig. 4, A and B). We investigated the signaling pathways downstream of TLR2 stimulation and found that Lyn−/− BMDCs showed increased phosphorylation of Syk, IKKα, ERK and p38 compared to WT cells (Fig. 4D and fig. S1), whereas double mutant Lyn−/−;Card9−/− BMDCs displayed WT levels of phosphorylation, except for Syk, which is upstream of Card9 (Fig. 4D). Consistent with cytokine production, Lyn−/− BMMs showed no increase in signaling after TLR2 stimulation (Fig. 4E). Because PKCδ is directly upstream of CARD9, and phosphorylates CARD9 (on serine and threonine residues) to activate CARD9 signaling in the C-type lectin pathways (5), we examined PKCδ phosphorylation. PKCδ was also hyper-phosphorylated in Lyn−/− BMDCs (Fig. 4F), but not BMMs (Fig. 4E), upon TLR2 stimulation. Hyper-phosphorylation of PKCδ was not reversed in Lyn−/−;Card9−/− double mutant BMDCs (fig. S2) . Consistent with hyper-activation of the upstream kinases (Syk and PKCδ), Lyn−/− BMDCs showed increased threonine phosphorylation of CARD9 after both TLR2 and TLR4 stimulation (Fig. 4G) which has been shown previously as a marker of the activation of signaling through CARD9 (5). These data demonstrate that Lyn deficiency resulted in hyper-activation of a Syk-PKCδ-CARD9 signaling pathway that increased signaling to MAPKs and NFκB, leading to increased TLR-stimulated cytokine production. This pathway was active in BMDCs but not BMMs. Deletion of CARD9 blocked downstream hyper-activation of IKKα, ERK1/2, and p38, but not the upstream Syk and PKCδ kinases, and this was sufficient to reverse the increased cytokine production in Lyn−/− BMDCs.
Fig. 4. CARD9 is required for the increased TLR-triggered signaling and cytokine production in Lyn-deficient BMDCs but not macrophages.

(A to C) BMDCs or BMMs from the indicated mice were stimulated with the indicated amount of Pam3CSK4 and LPS for 3 hours. Cell supernatants were analyzed for TNF-α and IL-6 by ELISA. (D to G) BMDCs or BMMs from the indicated mice were stimulated with 100 ng/ml Pam3CSK4 for the indicated time (panel G is from the 30 min time point). Cells were lysed and immunoblotted with the indicated antibodies. Densitometry analysis of the western blots is shown in fig. S1. Each western blot is representative of at least 3 independent experiments with cells pooled from 3 to 5 mice per group. * p<0.05, one way ANOVA with Tukey HSD test.
To determine whether Lyn deficiency resulted in non-specific elevation of all signaling pathways, we examined Dectin-1 signaling in Lyn−/− BMDCs. In contrast to TLR activation, Lyn−/− BMDCs showed reduced cytokine production and intracellular signaling after β-glucan stimulation (fig. S3, A and B). Similar results are reported in Lyn−/− BMMs (36). We have previously shown that Lyn-deficient BMDCs have wild type levels of other SFKs and no major alteration of surface integrins (CD11b and CD11c), FcγRs, MHCI or inhibitory receptors (PIR-B and Sirpα) (24). We also found no changes in protein levels of the signaling proteins in the CARD9 pathway (fig. S3C) nor in the counter regulatory phosphatase SHP1 in Lyn−/− BMDCs (fig. S3D).
To further test whether increased signaling through Syk and PKCδ lead to the increased cytokine production in Lyn−/− BMDCs following TLR stimulation, we used shRNA to knockdown expression of these upstream kinases in WT and Lyn−/− BMDCs. Knockdown of neither Syk nor PKCδ had any effect on WT cells, but reversed TNF-α hyper-production in Lyn−/− BMDCs following TLR2 stimulation (fig. S4).
Regulation of TLR-triggered signaling and cytokine production by Lyn is mediated by CD11b and SFKs Hck and Fgr.
The Src-family kinases Hck and Fgr are required for integrin signaling in myeloid leukocytes (37, 38), whereas Lyn is a known negative regulator of integrin outside-in signaling (39). However, SFKs are not required for TLR signaling in BMMs (40). More recently, integrin CD11b was implicated as a positive regulator of TLR signaling in DCs but not in macrophages (41). To investigate whether the influence of Lyn on CARD9-mediated TLR signaling occurred downstream of CD11b engagement, we compared signaling and cytokine production in BMDCs either in suspension or following adhesion. SFKs were phosphorylated upon TLR2-triggered signaling in attached WT BMDCs (Fig. 5A). This phosphorylation was significantly reduced in Lyn−/− cells and in CD11b-deficient BMDCs but not BMMs (Fig. 5, A to C). The data suggest that CD11b was required for TLR-stimulated SFK phosphorylation in BMDCs. Furthermore, the increased TLR-stimulated TNF-α production observed in Lyn-deficient BMDCs, relative to WT cells, only occurred when CD11b was engaged either by cellular attachment to the tissue culture wells or by antibody ligation (Fig. 5D). These data demonstrate that the increased cytokine production in Lyn-deficient BMDCs following TLR stimulation is dependent on a CD11b triggered pathway. In SFK triple-KO (Hck−/−;Fgr−/−;Lyn−/−) BMDCs, where signaling through CD11b is blocked, TLR stimulated TNF-α and IL-6 production is reduced compared to WT (Fig. 5, E and F), in contrast to what occurs in BMMs (40). Likewise, TLR activation of Syk and PKCδ downstream of TLR stimulation is lost in adherent SFK triple knockout BMDCs (fig. S5). As a result, downstream phosphorylation of CARD9 was also reduced in SFK triple-knockout BMDCs (Fig. 5G). Moreover, SFK triple-mutant mice were protected in the DSS colitis model (Fig. 5H), suggesting that reduced activation of DCs in vivo may be protective in this IBD model. These data suggest that Lyn kinase negatively regulates CD11b signaling in DCs in a pathway involving SFKs, Syk, and CARD9 to reduce TLR signaling.
Fig. 5. Regulation of TLR-triggered signaling and cytokine production by Lyn requires CD11b and Src-family kinases Hck and Fgr.
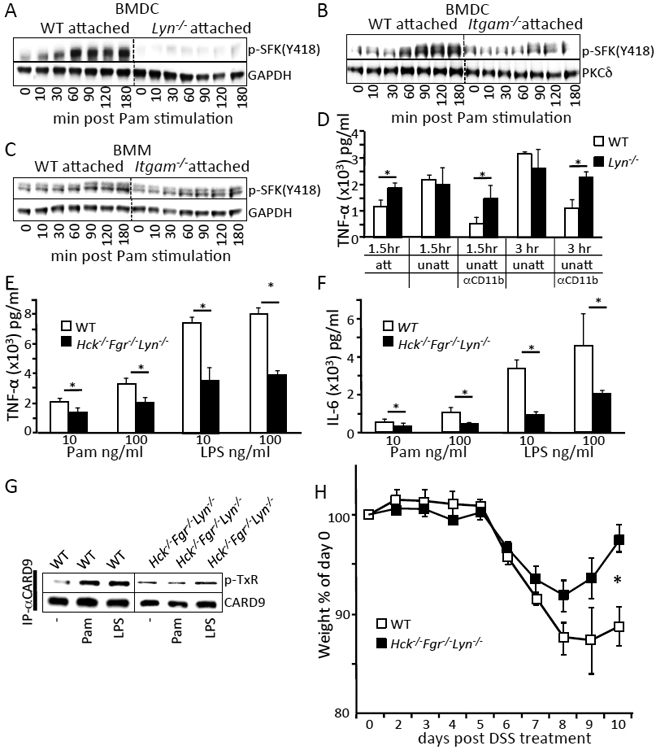
(A to C) BMDCs or BMMs from the indicated mice were stimulated with 100 ng/ml Pam3CSK4 for the indicated time. Cells were lysed and immunoblotted with the indicated antibodies. (D) BMDCs from the indicated mice were either centrifuged onto the bottom of tissue culture treated plates (att) or left in suspension (unatt) with or without the addition of antibody to CD11b (Mac-1) for 30 mins (αCD11b). Cells were stimulated with 100 ng/ml of Pam3CSK4 for the indicated times and supernatants were analyzed for TNF-α by ELISA. (E and F) BMDCs from the indicated mice were stimulated with the indicated amount of Pam3CSK4 and LPS for 3 hours. Cell supernatants were analyzed for TNF-α by ELISA. (G) BMDCs from the indicated mice were left untreated or stimulated with 100 ng/ml Pam3CSK4, 100 ng/ml LPS for 30 mins. Cells were lysed and immunoprecipitated with CARD9 antibody, followed by immunoblotting with phospho-threonine (P-TxR) antibody or CARD9 antibody. Densitometry analysis of the western blots are shown in fig. S1. Blots are representative of at least 3 independent experiments with cells pooled from 3 to 5 mice per group. (H) DSS-induced weight loss was monitored and analyzed as in Fig 1A. * p<0.05, one way ANOVA with Tukey HSD test for (D-F) or Student’s t-test (H).
CARD9 expression in DCs is critical for development of autoimmunity and colitis seen in DC-specific, Lyn-deficient mice.
We showed that global deletion of CARD9 can reverse autoimmune disease and intestinal inflammatory disease phenotypes in Lyn-deficient mice. We also found that CARD9 is a critical component of TLR-triggered hyperactivity in Lyn-deficient DCs and not macrophages. As we previously found, specific deletion of Lyn in DCs was sufficient to drive autoimmunity, as assessed by increased levels of serum antibodies against ssRNA and dsDNA, as well as high levels of splenic-activated CD4+ T cells and plasma cells, in 6-month-old mice (Fig. 6, A to F) (24). To investigate whether DC-specific Card9 expression is critical for the inflammatory diseases in Lyn−/− mice, we generated and compared single and double floxed Lyn and Card9 mice, crossed on to the Cd11c-Cre strain, which results in deletion primarily in convential and plasmacytoid DCs (42). Deletion of DC-specific Card9 resulted in complete reversal of spontaneous autoimmunity and reduced accumulation of splenic plasma cells observed in the Lynf/f;Cd11c-Cre animals(Fig. 6, A to F), and a trend towards reduction in splenic activated CD4+ T cells.
Fig.6. CARD9 expression in DCs is critical for development of autoimmunity and colitis in DC-specific lyn deficient mice.

(A to D) Serum was collected from the indicated mice at 6 months of age. The amount of antibody to ssRNA or dsDNA in the serum was analyzed by ELISA. (E and F) Cells were harvested from spleens of indicated mice at 3 months of age, stained with fluorescently labeled antibodies and analyzed by flow cytometry using the gating strategy shown in fig. S7. * p<0.05, **p<0.01, one way ANOVA with Tukey HSD test, each bar represents an n of 5-10 mice per group. Results are representative of at least 2 independent experiments.
To examine if the Lyn to CARD9 pathway in DCs also played a role in the colitis model, we exposed the Lynf/f;Cd11c-Cre and the Lynf/f;Card9f/f;Cd11c-Cre to 3.5% DSS. Deletion of Lyn in DCs alone resulted in a slight but significant weight loss and survival disadvantage when compared to control Lynf/f mice alone (fig. S6, A and B). This difference was not observed in the DC Lyn/Card9 double-deficient mice compared to the double flox controls (fig. S6, C and D). Together, these data indicate that the Lyn-to-CARD9 signaling pathway in DCs regulates both the spontaneous development of autoimmunity and the susceptibility to intestinal inflammatory disease.
Discussion
In this study, we report that CARD9 is a crucial link between the tyrosine kinase-driven CD11b integrin pathway and the TLR pathway in DCs. In the absence Lyn, CD11b signaling was increased which, through CARD9, amplified responses downstream of TLR2 and TLR4, leading to increased cytokine production by Lyn−/− DCs. Deletion of CARD9 in DCs alone reversed the development of autoimmunity and colitis in DC-specific Lyn-deficient mice. In conclusion, we have uncovered a previously unknown mechanism used by DCs to regulate the magnitude of TLR-mediated signaling using the adaptor protein CARD9.
We found that negative regulation of TLR signaling by Lyn is only detectable in splenic DCs and BMDCs but not in bone marrow-derived macrophages. This correlates with our observation that deletion of Lyn specifically in DCs (using CD11c-cre) but not in macrophages (using LysM-cre) resulted in inflammatory autoimmunity (24). Ling et al. recently reported that CD11b clustering only occurs during TLR4 activation in DCs but not in macrophages, which could explain a differential role for CD11b in DCs versus macrophages (41). CD11b also plays a role in Lyn activation in B cells during BCR stimulation (43). In the absence of CD11b, Lyn is poorly activated, resulting in loss of inhibitory signaling leading to hyper-reactive responses following BCR stimulation, which causes autoantibody production and Ig deposition in the kidneys in vivo. This observation is similar to our results observed in DCs lacking Lyn. These findings suggest that CD11b-dependent activation of Lyn kinase leading to the initiation of inhibitory signaling occurs in multiple immune cells. Further studies are required to elucidate the differential regulation of TLR signaling by Lyn kinase in DCs versus macrophages.
Our findings here demonstrated that Lyn possesses both activating (during Dectin-1 stimulation) and inhibitory (during CD11b engagement and TLR stimulation) functions in DCs. The molecular mechanism for this differential effect remains to be defined. We hypothesize that during Dectin-1 stimulation, the formation of the “phagocytic synapse” physically removes inhibitory receptors away from the activating signals therefore eliminating Lyn’s ability to phosphorylate ITIMs to initiate inhibitory responses. In this environment, Lyn’s positive regulatory role becomes dominant. Perhaps CD11b engagement during TLR stimulation does not segregate inhibitory receptors away from the activation signals, hence the activated Lyn kinase could have access to ITIM-containing inhibitory receptors, leading to enhanced recruitment of phosphatases (mainly SHP1 and SHIP1) that inhibit downstream pathways (25). In neutrophils, the main inhibitory receptors phosphorylated by Lyn to regulate integrin signaling are SIRP-α and PIR-B (39). Both SIRP-α and PIR-B have been shown to negatively regulate of TLR signaling, primarily by using knockdown approaches in cultured cells (44, 45). Both of these ITIM-containing receptors are known to recruit the phosphatase SHP1; deletion of SHP1 also leads to exaggerated TLR responses in various cell types (46, 47). Additionally, genetic deletion of SHP1 specifically in DCs leads to exaggerated TLR responses and the development of autoimmunity, mimicking the disease that develops in mice lacking Lyn kinase in DCs (48). DCs contain a large spectrum of ITIM-containing receptors that could co-aggregate with CD11b to become phosphorylated by Lyn and hence recruit SHP1 or other inhibitory phosphatases. Defining which DC receptors and downstream phosphatases are involved in the negative regulation of TLR signaling remains a future goal.
We showed that in the absence of Lyn, Hck and Fgr are necessary for the enhanced signaling in DCs in vitro and for the exaggerated gastrointestinal inflammation in the DSS colitis model. These data suggest that Lyn inhibits signaling by preventing Hck and Fgr from activating the Syk-PKCδ-CARD9 pathway during TLR stimulation. These observations suggest that Lyn has a higher affinity for phosphorylating ITIMs, whereas Hck and Fgr have higher affinity for phosphorylating ITAMs. Further investigation is needed to elucidate how Lyn interacts with Hck and Fgr and why they have different affinity for phosphorylating different targets.
Given our findings that loss of Lyn led to increased signaling through CARD9, which ultimately resulted in the development of autoimmunity, we hypothesize that mutations in CARD9 that could enhance activation of the NF-κB and MAPK pathways would also lead to the development of autoimmune and inflammatory disease. Indeed, Xu et al. found that the S12N mutation in Card9 leads to increased IL-5 and TH2 cell activity in mouse alveolar macrophages through increased activation of the non-canonical NF-κB factor RelB (15). Whether this mutation would also increase susceptibility to colitis in vivo remains to be determined. Given that dominant-negative mutations in CARD9 or agents that block formation of the CARD9-Malt1-Bcl10 complex result in reduced inflammatory responses, perhaps blocking CARD9 function would reduce the tyrosine kinase-to-TLR pathway crosstalk and potentially limit inflammatory responses (18, 19). This is consistent with our findings that deletion of CARD9 reverses the hyper-inflammatory state in Lyn−/− mice. This raises the potential that inhibition of CARD9 signaling, or of other members of the pathway for regulating TLR-mediated responses (including CD11b, Lyn, Hck, Fgr, Syk and PKCδ) may be potential treatment options for patients with IBD and AS who have polymorphisms in CARD9.
Materials and Methods
Mice
Lyn−/−, Lynf/f,Hck−/−Fgr−/−Lyn−/−, Il10−/− and CD11c-Cre mice were generated and described previously (24, 30, 33, 40), Card9−/− mice were a generous gift from David Underhill (Cedars-Sinai Medical Center, Los Angeles) and described previously (7). Card9f/f mice were a generous gift from Attila Mocsai (Semelweis University, Budapest) and described previously (32). Itgamtm1Myd (Itgam−/− or CD11b-deficient) mice were generous gifts from Katerina Akassoglou (UCSF). All mice were on the C57BL/6 background and kept in a specific pathogen-free facility at USCF. All animal experiments were performed in compliance with the Animal Welfare Act and Regulations, the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the Public Health Service Policy on the Humane Care and Use of Laboratory Animals and UCSF polies and guidelines.
Reagents
All antibodies used for biochemical analysis were purchased from Cell Signaling Technology except for α-GAPDH (Calbiochem) and α-PKCδ (BD Biosciences). All HRP-tagged secondary antibodies were purchased from Cell Signaling Technology. All antibodies used for flow cytometry were purchased from BD Biosciences or eBiosciences. Reagents used to stimulate cells were from the following sources: LPS from E.coli serotype O111:B4 (Alexis Biochemicals), Pam3CSK4 (Invivogen), biotinylated α-CD11b (clone M1/70, BD Biosciences), zymosan (Sigma-Aldrich). β-glucan were prepared from zymosan as described previously (4).
Cell culture
Bone marrow was flushed from the femurs and tibias of mice, and mononuclear cells were purified on a 68% Percoll (GE Healthcare Life Sciences) gradient as previously described (24). BMDCs were cultured in complete RMPI (Corning) (10% FBS (Gibco), 2 mM L-glutamine, 2mM nonessential amino acids, 2mM sodium pyruvate, 2mM HEPES (UCSF Cell Culture Facility) 50 μM 2-mercaptoethanol (Gibco) supplemented with 20 ng/ml recombinant mouse GM-CSF (PeproTech). BMMs were cultured similarly but supplemented with M-CSF from CMG conditioned media derived from a 3T3 cell line engineered to overexpress mouse M-CSF. Cells were used after 7-9 days in culture.
Cytokine measurement
BMDCs in suspension were counted (NucleoCounter NC-200) and centrifuged onto the bottom of 96 well tissue culture plates, or aliquoted into 1.5ml centrifuge tubes for experiments using non-adherent cells. BMMs were removed from plates using 2mM EDTA in PBS and plated as above. Cells were immediately stimulated with the indicated reagents for the indicated times. Supernatants were transferred into new 96 well plates and analyzed for TNF-α production by ELISA (eBioscience). All BMDCs experiments were performed in RPMI without GM-CSF.
Immunoblotting
Cells were counted and seeded as above. At the end of the stimulation, supernatants were removed and cells were directly lysed in NuPAGE LDS sample buffer (ThermoFisher Scientific) supplemented with 2-ME. Lysates were boiled at 95°C for 5 mins and ran on 4-12% Bis-Tris gels (ThermoFisher Scientific). Proteins were transferred onto PVDF membranes (Millipore) using a semi-dry transfer method. Membranes were blocked overnight in 4% BSA-TBS-T (Millipore) in 4°C and probed using the indicated antibodies. Membranes were incubated with SuperSignal chemiluminescent substrate (ThermoFisher Scientific) for 5 minutes and imaged using the Chemi-Doc Touch System (Bio-Rad).
Immunoprecipitation
Cells were counted and seeded as above. At the end of the stimulation, supernatants were removed and cells were washed with ice cold PBS. Cells were then lysed in NP-40 lysis buffer (1% NP40, 150mM NaCl, 20mM Hepes, 2mM EDTA) at 4°C. Lysates were cleared by centrifugation at 16,000xg for 10 mins. Supernatants were then incubated with antibody overnight at 4°C and then incubated with of Protein A/G plus agarose beads (Santa Cruz Biotech) for 2 hours at 4°C. Beads were washed with lysis buffer, then boiled in 1X NuPAGE LDS sample buffer and analyzed by immunoblotting.
ShRNA Knockdowns
ShRNAs targeting pkcd (TRCN0000280560) and targeting syk (TRCN0000023571) cloned into the pLKO.1 lentiviral vector (Sigma-Aldrich-mission shRNA) were transfected into LentiX 293T packaging cells. BMDCs were infected with the lentivirus expressing the shRNA for 24 hours (at day 4 of culturing). Cells were allowed to recover and selected by puromycin until day 7. The knockdown efficiency was analyzed by immunoblotting.
Splenic DC isolation and flow cytometry
Spleens were obtained from the indicated mice and mashed by using frosted microscope slides in Hanks’BSS (UCSF Cell Culture Facility) media supplemented with HEPES and 5mM EDTA (Fisher Scientific). Single Cell suspensions were prepared and counted. For flow cytometry, cells were blocked with anti-CD16/32 (clone 2.4G2) and mouse IgG (Sigma-Aldrich) for 10 mins, and incubated with the indicated antibodies for 1 hr. Data was collected on a Fortessa flow cytometer (Becton Dickinson) at the UCSF Flow Cytometry Core. FACS data was analyzed using FlowJo (FLowJo LLC). Gating strategies for the various immune cell types is shown in fig. S8. Splenic DCs were isolated using the MACS pan-DC negative selection kit (Miltenyi Biotech). CD11c+ cell purity following splenic DC selection was determined by flow cytometry. Splenic DCs were seeded and stimulated immediately in complete RPMI as above. Supernatants were analyzed for cytokine production by ELISA.
Histology and auto-antibody assessment of autoimmunity
Kidneys of the indicated mice were formalin fixed and stained with H&E by the UCSF Pathology Core. The representative photos were taken using a Nikon Eclipse 80i microscope at 100X magnification. Serum levels of anti-dsDNA IgG, IgM and anti-ssRNA IgG, IgM were determined by ELISA as previously described (24, 34).
DSS colitis
Female mice with similar weights and age at day 0 (17-19g) were given 3.5% or 2.5% dextran sulfate sodium (DSS) containing drinking water for 5 days, followed by untreated water for the remainder of the experiment. Animal weight and rectal bleeding/prolapse were measured daily. Mice that fell below 70% of their original weight and demonstrated limited mobility were sacrificed and counted as dead (IACUC guidelines (49)). Remaining mice were sacrificed at day 10 post DSS treatment or followed for survival. The colon lengths of mice were measured either when mice were found dead or sacrificed.
Quantification of immunoblots
Densitometry Units of each loaded immunoblot wells were measured using ImageJ. Densitometry units of the phosphorylated bands were normalized to its total protein control. Representative blots of three different experiments and the quantification of multiple blots is shown.
Statistical analysis
P value < 0.05 was considered statistically significant. All statistic tests used are indicated in the figure legends. Each in vitro and in vivo experiment was repeated at least three times and the representative results were reported. Data reported on aged mouse models for autoimmunity were representative of three individual cohorts of mice.
Supplementary Material
Figure S1. Densitometry quantification of immunoblots.
Figure S2. PKCδ phosphorylation in Lyn−/− and Lyn−/−;Card9−/− BMDCs.
Figure S3. Dectin-1 signaling in Lyn−/− BMMs and BMDCs.
Figure S4. Knockdown of Syk and PKCδ in Lyn−/− BMDCs.
Figure S5. TLR2 and TLR4 signaling in WT versus Hck−/−;Fgr−/−;Lyn−/− BMDCs.
Figure S6. DSS colitis development in DC-specific Lyn- and Lyn;CARD9-deficient mice.
Figure S7. Flow cytometry gating strategy.
Acknowledgements:
We thank A. DeFranco (UCSF) for advice throughout this project.
Funding: This project was supported by the NIH-NIAID (RO1AI113272 to C.L.). J.M. was supported by a post-doctoral fellowship from the Crohn’s and Colitis Foundation (ref #515747).
Footnotes
Competing interests: All authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes:
- 1.Colonna M, All roads lead to CARD9. Nat. Immunol 8, 554–555 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Drummond RA, Lionakis MS, Mechanistic insights into the role of C-type lectin receptor/CARD9 signaling in human antifungal immunity. Front. Cell. Infect. Microbiol 6, 39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodridge HS, Shimada T, Wolf AJ, Hsu YM, Becker CA, Lin X, Underhill DM, Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J. Immunol 182, 1146–1154 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, Forster I, Ruland J, Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Strasser D, Neumann K, Bergmann H, Marakalala MJ, Guler R, Rojowska A, Hopfner KP, Brombacher F, Urlaub H, Baier G, Brown GD, Leitges M, Ruland J, Syk kinase-coupled C-type lectin receptors engage protein kinase C-σ to elicit Card9 adaptor-mediated innate immunity. Immunity 36, 32–42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, Iwakura Y, Ohno N, Koseki H, Yoshida H, Penninger JM, Saito T, The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol 8, 619–629 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, Lin X, The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol 8, 198–205 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Alves de Medeiros AK, Lodewick E, Bogaert DJ, Haerynck F, Van Daele S, Lambrecht B, Bosma S, Vanderdonckt L, Lortholary O, Migaud M, Casanova JL, Puel A, Lanternier F, Lambert J, Brochez L, Dullaers M, Chronic and invasive fungal infections in a family with CARD9 deficiency. J. Clin. Immunol. 36, 204–209 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, Jamal S, Manguiat A, Rezaei N, Amirzargar AA, Plebani A, Hannesschlager N, Gross O, Ruland J, Grimbacher B, A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med 361, 1727–1735 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma X, Liu Y, Zhang H, Qiu R, Zhao H, Xin Q, Shan S, Dang J, Li J, Yang Z, Gong Y, Liu Q, Evidence for genetic association of CARD9 and SNAPC4 with ankylosing spondylitis in a Chinese Han population. J. Rheumatol 41, 318–324 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Pointon JJ, Harvey D, Karaderi T, Appleton LH, Farrar C, Stone MA, Sturrock RD, Brown MA, Wordsworth BP, Elucidating the chromosome 9 association with AS; CARD9 is a candidate gene. Genes Immun. 11, 490–496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhernakova A, Festen EM, Franke L, Trynka G, van Diemen CC, Monsuur AJ, Bevova M, Nijmeijer RM, van ‘t Slot R, Heijmans R, Boezen HM, van Heel DA, van Bodegraven AA, Stokkers PC, Wijmenga C, Crusius JB, Weersma RK, Genetic analysis of innate immunity in Crohn's disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet 82, 1202–1210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Momenzadeh P, Mahmoudi M, Beigy M, Garshasbi M, Vodjdanian M, Farazmand A, Jamshidi AR, Determination of IL1 R2, ANTXR2, CARD9, and SNAPC4 single nucleotide polymorphisms in Iranian patients with ankylosing spondylitis. Rheumatol. Int 36, 429–435 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D, Wijmenga C, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Cohen A, Colombel JF, Cottone M, Stronati L, Denson T, De Vos M, D'Inca R, Dubinsky M, Edwards C, Florin T, Franchimont D, Gearry R, Glas J, Van Gossum A, Guthery SL, Halfvarson J, Verspaget HW, Hugot JP, Karban A, Laukens D, Lawrance I, Lemann M, Levine A, Libioulle C, Louis E, Mowat C, Newman W, Panes J, Phillips A, Proctor DD, Regueiro M, Russell R, Rutgeerts P, Sanderson J, Sans M, Seibold F, Steinhart AH, Stokkers PC, Torkvist L, Kullak-Ublick G, Wilson D, Walters T, Targan SR, Brant SR, Rioux JD, D'Amato M, Weersma RK, Kugathasan S, Griffiths AM, Mansfield JC, Vermeire S, Duerr RH, Silverberg MS, Satsangi J, Schreiber S, Cho JH, Annese V, Hakonarson H, Daly MJ, Parkes M, Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet 42, 1118–1125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu X, Xu JF, Zheng G, Lu HW, Duan JL, Rui W, Guan JH, Cheng LQ, Yang DD, Wang MC, Lv QZ, Li JX, Zhao X, Chen CX, Shi P, Jia XM, Lin X, CARD9(S12N) facilitates the production of IL-5 by alveolar macrophages for the induction of type 2 immune responses. Nat. Immunol 19, 547–560 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Rosentul DC, Plantinga TS, Oosting M, Scott WK, Velez Edwards DR, Smith PB, Alexander BD, Yang JC, Laird GM, Joosten LA, van der Meer JW, Perfect JR, Kullberg BJ, Netea MG, Johnson MD, Genetic variation in the dectin-1/CARD9 recognition pathway and susceptibility to candidemia. J. Infect. Dis 204, 1138–1145 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaudoin M, Goyette P, Boucher G, Lo KS, Rivas MA, Stevens C, Alikashani A, Ladouceur M, Ellinghaus D, Torkvist L, Goel G, Lagace C, Annese V, Bitton A, Begun J, Brant SR, Bresso F, Cho JH, Duerr RH, Halfvarson J, McGovern DP, Radford-Smith G, Schreiber S, Schumm PL, Sharma Y, Silverberg MS, Weersma RK, Quebec IBDGC, Consortium NIG, International IBDGC, D'Amato M, Vermeire S, Franke A, Lettre G, Xavier RJ, Daly MJ, Rioux JD, Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet 9, e1003723–1003730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao Z, Conway KL, Heath RJ, Rush JS, Leshchiner ES, Ramirez-Ortiz ZG, Nedelsky NB, Huang H, Ng A, Gardet A, Cheng SC, Shamji AF, Rioux JD, Wijmenga C, Netea MG, Means TK, Daly MJ, Xavier RJ, Ubiquitin ligase TRIM62 regulates CARD9-mediated anti-fungal immunity and intestinal inflammation. Immunity 43, 715–726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leshchiner ES, Rush JS, Durney MA, Cao Z, Dancik V, Chittick B, Wu H, Petrone A, Bittker JA, Phillips A, Perez JR, Shamji AF, Kaushik VK, Daly MJ, Graham DB, Schreiber SL, Xavier RJ, Small-molecule inhibitors directly target CARD9 and mimic its protective variant in inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 114, 11392–11397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao L, Qin X, Peterson MR, Haller SE, Wilson KA, Hu N, Lin X, Nair S, Ren J, He G, CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J. Mol. Cell. Cardiol 92, 185–195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, Bridonneau C, Jegou S, Hoffmann TW, Natividad JM, Brot L, Taleb S, Couturier-Maillard A, Nion-Larmurier I, Merabtene F, Seksik P, Bourrier A, Cosnes J, Ryffel B, Beaugerie L, Launay JM, Langella P, Xavier RJ, Sokol H, CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med 22, 598–605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergmann H, Roth S, Pechloff K, Kiss EA, Kuhn S, Heikenwalder M, Diefenbach A, Greten FR, Ruland J, Card9-dependent IL-1β regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur. J. Immunol 47, 1342–1353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T, Fan C, Yao A, Xu X, Zheng G, You Y, Jiang C, Zhao X, Hou Y, Hung MC, Lin X, The adaptor protein CARD9 protects against colon cancer by restricting mycobiota-mediated expansion of myeloid-derived suppressor cells. Immunity 49, 504–514 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA, Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc. Natl. Acad. Sci. USA 110, E3311–3320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scapini P, Pereira S, Zhang H, Lowell CA, Multiple roles of Lyn kinase in myeloid cell signaling and function. Immunol. Rev 228, 23–40 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodie EJ, Infantino S, Low MSY, Tarlinton DM, Lyn, Lupus, and (B) lymphocytes, a lesson on the critical balance of kinase signaling in immunity. Front. Immunol 9, 401 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts ME, Bishop JL, Fan X, Beer JL, Kum WW, Krebs DL, Huang M, Gill N, Priatel JJ, Finlay BB, Harder KW, Lyn deficiency leads to increased microbiota-dependent intestinal inflammation and susceptibility to enteric pathogens. J. Immunol 193, 5249–5263 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Bishop JL, Roberts ME, Beer JL, Huang M, Chehal MK, Fan X, Fouser LA, Ma HL, Bacani JT, Harder KW, Lyn activity protects mice from DSS colitis and regulates the production of IL-22 from innate lymphoid cells. Mucosal Immunol. 7, 405–416 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W, Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 (1993). [DOI] [PubMed] [Google Scholar]
- 30.Scapini P, Lamagna C, Hu Y, Lee K, Tang Q, DeFranco AL, Lowell CA, B cell-derived IL-10 suppresses inflammatory disease in Lyn-deficient mice. Proc. Natl. Acad. Sci. USA 108, E823–832 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ermann J, Garrett WS, Kuchroo J, Rourida K, Glickman JN, Bleich A, Glimcher LH, Severity of innate immune-mediated colitis is controlled by the cytokine deficiency-induced colitis susceptibility-1 (Cdcs1) locus. Proc. Natl. Acad. Sci. USA 108, 7137–7141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemeth T, Futosi K, Sitaru C, Ruland J, Mocsai A, Neutrophil-specific deletion of the CARD9 gene expression regulator suppresses autoantibody-induced inflammation in vivo. Nat. Commun 7, 11004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan VW, Meng F, Soriano P, DeFranco AL, Lowell CA, Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 7, 69–81 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Lamagna C, Hu Y, DeFranco AL, Lowell CA, B cell-specific loss of Lyn kinase leads to autoimmunity. J. Immunol 192, 919–928 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ban T, Sato GR, Nishiyama A, Akiyama A, Takasuna M, Umehara M, Suzuki S, Ichino M, Matsunaga S, Kimura A, Kimura Y, Yanai H, Miyashita S, Kuromitsu J, Tsukahara K, Yoshimatsu K, Endo I, Yamamoto T, Hirano H, Ryo A, Taniguchi T, Tamura T, Lyn kinase suppresses the transcriptional activity of IRF5 in the TLR-MyD88 pathway to restrain the development of autoimmunity. Immunity 45, 319–332 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Freedman TS, Tan YX, Skrzypczynska KM, Manz BN, Sjaastad FV, Goodridge HS, Lowell CA, Weiss A, LynA regulates an inflammation-sensitive signaling checkpoint in macrophages. Elife 4, e09183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abram CL, Lowell CA, The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol 27, 339–362 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowell CA, Fumagalli L, Berton G, Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J. Cell. Biol 133, 895–910 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pereira S, Lowell C, The Lyn tyrosine kinase negatively regulates neutrophil integrin signaling. J. Immunol 171, 1319–1327 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Meng F, Lowell CA, Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med 185, 1661–1670 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ling GS, Bennett J, Woollard KJ, Szajna M, Fossati-Jimack L, Taylor PR, Scott D, Franzoso G, Cook HT, Botto M, Integrin CD11b positively regulates TLR4-induced signalling pathways in dendritic cells but not in macrophages. Nat. Commun 5, 3039–3044 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abram CL, Roberge GL, Hu Y, Lowell CA, Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, Xiang D, Nath S, Zhang HG, Ye H, Powell D, Yan J, Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat. Commun 4, 2813–2816 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Kong XN, Yan HX, Chen L, Dong LW, Yang W, Liu Q, Yu LX, Huang DD, Liu SQ, Liu H, Wu MC, Wang HY, LPS-induced down-regulation of signal regulatory protein {alpha} contributes to innate immune activation in macrophages. J. Exp. Med 204, 2719–2731 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakayama M, Underhill DM, Petersen TW, Li B, Kitamura T, Takai T, Aderem A, Paired Ig-like receptors bind to bacteria and shape TLR-mediated cytokine production. J. Immunol 178, 4250–4259 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Hardin AO, Meals EA, Yi T, Knapp KM, English BK, SHP-1 inhibits LPS-mediated TNF and iNOS production in murine macrophages. Biochem. Biophys. Res. Commun 342, 547–555 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Ramachandran IR, Song W, Lapteva N, Seethammagari M, Slawin KM, Spencer DM, Levitt JM, The phosphatase SRC homology region 2 domain-containing phosphatase-1 is an intrinsic central regulator of dendritic cell function. J. Immunol 186, 3934–3945 (201). [DOI] [PubMed] [Google Scholar]
- 48.Abram CL, Roberge GL, Pao LI, Neel BG, Lowell CA, Distinct roles for neutrophils and dendritic cells in inflammation and autoimmunity in motheaten mice. Immunity 38, 489–501 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, Gewirtz AT, Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Densitometry quantification of immunoblots.
Figure S2. PKCδ phosphorylation in Lyn−/− and Lyn−/−;Card9−/− BMDCs.
Figure S3. Dectin-1 signaling in Lyn−/− BMMs and BMDCs.
Figure S4. Knockdown of Syk and PKCδ in Lyn−/− BMDCs.
Figure S5. TLR2 and TLR4 signaling in WT versus Hck−/−;Fgr−/−;Lyn−/− BMDCs.
Figure S6. DSS colitis development in DC-specific Lyn- and Lyn;CARD9-deficient mice.
Figure S7. Flow cytometry gating strategy.