

Abstract
Psoriasis is a systemic inflammatory disease, associated with metabolic disorders, including high level of low-density lipoprotein. PCSK9, which promotes the degradation of low-density lipoprotein receptors and, there-fore, the increased concentration of circulating low-density lipoprotein, is also involved in inflammation. This study aims to examine the role of PCSK9 in psoriasis and to investigate the potential of topically applying small interfering RNA targeting Pcsk9 as a psoriasis treatment. We investigated the expression of PCSK9 in lesions of psoriasis patients and imiquimod-induced psoriatic reactions in Pcsk9-knockout and Pcsk9 small interfering RNA-treated mice, and we also used cultured human keratinocytes to investigate the role of PCSK9 in regulating cell proliferation and apoptosis. We found that PCSK9 is overexpressed in psoriatic lesions and that suppressing Pcsk9 can decrease the inflammatory reaction induced by imiquimod treatment and inhibit hyperproliferation of keratinocytes. We also found that suppressing PCSK9 can significantly alter the cell cycle and induce apoptosis of human keratinocytes. Taken together, our findings indicate that PCSK9 plays an important role in psoriasis and may be a therapeutic target.
INTRODUCTION
Psoriasis is a systemic inflammatory disease (Dreiher et al., 2008) that not only involves the skin but also coincides with several comorbidities, including cardiovascular disease and metabolic disorders (Davidovici et al., 2010). In addition, psoriasis has been shown to be associated with a higher level of low-density lipoprotein (LDL) (Santos-Juanes et al., 2015), and because of this, the American Journal of Cardiology recommends that all patients with moderate to severe psoriasis be screened for dyslipidemia (Friedewald et al., 2008). Accumulating evidence suggests that abnormal lipid metabolism and psoriasis share common pathogenic and molecular genetic mechanisms (Hansson and Libby, 2006; Rajpara et al., 2010; Shlyankevich et al., 2014). With regard to adaptive immunity, both diseases are strongly associated with a predominantly T helper (Th) type 1-cell subset producing IFN-γ, tumor necrosis factor-α, and Th17 cells, and both lack a significant proportion of Th2 cells and their corresponding regulatory/anti-inflammatory functions (Hansson and Libby, 2006; Rajpara et al., 2010). In addition, innate immune responses including toll-like receptoreinduced expression of proinflammatory cytokines are prominent in both psoriasis and atherosclerosis (Hansson and Libby, 2006; Rajpara et al., 2010). Furthermore, genome-wide association studies investigating the relationship between lipid metabolism and immune-mediated diseases suggest that, at a minimum, there is some degree of a shared genetic basis underlying the phenotypic associations (Andreassen et al., 2015). Statins, the drugs in hyper-cholesteremia treatment, have been hypothesized to decrease the risk of developing psoriasis and to augment the treatment efficacy of the current treatment (Andreassen et al., 2015; Rajpara et al., 2010) through their anti-inflammatory properties (Hansson and Libby, 2006).
PCSK9, which functions as a chaperone protein to the LDL receptor, is becoming a target for lowering blood cholesterol levels (McKenney, 2015) because it promotes the degradation of LDL receptors and thus increases plasma LDL concentration (Della Badia et al., 2016). PCSK9 is also involved in chronic inflammation (Ferri and Ruscica, 2016). For example, the NF-κB inflammatory pathway, which is induced by oxidized LDL, is down-regulated by blocking the expression of PCSK9 with small interfering RNA (siRNA), and in HepG2 cells, tumor necrosis factor-α can induce PCSK9 expression (Ruscica et al., 2016). Both NF-κB and tumor necrosis factor-α are key inflammatory cytokines involved in psoriasis and atherosclerosis, and they are also treatment targets for both diseases. In this study, we examined the role of PCSK9 in psoriasis and investigated the potential of developing a psoriasis treatment by topically applying siRNA to target Pcsk9.
RESULTS
Overexpression of PCSK9 in lesions of psoriasis patients
To investigate its potential role in psoriasis, we measured the expression of PCSK9 in lesions of psoriasis patients. Quantitative real-time PCR assays showed that at the mRNA level, PCSK9 expression is about 5 times higher in psoriatic plaques (n = 6) than normal healthy skin (n=4) (1.795 ± 0.4595 vs. 0.2426 ± 0.056, P < 0.05) (Figure 1a). Consistently, the Western blot results showed that at the protein level, PCSK9 is twice as high in psoriatic plaques than normal healthy skin (n = 4 and 2, respectively; 1.067 ± 0.0546 vs. 0.4946 ± 0.08819, P < 0.05) (Figure 1b and c). Immunohistochemistry (IHC) assay shows that psoriatic plaques have elevated expression of PCSK9 compared with normal healthy skin. In normal healthy skin (Figure 1d), few PCSK9-positive keratinocytes could be found in epidermis. In psoriatic plaques (Figure 1e), the elevated PCSK9 expression is found not only in the keratinocytes but also in the epithelial cells of the vessels in dermis. No false positive staining was detected by using rabbit IgG as false positive control (see Supplementary Figure S1 online).
Figure 1. Overexpression of PCSK9 in lesions of psoriasis patients.
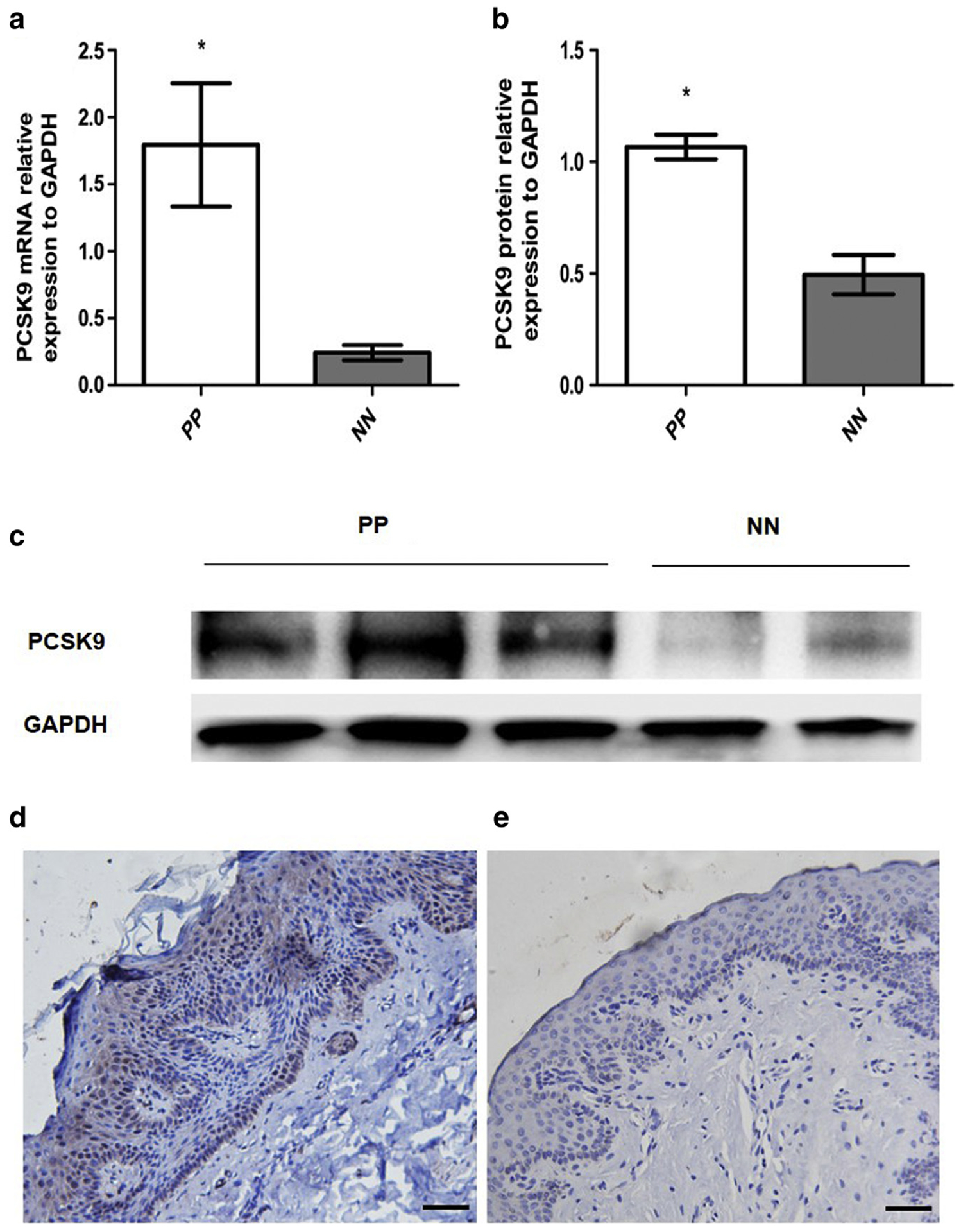
(a, b) Quantification of PCSK9 mRNA and protein in PP and NN. *P < 0.05, t test. (c) Western blot image. (d) Immunohistochemistry image of PP. Scale bar = 100 μm. (e) Immunohistochemistry image of NN. Scale bar = 100 μm. NN, normal healthy skin; PP, psoriatic plaque.
Imiquimod induces the expression of Pcsk9 in mouse skin
It is well known that imiquimod (IMQ) induces typical psoriatic reactions in animal models (van der Fits et al., 2009). We evaluated the expression of Pcsk9 in lesions of IMQ-treated B6 mice. Quantitative real-time PCR showed that the Pcsk9 mRNA expression was more than 5 times higher in the IMQ-treated skin than the controls (P < 0.05) (Figure 2a). IHC showed that in the normal skin, upper layers of the dermis had Pcsk9-positive fibroblasts, but there were no obvious positive cells in the epidermis (Figure 2b). In the IMQ-treated skin, with the typical psoriatic reaction, increased epidermal thickness was obvious. Pcsk9 is abundantly expressed in all epidermal layers (keratinocytes and melanocytes). Pcsk9 expression in the dermis is also elevated (Figure 2b). These results show that IMQ treatment up-regulates the expression of Pcsk9, suggesting that PCSK9 may play an important role in IMQ-induced psoriatic skin reactions.
Figure 2. IMQ induces the expression of Pcsk9 in mouse skin.
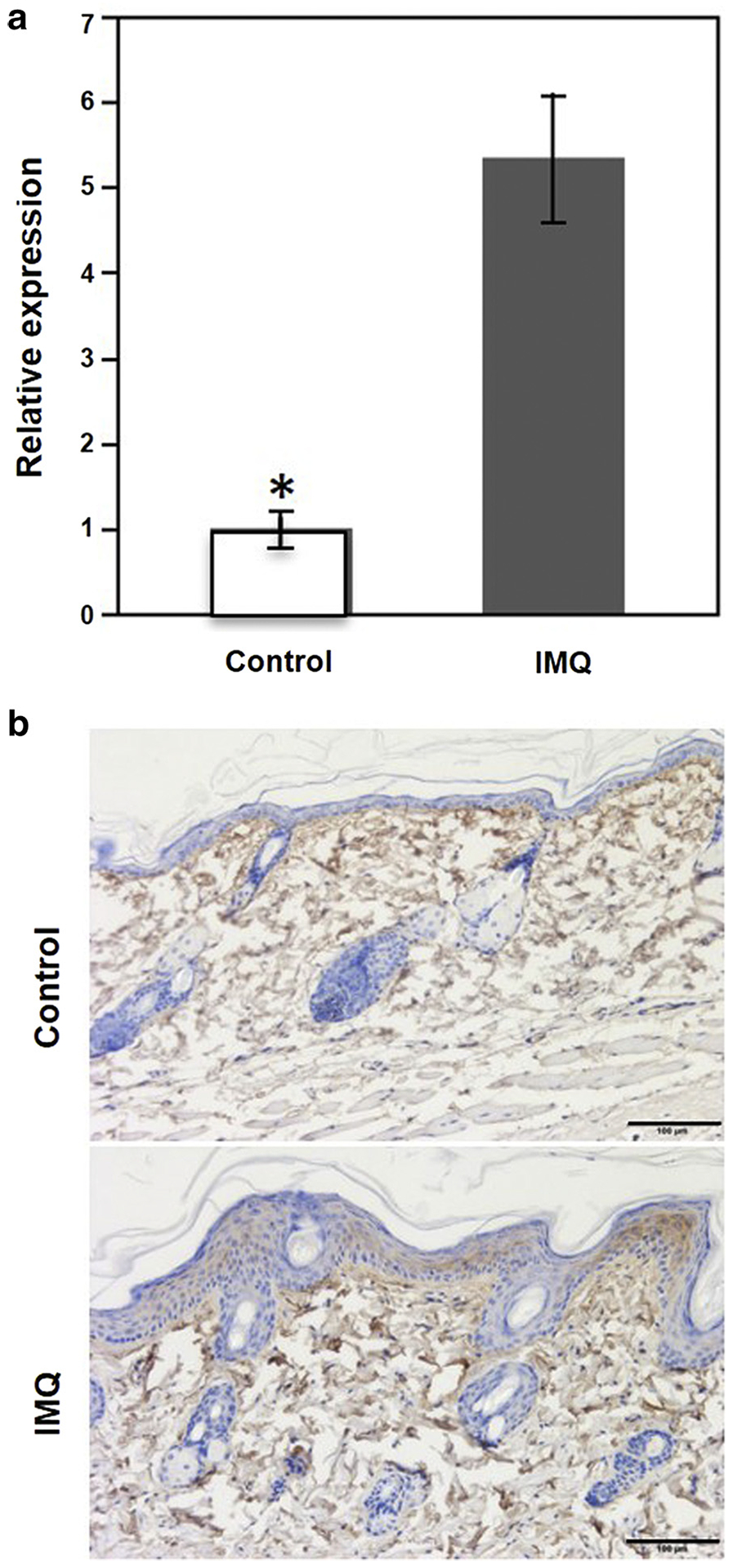
(a) Quantification of Pcsk9 mRNA expression in the IMQ-treated skin and the controls. *P < 0.05, t test. (b) Immunohistochemistry staining of normal skin and IMQ-treated skin by Pcsk9 antibody. Scale bars = 100 μm. IMQ, imiquimod.
The depletion of Pcsk9 suppresses IMQ-induced psoriasis-like reactions in vivo
Next, we tested the effects of IMQ on the skin of Pcsk9-homozygous knockout mice (Pcsk9−−) compared with wild-type (WT) mice (five mice/group). After IMQ treatment for 5 consecutive days, both groups developed lesions resembling psoriasis; however, except for scores of erythema on day 3, thickness on day 1, and scaling on days 4 and 5, at all other time points the scores in Pcsk9−/− mice were significantly weaker (P < 0.05) (Figure 3a and b). The cumulative scores of Pcsk−/− mice were significantly lower at all time points. Hematoxylin and eosin staining (Figure 3c) shows that IMQ-treated skin in WT mice developed typical characteristics of psoriasis, including hyperkeratosis, parakeratosis, acanthosis in epidermis, infiltration of inflammatory cells, and hyperproliferation of the vascular epithelia in dermis. Pcsk9−/− mice showed only limited epidermal thickening with mild hyperkeratosis but no significant parakeratosis, acanthosis, or infiltration of inflammatory cells. The thickness of epidermis in the treated area was significantly increased in both groups, but the increase was significantly suppressed in Pcsk9−/− mice (analysis of variance, P < 0.05) (Figure 3d).
Figure 3. The depletion of Pcsk9 suppresses IMQ-induced psoriasis-like reactions in vivo.
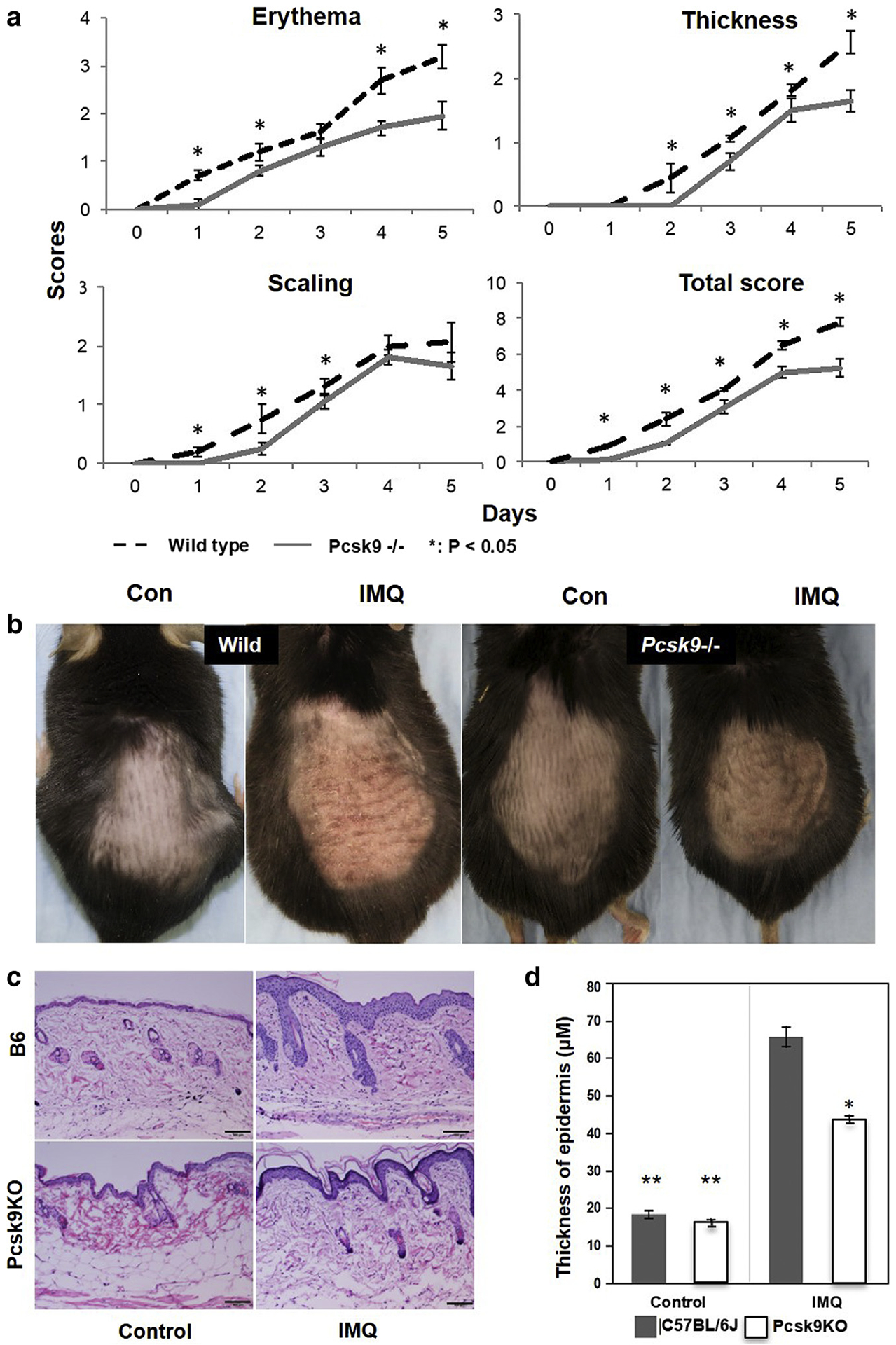
(a) Scores of erythema, scaling, thickness, and cumulative scores of WT mice and Pcsk9−/− mice during IMQ treatment. *P < 0.05, t test. (b) Representative photos of dorsal skin in WT and Pcsk9−/− mice with IMQ and vehicle treatments. (c) Hematoxylin and eosin staining of dorsal skin in WT and Pcsk9−/− mice with and without IMQ treatment. Scale bars = 100 μm. (d) Thickness of epidermis of normal skin and IMQ-induced skin in WT and Pcsk9−/− mice. *P < 0.05 comparing IMQ-treated skin between WT and Pcsk9−/− mice, **P < 0.05 comparing control skin and IMQ-treated skin in WT and Pcsk9−/− mice. Con, control; IMQ, imiquimod; KO, knockout; WT, wild type.
The depletion of Pcsk9 suppresses keratinocyte proliferation and inflammation
To further explore the mechanisms of suppressed psoriatic skin reaction in IMQ-treated Pcsk9−/−mice, we conducted IHC assays for cell proliferation marker Ki67 and an inflammatory cytokine regulator, NF-κB (p65) (Figure 4). Compared with WT mice, the increase of Ki67, p65, and phosphorylated p65 expression between untreated and treated skin was suppressed in Pcsk9−/− mice (Figure 4), although in both genotypes, Ki67, p65, and phosphorylated p65 were abundantly expressed after IMQ treatment. These results suggest that the depletion of Pcsk9 could suppress IMQ-induced keratinocyte hyperproliferation and inflammation, thus providing clues for further understanding the molecular mechanisms of the role of PCSK9 in psoriasis.
Figure 4. The depletion of Pcsk9 suppresses keratinocyte proliferation and inflammation induced by IMQ.
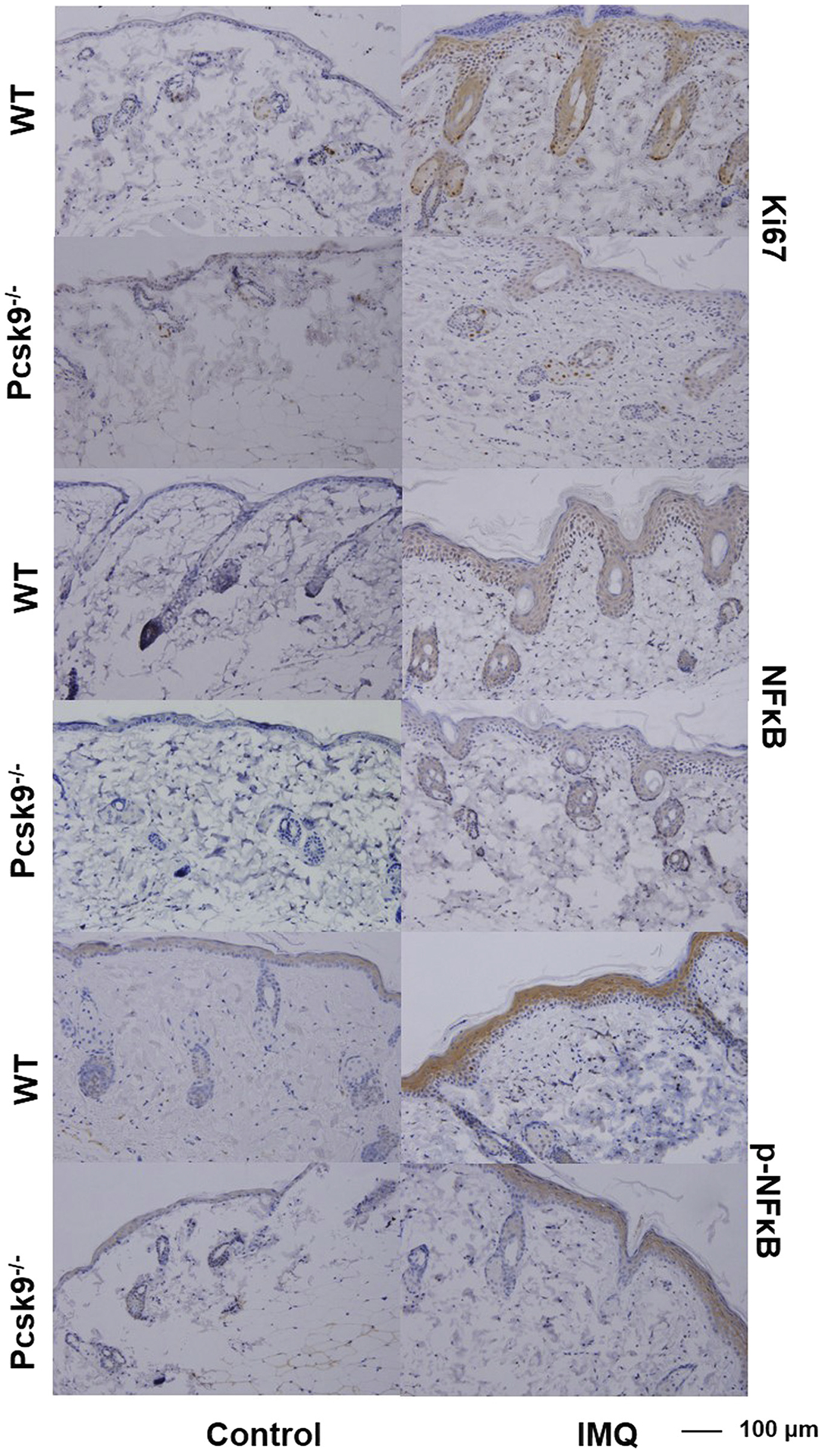
Immunohistochemistry staining of normal and IMQ-treated skin by Ki67, NF-κB (p65), and phosphorylated NF-κB (p-p65) antibodies. Scale bars = 100 μm. IMQ, imiquimod; WT, wild type.
Topical application of siRNA suppresses Pcsk9 and reduces IMQ-induced psoriasis-like reactions
To explore the potential of developing a therapy for psoriasis, we topically applied siRNA targeting mouse Pcsk9 (si-Pcsk9). At 48 hours after siRNA treatment, si-Pcsk9 significantly reduced mRNA expression of Pcsk9 in the skin (n ≥ 3, P < 0.05) (Figure 5a) compared with the control siRNA-treated skin (si-Con). Verified by Western blot, the knockdown efficiency was 63% (7.3 ± 0.8 vs. 2.7 ± 0.4 for si-Con vs. si-Pcsk9, respectively; P < 0.05) (Figure 5 b and c). After 5 consecutive days of siRNA and IMQ treatment, erythema, scaling, and thickness were much weaker in si-Pcsk9 mice compared with si-Con mice (Figure 5d). As showed in Figure 5e, the scaling score was continuously lower from the second day of IMQ treatment, the erythema score and cumulative score were significantly lower from the third day, and the thickness score was lower from the fourth day.
Figure 5. Topical application of siRNA suppresses Pcsk9 and reduces IMQ-induced psoriasis-like reactions.
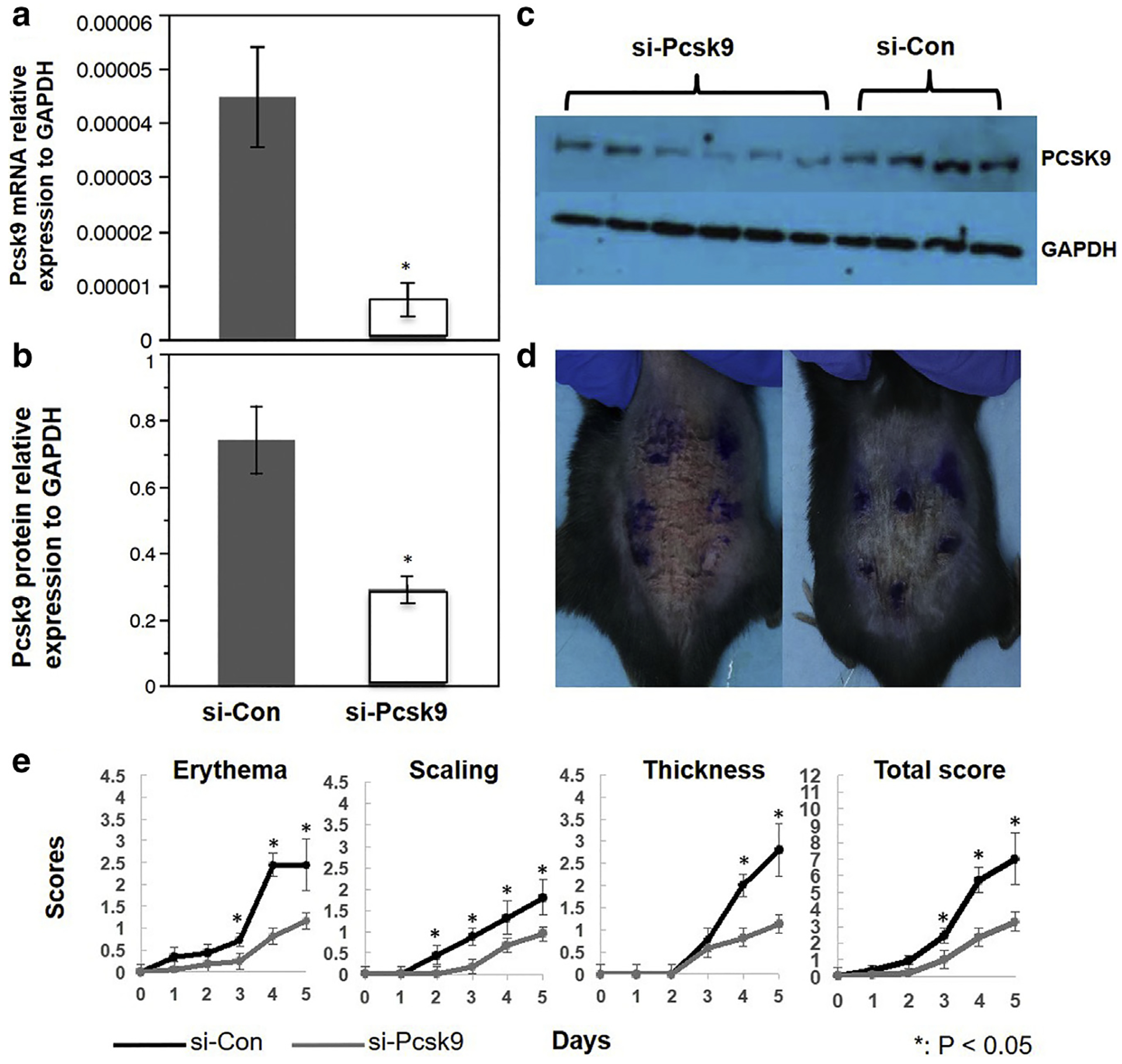
(a, b) Quantification of Pcsk9 mRNA and protein expression in the si-Pcsk9 transfected skin (48 hours after the treatment) and the controls. *P < 0.05, t test. (c) Western blot image. (d) Representative photos of dorsal skin in si-Con and si-Pcsk9 mice after 5 days of IMQ treatment. (e) Scores of erythema, scaling, thickness, and total scores of si-Con and si-Pcsk9 mice during IMQ treatment. *P < 0.05, t test. Con, control; IMQ, imiquimod; siRNA, small interfering.
Suppressing PCSK9 reduces cell viability, alters the cell cycle, and induces apoptosis of human keratinocytes in vitro
To further understand the effects of suppressing PCSK9 on keratinocytes, we carried out an in vitro study in which we verified that siRNA targeted to human PCSK9 (si-PCSK9) could effectively reduce the PCSK9 expression in cultured human keratinocytes (P < 0.05) (Figure 6a). The following 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (i.e., MTT) assay showed that compared with si-Con, si-PCSK9 significantly reduced keratinocyte viability at all three time points (24, 48, and 72 hours) after the si-PCSk9 treatment (Student t test, P < 0.001) (Figure 6b). Propidium iodide staining and flow cytometry assay (Figure 6c–e, and see Supplementary Figure S3 online) showed that the percentage of G0/G1 cells was significantly reduced (t test, P < 0.05) at the 24-hour and 72-hour time points and was suggestively reduced (P < 0.1) at the 48-hour time point in si-PCSK9etreated keratinocytes. However, compared with si-Con keratinocytes, the percentage of cells in S phase was significantly increased in si-PCSK9 keratinocytes at 24 hours and 72 hours (P < 0.05). The percentage of G2/M cells was suggestively increased (P < 0.1) at 48 hours in the si-PCSK9 keratinocytes. These results suggest that the suppression of PCSK9 could suppress the viability and alter the cell cycles of human keratinocytes in vitro. The propidium iodide staining showed that the si-PCSK9 treatment significantly (P < 0.05) increased the numbers of cells in the sub-G0/G1 stage at all three time points, suggesting an induction of apoptosis (Figure 6c–e).
Figure 6. The suppression of PCSK9 inhibits proliferation and induces apoptosis of human keratinocytes in vitro.
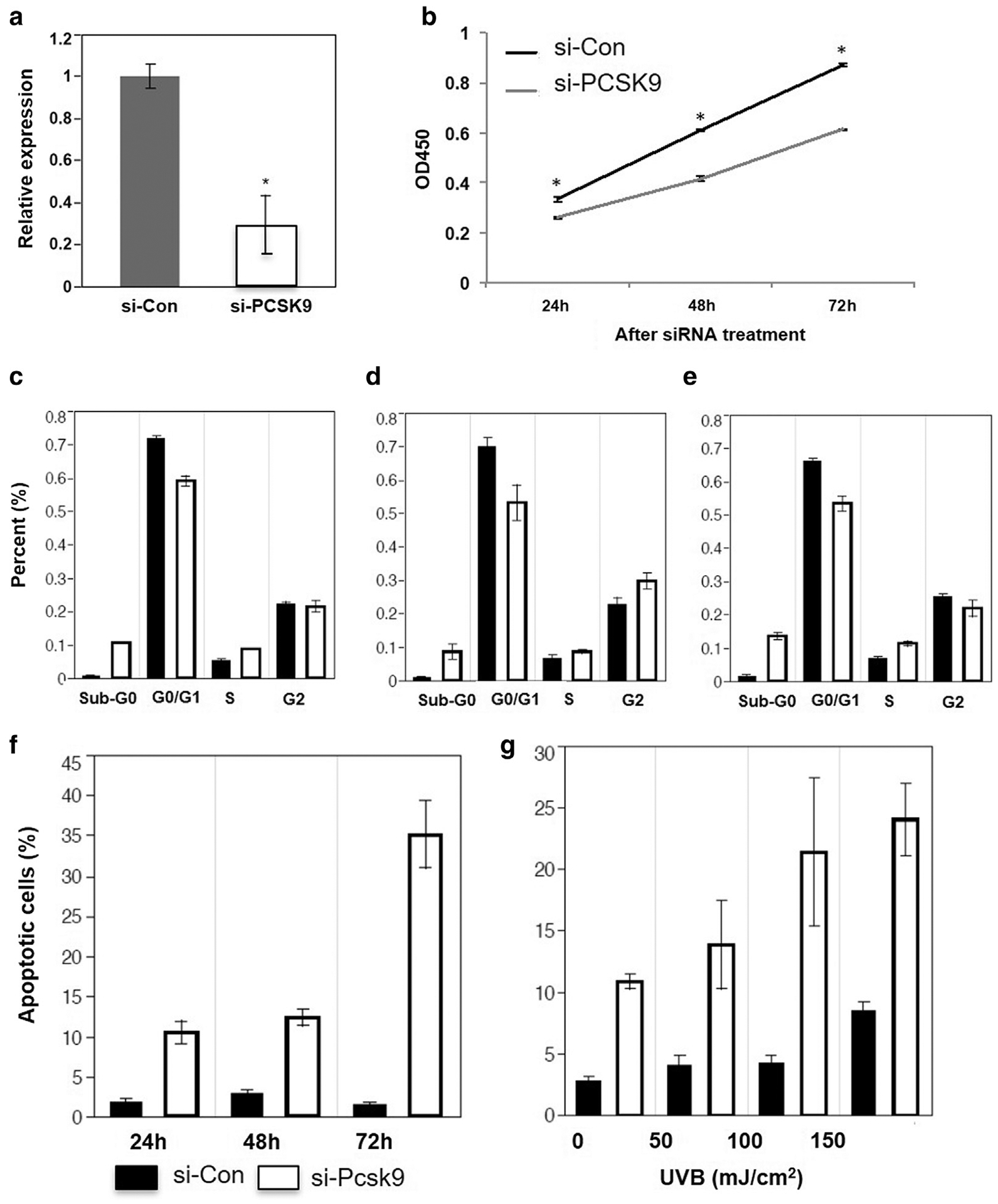
(a) Quantification of PCSK9 mRNA expression in cultured human keratinocytes transfected by si-Con and si-PCSK9. (b) MTT assay shows the viability of si-Con and si-PCSK9 transfected keratinocytes. (c–e) Percentages of G0/G1, S, and G2/M phase of keratinocytes transfected by si-Con and si-PCSK9 (24, 48, and 72 hours). (f) Percentage of apoptotic cells in keratinocytes transfected by si-Con and si-PCSK9 (24, 48, and 72 hours). (g) Percentage of apoptotic cells in si-Con– and si-PCSK9– transfected keratinocytes after exposure to narrow band–UVB in a concentration range from 0 to 150 mJ/cm2. *P < 0.05, t test. Con, control; h, hour; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; si, small interfering.
To verify this result, we applied annexin V assay and found that si-PCSK9 can significantly induce apoptosis in keratinocytes at all three time points (P < 0.01) (Figure 6f, and see Supplementary Figure S4 online). At the 72-hour time point, the number of apoptotic cells in the si-PCSK9 group was 22 times higher than in the si-Con group (35.1% ± 4.2% vs. 1.6% ± 0.22%, respectively). These results strongly suggest that PCSK9 is required for anti-apoptotic activity in keratinocytes.
UVB treatment induces apoptosis of keratinocytes and has been widely used in the clinical treatment of psoriasis (Lewis et al., 2003). We tested the effects of combining si-PCSK9 and UVB on apoptosis (Figure 6g, and see Supplementary Figure S5 online). Twenty-four hours after exposure to narrow-band UVB in a concentration range of 0–150 mJ/cm2, apoptosis was significantly elevated in si-PCSK9 keratinocytes compared with the si-Con cells (P < 0.01). As shown in Figure 6g, without UVB treatment, 10.9% cell death was found in si-PCSK9 only–treated human keratinocytes, whereas the UVB (150 mJ/cm2)-treated cells had 8.5% apoptosis without si-PCSK9 treatment. Interestingly, 24.0% cell death was found in the combined treatments of UVB (150 mJ/cm2) and si-PCSK9, which is 5.6% higher than the additive effect (19.4%). Analysis of variance suggests that there is a significant interactive effect between UVB and si-PCSK9 treatments (P < 0.05) (Table 1) at a dose of 150 mJ/cm2. No interactive effect is found when UVB doses were 50 and 100 mJ/cm2, suggesting that the interactive effect is UVB dose dependent.
Table 1.
siRNA and UVB induces apoptosis of human keratinocytes
| Nparm | DF | Sum of Square | F Ratio | Prob > F | |
|---|---|---|---|---|---|
| siRNA | 1 | 1 | 420.1 | 57.1 | <0.0001 |
| UVB | 1 | 1 | 267.0 | 36.3 | <0.0001 |
| siRNA × UVB | 1 | 1 | 41.8 | 5.7 | 0.044 |
Abbreviations: DF, degree of freedom; Nparm, number of parameters; Prob, probability; siRNA, small interfering RNA.
DISCUSSION
Association between metabolic disorders and psoriasis provides clues to identifying psoriasis-related genes
Psoriasis is a systemic, T-cell–mediated inflammatory disease (Nograles et al., 2010) associated with various comorbidities, including cardiovascular disease and metabolic disorders. Psoriasis and these comorbidities share a number of same inflammatory cytokines, such as Th1 and Th17 inflammatory molecules, which promote a proinflammatory state (Davidovici et al., 2010). The combination of our previous studies in metabolism and psoriasis have resulted in the identification of a psoriasis gene, NRIP1 (Luan et al., 2016). This study showed another candidate gene, PCSK9, which plays an important role in metabolism and has the potential to be a therapeutic target for treatment of psoriasis.
PCSK9 is a therapeutic target of psoriasis
PCSK9, a member of proprotein convertase family, is secreted by liver, intestine, and kidney. PCSK9 plays an important role in the elevation of plasma LDL cholesterol level, which is related to the development of cardiovascular disease. Also, high levels of PCSK9 are correlated to higher body mass index, higher triglyceride level, higher insulin and glucose levels, higher C-reactive protein level, and higher incidence of diabetes (Lakoski et al., 2009). Tumor necrosis factor-a and resistin, a proinflammatory adipokine, could induce PCSK9 expression (Melone et al., 2012; Nograles et al., 2010), suggesting that PCSK9 may also correlate with metabolic disorders and inflammation (Lakoski et al., 2009). Tang et al. (2012) reported that suppressing PCSK9 by using siRNA could significantly suppress inflammation in oxidized LDL-stimulated macrophages by inhibiting the activity of NF-κB.
Because PCSK9 is involved in cardiovascular disease, metabolic disorders, and low-grade inflammation, we hypothesized that PCSK9 may be involved in the pathogenesis of psoriasis. This hypothesis is strongly supported by the overexpression of PCSK9 in lesions of psoriasis patients and the up-regulated Pcsk9 expression in the IMQ-treated skin of B6 mice (Figures 1 and 2). Furthermore, our study showed that both genetically engineered global knockout and siRNA-induced local knockdown of Pcsk9 expression can significantly suppress IMQ-induced psoriatic reactions (Figures 3 and 5), thus establishing the important role of PCSK9 in psoriasis and suggesting that PCSK9 may be a therapeutic target for psoriasis treatment.
Suppressing PCSK9 inhibits IMQ-induced psoriatic reactions
Psoriasis is characterized by infiltration of inflammatory cells in both dermis and epidermis, as well as hyperproliferation of keratinocytes. To understand the underlying mechanisms of PCSK9 involved in psoriasis, we explored the role of PCSK9 on the inflammation in psoriasis. The erythema of skin reflects the severity of inflammation. As shown in Figure 3a and 5e, the scores of erythema of Pcsk9−/− mice and si-Pcsk9–treated skin are significantly lower than the control groups. The hematoxylin and eosin staining (Figure 3c) also shows much less inflammatory infiltration in lesions of the Pcsk9−/− mice. The IHC results show that the increases of p65, a subunit of NF-κB, and phosphorylated p65 induced by IMQ was suppressed in Pcsk9−/− mice. NF-κB is a key inflammation regulator in psoriasis, linking abnormal immune cell states and keratinocytes (Tsuruta, 2009) through its downstream cytokines (Goldminz et al., 2013). It has been shown that the IL-23/IL-17 axis plays a crucial role in the pathogenesis of psoriasis and that a monoclonal antibody against p40 (the subunit shared by IL-12 and IL-23) shows therapeutic efficacy in psoriasis (Krueger et al., 2007; Takata et al., 2011). Additionally, it has been shown that IMQ treatment-induced psoriasis lesions are also dependent on IL-23 and IL-17 (van der Fits et al., 2009). Our results suggest that both global depletion and topical suppression of Pcsk9 could decrease the inflammatory reaction induced by IMQ treatment.
Suppressing PCSK9 not only reduces inflammation but has also shown strong effects on inhibiting hyperproliferation of keratinocytes. As shown in Figure 6, the viability of human keratinocytes is significantly reduced and apoptosis is induced by si-PCSK9 transfection. These results suggest that the deletion of PCSK9 could decrease the growth and prompt apoptosis of human keratinocytes. Additionally, we found that the combination of narrow band–UVB and si-PCSK9 may have a synergistic effect on inducing apoptosis of keratinocytes. Narrow band–UVB treatment, as a treatment of psoriasis, could improve psoriasis symptoms by inducing apoptosis of keratinocytes (Lewis et al., 2003). Our data suggest that in the future, narrow band–UVB treatment and PCSK9 target therapy might be combined to increase the outcome of treatment. To test the proliferation of mice keratinocytes, Ki67, a cell proliferation marker, was detected. As shown in Figure 4, Ki67 is significantly suppressed in Pcsk9−/− mice compared with WT mice after IMQ treatment. The scores of scaling and thickness, also indicating the proliferation of keratinocytes, are significantly lower in Pcsk9−/− mice and si-Pcsk9 mice than in the controls (Figure 3a). Additionally, the hematoxylin and eosin-stained sections show that the increase of epidermis thickness is significantly suppressed in Pcsk9−/− mice (Figure 3c). Taken together, these results suggest that depletion of PCSK9 could suppress proliferation and induce apoptosis of keratinocytes in vitro and in vivo.
The results of this study strongly suggest that PCSK9 plays an important role in psoriasis. Suppressing PCSK9 can inhibit the hyperproliferation of keratinocytes and reduce the inflammation induced by IMQ via altering the expression of NF-κB. The identification of PCSK9 function in psoriasis may provide a treatment for psoriasis.
MATERIALS AND METHODS
Skin samples
The skin biopsy samples (5 mm) of 10 psoriatic lesions and 6 healthy controls were immediately stored in liquid nitrogen for quantitative real-time PCR and Western blot assay or fixed in formaldehyde (4%) for immunohistochemistry assay. All samples were collected in accordance with the ethical guidelines mandated by the ethical committee of Institute of Dermatology, Chinese Academy of Medical Sciences. Written informed patient consent was received from each patient/individual involved in this study.
Mice
B6;129S6-Pcsk9tm1Jdh/J (Pcsk9−/−, strain identification 005993) and C57BL/6J (B6, strain identification 000664) were purchased from The Jackson Laboratory (Bar Harbor, Maine; 04609). The Pcsk9−/− mice have been backcrossed to B6 in our laboratory for more than 10 generations and have been maintained by crossing heterozygous knockout mice (Pcsk9−/+). Female B6 mice (4–8 months old) were used in the IMQ treatment and the siRNA treatment study. All the mice were housed in Southern Illinois University School of Medicine, in accordance with National Institutes of Health guidelines. The animal protocol was approved by Laboratory Animal Care and Use Committee at the Southern Illinois University School of Medicine.
IMQ treatment
IMQ cream (5%; Sandoz, Princeton, NJ) was applied to both the back (50 μl) and the right ear (10 μl) of each mouse daily for 5 consecutive days, five mice/genotype group. Two experienced researchers gave the scores independently for erythema, scaling, and thickening on a scale from 0 to 4, and the cumulative score from 0 to 12 (van der Fits et al., 2009). The means of the scores are used in this report.
siRNA topical treatment
siRNA duplexes targeting mouse Pcsk9 (si-Pcsk9) and nonsense mouse control siRNA (si-Con) were designed (see Supplementary Table S2 online) and applied (Frank-Kamenetsky et al., 2008; Ritprajak et al., 2008). Duplexes were suspended in a 3:5 mixture of cream emulsifier (Johnson’s baby lotion; Johnson & Johnson, New Brunswick, NJ) and water at a final concentration of 12.5 μmol/L. The shaved region was treated with 20 ml si-Pcsk9 (n = 6), or si-Con (n = 4) once per day for 5 consecutive days. One hour after the siRNA treatment, IMQ treatment was applied.
Tissue collection
Animals were killed 24 hours after the final treatment. Skin from the si-Con and si-Pcsk9 treated regions were divided into three pieces to (i) be preserved in RNAlater (Sigma-Aldrich, St. Louis, MO) for RNA study, or (ii) flash frozen in liquid nitrogen for protein assay, or (iii) preserved in 10% formalin for histology studies.
Histology and IHC assay
Skin samples were embedded in paraffin. Sections (4 μm thick) of each specimen were cut for hematoxylin and eosin staining and IHC study. Tissue slides were deparaffinized and then rehydrated. After antigen retrieval and blocking, sections were incubated with primary antibodies at 4 °C overnight. HRP Detection System (Dako EnVision System HRP; Dako North America, Carpenteria, CA) was used for detection. Antibodies PCSK9 (SC-66996), NF-κB (SC-109), phosphorylated NF-κB (SC-33039), and Ki67 (NB600–1209) were used. After counterstaining with hematoxylin (Thermo Fisher Scientific, Waltham, MA), the sections were dehydrated and mounted. Because all the primary antibodies were raised in rabbit, rabbit IgG (catalog no. 10500C Invitrogen, Carlsbad, CA) was used as false positive control.
Western blot
Protein was separated by SDS-PAGE and transferred to poly-vinylidene difluoride membranes (NEN Life Science Products, Boston, MA). Primary antibody of PCSK9 (Ab-28770; Abcam, MA, and SC-66996; Santa Cruz Biotechnology, Dallas, TX) and GAPDH (no. 2118; Cell Signaling Technology, Danvers, MA), were incubated at 4 °C overnight. Primary antibodies were detected with goat anti-rabbit IgG, H & L Chain Specific Peroxidase Conjugate (no. 401315; Millipore Sigma, Burlington, MA), using SuperSignal West Pico Chemiluminescent Substrate (no. 34080, Thermo Fisher Scientific).
Cell lines
Secondary normal human keratinocytes isolated from 10 donors (FC-0064; Lifeline Cell Technology, Oceanside, CA) were cultured in serum-free keratinocyte culture medium (LL-0007, Lifeline Cell Technology) and incubated at 5% CO2 and 37 °C. Culture medium was changed every 2 days, and cells were passaged at 60%–70% confluence.
siRNA transfection of human keratinocytes
Human keratinocytes were seeded in six-well plates with 2 ml media per well without antibiotics. Cells were transfected at 60%–80% confluence with human PCSK9-specific siRNA (sc-45482, Santa Cruz Biotechnology) or nontargeting siRNA Control-A (si-Con) as the protocol of Lipofectamine 3000 (Life Technologies, Waltham, MA). After 24, 48, and 72 hours of siRNA transfection, the cells were harvested for real-time PCR, MTT assay, PE annexin V apoptosis assay and propidium iodide staining.
UVB radiation
A narrowband (311–312 nm) UVB lamp (TL 20W/01 RS; Philips, Amsterdam, the Netherlands) was used 24 hours after transfection. After rinsing with phosphate buffered saline twice, the keratinocytes in phosphate buffered saline were exposed to UVB, including 0, 50, 100, and 150 mJ/cm2 (flux = 1.25 mW/cm2, distance = 10 cm). Then phosphate buffered saline was replaced with serum-free keratinocyte culture medium for 24 hours.
MTT assay
At 24, 48, and 72 hours after transfection, 100 μl of 5 mg/ml 3-(4,5)-dimethylthiazol- 2-yl-2,5-diphenyltetrazolium bromide (i.e., MTT; Sigma Aldrich) and 1,900 ml serum-free culture medium were added to each well. After a 4-hour incubation, wells were aspirated, and 2 ml of DMSO was added to each well. Each well was divided into a 96-well plate, and the absorption at 540 nm was measured by spectrophotometry (PowerWave XS; BioTek, Winooski, VT).
Propidium iodide staining
Cells were trypsinized and resuspended in phosphate buffered saline with 0.1% bovine serum albumin. A total of 106 cells/ml were fixed in 25% ethanol overnight at 4 °C, then stained with propidium iodide (50 mg/ml) containing RNase A (0.7 mg/ml), and incubated at 37 °C. For each sample, 10,000 events were collected and examined by flow cytometry (BD Accuri C6; Becton Dickson, Franklin Lakes, NJ).
Annexin V staining
Cells were stained with PE annexin V and 7-amino-actinomycin (7-AAD-A) following the manufacturer’s instructions (BD Pharmingen, San Diego, CA) to detect early apoptosis cells (PE annexin V+/7-amino-actinomycin− events) and late apoptosis cells (PE annexin V+/7-amino-actinomycin+ events) and examined by flow cytometry (BD Accuri C6).
Quantitative real-time PCR
Total RNA was isolated by TRIzol reagent (Invitrogen, Carlsbad, CA). Complementary DNA was synthesized using the Verso cDNA synthesis kit (Thermo Fisher Scientific). An aliquot of 1 μg of total RNA was reverse-transcribed by verso reverse transcriptase using anchored oligo dT primers (Thermo Fisher Scientific). Real-time PCR was performed using the SensiFAST SYBR Hi-ROX Kit (Bioline, London, UK) on the ABI StepOnePlus Real-Time PCR machine (Applied Biosystems, Waltham, MA). Relative expressions of target genes were calculated with the 2−(ΔΔCT) method, using GAPDH as the reference gene. Sequences of primers are listed in Supplementary Table S3 online.
Statistical analysis
All data were analyzed with JMP10.0 (SAS Institute, Cary, NC) and expressed as means ± standard error. Student t test and one-way analysis of variance (ANOVA) were used for data analysis. P < 0.05 is considered as a significant difference.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by National Institutes of Health, United States, grants K01AG046432 and R03AG046605 (to RY) and Natural Science Foundation Jiangsu China (BK2016438) CAMS Innovation Fund for Medical Sciences (CIFMS-2017-I2M-1-017) (MC). Lisa Hensley kindly edited the manuscript. Division of Laboratory Animal Medicine of Southern Illinois University School of Medicine provides excellent environment for animal research.
Abbreviations:
- IHC
immunohistochemistry
- IMQ
imiquimod
- LDL
low-density lipoprotein
- si-Con
control small interfering RNA-treated skin
- siRNA
small interfering RNA
- Th
T helper
- WT
wild type
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at https://doi.org/10.1016/j.jid.2018.07.046.
REFERENCES
- Andreassen OA, Desikan RS, Wang Y, Thompson WK, Schork AJ, Zuber V, et al. Abundant genetic overlap between blood lipids and immune-mediated diseases indicates shared molecular genetic mechanisms. PloS One 2015;10(4):e0123057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovici BB, Sattar N, Prinz J, Puig L, Emery P, Barker JN, et al. Psoriasis and systemic inflammatory diseases: potential mechanistic links between skin disease and co-morbid conditions. J Invest Dermatol 2010;130: 1785–96. [DOI] [PubMed] [Google Scholar]
- Della Badia LA, Elshourbagy NA, Mousa SA. Targeting PCSK9 as a promising new mechanism for lowering low-density lipoprotein cholesterol. Pharmacol Ther 2016;164:183–94. [DOI] [PubMed] [Google Scholar]
- Dreiher J, Weitzman D, Davidovici B, Shapiro J, Cohen AD. Psoriasis and dyslipidaemia: a population-based study. Acta Derm Venereol 2008;88: 561–5. [DOI] [PubMed] [Google Scholar]
- Ferri N, Ruscica M. Proprotein convertase subtilisin/kexin type 9 (PCSK9) and metabolic syndrome: insights on insulin resistance, inflammation, and atherogenic dyslipidemia. Endocrine 2016;54:588–601. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A, et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci USA 2008;105:11915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedewald VE, Cather JC, Gelfand JM, Gordon KB, Gibbons GH, Grundy SM, et al. AJC editor’s consensus: psoriasis and coronary artery disease. Am J Cardiol 2008;102:1631–43. [DOI] [PubMed] [Google Scholar]
- Goldminz AM, Au SC, Kim N, Gottlieb AB, Lizzul PF. NF-κB: an essential transcription factor in psoriasis. J Dermatol Sci 2013;69:89–94. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nature Rev Immunol 2006;6:508–19. [DOI] [PubMed] [Google Scholar]
- Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med 2007;356:580–92. [DOI] [PubMed] [Google Scholar]
- Lakoski SG, Lagace TA, Cohen JC, Horton JD, Hobbs HH. Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab 2009;94:2537–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hurwitz SA, Spandau DF. UVB-induced apoptosis in normal human keratinocytes: role of the erbB receptor family. Exp Cell Res 2003;284: 316–27. [DOI] [PubMed] [Google Scholar]
- Luan C, Chen X, Hu Y, Hao Z, Osland JM, Chen X, et al. Overexpression and potential roles of NRIP1 in psoriasis. Oncotarget 2016;7:74236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney JM. Understanding PCSK9 and anti-PCSK9 therapies. J Clin Lipidol 2015;9:170–86. [DOI] [PubMed] [Google Scholar]
- Melone M, Wilsie L, Palyha O, Strack A, Rashid S. Discovery of a new role of human resistin in hepatocyte low-density lipoprotein receptor suppression mediated in part by proprotein convertase subtilisin/kexin type 9. J Am Coll Cardiol 2012;59:1697–705. [DOI] [PubMed] [Google Scholar]
- Nograles KE, Davidovici B, Krueger JG. New insights in the immunologic basis of psoriasis. Semin Cutan Med Surg 2010;29:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpara AN, Goldner R, Gaspari A. Psoriasis: can statins play a dual role? Dermatol Online J 2010;16(2):2. [PubMed] [Google Scholar]
- Ritprajak P, Hashiguchi M, Azuma M. Topical application of cream-emulsified CD86 siRNA ameliorates allergic skin disease by targeting cutaneous dendritic cells. Mol Ther 2008;16:1323–30. [DOI] [PubMed] [Google Scholar]
- Ruscica M, Ricci C, Macchi C, Magni P, Cristofani R, Liu J, et al. Suppressor of cytokine signaling-3 (SOCS-3) induces proprotein convertase subtilisin kexin type 9 (PCSK9) expression in hepatic HepG2 cell line. J Biol Chem 2016;291:3508–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Juanes J, Coto-Segura P, Fernandez-Vega I, Armesto S, Martinez-Camblor P. Psoriasis vulgaris with or without arthritis and independent of disease severity or duration is a risk factor for hypercholesterolemia. Dermatology 2015;230:170–6. [DOI] [PubMed] [Google Scholar]
- Shlyankevich J, Mehta NN, Krueger JG, Strober B, Gudjonsson JE, Qureshi AA, et al. Accumulating evidence for the association and shared pathogenic mechanisms between psoriasis and cardiovascular-related comorbidities. Am J Med 2014;127:1148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata A, Otsuka M, Kojima K, Yoshikawa T, Kishikawa T, Yoshida H, Koike K. MicroRNA-22 and microRNA-140 suppress NF-κB activity by regulating the expression of NF-κB coactivators. Biochem Biophys Res Commun 2011;411:826–31. [DOI] [PubMed] [Google Scholar]
- Tang Z, Jiang L, Peng J, Ren Z, Wei D, Wu C, et al. PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF-κB activation in THP-1-derived macrophages. Intl J Mol Med 2012;30:931–8. [DOI] [PubMed] [Google Scholar]
- Tsuruta D NF-κB links keratinocytes and lymphocytes in the pathogenesis of psoriasis. Recent Pat Inflamm Allergy Drug Discov 2009;3:40–8. [DOI] [PubMed] [Google Scholar]
- Van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 2009;182:5836–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.