

Abstract
Phakomatoses or Neurocutaneous syndromes are a heterogeneous group of disorders and have variable inheritance pattern. Currently, more than 30 entities are included in this group. These disorders primarily affect the central nervous system; however, skin, viscera, and other connective tissues can also be involved with variable clinical presentation. We will describe and illustrate the various radiological findings of the common entities through the iconography of the cases presented to our department.
Keywords: Central nervous system, magnetic resonance imaging, neurocutaneous, neurofibromatosis, phakomatoses
Introduction
The phakomatoses or neurocutaneous syndromes are a heterogeneous group of congenital disorders which primarily involve structures derived from the embryological neuroectoderm. These syndromes typically affect the central nervous system (CNS). Peripheral nerves, skin, connective tissue, and other organ systems may also be involved. Imaging plays an important role in diagnosis by early identification of pathognomonic findings in some cases. It is also important for screening of family members and monitoring of affected patients. The imaging technique of choice is magnetic resonance imaging (MRI) for the study of CNS. Computed tomography (CT) and ultrasonography (USG) are particularly useful for the study of abdominal manifestations.
1. Neurofibromatosis type 1 (NF1)
NF1 also known as von Recklinghausen disease is the most common of neurocutaneous and inherited tumor syndromes. It occurs in approximately 1 in 3000 to 1 in 4000 live births.[1,2] NF1 is inherited in an autosomal dominant pattern and is the result of the mutation of NF1 gene, located on chromosome 17q 11.2. The disease has a variable clinical presentation and affects the brain, skull, orbits, spine, musculoskeletal system, and skin. Café au lait spots are the earliest and most common clinical finding. Two or more criteria must be present for a diagnosis of NF1 [Table 1].
Table 1.
Diagnostic criteria of Neurofibromatosis type 1(NF1)
| Clinical diagnosis of NF1 is established when two or more of the following are seen in a patient: |
| i. Six or more cafe’ au lait macules with size over 5 mm in prepubertal individuals and over 15 mm in postpubertal individuals |
| ii. Two or more neurofibromas of any type or one plexiform neurofibroma |
| iii. Two or more Lisch nodules (iris hamartomas) |
| iv. Freckling in the axillary or inguinal region |
| v. Optic nerve glioma |
| vi. One or more distinctive osseous lesions like sphenoid dysplasia, pseudoarthrosis |
| vii. First degree relative with NF1 by the above criteria |
I. Intracranial manifestations, orbital and skull involvement
CNS lesions are seen in 15%–20% of cases and findings include both neoplastic lesions and hamartomas.[2,3] Optic pathway gliomas (OPG) are the most common CNS neoplasm associated with NF1, seen in approximately 15%–20% of patients.[2,4] Majority of them are juvenile pilocytic astrocytoma. They are seen as diffuse fusiform enlargement or elongation of one or both optic nerves [Figure 1]. The tumors may extend posteriorly into the optic chiasma, optic tracts and radiations. The tumors are isointense on T1W images and hyperintense on T2W images with variable post contrast enhancement.
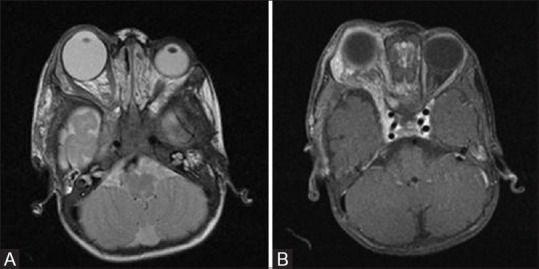
Figure 1 (A and B).
Axial T2 (A) and coronal FLAIR (B) MR images of a child with NF1 show enlargement and buckling of left optic nerve (white arrow) consistent with optic pathway glioma
The mesencephalic tectum is the commonest site of glioma after the optic pathways. These are typically iso- to hypointense on T1W and hyperintense on T2W MR images with no or minimal enhancement [Figure 2]. Hydrocephalus may be seen due to tectal plate glioma or associated aqueductal stenosis. Low-grade gliomas can also occur in the brainstem, cerebellum, and basal ganglia.
Figure 2 (A and B).
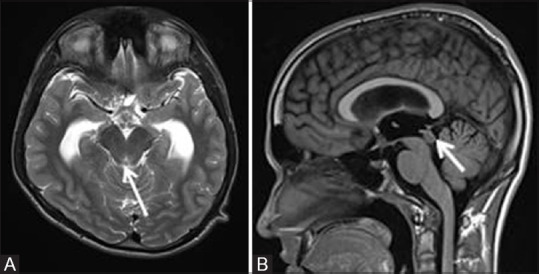
T2W axial MR image (A) of a 14-year male child with NF1 shows noncommunicating hydrocephalus with moderate dilatation of bilateral lateral and third ventricles with a small tectal plate glioma seen as T2 hyperintense lesion (white arrow). T1W sagittal MR image (B) showing the lesion to be isointense to hypointense (white arrow)
Nonneoplastic dysplastic white matter lesions are the most common neuroradiologic manifestations seen in approximately 70% of NF1 patients, usually in children.[2] These are known as foci of abnormal signal (FASI) or unidentified bright objects (UBO) and are seen as multifocal hyperintensities on T2/FLAIR images [Figure 3]. These represent regions of myelin vacuolization or hamartomas. Common locations are basal ganglia (globus pallidus), thalamus, cerebellar peduncles and dentate nuclei, centrum semiovale, and brainstem. They are typically iso- to minimally hypointense on T1W images with no mass effect or post contrast enhancement. They increase in size and number in the first decade of life followed by waning. Therefore, follow-up MRI helps to differentiate these lesions from low-grade neoplasms.
Figure 3 (A-C).
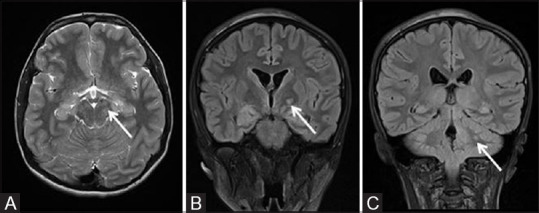
MR brain axial T2 and coronal FLAIR images of a 16-year-old male child with NF1 showing multiple areas of signal alternation (white arrows) appearing hyperintense on T2/FLAIR in brain stem (A), left ganglio-capsular region (B), and in left cerebellar hemisphere (C). These represent NF1 spots/Unidentified bright objects (UBO)
Neurofibromas (NFs) are commonly seen in NF1 patients. They may be localized discrete NFs or diffuse plexiform NFs (PNFs). NFs most often involve the scalp, orbits, neck, spine, and paraspinal soft tissues. They are hypointense on T1W and hyperintense on T2W MR images with strong heterogeneous post contrast enhancement. They may show the typical “target sign” with a hyperintense rim and relatively hypointense center on T2W images. PNFs are most common in the orbits and are seen as poorly marginated infiltrative masses involving the extra ocular muscles, orbital fat, and eyelid [Figure 4]. Involvement of skull base and adjacent neck spaces is commonly seen. Malignant peripheral nerve sheath tumors can arise in approximately 10% of PNFs and often difficult to distinguish from the parent tumor on imaging.[5]
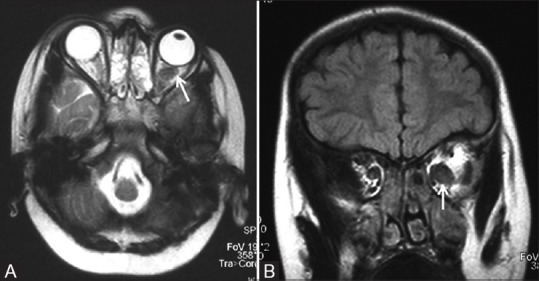
Figure 4 (A and B).
T2W axial (A) and T1 fat-saturated post contrast axial (B) MR images of a one-year-old child with NF1 showing right-sided ocular globe enlargement (Buphthalmos) and plexiform neurofibroma as enhancing T2 hyperintense soft tissue in right infratemporal region with involvement of extraconal orbital compartment and right cavernous sinus
Another characteristic finding of NF1 is bony dysplasia involving greater wing of sphenoid. Noncontrast CT scan demonstrates hypoplastic sphenoid wing with widening of orbital fissure and enlarged middle cranial fossa [Figure 5]. Exophthalmos may result from protrusion of anterior temporal lobe. Other orbital findings include buphthalmos (globe enlargement) and plexiform neurofibromas.
Figure 5 (A and B).
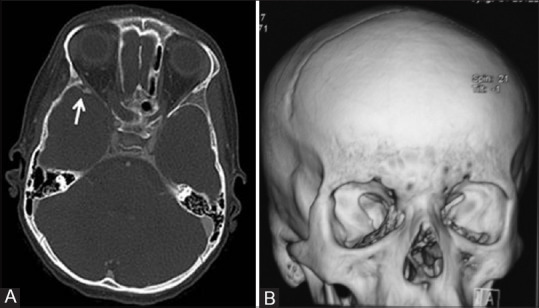
Axial CT image (A) in bone window setting of an 8-year-old girl child with NF1 reveals anterior displacement and thinning of the sphenoid bone on the right side (white arrow). 3D Volume rendered reconstructed CT image (B) of the same patient showing dysplasia of right sphenoid bone. Enlargement of the right middle cranial fossa as a result of greater sphenoid wing dysplasia is also visible
II. Spinal and other manifestations
Multiple bilateral intraspinal and paraspinal neurofibromas are commonly seen. Both intradural and extradural spinal lesions extend along the neural foramina with secondary enlargement of these foramina [Figure 6]. Other manifestations include kyphoscoliosis, dural ectasia with posterior vertebral body scalloping [Figure 6], and lateral thoracic meningoceles. Extra CNS manifestations include cutaneous neurofibromas, pseudoarthroses and bowing of the long bones of extremities [Figure 7], ribbon-like ribs, and overgrowth of all or a portion of a limb.
Figure 6 (A and B).
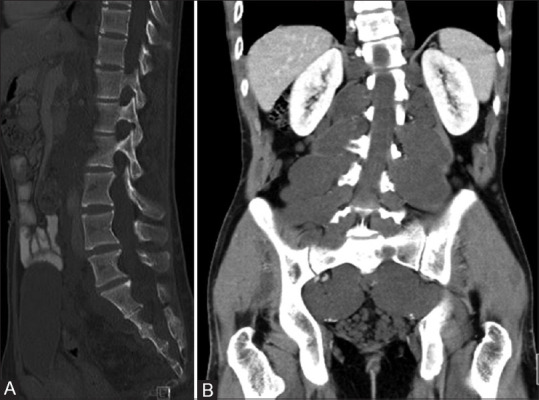
Sagittal bone window CT image (A) of a patient with NF1 showing posterior vertebral body scalloping due to dural ectasia. Post contrast coronal CT images (B) showing hypodense neurofibromas along spinal nerves in bilateral paraspinal location with widening of neural foramina
Figure 7.
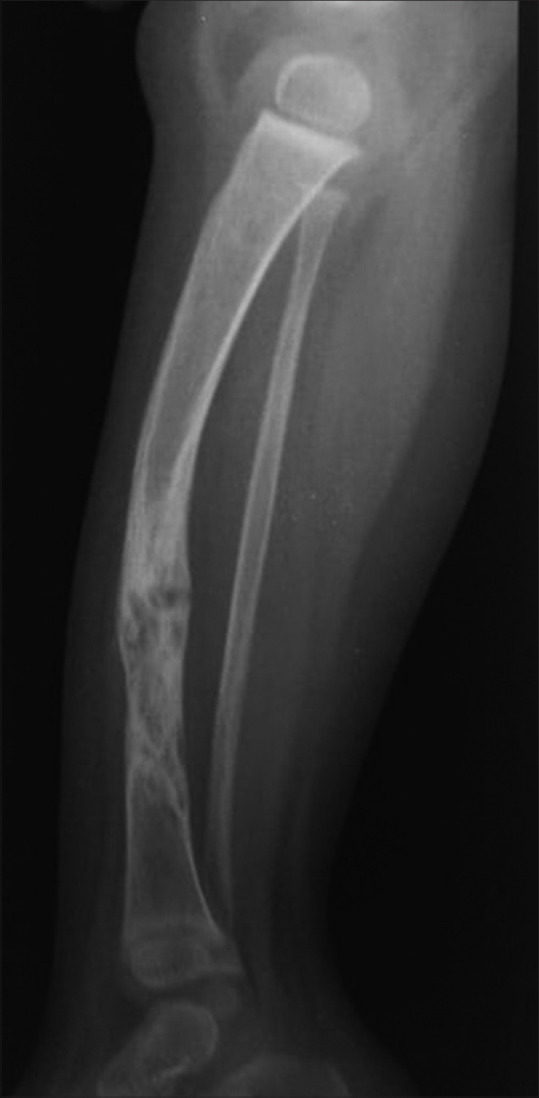
Lateral radiograph of right leg of a child showing the bowing of tibia with pseudoarthrosis formation
2. Neurofibromatosis type 2 (NF2)
Like NF1, NF2 is an autosomal dominant disorder with mutation of NF2 gene on chromosome 22q12. However, cutaneous manifestations are relatively uncommon in NF2. The prevalence of NF2 is much lower, about 1 in 25000–40,000.[2,6] Diagnostic criteria are described in Table 2.
Table 2.
Diagnostic criteria of Neurofibromatosis type 2 (NF2)
| A diagnosis of NF2 requires any one of the following two conditions: |
| i. Bilateral Vestibular schwannomas (VS) |
| ii. Family history of NF2 plus 1) Unilateral VS or 2) Any two of: meningioma, glioma, neurofibroma, schwannoma, or juvenile posterior subcapsular lenticular opacities |
I. Intracranial manifestations
CNS lesions are virtually present in all patients of NF2. The characteristic lesions are schwannomas of multiple cranial nerves most commonly vestibular nerve followed by trigeminal and oculomotor nerves. Bilateral vestibular schwannomas are pathognomonic of NF2. MR scan shows mass in cerebellopontine angle cistern with widening of internal auditory canal (Ice-cream cone appearance). They are characteristically bright on T2W, iso to hypointense on T1W MR Images with strong homogeneous contrast enhancement, although cystic changes are seen in some cases [Figure 8]. CISS or SPACE MR sequences are useful in better delineation of small tumors within the internal auditory canal.
Figure 8 (A and B).
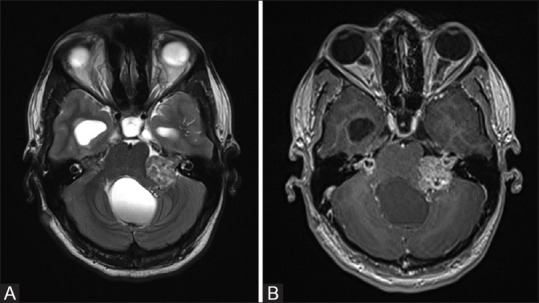
T2W axial MR image (A) of a patient with NF2 showing typical hyperintense bilateral vestibular schwannomas. There is also associated hydrocephalus. Post contrast image (B) shows relatively heterogeneous intense enhancement
Meningiomas occur in approximately 50% patients with NF2.[2] They are multiple and frequently located along falx cerebri and cerebral convexity. CT typically shows hyperdense extraxial mass lesions with areas of calcification and associated hyperostosis of adjacent skull. On MRI, they are typically isointense on both T1W and T2W images, with avid and homogeneous contrast enhancement [Figure 9]. Lateral ventricle is the common intraventricular location; they may be associated with choroid plexus enlargement and calcification [Figure 10].
Figure 9 (A and B).
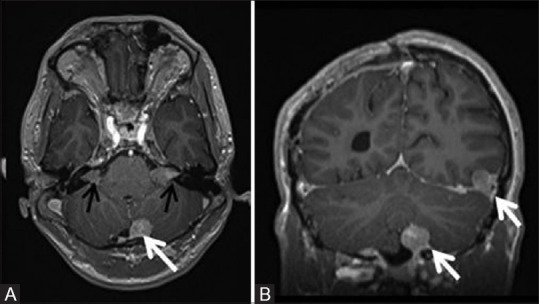
Post contrast axial (A) and coronal (B) T1W MR images of a 19-year-old patient with NF2 showing multiple dural based enhancing meningiomas (white arrows) and bilateral vestibular schwannomas (black arrows) with typical Ice cream cone appearance and widening of bilateral internal acoustic meatii
Figure 10 (A-C).
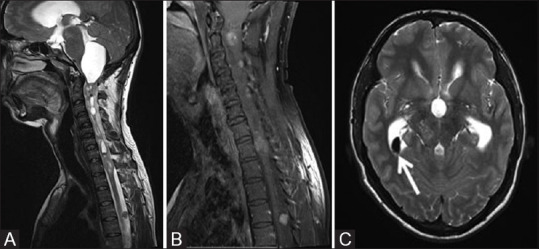
Sagittal T2W (A) and post contrast T1W (B) images of the cervicodorsal spine of the same patient as shown in Figure 8 showing intramedullary hyperintense ependymoma in cervicodorsal spinal cord with contrast enhancement with associated syrinx and large peritumoral cyst attenuating and deforming the fourth ventricle and upstream hydrocephalus. T2W axial MR image (C) of brain in the same patient showing small hypointense lesion in right lateral ventricle (white arrow) consistent with calcified choroid plexus meningioma
II. Spinal manifestations
Multiple schwannomas can occur along spinal nerve roots. These present as large dumbbell-shaped masses expanding the neural foramina with both intradural and extradural components. Multiple ependymomas are seen in spinal cord in the settings of NF2 especially in cervical cord and cervicomedullary junction. They typically present as well-circumscribed intramedullary enhancing masses appearing isointense on T1W and hyperintense on T2W images relative to the adjacent parenchyma [Figure 10].
3. Tuberous sclerosis (TS)
Tuberous sclerosis (TS) is a congenital multisystem disorder with prevalence of 1 in 6,000 to 10,000 and inherited in an autosomal dominant pattern.[2,6] Characteristic clinical presentation is triad of skin lesion (facial angiofibroma), mental retardation, and seizures. Hamartomatous growths are seen in multiple organ systems including CNS, eye, skin, kidneys, and lungs.
I. Intracranial manifestations
Cortical tubers or hamartomas are the most common intracranial lesions seen in 95% of cases.[3,7] They are found most commonly in gyri in frontal region. On CT, these tubers appear as hypodense area in widened gyri in younger patients which undergo calcification with progression of age. Similarly, MR appearance of these lesions also changes with age; however, MR delineates these lesions in all age groups. In neonates, the affected gyri appear enlarged and hyperintense relative to surrounding unmyelinated white matter on T1W images and hypointense on T2W MR images. With increasing age and progressive myelination, the lesions appear hypointense on T1W and hyperintense on T2W images [Figure 11].
Figure 11 (A and B).
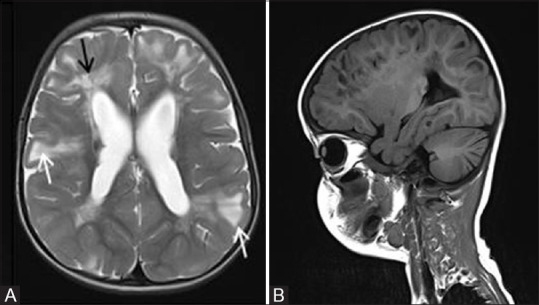
Axial T2-weighted image (A) of a 5-year-old boy with tuberous sclerosis complex shows cortical tubers as well-circumscribed areas of high signal intensity with widened gyri (white arrows). These areas appear hypointense on T1W sagittal image (B). Note made of radial migration line appearing as linear and curvilinear band of hyperintensity extending from the juxta-ventricular white matter to the cortex (black arrow)
Subependymal nodules or hamartomas are seen in 90% of the cases.[8] They are classically found along the ependymal surface of lateral ventricles in striothalamic groove between the caudate nucleus and the thalamus, just posterior to the foramen of Monro [Figures 12 and 13]. The CT and MR imaging appearances of these lesions alter with age, with progressive calcification on CT [Figure 12]. Noncalcified lesions appear hyperintense on T1-weighted and isointense to hyperintense on T2- weighted and FLAIR MR images with variable post contrast enhancement. The calcified lesions typically appear hypointense on T2 and show signal loss on susceptibility weighted MR images [Figure 13].
Figure 12 (A and B).
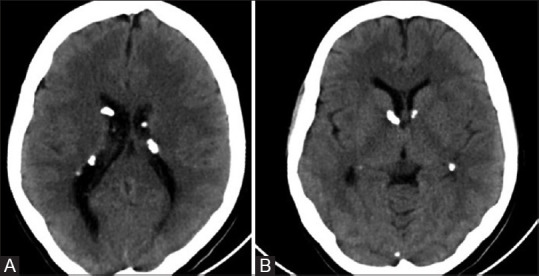
Noncontrast axial CT images (A and B) of a patient with TS showing multiple calcified subependymal nodules. Note made of typical location along ependymal surface of lateral ventricle in the strio-thalamic groove between caudate nucleus and thalamus, just posterior to the foramen of Monro
Figure 13.
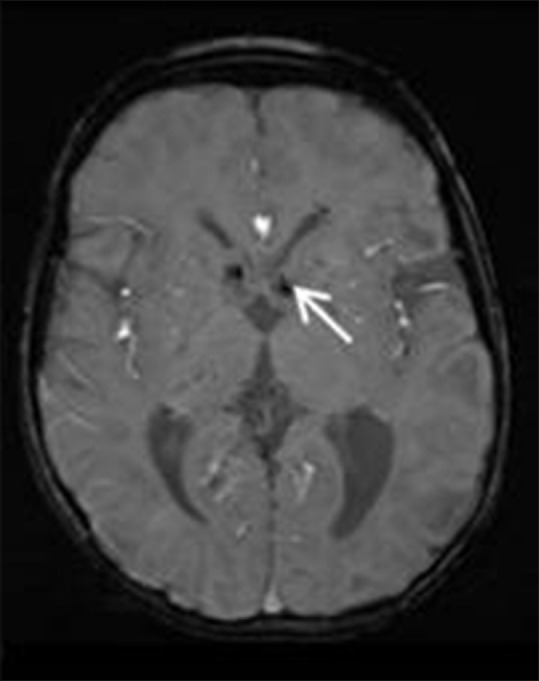
Susceptibility weighted axial MR image in a patient with TS showing multiple hypointense calcified subependymal nodules (white arrows) along the bilateral lateral ventricles near the foramen of Monro
Subependymal giant cell astrocytomas (SEGA) are seen in approximately 15% of TS patients.[8] They classically present as partially calcified enlarging masses at the foramen of Monro with associated hydrocephalus. They show mixed signal intensity on both T1W and T2W images and enhance intensely [Figure 14]. Increase in size during follow-up is the most important criteria to differentiate SEGA from subependymal nodules seen in TS patients. Neither their signal intensity nor contrast enhancement is confirmatory.
Figure 14 (A and B).
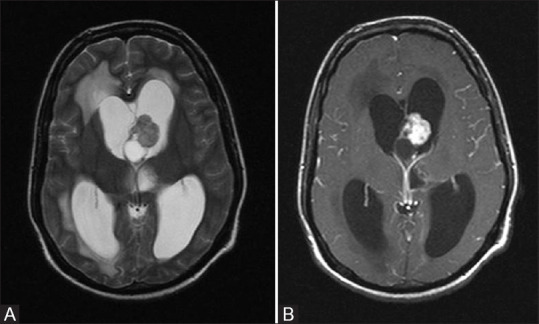
T2W axial (A) and post contrast T1 fat-saturated axial (B) images in a patient with TS show a mixed signal intensity solid cystic lesion near foramen of Monro with intense post contrast enhancement. This is classic appearance and location of Giant cell astrocytoma. Note made of associated hydrocephalus with periventricular transependymal edema
White matter lesions known as radial migration lines are seen in 90% of cases.[8] On T2W MR images, they appear as linear or curvilinear hyperintense area in the white matter, extending from subependymal nodules to cortical tubers [Figure 11].
II. Other manifestations
Pulmonary lymphangioleiomyomatosis (LAM) is a rare disease, which can be seen in patients with TS and typically affects females. CT chest classically shows multiple well defined thin walled cystic lesions diffusely distributed in lung parenchyma. Almost 70%–80% of patients with TS develop renal angiomyolipoma (AML).[7,8,9] Diagnosis is relatively straightforward on CT and MR imaging showing intralesional fat [Figure 15]. AMLs are frequently associated with intratumoral aneurysms which can undergo rupture. Unlike AML, renal cell carcinoma (RCC) is a relatively rare manifestation of TS and imaging findings depend on RCC subtypes.
Figure 15.
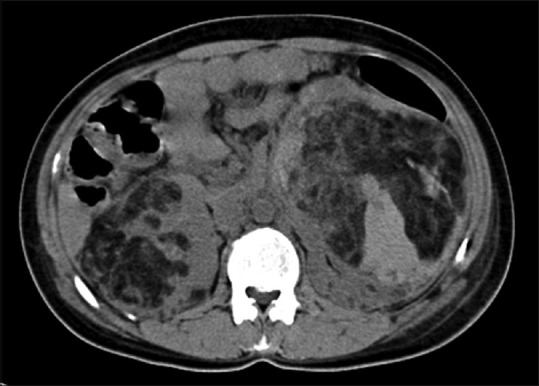
Noncontrast axial CT image in a patient with TS showing large AML involving bilateral kidneys with intratumoral fat attenuation areas
4. Sturge weber syndrome (SWS)
This is a rare congenital disorder with an incidence of about 1 in 40,000 to 50,000[6,10] and affects both sexes equally. The disease affects vasculature of face, eye, and leptomeninges. The facial angioma (port wine stain) is usually present at birth and is typically seen along the distribution of ophthalmic division of the trigeminal nerve. Leptomeningeal angioma involving the pia mater in one cerebral hemisphere is the major intracranial abnormality, which leads to paucity of superficial cortical venous drainage with chronic venous ischemia.
Post contrast T1W MR images best demonstrate the leptomeningeal angioma. T2W images show low signal in the affected gyri and adjacent white matter likely due to calcification, or accelerated myelination. Associated cerebral cortical atrophy and calcification can be seen as early as one year of age [Figure 16]. Skull radiographs may show the classical parallel tram-track calcification. Contrast- enhanced CT and MR also show enlargement and calcification of the ipsilateral choroid plexus [Figure 17]. Enlarged vessels can be seen deep in the affected hemisphere representing prominent deep medullary veins. MR venography typically reveals relative paucity of superficial cortical veins in affected cerebral hemisphere [Figure 17].
Figure 16.
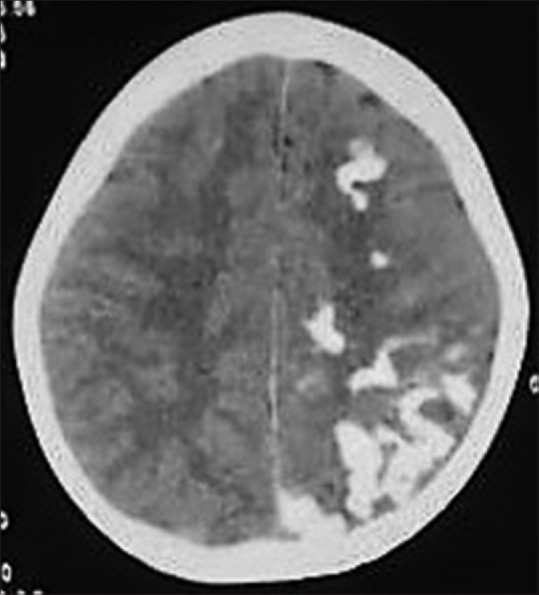
Axial noncontrast CT image of a 7-year-old child with SWS showing typical curvilinear cortical gyral calcification in left cerebral hemisphere with secondary left cerebral atrophy
Figure 17 (A-D).
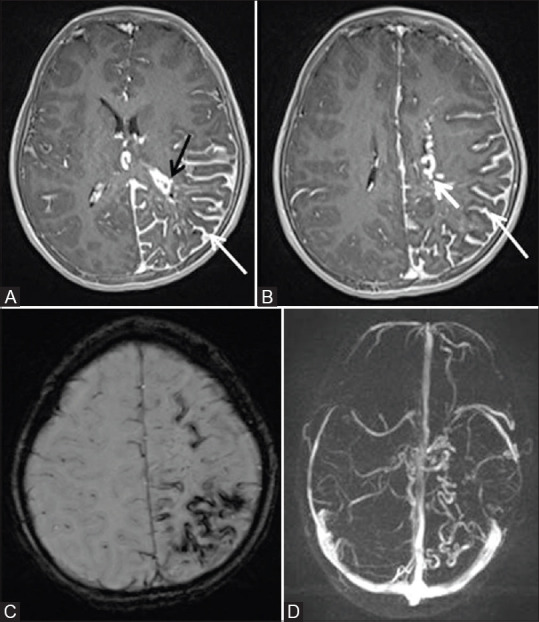
Post contrast fat sat T1W axial (A and B) MR images of the same child as shown in Figure 16, showing diffuse pial angioma (thin white arrow) in left cerebral hemisphere with atrophy of underlying cortex. Note made of enlargement of choroid plexus in left lateral ventricle (black arrow) and enlarged deep medullary veins on left side (thick white arrow). Susceptibility weighted axial MR image (C) showing curvilinear areas of blooming noted in left cerebral hemisphere consistent with calcification. MR Venography reconstructed image in axial plane (D) showing relative paucity of superficial cortical veins with prominent deep medullary veins in the affected left cerebral hemisphere
5. Von Hippel-Lindau syndrome (VHL)
VHL is inherited in autosomal dominant fashion with an incidence of approximately 1 in 36,000.[6] Characteristic manifestations include multiple neoplasms involving central nervous system and abdominal visceral organs. Hemangioblastomas are typical CNS tumors associated with this condition and can affect both brain and spinal cord. Most of the intracranial hemangioblastomas involve the cerebellum (65%), followed by brain stem (20%).[3] Cyst with a solid enhancing mural nodule is the most common imaging appearance of these lesions. The solid nodule appears hyperintense on T2W and isointense to brain on T1W MR Images. The nodules may also show flow voids within. On post contrast study, the mural nodule enhances avidly, whereas the cyst wall rarely enhances [Figure 18]. Relatively uncommon imaging appearance of hemangioblastoma includes homogenously enhancing completely solid lesion with intermediate to high signal on T2W MRI. When present in spine, there may also be associated syrinx within the cord [Figure 19].
Figure 18 (A-C).
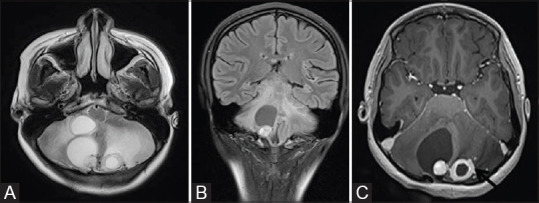
T2W axial (A) and FLAIR coronal (B) MR images of a patient with VHL show multiple hemangioblastomas in both cerebellar hemisphere with typical cyst with nodule appearance. Post contrast axial (C) image shows intense enhancement in the nodule. Thick ring-like enhancement is seen in left-sided lesion (black arrow)
Figure 19 (A and B).
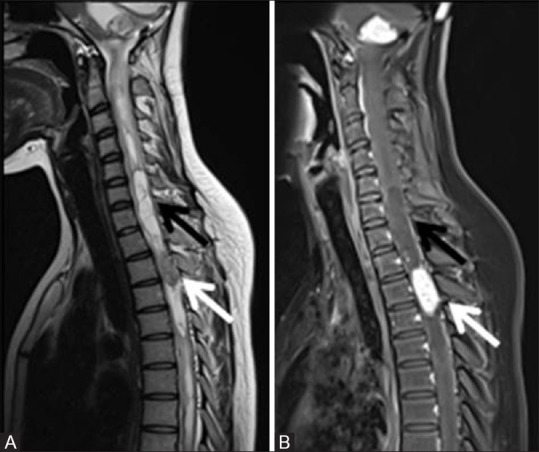
T2W (A) and post contrast fat sat T1W (B) sagittal MR images of cervicodorsal spine of the same patient as shown in Figure 18 showing a spinal intramedullary hemangioblastoma as a solid nodular enhancing lesion (white arrow) in upper thoracic spinal cord with associated syrinx formation (black arrow). The lesion is hypointense on T1 and iso to hyperintense on T2W image
Patients with VHL show relatively increased incidence of various benign and malignant neoplasms of abdominal organs. The most common findings are pancreatic cysts seen in 50%–91% patients followed by renal cysts, renal cell carcinoma, pheochromocytoma, neuroendocrine tumors of pancreas, and serous cystadenoma of pancreas in that order.[11] Contrast-enhanced CT or MR imaging of the abdomen shows the characteristic imaging findings in these lesions [Figure 20].
Figure 20.
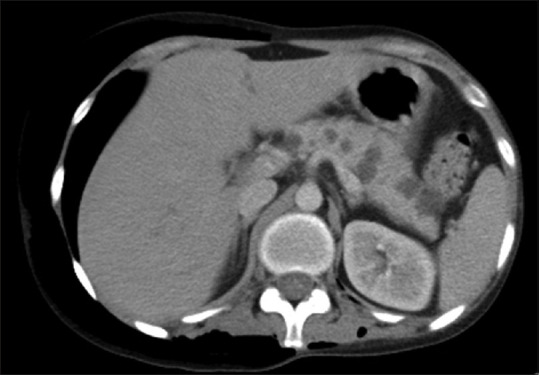
Contrast-enhanced axial CT image of a patient with VHL showing multiple variable-sized cysts involving pancreas
6. Uncommon phacomatoses
In addition to the common neurocutaneous syndromes described above, a large number of relatively uncommon phacomatoses have been described in literature. They can be broadly divided into vascular phacomatoses, melano-phacomatoses, and organoid phacomatoses. Some of these entities will be briefly discussed in the following section that may be encountered in clinical practice.
i. Ataxia telangiectasia
Ataxia telangiectasia (AT) is a rare autosomal recessive progressive neurodegenerative disorder. It is the most common cause of cerebellar ataxia in children younger than 5 years of age.[12] It is clinically characterized by multiple telangiectasias, cerebellar ataxia, pulmonary infections, and immunodeflciency. AT is classically associated with high and progressive serum levels of alpha-fetoprotein, which may serve as a diagnostic clue.[12]
Neuroimaging reveals early onset of progressive cerebellar atrophy, sometimes predominantly affecting the vermis without signal intensity abnormalities of the cerebellar cortex along with compensatory enlargement of fourth ventricle [Figure 21]. In addition to cerebellar volume loss, cerebral white matter changes in the form of T2 and FLAIR hyperintense areas have also been described on MRI, probably attributed to demyelination and gliosis. MR imaging commonly shows multiple capillary telangiectasias with focal signal hypointensities on susceptibility weighted imaging (SWI) or T2*-weighted sequences and faint focal enhancement on contrast-enhanced T1-weighted sequences.[13]
Figure 21 (A and B).
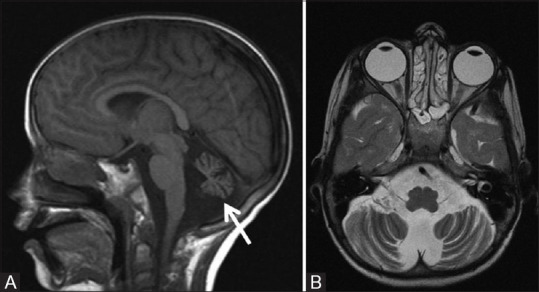
A 10-year-old male child with ataxia telangiectasia. Sagittal T1W (A) and axial T2W (B) MR images show diffuse atrophy of cerebellum involving the vermis and cerebellar hemispheres with prominent cerebellar folial pattern without any signal alternation in cerebellar cortex. Note made of associated dilatation of fourth ventricle
ii. Neurocutaneous melanosis
Neurocutaneous melanosis (NC) is a rare disorder that presents with congenital pigmented nevi (often large and multiple) and CNS involvement characterized by leptomeningeal or parenchymal melanin deposition (melanosis or melanoma). The cutaneous lesions are the hallmark of the diagnosis of NC; however, MRI plays a critical role in demonstrating CNS melanin deposition and should be performed in the first 6 months of life.[12]
CNS melanosis appears as parenchymal nodular or leptomeningeal linear hyperintense lesions on nonenhanced T1-weighted MR images. The brainstem, cerebellum, meninges, and anterior temporal lobe (especially amygdala) are most commonly affected structures. Progressive growth of the melanotic lesions, surrounding edema, mass effect, central necrosis, and hemorrhage raise suspicion for transformation to malignant melanoma. Nodular or thick plaque-like meningeal contrast enhancement is also concerning for malignant degeneration as normal melanin deposits usually do not enhance.[13]
iii. Epidermal nevus syndrome
Epidermal nevus syndrome represents a group of distinct disorders characterized by hamartomatous epidermal lesions and abnormalities of the CNS, eyes, and/or skeletal system. Epidermal nevi are usually present at birth and often occur on the face or scalp as well as on the trunk or the limbs.
The neuroimaging findings are variable depending on the underlying abnormality. Hemimegalencephaly is a relatively prominent neuroimaging feature. Other malformations of cortical development, hemiatrophy of the cerebral hemispheres, white matter signal alterations, intracranial or intra spinal lipomas, and vascular malformations may also occur.[13]
iv. Incontinentia pigmenti
Incontinentia pigmenti is a very rare X linked condition that primarily affects the skin but may also involve many other organ systems, including the brain, teeth, bones, and eyes.
Neuroimaging findings are variable; both hemorrhagic and ischemic lesions have been described. The most common imaging features are brain infarction, lesions with atrophy involving the corpus callosum, and T2/FLAIR white matter signal hyperintensity. In neonates with acute symptoms, diffusion weighted images (DWI) typically show patchy, punctate, and confluent areas of restricted diffusion in the periventricular and subcortical white matter, basal ganglia, thalami, corpus callosum, and cerebellum. Hypointense foci on susceptibility weighted imaging (SWI), indicating micro-hemorrhages, may overlap with areas of acute injury.[12]
v. Hypomelanoisis of Ito
Hypomelanosis of Ito, also known as incontinentia pigmenti achromians, most commonly affects the skin, the central nervous system, and the musculoskeletal system. It is characterized by the distinctive pattern of the skin involvement in the form of hypomacular zones with irregular borders along the skin cell developmental lines of Blaschko.[12]
Neuroimaging findings are nonspecific and variable. MRI usually shows T2/FLAIR signal abnormalities involving both the subcortical and periventricular white matter of the parietal lobes. Cystic lesions and prominent perivascular spaces within the white matter signal abnormalities may be present. Diffuse brain atrophy with cerebellar hypoplasia may also be noted. Disorders of cortical development are also commonly found including polymicrogyria, heterotopia, lissencephaly, or hemimegalencephaly.[12,13]
vi. Basal cell nevus syndrome
Basal cell nevus syndrome (BCNS), also known as Gorlin–Goltz syndrome, is a rare disorder characterized by the presence of basal cell epitheliomas and carcinomas, odontogenic keratocysts, palmar/plantar pits, and ectopic calcification of the falx cerebri.
The most typical imaging findings of BCNS are odontogenic keratocysts [Figure 22] and cerebral falx calcifications. In addition, pronounced calcifications of the pachymeninges including the tentorium, choroid plexus, and basal ganglia may be detected readily on CT scan. Other abnormalities include macro- and microcephaly, skeletal abnormalities such as bifid ribs and kyphoscoliosis, and abdominal lesions such as hamartomas and ovarian fibromas. Neuroimaging surveillance is needed due to higher risk of development of intracranial tumors, especially meningiomas and medulloblastomas.[12]
Figure 22.
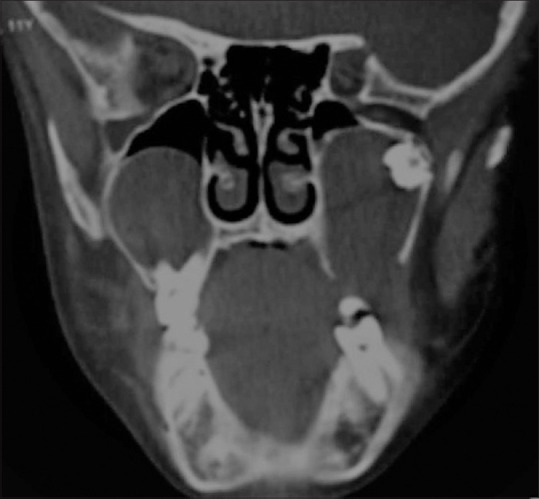
A 11-year-old boy with clinical suspicion of Basal cell nevus syndrome presenting with multiple basal cell nevi and carcinomas. CT coronal image of the maxillo mandibular region of the child shows multiple odontogenic keratocysts involving maxilla and mandible. These appear as well circumscribed unilocular cystic lesions containing unerupted tooth
vii. PHACES syndrome
PHACES syndrome has strong female preponderance and represents a constellation of abnormalities; classically described as the association of Posterior fossa malformation, facial Hemangioma, Arterial anomalies, Cardiac defects/coarctation of the aorta, Eye abnormalities, and Sternal defects.
Typical neuroimaging findings include posterior fossa abnormalities, classically unilateral cerebellar hypoplasia and Dandy-Walker malformation, and arterial abnormalities, most commonly internal carotid artery (ICA) dysgenesis and persistence of embryonic arteries. Affected patients are at an increased risk to develop intracranial aneurysms and should be screened by MR angiography. Patients may also have malformations of cortical development such as polymicrogyria or heterotopia as well as anomalies of the corpus callosum or septum pellucidum.[13]
viii. Encephalocraniocutaneous lipomatosis
Encephalocraniocutaneous lipomatosis (ECCL) is a rare congenital disorder characterized by ipsilateral scalp, eye, and brain abnormalities. Approximately 90% of the affected patients present with the characteristic skin manifestation, the nevus psiloliparus, focal area of alopecia overlying a scalp lipoma.[12] Ocular abnormalities are variable, scleral choristomas being the most common among them.
The hallmark neuroimaging finding is unilateral cerebral atrophy ipsilateral to a scalp nevus psiloliparus [Figure 23]. Other findings include intracranial and intraspinal lipomas, arachnoid cysts [Figure 23], deficiency of the falx, hydrocephalus, corpus callosum dysgenesis, ipsilateral diffuse leptomeningeal angiomatosis, inner ear abnormalities, and cortical malformations, most typically hemimegalencephaly and associated facial hypertrophy.[12] Cortical calcifications [Figure 23] are easily identified on noncontrast CT and susceptibility weighted MR images.
Figure 23 (A and B).
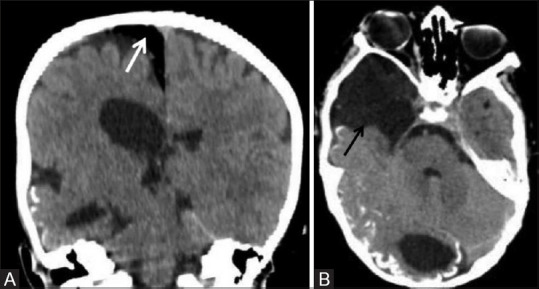
A 5-year-old male child with encephalocraniocutaneous lipomatosis presented with characteristic skin manifestation, the nevus psiloliparus and typical ocular abnormalities in the form of multiple scleral choristomas. Noncontrast coronal (A) and axial (B) CT images showing unilateral right-sided cerebral atrophy, cortical calcification in right occipital parenchyma, arachnoid cyst in right temporal region (black arrow), and extraxial intracranial lipoma (white arrow) in right para-falcine location
ix. Parry–Romberg syndrome
Parry–Romberg syndrome, also known as progressive facial hemiatrophy, is a rare progressive craniofacial disorder. Characteristic for this disease is a slowly progressive hemifacial atrophy involving the skin, underlying soft tissues, cartilaginous structures, and bone. If patients are neurologically symptomatic, they usually present with seizures, which prompts neuroimaging.
CT and MR imaging usually demonstrates the characteristic hemiatrophy of the face that tends to include the subcutaneous tissues, bones, and calvarium [Figure 24]. In addition, there may be cortical calcifications as well as white matter signal alterations, microhemorrhages, and enlargement of the ipsilateral lateral ventricle.[13]
Figure 24.
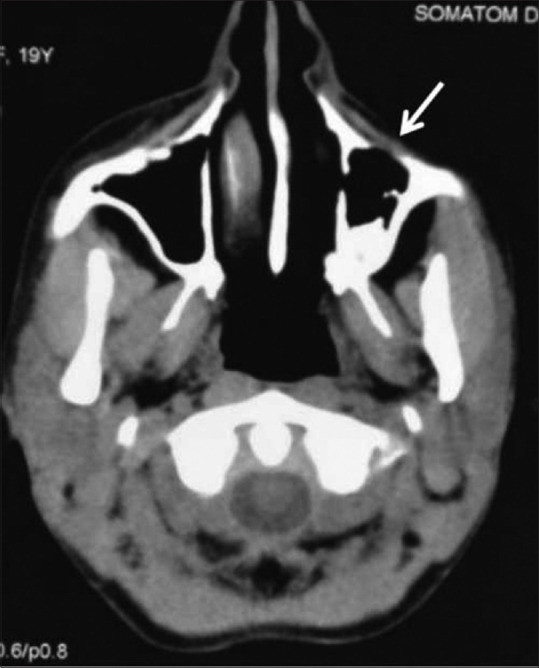
Axial noncontrast CT image in a 19-year-old female with Parry–Romberg syndrome showing hemiatrophy of the left side of the face involving the subcutaneous tissues and underlying maxillary bone
x. Cowden's syndrome
Cowden's syndrome, also called multiple hamartoma syndromes, is a rare autosomal dominant disorder, which predisposes to multiple hamartomatous neoplasms affecting almost all parts of the body. The term “COLD syndrome” refers to the association of Cowden's syndrome plus Lhermitte–Duclos disease. There is a relative predilection for central nervous system, skin, and mucosal linings of gastrointestinal and genitourinary tract. Diagnosis is based on genetic testing and the criteria established by the International Cowden's Syndrome Consortium.
Lhermitte–Duclos disease (LDD) also known as Dysplastic cerebellar gangliocytoma is a rare hamartomatous disorder. Recent studies have confirmed the relationship of Lhermitte–Duclos disease with Cowden's syndrome. At present, it is classified as a benign slowly progressive WHO Grade-I tumor, with potential for recurrence. MR imaging shows expansile cerebellar mass with typical striated, enlarged folial pattern consisting of alternating bands. These bands appear hyper to isointense on T2-weighted images and iso to hypointense on T1-weighted images [Figure 25]. SW images show prominent venous channels within the lesion which shows striking linear enhancement on post contrast image, though the mass itself usually does not enhance. Classical neurological imaging findings are sufficient for diagnosis and biopsy is usually not required.[14]
Figure 25 (A-C).
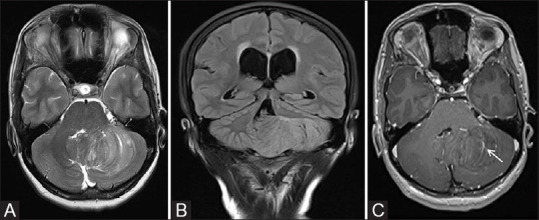
MR images of a 38-year-old female patient with LDD showing a nonenhancing expansile left cerebellar mass with thickened folia and pathognomonic “Tiger stripe” pattern on axial T2W image (A). Note made of mass effect on fourth ventricle with upstream hydrocephalus as seen on coronal FLAIR image (B). Post contrast axial image (C) shows linear enhancing prominent venous channels within the lesion (white arrow)
Testicular lipomatosis is an important diagnostic criterion because of the frequency of its occurrence in Cowden's syndrome.[15] The ultrasound appearance of testicular lipomatosis is pathognomonic. At USG, testicular lipomatosis is seen as multiple hyperechogenic foci (about 40 foci per testis) approximately 1–6 mm in diameter in the parenchyma of testes.[15] The lesions are randomly scattered and do not demonstrate posterior acoustic shadowing or vascularity [Figure 26]. Differential diagnoses include testicular microlithiasis and testicular involvement from lymphoma, sarcoidosis, and metastasis.
Figure 26.
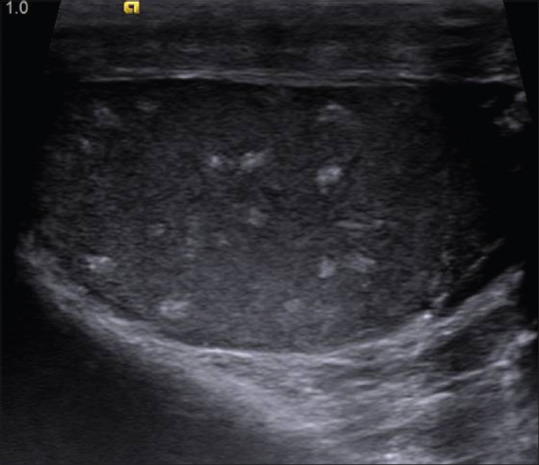
Ultrasonographic image of right testes in a patient with Cowden's syndrome showing multiple small well defined hyperechoic intratesticular masses with no shadowing. Similar findings were also seen in left testis. Testicles are normal in size and contour
Conclusion
The Phakomatoses group includes almost 30 or more individual syndromes. Although genetic testing is available, the manifestations of these syndromes cover a wide range. Imaging plays an important role in screening, early identification of abnormalities, and follow-up of lesions in a previously diagnosed case. Radiologists should be familiar with these syndromes to guide appropriate treatment and prognosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Rodriguez D, Young Poussaint T. Neuroimaging findings in neurofibromatosis type 1 and 2. Neuroimaging Clin N Am. 2004;14:149–70. doi: 10.1016/j.nic.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Osborn AG. Neurocutaneous Syndromes. Osborn's Brain: Imaging, Pathology and Anatomy. 1st ed. Canada: Amirsys; 2013. pp. 1131–70. [Google Scholar]
- 3.Herron J, Darrah R, Quaghebeur G. Intra-cranial manifestations of the neurocutaneous syndromes. Clin Radiol. 2000;55:82–98. doi: 10.1053/crad.1999.0328. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien WT. Neuroimaging manifestations of NF1-A pictorial review. J Am Osteopath Coll Radiol. 2015;4:16–21. [Google Scholar]
- 5.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–4. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin DD, Barker PB. Neuroimaging of phakomatoses. Semin Pediatr Neurol. 2006;13:48–62. doi: 10.1016/j.spen.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Von Ranke FM, Faria IM, Zanetti G, Hochhegger B, Souza AS Jr, Marchiori E. Imaging of tuberous sclerosis complex: A pictorial review. Radiol Bras. 2017;50:48–54. doi: 10.1590/0100-3984.2016.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manoukian SB, Kowal DJ. Comprehensive imaging manifestations of tuberous sclerosis. AJR. 2015;204:933–43. doi: 10.2214/AJR.13.12235. [DOI] [PubMed] [Google Scholar]
- 9.Umeoka S, Koyama T, Miki Y, Akai M, Tsutsui K, Togashi K. Pictorial review of tuberous sclerosis in various organs. Radiographics. 2008;28:e32. doi: 10.1148/rg.e32. [DOI] [PubMed] [Google Scholar]
- 10.Agrawal PJ, Kharat AT, Kuber R, Shewale S. Spectrum of CT and MR findings in Sturge-Weber syndrome: A case report. Med J DY Patil Univ. 2014;7:497–501. [Google Scholar]
- 11.Leung RS, Biswas SV, Duncan M, Rankin S. Imaging features of von Hippel-Lindau disease. Radiographics. 2008;28:65–79. doi: 10.1148/rg.281075052. [DOI] [PubMed] [Google Scholar]
- 12.Barros FS, Marussi VH, Amaral LL, da Rocha AJ, Campos CM, Freitas LF, et al. The rare neurocutaneous disorders update on clinical, molecular, and neuroimaging features. Top Magn Reson Imaging. 2018;27:433–62. doi: 10.1097/RMR.0000000000000185. [DOI] [PubMed] [Google Scholar]
- 13.Pfahler V, Ertl-Wagner B. Phacomatoses neuroimaging and clinical findings. Clin Neuroradiol. 2019:1677–703. [Google Scholar]
- 14.Shinagare A, Patil N, Sorte S. Case 144: Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) Radiology. 2009;251:298–303. doi: 10.1148/radiol.2511071390. [DOI] [PubMed] [Google Scholar]
- 15.Venkatanarasimha N, Hilmy S, Freeman S. Case 175: Testicular lipomatosis in Cowden disease. Radiology. 2011;261:654–8. doi: 10.1148/radiol.11091151. [DOI] [PubMed] [Google Scholar]