

Abstract
BACKGROUND
Rheumatoid arthritis, like many inflammatory diseases, is characterized by episodes of quiescence and exacerbation (flares). The molecular events leading to flares are unknown.
METHODS
We established a clinical and technical protocol for repeated home collection of blood in patients with rheumatoid arthritis to allow for longitudinal RNA sequencing (RNA-seq). Specimens were obtained from 364 time points during eight flares over a period of 4 years in our index patient, as well as from 235 time points during flares in three additional patients. We identified transcripts that were differentially expressed before flares and compared these with data from synovial single-cell RNA-seq. Flow cytometry and sorted-blood-cell RNA-seq in additional patients were used to validate the findings.
RESULTS
Consistent changes were observed in blood transcriptional profiles 1 to 2 weeks before a rheumatoid arthritis flare. B-cell activation was followed by expansion of circulating CD45−CD31−PDPN+ preinflammatory mesenchymal, or PRIME, cells in the blood from patients with rheumatoid arthritis; these cells shared features of inflammatory synovial fibroblasts. Levels of circulating PRIME cells decreased during flares in all 4 patients, and flow cytometry and sorted-cell RNA-seq confirmed the presence of PRIME cells in 19 additional patients with rheumatoid arthritis.
CONCLUSIONS
Longitudinal genomic analysis of rheumatoid arthritis flares revealed PRIME cells in the blood during the period before a flare and suggested a model in which these cells become activated by B cells in the weeks before a flare and subsequently migrate out of the blood into the synovium. (Funded by the National Institutes of Health and others.)
RHEUMATOID ARTHRITIS SYMPTOMS ARE highly dynamic, with periods of stability interrupted by unpredictable flares of disease activity. Such waxing and waning clinical courses are characteristic of many autoimmune diseases, including multiple sclerosis,1 systemic lupus erythematosus,2 and inflammatory bowel disease,3,4 underscoring a need to develop approaches to understand the factors that trigger transitions from quiescence to autoimmune flare.
Microarray studies of blood specimens from sparse time-series data have identified few genes that are significantly associated with rheumatoid arthritis activity.5–8 To look for molecular changes in blood that anticipate clinical flares, we sought to optimize methods by which the patients themselves could obtain fingerstick blood specimens for RNA sequencing (RNA-seq), facilitating weekly blood sampling for periods of months to years.
We explored the pathophysiology of rheumatoid arthritis using longitudinal, prospective analysis of blood transcriptional profiles from individual patients over time. We analyzed patient reports of clinical disease activity and RNA-seq data from patients across multiple clinical flares. Collecting samples longitudinally enabled a search for transcriptional signatures that preceded clinical symptoms, and comparison of these blood RNA profiles with data from synovial single-cell RNA-seq (scRNA-seq)9 was used to determine whether biologically coherent sets of transcripts were identifiable in the blood before symptom onset and as patients began to have symptoms.
METHODS
PATIENTS
We enrolled patients who met American College of Rheumatology–European League Against Rheumatism 201010,11 criteria for having rheumatoid arthritis and who were seropositive for cyclized citrullinated protein antibody (CCP). Disease activity was assessed from home each week, or up to four times daily during escalation of flares, with the Routine Assessment of Patient Index Data 3 (RAPID3) questionnaire.12 Disease activity was also assessed at clinic visits, each month, and during flares with the use of both RAPID3 and the Disease Activity Score 28 (DAS28), which incorporates tenderness and swelling from 28 joints, erythrocyte sedimentation rate, and patient global assessment of disease activity. Complete blood counts, including counts of white cells, neutrophils, monocytes, lymphocytes, and platelets, were assessed by the Memorial Sloan Kettering Cancer Center clinical laboratory. We also used fluorescence-activated cell sorting to assess peripheral blood mononuclear cells (PBMCs) from 19 additional seropositive patients with rheumatoid arthritis (irrespective of disease activity) and 18 age- and sex-matched participants without rheumatoid arthritis.
The study was approved by the institutional review board of Rockefeller University. All the patients in the study provided written informed consent before participation. The authors vouch for the accuracy and completeness of the data presented in this report.
RNA PREPARATION FROM FINGERSTICK BLOOD SPECIMENS
Patients performed fingerstick collection of three drops of blood at home; the blood was placed into a microtainer tube prefilled with fixative, and the specimens were mailed overnight each week to Rockefeller University. RNA was extracted with the PAXgene RNA kit and was purified in accordance with the manufacturer’s protocols, with the exception of the volume of all washes and elutions being decreased to 25% of the volume recommended by the manufacturer. RNA was assessed with the Agilent BioAnalyzer for quantity and quality. For library preparation, we used the Globin-Zero kit (EpiCentre) and the Illumina Truseq mRNA Stranded Library kit, with 11 to 12 polymerase-chain-reaction (PCR) cycles for 5 to 8 nmol per liter input, and sequencing was performed on a HiSeq2500 system (Illumina) with 150-base paired-end reads. Reads were aligned to the human reference genome (hg19) with Gencode, version 18, as the reference gene annotation with the use of STAR software, version 2.3.1z,13 and were quantified with featureCounts software, version 1.5.0-p2.14 Samples with at least 4 million paired-end reads were retained for analysis.
COMPARISON OF DISEASE-ACTIVITY MEASURES
To describe the bivariate relationship between clinical assessments of disease activity and disease activity as assessed with RAPID3, we used the locally weighted scatterplot smoothing (LOWESS) technique. R2 values were calculated to assess correlations of complete blood counts inferred from CIBERSORTx and counts measured by clinical laboratories. Inferred CIBERSORTx lymphocyte counts were calculated as the sum of the counts of naive B cells, memory B cells, CD8 T cells, naive CD4 T cells, resting memory CD4 T cells, and activated memory CD4 T cells. The counts of monocytes, M0 macrophages, M1 macrophages, and M2 macrophages were summed to infer CIBERSORTx monocyte counts. One-way analysis of variance was used to test for significant differences among various clinical features according to disease-activity state.
ANALYSES OF DIFFERENTIAL EXPRESSION AMONG PATIENTS
Before gene-expression analysis, specimens were labeled as baseline (i.e., obtained during a period of stable RAPID3 scores), flare (obtained during a period in which the RAPID3 score was >2 SD above the baseline mean), or glucocorticoid (obtained during a period when the patient was taking glucocorticoids). We used edgeR software, version 3.24.3,15 to analyze differential gene expression in flare specimens as compared with baseline specimens. Permutation testing (1×106 samples) was used to test for significance in the overlap between the groups of genes that had decreased expression during flares in each of the four patients. Analysis of functional groups of genes, as defined by the Gene Ontology Consortium (with goana, from the limma software package, version 3.38.3),16 was used to identify enriched pathways among the genes that had significant differential expression during a flare relative to baseline in the index patient (those with a false discovery rate of <0.1) and that were consistent in the direction of expression in both the index patient and the three additional patients with rheumatoid arthritis who were included in the longitudinal study (i.e., log2 relative expression [flare:baseline ratio] either both positive or both negative).
TIME-SERIES ANALYSIS OF GENE EXPRESSION IN THE INDEX PATIENT
We performed an analysis of longitudinal data from the index patient, using ImpulseDE2 software, version 1.8.017 (see the Methods section in Supplementary Appendix 1, available with the full text of this article at NEJM.org). Specimens from 8 weeks before a flare to up to 4 weeks after the start of a flare were analyzed (excluding 65 specimens obtained while the patient was taking glucocorticoids). The date of library preparation was included in the model for batch correction, and the genefilter software package, version 1.64.0,18 was used to filter out genes with low levels of expression.
IDENTIFICATION AND CHARACTERIZATION OF COEXPRESSED GENE CLUSTERS
We hierarchically clustered the mean expression of significantly differentially expressed genes that were identified in the ImpulseDE2 analysis according to week relative to flare initiation (batch-corrected expression values [expressed as log2 reads per kilobase million] were calculated with edgeR) and identified five coexpressed gene clusters (clusters 1 through 5). We analyzed these five gene clusters for Gene Ontology term enrichment (with the use of goana). To further characterize expression patterns in gene clusters over time, for each cluster the mean expression level for each gene was calculated across flares per week, then normalized across weeks. ABIS19 and CIBERSORTx20 were used to deconvolute gene-expression data. To aggregate a given gene cluster or cell type with gene markers, the mean of standardized gene-expression scores or de-convoluted cell-type scores, respectively, within each week was plotted. To identify synovial scRNA-seq cluster–specific marker-gene signatures, we used a previously published data set20 to compare the cells from one scRNA-seq cluster with cells from all the other scRNA-seq clusters, using the scRNA-seq log2(counts per million + 1) matrix. We generated lists of the top 200 marker genes for each scRNA-seq cluster using the criteria of a log2 expression ratio greater than 1 (relative to other scRNA-seq clusters), an area under the curve greater than 0.6, and a percentage of expressing cells greater than 40%. We used Fisher’s exact test to evaluate enrichment of synovial cell subtype marker genes in the five coexpressed gene clusters. P values were corrected for multiple hypothesis testing with the use of the Benjamini–Hochberg procedure.
FLOW CYTOMETRY AND SORTING
To assess percentages of cells of interest among PBMCs, specimens were stained with antibodies to CD31-APC (clone WM59), mouse IgG1-APC (MOPC-21), PDPN-PerCP (NZ1.3), rat IgG2a (eBR2a)-PerCP, CD45-PE (HI30), mouse IgG1-PE (MOPC-21), and TO-PRO-3 and analyzed on the BD FACSCalibur cell sorting and analysis platform with the use of FlowJo, version 10.6.1 (BD). To flow sort and sequence cells of interest, 20 million to 100 million cells from CD14-depleted products of leukapheresis were stained with CD31-APC (WM59), mouse IgG1-APC (MOPC-21), PDPN-PerCP (NZ1.3), rat IgG2a-PerCP (eBR2a), CD45-FITC (HI30), mouse IgG1-FITC (MOPC-21), and DAPI (4′,6-diamidino-2-phenylindole, dihydro-chloride) and sorted on BD FACSAriaII. The Illumina Stranded TruSeq library kit was used to generate complementary DNA libraries that were sequenced with the MiSeq sequencing platform (Illumina). DESeq2 software, version 1.24.0,21 was used for differential expression analysis.
RESULTS
CLINICAL PROTOCOL DEVELOPMENT
We developed strategies for home blood collection that would provide high-quality RNA in large quantities for sequencing (15 to 50 ng of RNA; mean [±SD] RNA integrity score, 6.9±1.7 [scores range from 1 to 10, with higher scores indicating less degradation and higher integrity]) (Figs. S1 through S7 in Supplementary Appendix 1; note that all supplementary figures and tables are located in Supplementary Appendix 1). The study patients also documented disease activity with the RAPID3 questionnaire. Four patients (Table S1) were followed for 1 to 4 years with weekly home collection of fingerstick blood specimens coupled with completion of RAPID3 questionnaires and monthly clinic visits, during which DAS28 data were collected (Fig. 1A). RNA was sequenced from a total of 189 fingerstick blood specimens from the four patients; 162 (87%) of the specimens passed quality-control filtering. In the index patient, we assessed 364 time points by RAPID3 during eight flares over a period of 4 years and analyzed specimens from 84 time points by RNA-seq. We collected data on disease activity from 43 clinic visits from the index patient and from 25, 14, and 12 clinic visits for the other three patients who were included in the longitudinal study.
Figure 1. Study Overview and Validation of In-Home Assessments of Disease Activity and Gene Expression.
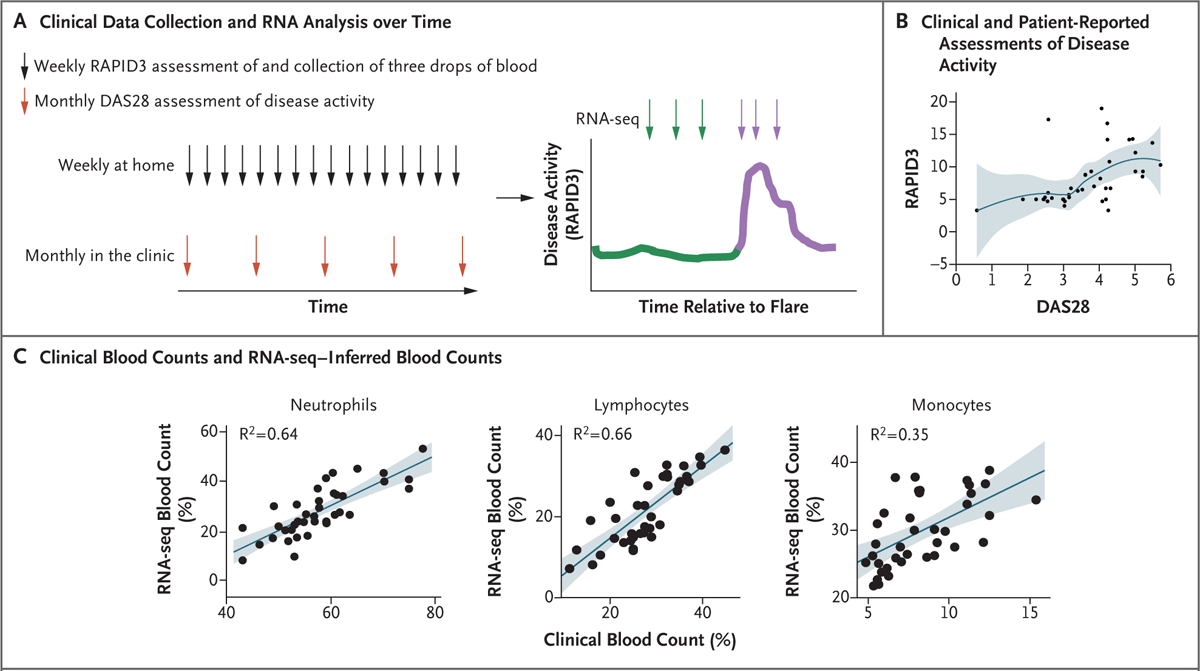
Panel A shows an overview of the collection of clinical data and specimens over time. The Routine Assessment of Patient Index Data 3 (RAPID3) questionnaire and the Disease Activity Score 28 (DAS28) were used to assess disease activity weekly at home and monthly in the clinic, respectively. Green indicates the time leading up to a flare, and purple the time during a flare. Panel B shows a scatterplot (with locally weighted smoothing) of the relationship between the change in scores on the RAPID3 questionnaire and the DAS28 in the index patient. DAS28 scores range from 2 to 10, with higher scores indicating more disease activity. RAPID3 scores range from 0 to 30, with higher scores indicating more severe disease. The blue line represents the point estimates, and the shaded area represents the 95% confidence interval. Panel C shows neutrophil, lymphocyte, and monocyte counts in paired clinical complete blood counts conducted with blood drawn by venipuncture as compared with the CIBERSORTx-inferred blood counts from RNA sequencing (RNA-seq) data obtained with the use of fingerstick blood specimens (38 paired specimens). The shaded areas represent the 95% confidence intervals.
Patient-reported assessments of disease activity (with RAPID3) were positively correlated with clinician assessments of disease activity (with DAS28) (Fig. 1B and S8). RNA-seq–inferred white-cell counts were also positively correlated with clinical laboratory measurements of complete blood counts (Fig. 1C), which suggested that RNA-seq of fingerstick blood was of sufficient quality to provide information that correlated with criterion-standard clinical measurements of blood counts. Taken together, these data indicate that patient reports of disease activity paired with analysis of fingerstick blood specimens may provide a high-quality and robust means by which patients can participate in longitudinal clinical research studies.
CLINICAL AND MOLECULAR FEATURES OF FLARES
Flares were associated with increases in objective clinical and laboratory measures of rheumatoid arthritis disease activity in the index patient (Fig. 2A and Fig. S9). RNA-seq analysis of fingerstick blood specimens identified 2613 genes that were differentially expressed at the time of a flare as compared with baseline (false discovery rate, <0.1); the expression of 1437 of these genes was increased during a flare (Fig. 2B and Supplementary Appendix 2). Pathway analysis identified enrichment in myeloid, neutrophil, Fc receptor–signaling, and platelet-activation genes (Fig. 2C and Supplementary Appendix 3), findings consistent with clinical blood-count measurements during flares (Fig. S9). The expression of 1176 genes was significantly decreased during flares, and pathway analysis of these genes indicated enrichment for extracellular matrix, collagen, and connective-tissue development (Fig. 2D and Supplementary Appendix 3).
Figure 2. Clinical and Transcriptional Characteristics of Rheumatoid Arthritis Flares in the Index Patient.
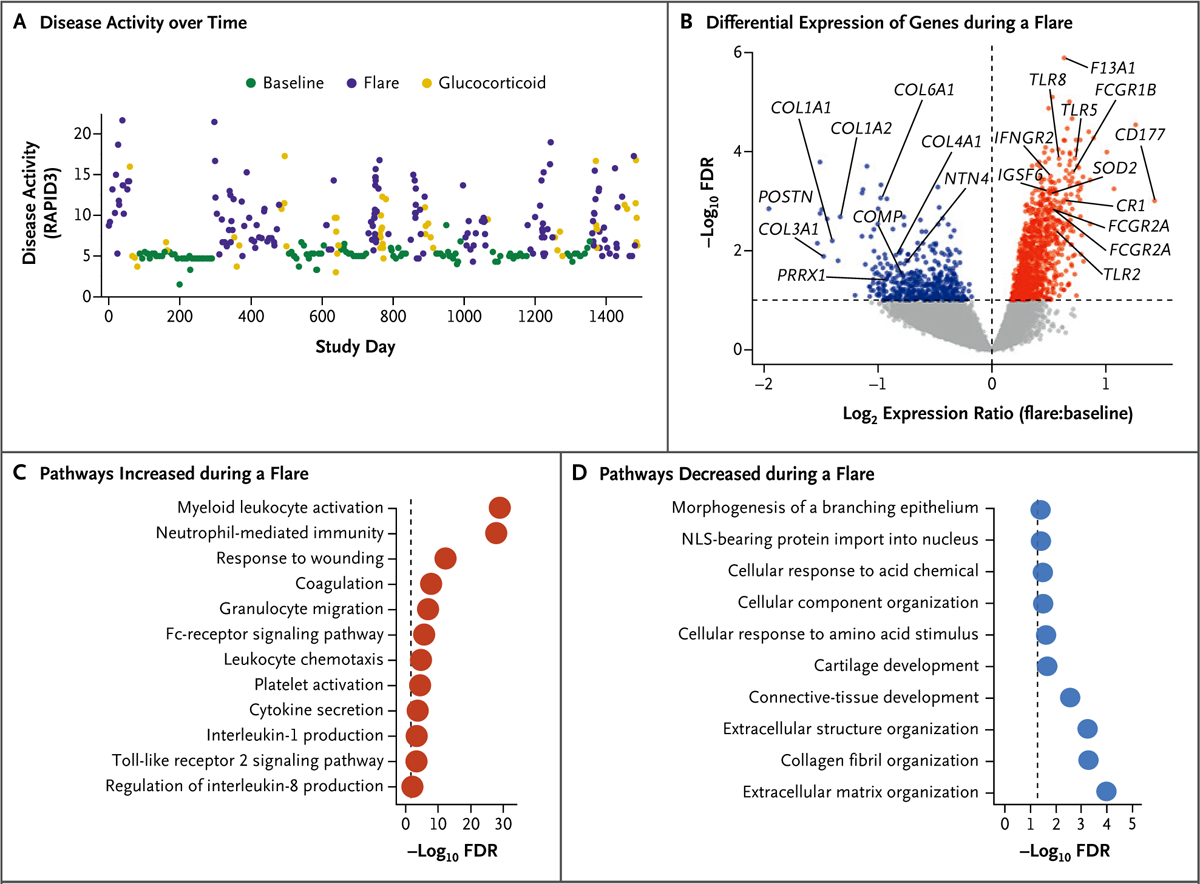
Panel A shows disease activity (measured with the RAPID3 questionnaire; 356 scores included in the analysis) over the course of 4 years in the index patient. Panel B shows a volcano plot of differential gene expression during flares (46 specimens) and during baseline (33 specimens), with significance (−log10 false discovery rate [FDR]) plotted against the log2 relative expression (flare:baseline ratio). Gray points indicate genes with no significant difference in expression between flares and baseline (with FDR >0.1), red indicates genes with significantly increased expression during a flare (FDR <0.1 and log2 expression ratio >0), and blue indicates genes with significantly decreased expression during a flare (FDR <0.1 and log2 expression ratio <0). Panels C and D show pathways enriched among genes with significantly increased (Panel C) or decreased (Panel D) expression during a flare relative to baseline. The dashed line represents the threshold for significance (FDR <0.05, or −log10 FDR >1.3). NLS denotes nuclear localization signal.
TIME-SERIES ANALYSIS OF MOLECULAR EVENTS LEADING TO FLARES
To analyze the trajectories of gene expression over time and identify potential antecedents to flares, we performed time-series analysis of the RNA-seq data (Fig. 3A). Disease-activity scores in the weeks just before a flare were the same as the baseline scores 2 months before a flare, underscoring the challenges of identifying both a time frame and a gene-expression signature that precede a flare. We focused the analysis on 65 specimens that were acquired 8 weeks before a flare and 4 weeks after flare initiation, binning samples according to the week in which they were drawn. With this method, we identified 2791 genes with significant differential expression over the time leading up to and during a flare (false discovery rate, <0.05), and with hierarchical clustering of gene expression we identified five clusters (Fig. 3B and Supplementary Appendix 4). Cluster 1 represented a group of genes that had increased expression after symptom onset (Fig. 3C and 3D) and had a high degree of overlap (Fig. 3E) with genes that had higher expression during flares than at baseline (Fig. 2B). These gene-expression clusters were reproducibly altered in five separate clinical flare events (Fig. S10).
Figure 3. Transcriptional Characteristics of Immune Activation before Symptom Onset in Rheumatoid Arthritis Flares.
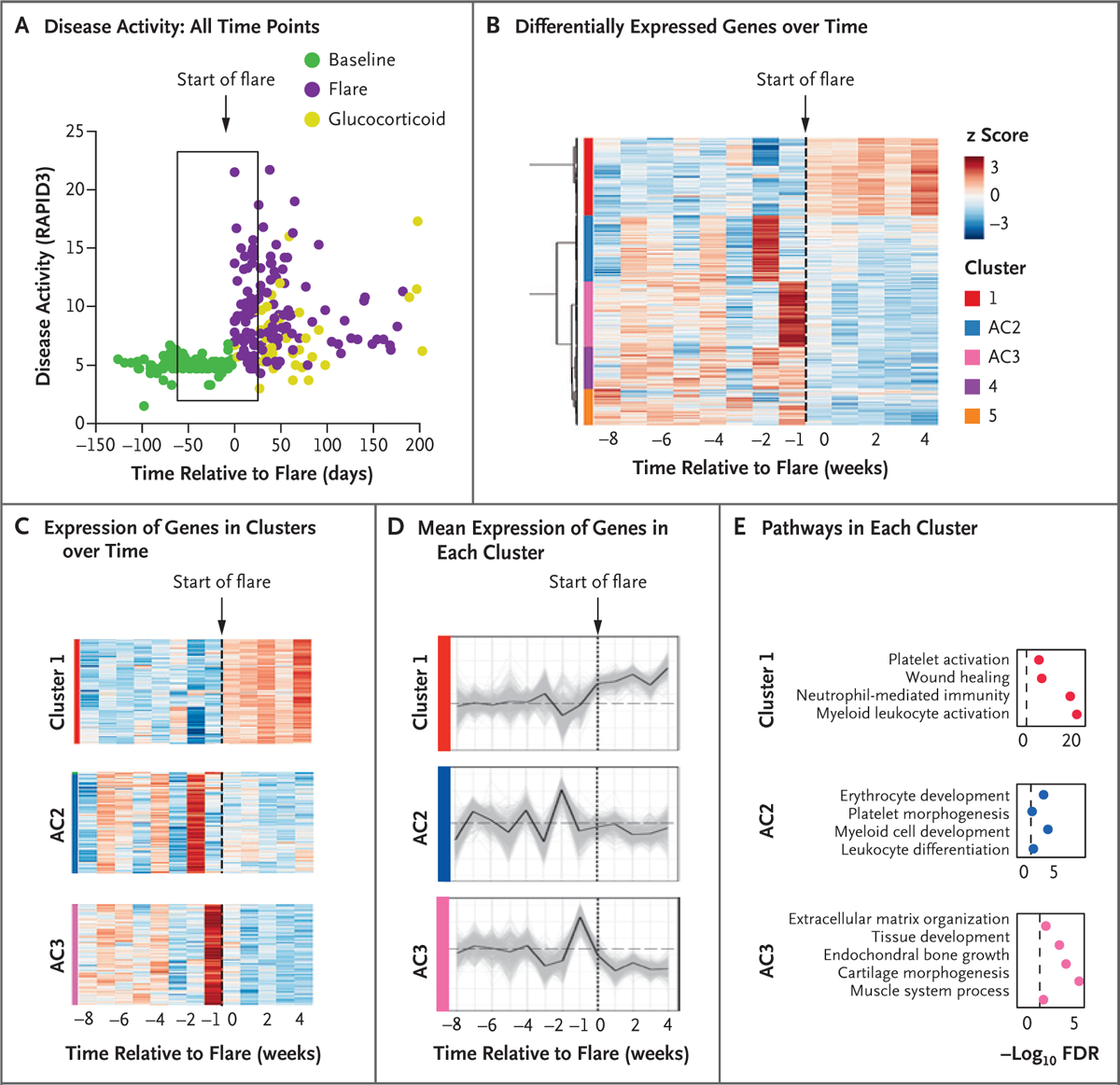
Panel A shows RAPID3 disease-activity scores over time (measured in days). The box represents disease activity from day −56 to day 28 relative to the start of a flare (day 0). Panel B shows hierarchical clustering of z scores of 2791 significantly differentially expressed genes over time. Significant clusters are labeled by color. Antecedent cluster 2 (AC2) and AC3 refer to clusters of genes with expression that changed before flares. Panel C shows a detailed representation of the cluster 1, AC2, and AC3 genes depicted in Panel B over the time to a flare. Panel D shows the mean standardized cluster gene expression over the time to a flare. Light gray lines represent expression of individual genes in the cluster. The dashed horizontal line represents the mean baseline gene expression (weeks −8 to −4). Panel E shows pathways enriched in cluster 1, AC2, and AC3. The dashed vertical line represents the threshold for significance (FDR <0.05, or −log10 FDR >1.3).
We further focused on two clusters of genes that were differentially expressed before a flare (Fig. 3C and 3D). Antecedent cluster 2 (AC2) transcripts increased 2 weeks before a flare and were enriched with developmental pathways for naive B cells and leukocytes. Two additional means of deconvoluting the RNA-seq data — the CIBERSORTx and ABIS software packages — independently confirmed evidence of increases in naive B-cell populations before a flare, and all analyses showed evidence of innate inflammatory signatures (neutrophils and monocytes) during a flare (Figs. S11 and S12).
Antecedent cluster 3 (AC3) transcripts increased during the week before a flare and then decreased for the duration of the flare (Fig. 3C and 3D). AC3 was enriched for pathways that were not typical of blood specimens, including cartilage morphogenesis, endochondral bone growth, and extracellular matrix organization (Fig. 3E and Supplementary Appendix 5), suggesting the presence of a mesenchymal cell.
TIME-SERIES ANALYSIS OF SYNOVIAL CELL MARKER GENES IN FLARES
To better characterize the clusters identified by the time-series analysis of fingerstick blood and their relevance to synovitis (Fig. 3C), we examined them for enrichment in synovial cell sub-types characterized by scRNA-seq. This analysis of 5265 single synovial cells from patients with rheumatoid arthritis and patients with osteo-arthritis identified four fibroblast, four B-cell, six T-cell, and four monocyte subpopulations (Fig. 4A). We identified approximately 200 marker genes that best distinguished each of 18 synovial cell types. AC2 was enriched with naive B-cell genes (Fig. 4A and Fig. S12), and AC3 was enriched with three sublining fibroblast genes (CD34+, HLA-DR+, and DKK3+) (Fig. 4A). Two of these fibroblast subsets, CD34+ and HLA-DR+, are more abundant in inflamed synovium than in uninflamed synovium.22 We plotted the expression of those transcripts that were common to both synovial sublining fibroblasts and AC3 over time; again, we noted their increased expression in blood 1 week before a flare and decreased expression during a flare (Fig. 4B, Fig. S13, and Supplementary Appendix 6).
Figure 4. Expression of AC3 Genes in PRIME Cells.

Panel A shows synovial cell subtype marker genes in clusters identified in blood. Enrichment scores of 200 single-cell RNA-seq (scRNA-seq) marker genes from 18 synovial subset cell types are shown. The dashed vertical line represents the threshold for significance (FDR <0.05, or −log10 FDR >1.3). ABC denotes age-associated B cell, CTL cytotoxic T lymphocyte, IFN interferon, Tph/fh T peripheral helper/follicular helper, Treg T regulatory. Panel B shows the means of the standardized expression of genes that are common to synovial sublining fibroblasts (CD34+DKK+HLA−DRA+ fibroblasts) and AC3 in blood over the time to a flare (the dashed vertical line represents the start of a flare); I bars represent 95% confidence intervals. For standardization, the mean expression level for each gene was calculated across flares per week and then normalized across weeks. Panel C shows a Venn diagram of the numbers of AC3 genes that decreased during flares in the index patient (Patient 1) and the three additional patients (Patients 2, 3, and 4). Shading is used to highlight overlap between the index patient and the other patients. Panel D shows the log2 relative expression ratio for AC3 genes in PRIME cells (flow-sorted CD45−CD31−PDPN+ cells) as compared with hematopoietic cells (flow-sorted CD45+) and in stained peripheral blood mononuclear cells (not flow sorted) as compared with hematopoietic cells (flow-sorted CD45+) as a technical control for the stress of flow sorting. The box-and-whisker plots indicate the median, interquartile range, and 1.5 times the interquartile range; the P value is from a Mann–Whitney U test.
Overall, 622 of 625 AC3 genes decreased in expression during a flare in the index patient, and a subset (194 genes) also decreased in expression during flares in at least three of four patients (22 genes decreased in expression in all four patients) (Fig. 4C and Supplementary Appendix 7); a permutation test indicated that this overlap was greater than would have been expected by chance (P<0.001). Pathway analysis of the subset of 194 overlapping genes again showed that it was enriched for genes associated with extracellular matrix and secreted glycoprotein.
We further tested whether cells that expressed surface markers of synovial fibroblasts were detectable by flow cytometry in blood from patients with rheumatoid arthritis. CD45−CD31−PDPN+ cells were more common in blood from 19 additional patients with rheumatoid arthritis than in blood from healthy controls (Figs. S14 and S15). RNA-seq of CD45−CD31−PDPN+ cells sorted by fluorescence-activated cell sorting confirmed that they were enriched for AC3 cluster genes (Fig. 4D) and for synovial fibroblast genes and expressed classical synovial fibroblast genes such as FAP, DKK3, CDH11, as well as collagens and laminins (Figs. S16 through S18 and Table S2). Given their expression of classical mesenchymal surface markers and genes, we refer to these cells as preinflammatory mesenchymal cells, or PRIME cells. Taken together, our observations suggest a model in which sequential activation of B cells activates PRIME cells just before flares; these cells are then evident during flares in inflamed synovium as inflammatory sublining fibroblasts (Fig. 5).
Figure 5. Model of Blood and Synovial Gene-Expression Changes before and during Rheumatoid Arthritis Flares.
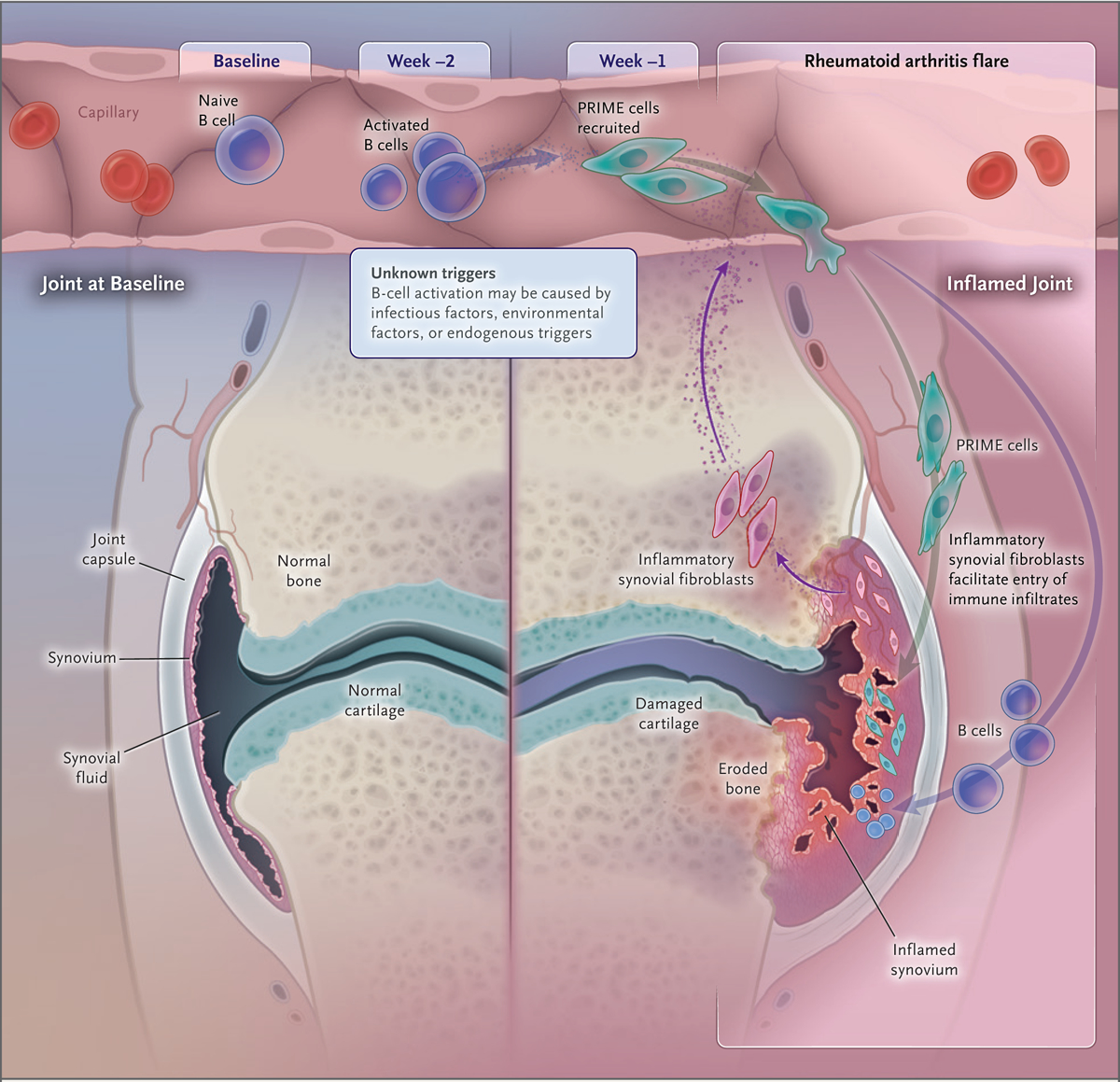
Inflammatory signals activate naive B cells (AC2), which in turn activate PRIME cells (AC3); these cells harbor the signature of synovial sublining fibroblast genes. According to this model, PRIME cells demarginate and are increased in blood before a flare and then decrease just after symptom onset; these cells or their progeny are increased in inflammatory synovium in patients with rheumatoid arthritis, where they contribute to and may be sufficient to cause joint inflammation.
DISCUSSION
We present longitudinal genomics as a strategy to study the antecedents to rheumatoid arthritis flares that may be generalizable to autoimmune diseases associated with waxing and waning clinical courses. We developed tools for patients to acquire both data on clinical symptoms and molecular data at home over the course of many years. This allowed us to capture data before the onset of flares and identify different RNA signatures (AC2 and AC3) evident in peripheral blood 1 to 2 weeks before a flare.
The RNA signature of AC3 and sorted CD45−CD31−PDPN+ circulating cells revealed enrichment for pathways including cartilage morphogenesis, endochondral bone growth, and extracellular matrix organization (Fig. 3E) and strongly overlapped with the RNA signatures of synovial sublining fibroblasts. We therefore propose that antecedent PRIME cells are the precursors to inflammatory sublining fibroblasts that have previously been found adjacent to blood vessels in inflamed synovium in patients with rheumatoid arthritis.23
Inflamed sublining fibroblasts have been shown to be pathogenic in an animal model of arthritis.24 Our finding that AC3 genes in humans share molecular characteristics of sublining fibroblasts, together with the observation that these cells spike before a flare but are less detectable in blood during a flare (Figs. 2 and 4) support a model in which PRIME cells immigrate from blood to the synovium, where they contribute to the inflammatory process (Fig. 5). This model is consistent with the observation that in rheumatoid arthritis, synovial fibroblasts can traffic to cartilage implants and are sufficient to passively transfer synovial inflammation in mice.25 Together, our data suggest the mesenchymal signal detected in AC3 before flares represents a previously uncharacterized type of trafficking fibroblast that circulates in blood.
In addition, we observed a second RNA signature, AC2, that was activated in blood before the spike in AC3. AC2 bears RNA hallmarks of naive B cells. This finding is reminiscent of those in recent studies showing that autoreactive naive B cells are activated in patients with rheumatoid arthritis.26 Although the triggers of this B-cell activation are unknown, infectious factors (e.g., bacterial or viral antigens), environmental factors, or endogenous toxins27–29 could either provide a source of specific antigens or activate pattern-recognition receptors.
We developed methods for collecting frequent (weekly) longitudinal clinical and gene-expression data that can be used to identify changes in transcriptional profiles in blood weeks before the onset of symptoms in patients with rheumatoid arthritis. This approach led to the identification of PRIME cells, which bear hallmarks of synovial fibroblasts, are more common in patients with rheumatoid arthritis than in healthy controls, and increase in blood just before flares. We suggest that before a clinical flare, B-cell immune activation (detected as AC2) acts on PRIME cells, which traffic to the blood (detected as AC3) and subsequently to the synovial sublining during flares of disease activity. More generally, this study of rheumatoid arthritis provides an example of an approach to the study of waxing and waning inflammatory diseases.
Supplementary Material
Acknowledgments
Supported by grants from the National Institutes of Health (NIH) (T32 HG003284, to Dr. Yao; UH2AR067677 and U01 HG009379, to Dr. Raychaudhuri; R01 GM071966, to Dr. Troyanskaya; and NS034389, NS081706, NS097404, and 1UM1HG008901, to Dr. Darnell), the Simons Foundation (SFARI 240432, to Dr. Darnell), the Robertson Foundation (to Drs. Darnell and Orange), Rockefeller University (UL1 TR001866 from the National Center for Advancing Translational Sciences), the NIH Clinical and Translational Science Award Program, a Rheumatology Research Foundation Bridge to K (to Dr. Orange), the Bernard and Irene Schwartz Foundation (to Dr. Orange), the Iris and Junming Le Foundation (to Dr. Orange), and a Rockefeller Clinical and Translational Science Award Program Pilot Award (to Dr. Orange). Dr. Troyanskaya is a senior fellow of the Genetic Networks program of the Canadian Institute for Advanced Research. Dr. Darnell is an Investigator of the Howard Hughes Medical Institute.
We thank the staff of Rockefeller University Hospital and the Hospital for Special Surgery, particularly Rhonda Kost, Arlene Hurley, Caroline Jiang, and Weijia Yuan.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
REFERENCES
- 1.Steinman L Immunology of relapse and remission in multiple sclerosis. Annu Rev Immunol 2014; 32: 257–81. [DOI] [PubMed] [Google Scholar]
- 2.Fava A, Petri M. Systemic lupus erythematosus: diagnosis and clinical management. J Autoimmun 2019; 96: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braun J, Wei B. Body traffic: ecology, genetics, and immunity in inflammatory bowel disease. Annu Rev Pathol 2007; 2: 401–29. [DOI] [PubMed] [Google Scholar]
- 4.Braun J, Baraliakos X, Listing J, et al. Differences in the incidence of flares or new onset of inflammatory bowel diseases in patients with ankylosing spondylitis exposed to therapy with anti-tumor necrosis factor alpha agents. Arthritis Rheum 2007; 57: 639–47. [DOI] [PubMed] [Google Scholar]
- 5.Sellam J, Marion-Thore S, Dumont F, et al. Use of whole-blood transcriptomic profiling to highlight several pathophysiologic pathways associated with response to rituximab in patients with rheumatoid arthritis: data from a randomized, controlled, open-label trial. Arthritis Rheumatol 2014; 66: 2015–25. [DOI] [PubMed] [Google Scholar]
- 6.Sanayama Y, Ikeda K, Saito Y, et al. Prediction of therapeutic responses to toci lizumab in patients with rheumatoid arthritis: biomarkers identified by analysis of gene expression in peripheral blood mononuclear cells using genome-wide DNA microarray. Arthritis Rheumatol 2014; 66: 1421–31. [DOI] [PubMed] [Google Scholar]
- 7.Tanino M, Matoba R, Nakamura S, et al. Prediction of efficacy of anti-TNF biologic agent, infliximab, for rheumatoid arthritis patients using a comprehensive transcriptome analysis of white blood cells. Biochem Biophys Res Commun 2009; 387: 261–5. [DOI] [PubMed] [Google Scholar]
- 8.Teixeira VH, Olaso R, Martin-Magniette M-L, et al. Transcriptome analysis describing new immunity and defense genes in peripheral blood mononuclear cells of rheumatoid arthritis patients. PLoS One 2009; 4(8): e6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang YJ, Ioerger TR, Huttenhower C, et al. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog 2012; 8(9): e1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62: 2569–81. [DOI] [PubMed] [Google Scholar]
- 11.Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2010; 69: 1580–8. [DOI] [PubMed] [Google Scholar]
- 12.Pincus T, Swearingen CJ, Bergman M, Yazici Y. RAPID3 (Routine Assessment of Patient Index Data 3), a rheumatoid arthritis index without formal joint counts for routine care: proposed severity categories compared to disease activity score and clinical disease activity index categories. J Rheumatol 2008; 35: 2136–47. [DOI] [PubMed] [Google Scholar]
- 13.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014; 30: 923–30. [DOI] [PubMed] [Google Scholar]
- 15.Robison EH, Mondala TS, Williams AR, Head SR, Salomon DR, Kurian SM. Whole genome transcript profiling from fingerstick blood samples: a comparison and feasibility study. BMC Genomics 2009; 10: 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43(7): e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer DS, Theis FJ, Yosef N. Impulse model-based differential expression analysis of time course sequencing data. Nucleic Acids Res 2018; 46(20): e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bourgon R, Gentleman R, Huber W. Independent filtering increases detection power for high-throughput experiments. Proc Natl Acad Sci U S A 2010; 107: 9546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monaco G, Lee B, Xu W, et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep 2019; 26(6): 1627.e7–1640.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019; 37: 773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang F, Wei K, Slowikowski K, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol 2019; 20: 928–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizoguchi F, Slowikowski K, Wei K, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun 2018; 9: 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Croft AP, Campos J, Jansen K, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019; 570: 246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefèvre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med 2009; 15: 1414–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu DR, McDavid AN, Kongpachith S, et al. T cell-dependent affinity maturation and innate immune pathways differentially drive autoreactive B cell responses in rheumatoid arthritis. Arthritis Rheumatol 2018; 70: 1732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikuls TR, Payne JB, Yu F, et al. Periodontitis and porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol 2014; 66: 1090–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konig MF, Abusleme L, Reinholdt J, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med 2016; 8: 369ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eriksson K, Nise L, Alfredsson L, et al. Seropositivity combined with smoking is associated with increased prevalence of periodontitis in patients with rheumatoid arthritis. Ann Rheum Dis 2018; 77: 1236–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.