

Abstract
Aim:
Autologous hematopoietic stem cell transplantation (ASCT) is the standard-of-care curative treatment for relapsed or refractory diffuse large B-cell lymphoma (RR-DLBCL), but the relapse rate is usually high.
Materials & methods:
In this study, we treated 14 RR-DLBCL patients by combining ASCT and anti-CD19 chimeric antigen receptor T-cell therapy.
Results:
Eleven (78.57%) patients achieved complete or partial remission. Median duration of progression-free survival was 14.82 months (95% CI: 0.00–31.20 months) with 6-month progression-free survival rate of 64.29% (95% CI: 39.18–89.40%). Median overall survival was not achieved, with 1-year overall survival rate of 65.48% (95% CI: 36.00–94.96%). No neurotoxicity was observed.
Conclusion:
Our study demonstrated safety and feasibility of ASCT and anti-CD19 chimeric antigen receptor T-cell treatment for RR-DLBCL patients.
Keywords: : autologous hematopoietic stem cell transplantation, chimeric antigen receptor T-cell therapy, diffuse large B-cell lymphoma, relapsed or refractory, safety
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL), accounting for 30–40% of newly diagnosed NHL [1]. For DLBCL patients, rituximab combined with chemotherapy is the standard aggressive therapy, with more than 70% patients achieving complete remission (CR) [2–6]. However, 20–50% DLBCL patients are primary refractory to chemoimmunotherapy or relapse after achieving CR [7,8]. For the relapsed or refractory (RR) patients, only 30–40% patients respond to salvage chemotherapy and usually proceed to undergo autologous hematopoietic stem cell transplantation (ASCT) [9–12]. Even among the patients who are able to receive ASCT, 50% patients eventually relapse [13,14]. For patients that relapse after ASCT within 12 months, only 26% patients respond to salvage chemotherapy and the median survival is only 6.3 months [15]. Some RR-DLBCL patients received allogeneic hematopoietic stem cell transplantation (Allo-HSCT), however, Allo-HSCT could induce graft-versus-host disease. For economic and safety reason, many patients choose ASCT, but ASCT could not benefit them a lot. Thus, there is a poor outlook for patients with RR-DLBCL, and until now, there are no curative treatment options.
Chimeric antigen receptor (CAR) T-cell therapy has achieved marked success in treating hematological malignancies, especially in B-cell acute lymphoblastic leukemia. Clinical trials with CAR T-cell therapy have shown CR rates of 70–90% in RR B-cell acute lymphoblastic leukemia [16–23]. Anti-CD19 CAR T-cell therapy has also been widely investigated in B-cell NHL treatment since the first advanced follicular lymphoma patient received CAR T-cell therapy in 2010 [24]. In previous clinical studies, DLBCL patients who received CAR T-cell therapy treatment showed objective response rates (ORRs) of 52–82%; however, only 40–65% maintained survival without relapse after CAR T-cell therapy during the first 1–2 years [25–27]. Thus, pursuing higher response rate and durable remission are the main challenge for CAR T-cell therapy in lymphoma.
In the current clinical trial, we designed a protocol to treat 14 patients with RR-DLBCL by sequential infusing ASCT and anti-CD19 CAR T-cell, aiming to improve survival of RR-DLBCL patients. With this therapy, 78.57% patients obtained remission, with no severe toxic effects.
Materials & methods
Study design
The clinical trial (ChiCTR-OIN-15007668) is an open-label, uncontrolled, single-arm pilot study approved by the institutional review boards of Second Military Medical University and Changhai Hospital. This trial has been conducted at Changhai Hospital. Written informed consents from patients have been obtained according to the Declaration of Helsinki protocol.
Eligible patients underwent hematopoietic stem cell and peripheral blood mononuclear cells (PBMNCs) apheresis before ASCT conditioning therapy. The ASCT conditioning regimen was semustine, cytarabine, etoposide and cyclophosphamide, including oral semustine (250 mg/m2) on day -6, followed by cytarabine (300 mg/m2) infusion for 1 h every 12 h, etoposide (100 mg/m2) infusion for 3 h and cyclophosphamide (1.0 g/m2) on days -5 through -2. Autologous hematopoietic stem cells were infused on day 0 and CAR T-cell was given on day 6 after ASCT.
CAR T-cell production
T cells were collected from patients’ PBMNCs by leukapheresis and activated with anti-CD3 (1000 µg/ml) and anti-CD28 (500 µg/ml) microbeads. The CD19-specific scFv was designed and produced by HuaDao Biopharma (Shanghai) Limited Corporation. The CD19-4-1BB transgenes were transduced into T cells with lentiviral vector, and the cells expanded with the use of IL-2 (600 IU/ml) for 10–14 days. Sterility, transduction efficiency, phenotype, cytotoxicity and cytokine release were routinely examined in vitro. Sample processing, CAR T-cell manufacturing and laboratory analyses were mainly conducted by HuaDao Biopharma (Shanghai) Limited Corporation.
CAR T-cell examinations ex vivo & in vivo
Tumor lysis activity of CAR T-cell ex vivo was analyzed using lactic acid dehydrogenase (LDH) release assay. In brief, CAR T-cell were co-incubated with target cells (CD19-K562) or non-target cells (K562) for 16 h, and supernatants were collected for LDH examination. Abundance of LDH was proportional to the number of target or non-target cells lysed, reflecting CAR T-cell tumor lysis activity. CAR T-cell cytokine secretion ex vivo was detected by Cytometric Bead Array. CAR T-cell sub-population ex vivo was examined by flow cytometer. In vivo CAR T-cell cytokine secretion at a series of time points was analyzed by the Immulite 1000 Immunoassay System (Siemens, Berlin, Germany). CAR T-cell expansion and persistence in vivo was analyzed by quantitative-PCR and flow cytometry. The details of examination procedures and regents were described in our previous work [28].
Disease & toxicity assessment
Disease assessment with positron-emission tomography and computed tomography (PET-CT) or CT alone were performed before ASCT and 1 month, 3 months, 6 months, 9 months and 1 year after ASCT infusion. CR was defined as complete resolution of all areas of disease on PET-CT assessments, while partial remission (PR) was defined as ≥50% reduction in the greatest diameter of all sites of known disease and no new sites of disease. Stable disease (SD) was defined as <50% reduction in the diameter of all disease sites. Progressive disease (PD) was defined as enlargement of all disease site, or development of new disease site. Toxicity assessment was based on NCI Common Terminology Criteria for Adverse Events 4.02. Cytokine-release syndrome (CRS) assessment was conducted using the University of Pennsylvania Cytokine Release Syndrome Grading System [29].
Progression-free survival (PFS) was defined as the length of the time from ASCT to relapse, death or the last follow-up point from CR or PR. Overall survival (OS) represented the length of time from ASCT to death due to any cause or the last follow-up visit.
Statistical analysis
The survival probability of PFS and OS was analyzed using the Kaplan–Meier method. p-value < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 6.0.
Results
Patient characteristics
From December 2016 to December 2018, a total of 14 RR-DLBCL patients were enrolled in the current study. Among these patients, five (35.71%) were females and nine (64.29%) were males. The median age was 39 (range: 19 to 68 years) years old. At baseline, after received first- and second-line therapy, while no patient had a prior ASCT among the 14 patients, seven (50.00%) patients had SD, six (42.86%) patients had PD and one (7.14%) patients relapsed. Three patients received six courses first-line therapy and disease progressed, so we assessed it as primary refractory. After enrollment, patients were treated with sequential ASCT and CAR T-cell treatment as consolidation therapy. Patients received semustine, cytarabine, etoposide and cyclophosphamide conditioning chemotherapy before ASCT. The infused median MNC dose of ASCT was 4.89 × 108/kg (range: 3.51 to 8.72 × 108/kg), and median CD34-positive cells dose of ASCT was 3.44 × 106/kg (range: 0.44 to 12.26 × 106/kg). CAR T-cell were infused at 6 days after ASCT, and the median CAR T-cell dose was 2.43 × 106/kg (range: 0.74 to 7.89 × 106/kg). The patients’ characteristics were described in Table 1.
Table 1. . Clinical characteristics of 14 relapsed or refractory diffuse large B-cell lymphoma patients.
| n | Gender | Age | Diagnosis | Disease status before ASCT | Sites of active disease | Conditioning therapy | MNC cell dose (×108/kg) |
CD34 cell dose (×106/kg) |
CAR T-cell dose (×106/kg) |
CRS max grade | Best response | Duration of best response (month) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 47 | DLBCL | SD | Multiple LN | CEAC | 4.60 | 0.44 | 0.74 | 1 | CR | 2.43 |
| 2 | F | 52 | DLBCL | SD | Multiple LN | CEAC | 4.61 | 1.15 | 3 | 1 | CR | 8.31+ |
| 3 | M | 32 | DLBCL | PD | Pelvic LN | CEAC | 7.14 | 1.14 | 1.2 | 2 | PD | 1.35 |
| 4 | M | 68 | DLBCL | SD | Abdomen LN | CEAC | 5.94 | 2.37 | 2.14 | 1 | CR | 20.4+ |
| 5 | M | 44 | DLBCL | PD | Abdomen LN | R-CEAC | 8.45 | 12.26 | 0.88 | N | PD | 3.65 |
| 6 | M | 28 | DLBCL | Relapse | Multiple LN | CEAC | 5.06 | 5.06 | 7.89 | 2 | CR | 16.07+ |
| 7 | F | 56 | DLBCL | PD | Breast | CEAC | 3.60 | 4.11 | 2.37 | 2 | PR | 14.82 |
| 8 | M | 22 | DLBCL | SD | Cervical LN | R-CEAC | 4.72 | 1.65 | 1.5 | 1 | PD | 5.62 |
| 9 | F | 44 | DLBCL | SD | Abdomen LN | R-CEAC | 4.70 | 12 | 2.49 | N | PR | 9.88+ |
| 10 | M | 27 | DLBCL | PD | Mediastinum | R-CEAC | 4.50 | 7 | 1.72 | 2 | PR | 11.4+ |
| 11 | F | 37 | DLBCL | SD | Abdomen LN | R-CEAC | 3.51 | 2.12 | 5 | 3 | CR | 10.91+ |
| 12 | M | 19 | DLBCL | PD | Multiple LN | CEAC | 7.54 | 2.76 | 6.32 | 2 | CR | 7.75+ |
| 13 | M | 39 | DLBCL | SD | Abdomen LN | CEAC | 5.69 | 6.67 | 3.59 | 1 | PR | 6.6+ |
| 14 | M | 56 | DLBCL | PD | Mediastinum | R-CEAC | 8.72 | 5.6 | 3 | 2 | PR | 1.81 |
ASCT: Autologous hematopoietic stem cell transplantation; CAR: Chimeric antigen receptor; CEAC: Semustine, cytarabine, etoposide and cyclophosphamide; CR: Complete remission; CRS: Cytokine-release syndrome; DLBCL: Diffuse large B-cell lymphoma; F: Female; LN: Lympho nod; M: Male; MNC: Mononuclear cell; N: No CRS; PD: Progressive disease; PR: Partial remission; R: Rituximab; SD: Stable disease.
Quality control analyses for CAR T-cell products
All CAR T-cell products met quality control criteria. The expression of CD3, CD4, CD8, CD45RA, C-C chemokine receptor-7 (CCR7) on the T-cell surface were analyzed to depict CD4-positive, CD8-positive, naive T cell (CD45RA + CCR7+), memory T cell (CD45RA-CCR7+/CCR7−) or effector T cell (CD45RA + CCR7-) proportions in total T cells (Figure 1A). On average, CD8-positive T cells accounted for 60% of total T cells and the majority were T memory cells. To measure CAR T-cell antitumor activity, we examined CAR T-cell lysis activity when combined with a CD19-positive tumor cell line (K562-CD19) and K562 cell line by LDH release assay. The results showed that CAR T cell expressed robust cytotoxicity against CD19-positive cells but had little impact on K562 cell line (Figure 1B). Cytokines levels of IL-2, TNF-α and IFN-γ in the medium markedly increased when CAR T cells encountered the CD19-positive tumor cell line (Figures 1C–E). The results indicated that CAR T-cell could specifically recognize target cells and possess potent antitumor activities in vitro.
Figure 1. . Chimeric antigen receptor T-cell subpopulations and antitumor activity ex vivo.
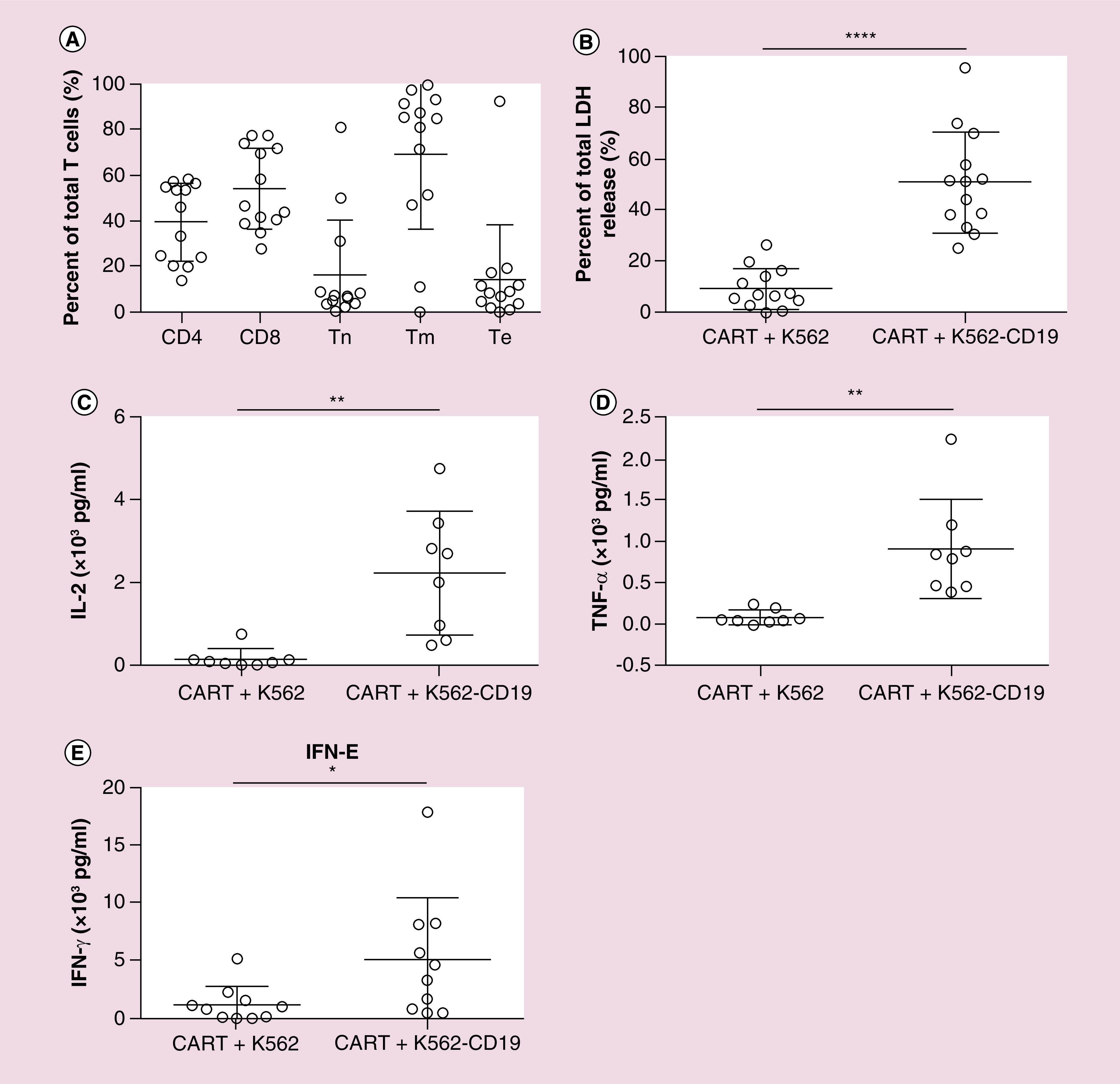
(A) Percentages of T-cell subpopulations in CAR T-cell products detected by flow cytometer. Tn express markers of CD45RA and CCR7, Tm do not express marker of CD45RA, Te express marker of CD45RA and do not express CCR7. (B) CAR T-cell tumor lysis activity. CAR T cell were co-incubated with target cell (K562-CD19) or nontarget cell (K562) at E:T of 4:1 for 16 h. LDH level in the supernatant was determined. CAR T cell robustly killed target cell but had little impact on nontarget cells. ****p < 0.0001. (C–E) IL-2, TNF-α and IFN-γ secretion in coculture system of CAR T cell and target or nontarget cells. CAR T cell secreted high level of IL-2 and TNF-α only when counteracted target cells. *p < 0.05; **p < 0.01.
CAR: Chimeric antigen receptor; CCR7: C-C chemokine receptor-7; ET: Effector-to-target cell ratio; LDH: Lactic acid dehydrogenase; Te: Effector T cells; Tm: Memory T cells; Tn: Naive T cells.
Patient responses
Among 14 patients, five patients had PET-CT and nine patients had CT evaluation at 1 month after ASCT and CAR T-cell therapy. Moreover, 11 of 14 (78.57% ORR) patients responded to ASCT and CAR T-cell sequential therapy. For 14 patients, six (42.86%) patients achieved CR, five (35.71%) patients obtained PR and three (21.43%) patients remained PD. And the assessment at 3 months after treatment, remission rate was 71.43%. There was no association between patients’ responses with CAR T cell and MNC doses of ASCT infused (Figures 2A–B). CAR T-cell lentivirus vector specific gene regions were detected in patients 15 days after infusion, indicating expansion and persistence of the CAR T cell in vivo (Figure 2C). There was no significant difference of CAR T-cell expansion in vivo between PR/CR and PD patients (Figures 2D–E). Responses were not associated with subgroups categorized according to patients’ age, disease states, cell of origin or treatment history (Figure 2F).
Figure 2. . Patient response and possible relevant factors.
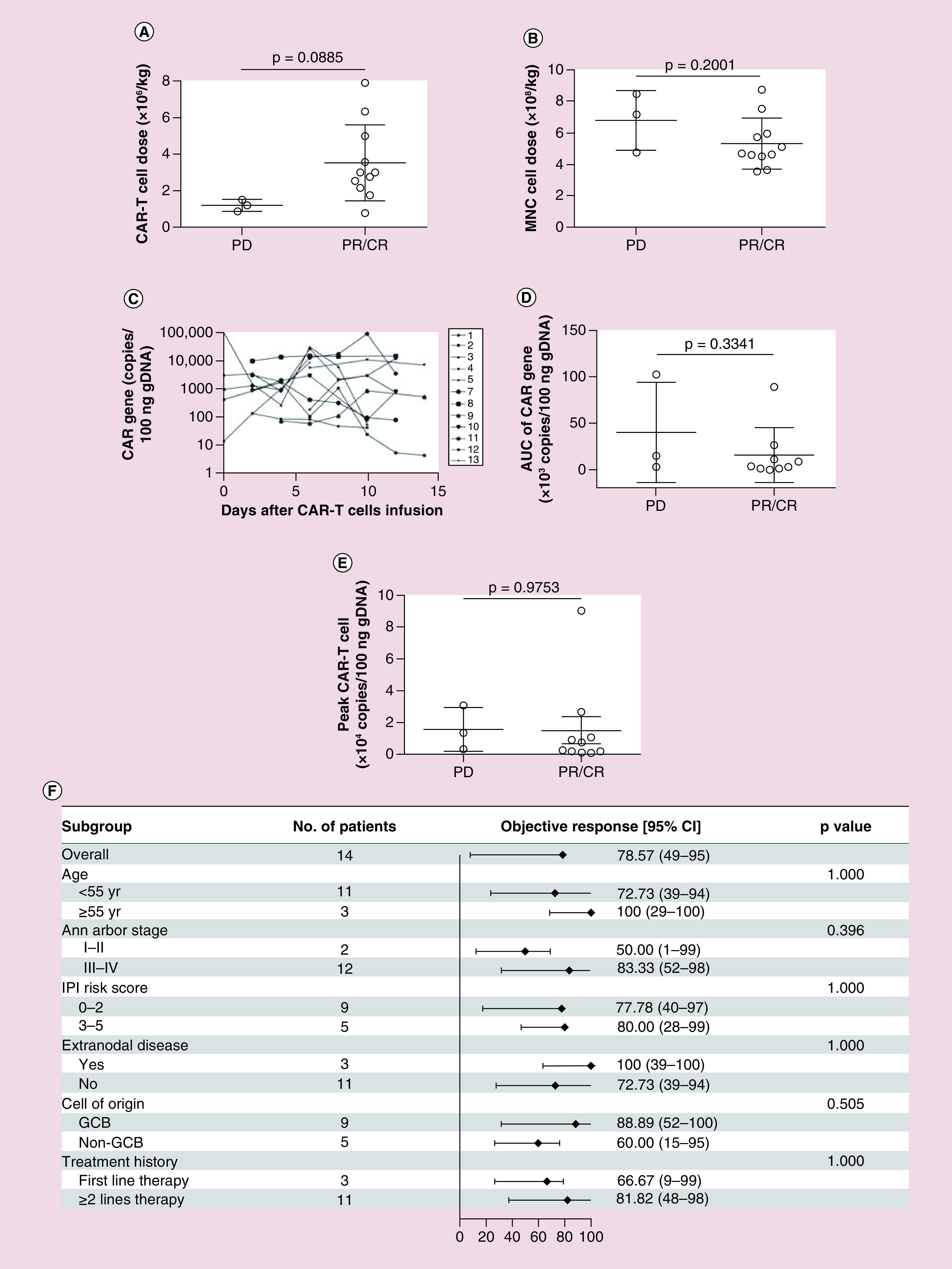
(A) Relationship of patient responses of PR/CR or PD to CAR T-cell dosage. (B) Relationship of patient responses of PR/CR or PD with infused MNC dosage. (C) CAR T-cell expansion detected by qPCR from day 1 through day 14 after T-cell infusion. (D) Relationship of patient response to CAR expansion in vivo AUC. (E) Relationship of patient responses of PR/CR or PD to CAR T-cell peak/expansion level in vivo. (F) The rate of objective response (CR + PR) and 95% CI according to patient baseline clinical characteristics including the age, Ann Arbor stage, IPI risk score extranodal disease, cell of origin and treatment history. Solid diamond indicates the observed objective response rate. Line extended from diamond means 95% CI.
AUC: Area under curve; CAR: Chimeric antigen receptor; CR: Complete remission; GCB: Germinal center B cells; IPI: International prognostic index; MNC: Mononuclear cell; PD: Progressive disease; PR: Partial remission; qPCR: Quantitative-PCR.
Adverse effects
CRS has been reported as the most common adverse effects in CAR T-cell therapy [30]. In this study, we observed that seven (50.00%) patients showed no or grade 1 CRS, seven (50.00%) patients had grade 2–3 CRS, with symptoms included fever (>38°C) and hypotension after CAR T-cell infusion. Two patients were treated with anti-IL-6 receptor monoclonal antibody and dexamethasone. CRS was positively associated with the level of serum soluble IL-2 receptor and IL-10 (Figures 3A–B); while there was no significant differences of serum IL-6 and C-reactive protein levels between patients with no/mild CRS and patients with moderate CRS (Figures 3C–D). No patients had manifestations of neurotoxicity. The median time of neutrophil and platelet engraftment was 11 days (range: 9 to 12 days) and 14 days (range: 11 to 24 days), respectively. Adverse effects included one patient developed lasted B-cell aplasia for nearly 1 month, and three patients reported diarrhea. Infection complications including upper respiratory tract infection, pneumonia and septicemia were observed in three patients who were then managed by supportive care and antibiotics.
Figure 3. . Serum cytokine levels and cytokine-release syndrome severity.
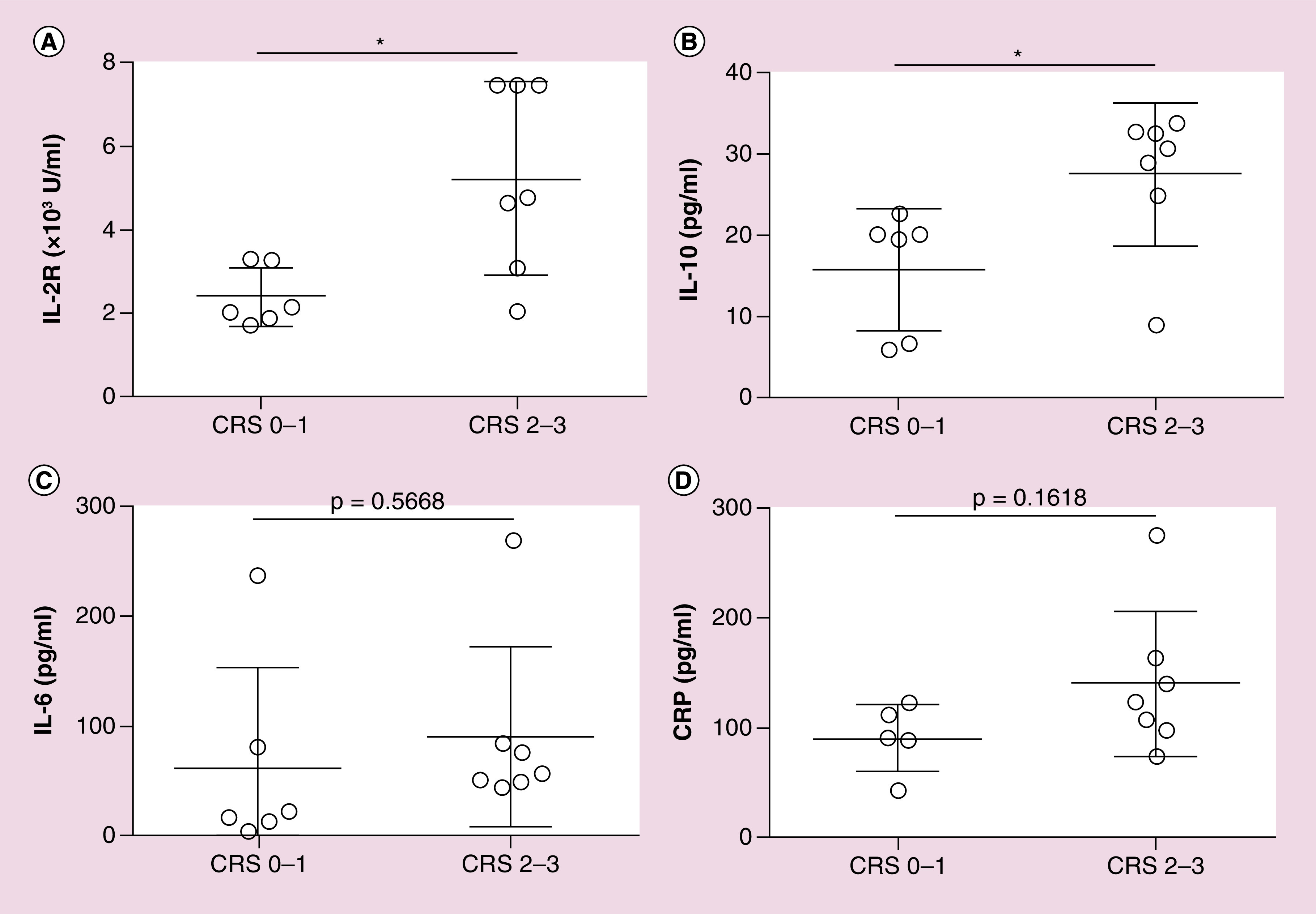
(A & B) sIL-2R and IL-10 levels were significantly higher in grade 2 and 3 CRS than those in grade 0 and 1 CRS. (C & D) Serum IL-6 and CRP showed no difference between grade 0 and 1 CRS and grade 2 and 3 CRS.
CRP: C-reactive protein; CRS: Cytokine-release syndrome; sIL-2R: Serum soluble IL-2 receptor.
Survival
Median follow-up was 10.28 months (range: 2.43 to 20.4 months) after ASCT and CAR T-cell infusion. To the observation end point, five of the six CR patients remained in remission without further treatment, and the other died of food poisoning; two of the five PR patients relapsed in 1.8 and 14.8 months, and one died. Three PD patients were alive. The median duration of PFS was 14.82 months (95% CI: 0–31.20 months), with PFS rate of 64.29% (95% CI: 39.18–89.40 %) at 6 months. The median OS of the patients was not reached, with 1-year OS rate of 65.48% (95% CI: 36.00–94.96%) (Figures 4A–B). The first patient in Table 1 died from food poisoning. She accepted ASCT and anti-CD19 CAR T-cell therapy in 9 January 2017. During anti-CD19 CAR T-cell therapy, the patient had grade 1 CRS and lasted for a week. 1 month later, PET-CT results showed the patient reached CR and laboratory test showed that the patient haemogram recovery. However, the patient sought a folk remedy of traditional Chinese medicine called ‘cinobufagin’ as a diet therapy for consolidation. Due to improper dispensing and over dose, the patient developed multiple organ failure on 20 March 2017 and died on 24 March 2017.
Figure 4. . Probability of patient progression-free survival and overall survival.
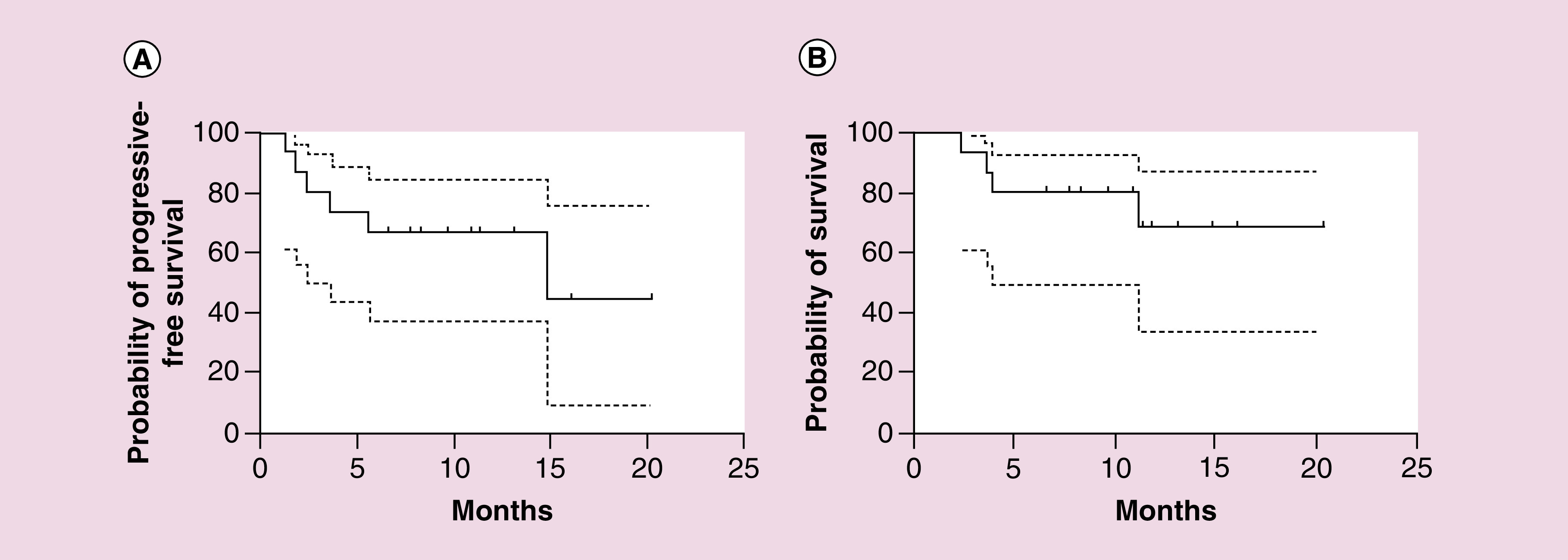
(A) Patient median PFS was 14.82 months (95% CI: 0–31.20 months) after ASCT and CAR T-cell sequential treatment. PFS rate at 6 months was 64.29% (95% CI: 39.18–89.40%). (B) Median OS was not reached. OS rate at 1 year was 65.48% (95% CI: 36.00–94.96%). In both (A & B), dotted lines represent 95% CI.
ASCT: Autologous hematopoietic stem cell transplantation; CAR: Chimeric antigen receptor; OS: Overall survival; PFS: Progression-free survival.
Discussion
This study showed a high response rate to ASCT and anti-CD19 CAR T-cell sequential therapy in RR-DLBCL patients. The ORR was 78.57% with median duration of PFS 14.82 months (95% CI: 0–31.20 months), with PFS rate of 64.29% (95% CI: 39.18–89.40%) at 6 months. The median OS of the patients was not reached, with OS rate of 65.48% (95% CI: 36.00–94.96%) at 1 year. Our results indicated that ASCT and CAR T-cell sequential therapy compared favorably to ASCT or CAR T-cell therapy alone for RR-DLBCL. Tisagenlecleucel CAR T-cell therapy for RR-DLBCL showed best overall response rate of 52% (42% CR, 12% PR) [27]. Axicabtagene Ciloleucel CAR T-Cell therapy showed an objective response of 82%, however, patients PFS rates at 6 months was only 49% (95% CI: 39–58%), and OS rate at 1 year was 59% (95% CI: 49–68%) [26]. A Phase I clinical trial on central memory-derived CAR T cell following ASCT in B-cell NHL has reported 13 of 16 (81.25%) patients achieved CR or continuing CR, and 50–75% patients were progression free at 1 year, but it is hard to assess disease response by CAR T cell or ASCT alone, as nine of 16 patients were CR before ASCT. However, this clinical trial has demonstrated the safety and feasibility of successively administering ASCT and CAR T-cell therapy for lymphoma patients [31]. In the current study, all 14 DLBCL patients were in PD, SD or relapsed after remission, and six patients obtained CR and five patients PR after ASCT and CAR T-cell sequential therapy. Thus, our findings suggest that ASCT and CAR T-cell sequential therapy is promising in improving outcomes of RR-DLBCL patients.
ASCT and CAR T-cell sequential therapy could benefit lymphoma patients in three ways. First, ASCT benefits anti-CD19 CAR T-cell by debulking active disease. Anti-CD19 CAR T-cell has inferior efficacy in lymphoma and solid tumors. We performed conditioning therapy and ASCT to kill lymphoma cells, reduce tumor volume and destroy immunosuppressive microenvironment before anti-CD19 CAR T-cell infusion, which could enhance anti-CD19 CAR T-cells’ function and persistence. Second, ASCT and its conditioning chemotherapy could deeply deplete lymphocytes that inhibit CAR T-cell function [32]. Several preclinical studies on mouse suggested that the recipient lymphocyte depletion could improve CAR T-cell efficacy by depleting regulatory T cells [33,34]. Previous studies reported that lympho-depleting conditioning could improve CAR T-cell antitumor effect, expansion and persistence, and thus, enhance disease-free survival [35,36]. Third, high-dose chemotherapy and ASCT aggravated leukopenia that could alleviate CRS [16,30,36]. Several studies reported that myeloid cell derived cytokines, such as IL-1, IL-6 and GM-CSF, were the major sources of CRS [37–39]. Our results showed that most patients in this study developed mild or moderate CRS, which might be attributing to myeloid cells and cytokines reduction by high-dose chemotherapy and ASCT.
There was a wide range of CAR T-cell dose infusion, from 0.74–7.89 × 106/kg. As these patients were RR-DLBCL patients who had accepted several courses of first- and second-line chemotherapy, patients developed different level of bone marrow suppression and lymphocyte damage. Therefore, some patients could not obtain enough PBMNCs and some patients had CAR T-cell expansion insufficient. Though we expected all of the patients obtained enough CAR T cells for therapy, these clinical factors restricted our therapy and lead to a discrepancy in CAR T-cell dose infusion. In this trial, we think some patients did not respond to ASCT and CAR T-cell therapy for two reasons. First, patients were in PD state before ASCT and CAR T-cell therapy. Lymphoma was rapidly progress during therapy, which offset ASCT and CAR T-cell efficacy. Second, lymphoma bulky mass cannot be cleared by ASCT and CAR T-cell and the complex immunosuppressive microenvironment suppressed CAR T-cell function, the remains of lymphoma developed rapidly. Due to the reasons above, in clinical practice, it would be better to give high-dose chemotherapy in PD or SD patients to obtain CR or PR, as PD or SD patients could not get a high possibility of remission in ASCT and CAR T-cell therapy.
Higher serum soluble IL-2 receptor levels correlated with more severe CRS patients observed in the current study, consistent with earlier reports [17,40]. Besides, we found that IL-10 elevation was also associated with severe CRS, which has not been well documented. IL-10 was known as an anti-inflammatory cytokine produced by activated immune cells and mainly target on macrophages [41]. Thus, we assumed that macrophages may be a key element in CRS development and that increased IL-10 in severe CRS patients might play a protective role by inhibiting macrophages activation. We did not find any correlation of severe CRS with elevated IL-6 or C-reactive protein, which were reported in previous studies [17,23,26,42,43].
In this therapy, we combined CAR T cell with ASCT to enhance patients’ remission and prolong duration of remission. And we are conducting another clinical trial to compare ASCT and CAR T-cell therapy with ASCT alone in treating RR-DLBCL patients. We hope this trial will give us a clear answer to the question that if patients with RR-DLBCL could benefit from CAR T cell and ASCT therapy.
Conclusion
The current study demonstrates that ASCT and anti-CD19 CAR T-cell sequential treatment could induce a high remission rate in RR-DLBCL patients. Our findings support the notion that ASCT and anti-CD19 CAR T-cell sequential treatment is an effective therapeutic option for RR lymphoma patients.
Summary points.
Relapsed or refractory diffuse large B-cell lymphoma (RR-DLBCL) patients have poor prognosis. High-dose chemotherapy and Autologous hematopoietic stem cell transplantation (ASCT) are the main salvage therapy for RR-DLBCL. However, the remission rate is not very high and half of the patients obtained remission after ASCT finally relapsed. Chimeric antigen receptor (CAR) T-cell therapy has achieved high response rate in RR-DLBCL, but the duration of remission is not long. To pursue high remission rate and long-term remission for RR-DLBCL, we have designed the clinical protocol by sequentially infusing ASC and CAR T-cell.
Fourteen RR-DLBCL were enrolled in this study. Patients received CEAC (semustine, cytarabine, etoposide and cyclophosphamide) regimen as conditioning therapy and accepted ASCT. During this process, CAR T cells were prepared and CAR T-cells infusion were scheduled at 6 days after ASCT. Disease assessment were arranged before ASCT and at 1 month, 3 months, 6 months, 9 months and 1 year after ASCT infusion by PET/computed tomography or computed tomography.
A total of 78.57% (11/14) and 71.43% (10/14) patients obtained complete remission or partial remission on 1 and 3 months assessment, respectively. However, 50.00% patients showed grade 2–3 cytokine-release syndrome (CRS) and no life-threatening adverse effects were observed. Patients’ median duration of progression-free survival was 14.82 months (95% CI: 0–31.20 months), with progression-free survival rate of 64.29% (95% CI: 39.18–89.40 %) at 6 months. The median overall survival of the patients was not reached, with 1-year overall survival rate of 65.48% (95% CI: 36.00–94.96%).
Sequentially infusion of ASCT and CAR T cell could destroy immunosuppressive microenvironment of lymphoma, improve CAR T-cell expansion and persistence, and thus, induce deeper remission. Besides, conditioning therapy before ASCT could alleviate CAR T-cell caused CRS by depleting myeloid cells and reducing cytokines release, such as IL-1, IL-6 and GM-CSF.
IL-2R and IL-10 were associated with severity of CRS. Grade 2–3 patients showed higher serum levels of IL-2R and IL-10.
This study demonstrates efficacy and feasibility of ASCT and anti-CD19 CAR T-cell sequential treatment for RR-DLBCL patients. RR-DLBCL patients showed high response rate. In the near future, ASCT and anti-CD19 CAR T-cell sequential treatment will be an effective therapeutic option for RR-DLBCL patients.
Footnotes
Financial & competing interests disclosure
J Yang received funding from the National Natural Science Foundation of China (NSFC) (81470322, 81770209) and the National Science and Technology Major Project (2017ZX09304030). The remaining authors received no specific funding for this work. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing assistance was utilized in the production of this manuscript. The authors thank J Chen, B Sellers and Z Wu for their kind help with the manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board from the Shanghai Changhai Hospital Ethics Committee and have followed the principles outlined in the Declaration of Helsinki for all human experimental investigations. In addition, informed consent was obtained from the participants involved.
Data sharing statement
The authors certify that this manuscript reports original clinical trial data, Clinical trial registration: ChiCTR-OIN-15007668. Individual participant data that underlies the results reported in the article, after de-identification (text, tables, figures and supplement) are available along with the study protocol and statistical analysis plan. The data will be available until the trial ends. Proposals to access data should be directed to Tao Wang using the following email address chwangtao18@163.com.
References
- 1.Armitage JO, Gascoyne RD, Lunning MA, Cavalli F. Non-Hodgkin lymphoma. Lancet 390(10091), 298–310 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Mounier N, Briere J, Gisselbrecht C. et al. Rituximab plus CHOP (R-CHOP) overcomes bcl-2--associated resistance to chemotherapy in elderly patients with diffuse large B-cell lymphoma (DLBCL). Blood 101(11), 4279–4284 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Coiffier B, Thieblemont C, Van Den Neste E. et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood 116(12), 2040–2045 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coiffier B, Lepage E, Briere J. et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 346(4), 235–242 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Pfreundschuh M, Trumper L, Osterborg A. et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol. 7(5), 379–391 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Feugier P, Van Hoof A, Sebban C. et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d'Etude des Lymphomes de l'Adulte. J. Clin. Oncol. 23(18), 4117–4126 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Thompson CA, Ghesquieres H, Maurer MJ. et al. Utility of routine post-therapy surveillance imaging in diffuse large B-cell lymphoma. J. Clin. Oncol. 32(31), 3506–3512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sehn LH, Berry B, Chhanabhai M. et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood 109(5), 1857–1861 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Kewalramani T, Zelenetz AD, Hedrick EE. et al. High-dose chemoradiotherapy and autologous stem cell transplantation for patients with primary refractory aggressive non-Hodgkin lymphoma: an intention-to-treat analysis. Blood 96(7), 2399–2404 (2000). [PubMed] [Google Scholar]
- 10.Stiff PJ, Unger JM, Cook JR. et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N. Engl. J. Med. 369(18), 1681–1690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haioun C, Lepage E, Gisselbrecht C. et al. Survival benefit of high-dose therapy in poor-risk aggressive non-Hodgkin’s lymphoma: final analysis of the prospective LNH87-2 protocol--a groupe d'Etude des lymphomes de l'Adulte study. J. Clin. Oncol. 18(16), 3025–3030 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Philip T, Guglielmi C, Hagenbeek A. et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N. Engl. J. Med. 333(23), 1540–1545 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Hamadani M, Hari PN, Zhang Y. et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 20(11), 1729–1736 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gisselbrecht C, Schmitz N, Mounier N. et al. Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: final analysis of the collaborative trial in relapsed aggressive lymphoma. J. Clin. Oncol. 30(36), 4462–4469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crump M, Neelapu SS, Farooq U. et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 130(16), 1800–1808 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brudno JN, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 15(1), 31–46 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Maude SL, Frey N, Shaw PA. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371(16), 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davila ML, Bouhassira DC, Park JH. et al. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int. J. Hematol. 99(4), 361–371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shank BR, Do B, Sevin A, Chen SE, Neelapu SS, Horowitz SB. Chimeric antigen receptor T cells in hematologic malignancies. Pharmacotherapy 37(3), 334–345 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Brentjens RJ, Riviere I, Park JH. et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118(18), 4817–4828 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davila ML, Riviere I, Wang X. et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 6(224), 224–225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee DW, Kochenderfer JN, Stetler-Stevenson M. et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a Phase I dose-escalation trial. Lancet 385(9967), 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turtle CJ, Hanafi LA, Berger C. et al. CD19 CAR-T cells of defined CD4+: CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 126(6), 2123–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kochenderfer JN, Wilson WH, Janik JE. et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116(20), 4099–4102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locke FL, Neelapu SS, Bartlett NL. et al. Phase I Results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol. Ther. 25(1), 285–295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neelapu SS, Locke FL, Bartlett NL. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377(26), 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuster SJ, Bishop MR, Tam CS. et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380(1), 45–56 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Wang T, Gao L, Hu X. et al. Chimeric antigen receptor-modified donor lymphocyte infusion improves the survival of acute lymphoblastic leukemia patients with relapsed diseases after allogeneic hematopoietic stem cell transplantation. J. Immunother. 42(3), 81–88 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Dueck AC, Mendoza TR, Mitchell SA. et al. Validity and reliability of the US National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). JAMA Oncol. 1(8), 1051–1059 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127(26), 3321–3330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Popplewell LL, Wagner JR. et al. Phase I studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 127(24), 2980–2990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 155(4), 1063–1074 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348(6230), 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gattinoni L, Finkelstein SE, Klebanoff CA. et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 202(7), 907–912 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gardner RA, Finney O, Annesley C. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 129(25), 3322–3331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hay KA, Hanafi LA, Li D. et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 130(21), 2295–2306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Norelli M, Camisa B, Barbiera G. et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 24(6), 739–748 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Sterner RM, Sakemura R, Cox MJ. et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 133(7), 697–709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giavridis T, van der Stegen SJC, Eyquem J. et al. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 24(6), 731–738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porter D, Hwang W, Frey N. et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 7(303), 303–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ip WKE, Hoshi N, Shouval DS. et al. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356(6337), 513–519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teachey DT, Lacey SF, Shaw PA. et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 6(6), 664–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee DW, Kochenderfer JN, Stetler-Stevenson M. et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a Phase I dose-escalation trial. Lancet 385(9967), 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]