

Abstract
Current in vitro anti-tumor drug screening strategies are insufficiently portrayed lacking true perfusion and draining microcirculation systems, which may post significant limitation in reproducing the transport kinetics of cancer therapeutics explicitly. Herein, we report the fabrication of an improved tumor model consisting of bioprinted hollow blood vessel and lymphatic vessel pair, hosted in a three-dimensional (3D) tumor microenvironment-mimetic hydrogel matrix, termed as the tumor-on-a-chip with bioprinted blood and lymphatic vessel pair (TOC-BBL). The bioprinted blood vessel was perfusable channel with opening on both ends while the bioprinted lymphatic vessel was blinded on one end, both of which were embedded in a hydrogel tumor mass, with vessel permeability individually tunable through optimization of the composition of the bioinks. We demonstrated that systems with different combinations of these bioprinted blood/lymphatic vessels exhibited varying levels of diffusion profiles for biomolecules and anti-cancer drugs. Our TOC-BBL platform mimicking the natural pathway of drug-tumor interactions would have the drug introduced through the perfusable blood vessel, cross the vascular wall into the tumor tissue via diffusion, and eventually drained into the lymphatic vessel along with the carrier flow. Our results suggested that this unique in vitro tumor model containing the bioprinted blood/lymphatic vessel pair may have the capacity of simulating the complex transport mechanisms of certain pharmaceutical compounds inside the tumor microenvironment, potentially providing improved accuracy in future cancer drug screening.
Keywords: bioprinting, tumor-on-a-chip, blood vessel, lymphatic vessel, diffusion
Graphical Abstract
1. Introduction
The two-dimensional (2D) cancer cell cultures are restricted in serving as adequate models for drug screening and drug development[1, 2], due to challenges associated with for example, hypersensitivity to anti-cancer drugs[3], and inability to recapitulate the complex intricate interactions within a tumor microenvironment (TME)[4]. The TME is capable of acting as both a potential barrier and facilitator of tumor proliferation by affecting various processes including local growth resistance, immune system interactions, and the formation of distant metastases[5]. During cancer drug screening, 2D cell models sometimes fail to assist when exposed to harsh microenvironments for prolonged durations[6]. To address the limitations of 2D monolayer cell cultures, there has been an increasing interest in developing three-dimensional (3D) cancer models, which replicate the natural physiology of tumors more effectively with higher precision in predicting drug efficacy. Earlier examples were primarily based on tumor spheroids, which were inadequate due to their simplicity and reproducibility[7]. More recently, tumor cells have been seeded within 3D scaffolds or matrices, and similar to an in vivo tumor, they freely extend[8], polarize[9], migrate, and cluster[10, 11] within the matrix. Moreover, these 3D tumor models display advantages in re-establishing cell functions[12], signaling pathways[13, 14], and drugs responses[15] as compared to traditional 2D cultivation models, hence becoming more favorable in screening of pharmaceutical compounds[16–18]. For example, we and colleagues have reported an approach to generate a glioblastoma microenvironment featuring a co-culture of macrophages and glioblastoma cells in a mini-brain model, where it was shown that the glioblastoma cells were able to actively recruit the macrophages and then prime them to glioma-associated macrophages (GAMs), similar to the in vivo-scenario[19]. Nevertheless, while these enhanced models reproduce the complex structures of the tumor masses to a large extent, they typically lack the dynamic microenvironments that the tumors experience, such as the blood flows that carry the drug molecules to the tumor sites.
To this end, numerous microfluidics-enabled 3D systems, namely the organ-on-a-chip systems, have been developed in the past couple decades to introduce the dynamic microenvironmental cues besides the 3D structures of engineered tissue models, including the tumor models[20–22]. While the integration of perfusable blood vessels are becoming increasingly common[23–25], to date only few in vitro models have been reported to simultaneously re-establish both blood and lymphatic vessels within the tumor masses. In fact, the need for lymphatic drainage is an important yet overlooked aspect in designing such tumor models. In the mammalian body, the lymphatic vessels function in facilitating lymphatic collection and eventual removal of desired substances[26]. In addition, homeostasis of the interstitial fluid is maintained by the microcirculation system, based on permeation from the blood vessels and subsequent absorption by the lymphatic vessels[27]. More relevant to cancer, the lymphatic draining system not only plays a critical role in metastasis[28], but it also provides a preferential recycling route to most administered anti-tumor drugs in vivo[29]. Therefore, designing an in vitro tumor model exhibiting the microcirculation network to represent the functions of blood vessels as well as lymphatic vessels, is indispensable. Such models tend to provide useful information regarding the role of lymphatics in cancer (or chronic inflammation pathological conditions) and to further support the development of new and improved therapeutic strategies[30].
To this end, numerous reports have been focused on uncovering the active roles of lymphatic endothelium in tumor cell transport and modulation of antitumor immune responses[31–33]. Kamm and co-authors modeled tumorigenesis featuring both lymphatic angiogenesis and vascular angiogenesis[34]. Jeon and co-authors described a microfluidic platform for the reconstitution of lymphangiogenesis on-chip to achieve drug screening[35]. While such expositions have successful modeled the in vivo scenarios, the sole reliance on self-sprouting to generate the vessels inevitably prolongs the time needed for constructing the devices. Among the various strategies to generate the hollow vessel-like structures, 3D bioprinting is perhaps one that allows precise yet convenient control over the deposition of hydrogel biomaterials to obtain complex biomimetic patterns otherwise not attainable by traditional microfabrication techniques[36]. Indeed, 3D bioprinting has provided a set of enabling tools for tissue biofabrication in tissue modeling and related applications[37, 38]. Here we report the establishment of a tumor-on-a-chip platform possessing the blood and lymph vessel pair, embedded in a microfluidic bioreactor controlled by a micro-flow system, to achieve a dynamic microenvironment for cultured MCF-7 breast cancer cells in a 3D hydrogel matrix based on gelatin methacryloyl (GelMA)[39, 40]. The perfusable hollow blood vessel and the one end-blinded hollow lymph vessel were bioprinted separately with individually tunable permeability profiles matching those of their native counterparts.
Anti-cancer drug transport profiles were subsequently investigated with regard to different combinations of the blood and lymphatic vessels and tumor cell arrangements. It is anticipated that this unique approach will likely provide a viable in vitro platform to analyze the dynamics and interactions between blood and lymph vessels in a tumor microenvironment during anti-cancer drug screening.
2. Results and Discussions
2.1. Bioprinting setup and bioink properties
Although 2D models have been extensively applied for drug screening[41], it is not straightforward to estimate the transport of drug molecules in bulk tissues and interpolate to in vivo conditions due to the complex TME (Figure 1A). Even with the majority of the existing 3D models, it is still difficult to recapitulate the drug diffusion effects without integrating a microcirculation system containing not only perfusable blood vessels but also importantly, the draining lymphatic vessels[42]. As a result, many in vitro false positive-screened compounds are proceeded for clinical trials leading to a high dropout rate, further causing unnecessary waste of capital and time[43]. Consequently, several chip-based systems have been reported containing such blood/lymphatic vessel pair[44–46], which generally are not conveniently tunable in vascular parameters such as their permeability.
Figure 1.

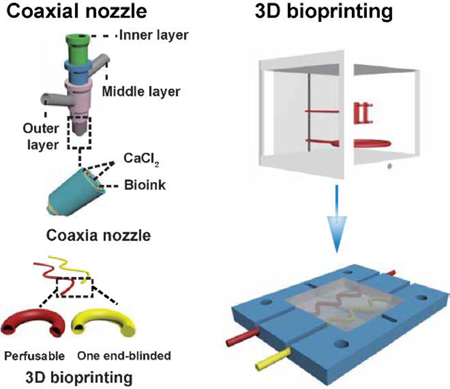
Schematic showing the biomimetic in vitro model of tumor-on-chip with bioprinted blood and lymphatic vessel pair (TOC-BBL). A) The complex tumor in vivo structure. B) The simplified model of blood and lymphatic vessel pair in the tumor microenvironment. C) Schematic of the TOC-BBL containing the pair of bioprinted blood/lymphatic vessel pair. The red-colored perfusable hollow tube was bioprinted to privide blood fluid flow, the yellow-colored one end-blinded hollow tube was bioprinted to enable lymphatic drain flow, and the chamber was seeded with tumor cells embedded in an ECM-like hydrogel. D) The composition of the bioinks used for bioprinting the blood vessel and the lymphatic vessel. E) The setup of a multi-layer, concentric, coaxial nozzle for co-delivery of the bioink and the crosslinking agent. F) The bioprinting of the two different hollow tubes using the perfusable hollow tube mimicking the blood vessel and the one end-blinded hollow tube mimicking the lymphatic vessel.
Alternatively, we have designed a bioprinting-enabled system featuring a pair of perfusable hollow blood vessel and single-outlet lymphatic vessel, with the tumor cells seeded within a hydrogel niche in between the microcirculation network (Figure 1B), termed as the tumor-on-a-chip with bioprinted blood and lymphatic vessel pair (TOC-BBL, Figure 1C). The TOC-BBL comprised of four layers, i.e., the two polydimethylsiloxane (PDMS) layers in the middle and the two external poly(methyl methacrylate) (PMMA) layers tightened using screw/bolt sets to ensure sealing. The chamber of the lower PDMS layer was used to house the bioprinted vascular/lymphatic vessels and the tumor cells in a GelMA matrix, while the chamber of the upper PDMS layer served as the reservoir of tumor cell culture medium. Within the bioprinter, curve-shaped hollow vascular (perfusable) and lymphatic (one end-blinded) tubes were precisely deposited, which were intended to emulate the complex geometry of their native counterparts as compared to the models generated using simple methods such as needle templating[47, 48]. The fabricated bioreactor was connected to a peristaltic pump to generate different flow rates for the vascular and lymphatic vessels.
After systemic administration in a patient, the drug first reaches the tumor via blood-borne circulation along the blood vessel, diffuse (or in some cases be actively transported) across the vascular wall, go through interstitial transport across the tumor mass, and is picked by the tumor cells[49, 50]. The excessive drug may be further drained to lymphatic vessels and in return, the presence of lymphatic vessels with constant drainage may affect the transport rate of the drug molecules in the tumor tissue. Indeed, the microfluidic technology has enabled precise control of flow at microscales[51], and our developed TOC-BBL platform featuring the pair of bioprinted perfusable blood vessel and drainage lymphatic vessel, could be potentially analogous to the vital in vivo conditions and structures. However, its physiological relevance in comparison with the in vivo diffusion profiles remains to be carefully examined.
2.2. Fabrication and characterization of bioprinted vessel constructs
In human body, the permeability of lymphatic vessels is much higher than that of the blood vessels. As reported, the typical permeability values of bovine serum albumin (BSA) for blood vessels in a tumor region are in the range of 1–8× 10−7 cm·s−1[52–55], while 0.1–5× 10−6 cm·s−1 for lymphatic vessels[56–58]. To obtain different permeability to match that of the blood and lymphatic vessels, we fabricated these vessels with meticulously optimized bioinks. In our previous studies, it was reported that the increased branching of polyethylene glycol (PEG) molecules could improve bimolecular diffusion and pore size of the hydrogels when compared with linear PEG[59, 60]. Therefore, PEG molecules with straight and branched structures were adopted as the bioink components to tune the permeability of pir bioprinted vessels.
Specifically, we included PEG diacrylate (PEGDA) and 8-arm polyethylene glycol-octaacrylate (PEGOA) to formulate blood vessels and lymphatic vessels, respectively, in our basic bioink containing GelMA, photoinitiator (PI), and alginate (Figure 1D)[59, 61–66]. Bioink flow was dispensed from the middle layer of the coaxial nozzle and calcium chloride (CaCl2) solution from both the inner and outer needles, inducing immediate physical crosslinking of the alginate component within the bioink modeling hollow tubular vessel structures[67] (Figure 1E).
The bioprinted hollow microfibers were further photocrosslinked for the GelMA and PEGDA/PEGOA components under UV exposure[68]. In particular, the lymphatic vessels were obtained by discontinuing the flow of CaCl2 in the inner needle upon bioprinting, while the CaCl2 flow was maintained continuous from the outer needle to facilitate the crosslinking of the bioink as well as maintaining a smooth exterior surface of the vessels. As such, our bioprinting method utilizing the multi-layered coaxial, concentric nozzle could easily allow the fabrication of perfusable hollow tubes and one end-blinded tubes having different wall thicknesses by adjusting the flow rates of both the bioink phase and the CaCl2 phase (Figure S1). Finally, GelMA hydrogel containing MCF-7 breast tumor cells was added into the chamber enclosing the bioprinted vessel pair, and the stromal GelMA was photocrosslinked forming the TOC-BBL system (Figure 1F).
2.3. Mechanical properties of bioinks and bioprinted structures
To optimize the mechanical properties of the bioink, various aspects of GelMA, alginate, and PEGA/PEGOA were examined, including the printability, elastic modulus, and rheological behaviors. Evaluation of the printability of the bioinks of different compositions was performed by a continuous flow of fluids using coaxial nozzles. Briefly, 2% w/v or 3% w/v alginate combined with 5% w/v or 7% w/v of GelMA could be bioprinted smoothly (Figure 2A). The hollow tubes could be bioprinted continuously when the flow rate of the inner CaCl2 solution (0.3 M) was 1.5 folds (35 μL·min−1) that of the middle bioink phase (52.5 μL·min−1), while the flow rate of the outer CaCl2 solution was set at half (17.5 μL·min−1).
Figure 2.

The mechanical properties of the bioinks. A) Printability mapping of the bioinks; o: printable, × : non-printable. B) Elastic moduli of bioinks composed of 3, 5, and 7% w/v GelMA, 2 and 3 % w/v alginate, as well as the different concentration of PEGDA/PEGOA. C) Viscosity measurements and rheological behaviors of 5% w/v GelMA, 3% w/v alginate, and 4% w/v PEGDA/PEGOA alone, and the blend bioink at various concentrations. All measurements were performed at room temperature.
The viscosities of the bioinks were further analyzed by rheological measurements (Figure 2C). The results showed that the bioink viscosities increased with higher concentration of alginate, and higher concentration of PEGDA/PEGOA also made the bioink more viscous. Moreover, the viscosities of the bioinks decreased when the shear rate increased indicating their shear-thinning properties, which is comparable to our earlier studies[59].
To measure the mechanical properties of the crosslinked bioinks, the compression tests were performed. Air bubbles trapped inside the bioink were removed by vacuum decompression before crosslinking, as entrapped air could create defects[69]. The results showed that the compressive moduli of the crosslinked bioinks were enhanced by increased concentration of PEGDA or PEGOA (Figure 2B). The enhanced mechanical properties of the hydrogels could sufficiently support perfusable hollow or one end-blinded tubular constructs when internal and external pressures are applied.
Based on the mechanical tests, we selected 7% w/v GelMA together with 3% w/v alginate, and ≤4% w/v PEGDA/PEGOA as the bioink for bioprinting the vessels in subsequent experiments.
2.4. Permeability evaluations of the bioprinted hollow tubes
The permeability of blood and lymphatic vessels inside a tumor tissue is a fundamental property that affects drug transport and efficacy[70–72]. The perfusable tubes were bioprinted in a microfluidic bioreactor and embedded in the GelMA matrix (Figure 3A), and different formulations of the bioinks and the GelMA matrices were examined. The permeability of the vessels was measured by FITC-BSA diffusion at different times. The diffusional permeability of BSA (PBSA) of bioprinted hollow tubes made with bioinks containing PEGDA or PEGOA were calculated (Figure 4A). We observed that for the bioink formulation containing 4% w/v PEGDA, 3% w/v alginate, and 7% w/v GelMA, while 5% w/v GelMA in the tumor chamber, the PBSA was calculated as 5.07 ± 0.407 × 10−7 cm·s−1. Contrarily, for the bioink having 4% w/v PEGOA, 3% w/v alginate, and 5 % w/v GelMA, with 5% w/v GelMA in the chamber, the PBSA was 1.76 ± 0.623 × 10−6 cm·s−1 (Figure 4B). Based on these results with permeability values matching those of the native blood and lymphatic vessels that had been previously measured in vivo[58], we selected the bioink containing 3% w/v alginate, 4% w/v PEGDA, and 7% w/v GelMA to for bioprinting the perfusable blood vessels, the bioink made of 3% alginate, 4% w/v PEGOA, and 5% w/v GelMA to bioprint one end-blinded lymphatic vessels, and 5% w/v pure GelMA as the tumor matrix for the subsequent experiments. However, it should be noted that these permeability values represent those in the bioprinted hollow channels only and not when encapsulated in the hydrogel matrices. The permeability of the latter can be directly calculated using our recently developed model[73].
Figure 3.

The chip design for perfusable detection. A) Schematic diagrams of the permeability test. The perfusable hollow tube was bioprinted and embedded in the chamber, and the chamber was filled with GelMA and crosslinked under UV. After the two middle PDMS layers and the two outside PMMA layers were fixed with screws and nuts, the test molecule solution was injected by a syringe pump at 1 ml min−1, and the time-lapse images were acquired under fluorescence microscope and analyzed for fluorescence intensities. B) Schematic diagram of the diffusion test. The bioprinted perfusable blood vessel (red) and the one end-blinded lymphatic vessel (yellow) were embedded in GelMA with/without tumor cells in the lower half of the chamber. The medium for the tumor cells was filled in the upper half of the chamber. After the device was assembled, a solution of fluorescent molecules or DOX was perfused by a peristaltic pump from the blood vessel and diffusion measured at the different times, in the presence or absence of the lymphatic vessel. When the lymphatic vessel was present, a draining flow was applied.
Figure 4.

The permeability of BSA in different vessels. A) Fluorescence images of BSA-FITC perfused in the hollow tubes bioprinted with different bioinks at different time points. B) The different permeability of BSA measured for the hollow tubes bioprinted using bioinks containing various concentrations of PEGDA/PEGOA and different concentrations of GelMA as the embedding matrix.
2.5. Integration with tumor cells
The structure of the TOC-BBL model and its operational procedures are shown in Figure 3B. TOC-BBL was designed to be a microfluidic platform with two types of fluid flows, i.e., those in the blood vessel (perfusable tube) and the lymphatic vessel (one end-blinded). These microchannels were configured in a 3D geometry by embedding the bioprinted vessel pair in a GelMA matrix inoculated with MCF-7 breast cancer cells. In Figure 3B, the red channel in the schematic indicates the perfusable tube simulating the blood vessel (GelMA/alginate/PEGDA; PBSA = 5.07 ± 0.407 × 10−7 cm·s−1), while the yellow channel represents the lymphatic vessel (GelMA/alginate/PEGOA, PBSA = 1.76 × 0.623 10−6 cm·s−1).
2.6. Transport of drug molecules in the TOC-BBL system
One of our primary aims was to develop an in vitro anti-tumor drug screening system reproducing the in vivo physiology of the tumor microenvironment for cancer research and drug development. We first compared the diffusion of fluorescein isothiocyanate (FITC) in one- and two-channel configurations with or without cells to examine the drug transport rates at different times in our TOC-BBL (Figure 5). It was observed that when the TOC-BBL tumor chamber contained the MCF-7 (5× 107 cells·mL−1) cells encapsulated in GelMA for 3 days, the transport of FITC was slower as compared to the tumor chamber only having pure GelMA without cells. The delayed transport of FITC was possibly due to the high density of tumor cells (MCF-7) in the chamber slowing down the transport of drugs, and perhaps also uptake of the molecules by the cells[74]. Indeed, it was revealed that increased density of the cells in the GelMA matrix further induced monotomic decrease in the transport of FITC. Meanwhile, the microcirculation system containing not only the blood vessel but also the draining lymphatic vessel accelerated the diffusion of FITC (Mw = 389.38 D).
Figure 5.

FITC transport in the TOC-BBL. A) Diffusions of FITC were examined in one-/two-channel configurations with different concentrations of tumor cells. B) The comparisons of diffusion constants of various biomolecules and DOX measured in the TOC-BBL. * P<0.05.
To better understand the performance of our TOC-BBL, we further examined molecules with different molecular weights to observe their differential permeability. Moreover, diffusion profiles of FITC-BSA (Mw ~68 kDa) and FITC-dextran (Mw ~10 kDa) in the TOC-BBL platforms were monitored for 24 h (Figure 6A). Doxorubicin (DOX, Mw = 579.98 Da) diffusion was further evaluated, to verify the performance of the TOC-BBL system in anti-cancer drug transport, where we experimented different conditions in one-/two-channel configurations with/without tumor cells (Figure 6B). The diffusion rate of these molecules and drugs showed that the TOC-BBL platform could serve as an effective in vitro drug screening model. Analyzing the results, we noticed much faster DOX accumulation than FITC-dextran under similar conditions (P<0.05). The diffusion profiles in the one-channel (only blood vessel) configuration were quite different from the TOC-BBL systems encompassing the blood vessel-and-lymph drainage microcirculation system, where the diffusion rates were much higher for all molecules/drugs assessed as compared to those when the chips only contained single blood vessels (Figure 6C). Drug diffusion in vivo depends on the following three aspects: physical and chemical properties of drugs[75] and the structure and physiological function of the absorptive sites[76]. Therefore, it is very important to study drug diffusion behaviors in a biomimetic tumor model where the various microenvironmental properties can be precisely controlled[77]. To this end, we bioprinted the hollow tubular blood and lymphatic vessels with different permeability by adjusting the proportions of PEGDA/PEGOA in the bioink and embedded in the TOC-BBL platform containing well-defined GelMA matrix, to tune the diffusion coefficients for various biomolecules and drugs using a purely biomaterials approach by effectively regulating the density of the hydrogels[78].
Figure 6.

Biomolecule and drug transport in the TOC-BBL. A) FITC with different molecular weights were examined for their diffusion profiles in the TOC-BBL under the two-channel configuration. B) The diffusions of DOX were examined in one-/two-channel configurations in the TOC-BBL. C) The comparisons of diffusion constants of various biomolecules and DOX measured in the TOC-BBL. * P<0.01.
2.7. Effects of DOX delivery on cell viability
The effects of DOX perfusion on the MCF-7 breast cancer cells embedded in the GelMA matrix in the TOC-BBL platform was investigated at different time intervals. The cell viability was then assessed using live/dead staining. As shown in Figure 7A and B, post-cultivation in the bioreactor the viability of the MCF-7 cell was more than 90% measured at all time points. Cytochemical analysis with F-actin/4’, 6-diamidine-2-phenylindole (DAPI) staining was conducted to observe spreading and proliferation of the encapsulated cells at different times (Figure 7A), where confluence of the cells was achieved at Day 5.
Figure 7.

Anti-cancer drug effects in the TOC-BBL. Anti-cancer drug effects in the TOC-BBL. A) Fluorescence micrographs showing viability and spreading of MCF-7 cells in the GelMA matrix in the TOC-BBL at different time points. B) Quantification of cell viability in the TOC-BBL. C) Fluorescence micrographs showing cell viability in the TOC-BBL after DOX delivery (0.5 µM) at 24 h in one-/two-channel configurations. D) Quantification of cell viability. E) Dose-response curves of 2D and 3D-cultured MCF-7 cells to DOX. F) Fluorescent micrographs showing viability of 2D- and 3D-cultured MCF-7 at different concentrations of DOX. * P<0.01.
We further compared the therapeutic effects of DOX delivery on the cells in cases of one channel (blood vessel-only) or two channels (blood and lymphatic vessel pair) in our TOC-BBL. Similar to the dye molecules, we found a comparatively reduced transport rate of DOX in the presence of MCF-7 cells compared to when there were no cells in the GelMA hydrogel matrix (P<0.01, Figure 7C, D), indicating the possibly blockage and uptake of the DOX by the cells on the path. Moreover, the cell viability was greatly increased in the presence of two channels (blood/lymphatic) in comparison with only one (blood) channel (Figure 7C, D). The high cell viability in the two-channel system might be attributed to the timely elimination of DOX by the lymph drainage system, while in the case of only single perfusable channel the drug molecules would accumulate in the tumor mass, causing higher dosage. The dose-response curves for 2D/3D cultured MCF-7 cells are shown in Figure 7C, and the IC50 values of DOX in MCF-7 were 0.30 μM in 2D while 6.2 μM in 3D. The dose-responses and morphologies of cells presented the significant differences between 2D and 3D cell cultures (Figure 7E, F). The diffusion profiles of these molecules and drugs showed that the TOC-BBL platform could potentially serve as an effective in vitro drug screening model in the future with further optimizations. Our results indicated the importance of including a draining lymphatic vessel in the TOC-BBL system to tune the transport profiles of anti-cancer drugs, and by varying the bioink composition the permeability values could be easily modulated possibly relating to that of different vessel types inside a tumor volume.
Furthermore, we studied the potential effects of cells on drug transport behaviors. Specifically, human umbilical vein endothelial cells (HUVECs) and human lymphatic endothelial cells (HLECs) were seeded onto the surfaces of the bioprinted blood vessels and lymphatic vessels, respectively. It was shown that both of the two types of endothelial layers could be formed with cells strongly expressing junction biomarker CD31 (Figure S2). We subsequently compared the diffusion rates in groups with and without these endothelial cells. We found that, as anticipated, the diffusion rates in the groups containing HUVECs and HLECs were slower than the respective groups without cells (Figure S3), potentially due to the formation of tight junctions by these cells that served as another layer of barriers on top of those formed by the materials themselves. Indeed, the inclusion of the endothelial cells would make the model more biologically relevant; however, the improved barrier function, as brought by these cells, could still be realized through adjustment our bioink formulations, while the latter is more convenient to achieve.
3. Conclusions
We have reported the bioprinting of blood and lymph vessel pair in a TOC-BBL platform reproducing the microcirculation featuring both delivery and drainage routes to better mimic the transport kinetics of biomolecules and drugs. The platform although in its current form is not high-throughput, it could still offer a convenient way for in vitro drug screening by taking into consideration the microcirculation based on the bioprinted blood and lymphatic vessel pair. In addition, while both the perfusable blood vessels and lymphatic vessels we utilized in the current work were cell-free and lacked endothelial cell lining and active transport capacity, our TOC-BBL is anticipated to provide understanding of the kinetics of drug and biomolecule transport when diffusion is the primary factor. We observed that the permeability parameters of the bioprinted blood and lymphatic vessels could be controlled by precisely tuning the composition of the bioink, which could meet the different biological needs for delivery and drainage channels under various scenarios. Nevertheless, studies on tumor metastasis might be limited using our platform, due to the need for denser materials for tuning the permeability of our bioprinted vessels. Moreover, since in the present study only one pair of relatively simple blood and lymph vessels was analyzed, extensive research using more complex network of blood and lymphatic vessels may be required to delineate explicit knowledge of drug diffusion and removal phenomenon in the complex tumor microenvironments featuring hierarchically arranged blood and lymphatic vessels. Further research using more complex ECM protein compositions and various stromal cells is also required.
4. Experimental Section
Cells and materials
MCF-7 human breast cancer cells (HTB-22, ATCC, USA) were cultured using Dulbecco’s modified Eagle medium (DMEM, ThermoFisher, MA, USA) supplemented with 10% v/v fetal bovine serum (FBS, ThermoFisher). HUVECs (cAP-0001, Angio-Proteomie, USA) and HLECs (CC-2810, Lonza) were cultured in their respective recommended media by the vendors. CaCl2, ethylene diamine tetraacetic acid (EDTA), and PI (2-hydroxy-4’-(2-hydroxyethoxyethoxy)-2-methypropiophenone), FITC, FITC-BSA, and FITC-dextran were purchased from Sigma-Aldrich (USA). PEGDA and PEGOA were purchased from JenKem Technology, USA. Phosphate-buffer saline (PBS), paraformaldehyde (PFA), Live/Dead Viability/ Cytotoxicity kit, Alexa 488-phalloidin, and DAPI were purchased from ThermoFisher, USA. Ultrapure water with an electrical resistivity of 18.2 mΩ was obtained from the Mili-Q purification system (Millipore Corporation, USA). Needles of different sizes (14G, 18G, and 25G) were purchased from BD Biosciences, USA.
Preparation of GelMA
GelMA with moderate degree of methacryloyl substitution was synthesized as reported previously[59]. To determine the degree of functionalization (DoF) value, we obtained the 1H NMR spectra of GelMA in D2O. 3-(Trimethylsilyl)-1-propanesulfonic acid sodium salt was used as the internal standard with a known concentration (20 mg mL−1) and pristine gelatin was used as the control. Two peaks at chemical shifts δ of ~5.4 and ~5.6 ppm were attributed to the protons in the methacryloyl groups, while δ = 0 ppm showed the -CH3 groups in the trimethylsilyl group (Figure S4). Based on the integral area of these peaks, we could calculate the DoF value of GelMA as 39%. The fluoroaldehyde assay method showed a similar value that was comparable to that obtained from the 1H NMR spectra.
Preparation of bioinks and the bioprinting procedure
The bioinks were prepared by mixing different concentrations of GelMA, alginate, and PEGDA or PEGOA in ultrapure water, finally 0.5% w/v PI was also added in the prepared solution. Later, all the solutions were filtrated with 0.2 μm syringe filters, kept at 4 °C (for storage), while warmed at 37 °C for 0.5 h prior using.
A custom-made coaxial nozzle device containing three injection channels with different needle sizes was manufactured (Figure 1E). Briefly, needles of 25G, 18G, and 14G were utilized for making inner core, middle layer, and outer sheath, respectively. Junction points were sealed permanently using epoxy glue (Devcon, USA). A commercial bioprinter (Allevi 2, Philadelphia, PA, USA) was utilized to host this custom-made nozzle device for bioprinting. The needles were connected to three separate pumps for extrusion via polyvinyl chloride (PVC) tubes. The bioink was delivered at 35 μL·min−1 through the middle layer (20–50 μL·mL−1) while the CaCl2 solutions (0.3 M) were ejected from both the inner (52.5 μL·min−1) and outer (17.5 μL·min−1) layers to immediately crosslink the alginate (component within the bioink) and to obtain the tubular structures. UV light was illuminated 6.9 mW·cm−2 of UV light for 40 s at the outlet of the nozzle device for covalent crosslinking of the GelMA and PEGDA/PEDTA components, followed by immersion in a 20 mM EDTA solution for 5 min to remove the alginate[79]. Alginate in the bioink served as a sacrificial ingredient, removed later on, and hence afford more permeable hydrogel.
The bioink optimization: mechanical properties
The mechanical properties of the bioinks were determined via printability, rheology, and compress tests. The measurement of printability of bioinks is based on if the bioinks could be smoothly extruded to form uniform tubular structures via the coaxial nozzle. The measurement of printability was based on if the bioinks could be smoothly extruded to form uniform tubular structures via the coaxial nozzle. To prepare the test samples, different concentrations of GelMA (3% w/v, 5% w/v, and 7% w/v), alginate (1% w/v, 2% w/v, 3% w/v, and 4% w/v), and PEGOA or PEGDA (1% w/v, 3% w/v, 5% w/v, 7% w/v, and 10% w/v) were dissolved in ultrapure water. The different formulations of bioinks were extruded, and the bioprinted tubular structures were analyzed.
The rheological properties of bioink with different components were measured using a rotational rheometer (Anton Paar RHEOPLUS-32, USA). Various concentrations of GelMA, PEG, and alginate in the blend bioink was measured as reported in previous studies[80, 81].
For the compression test of the samples, bioink was both ionically and photo crosslinked. The bioink was pipetted to a prepared cubic PDMS mold (4 × 4 × 4 mm3) and soaked in 0.3 M CaCl2 solution for 2 min, followed by exposing to 6.9 mW·cm−2 of UV light for 40 s, immersion with 20 mM EDTA solution for 5 min, and washing with ultrapure water for three time at 37 °C before testing. During the testing, the compressive strength of the samples was assessed at a cross speed of 30 mm·s−1 and a 60% strain level according to previously reported procedures[68] using a mechanical testing machine (Instron model 5542, Norwood, MA, USA) at 37 °C.
The Bioink optimization: permeability of the hollow tubes
Permeability was tested in the microfluidic bioreactor (Figure 3A). The designed bioreactor consisted of two hemi-chambers and arranged by a pair of rigid supports made of PMMA and two complementary PDMS micro-fabricated gaskets. Both PMMA layers were rectangular in shape (5 × 1.5 × 0.4 cm3) and contained 4 clearance holes, allowing mechanical sealing of PDMS (5 × 1.5 × 0.8 cm3), sandwiched in between using sets of screw and nuts guaranteeing the hydraulic tightness, resulted thickness of the bioreactor chamber was 1.6 cm. The main chamber of the bioreactor was squared (4 × 4 mm2), tightened sets of screws fixed the hydrogel and the bioprinted vessel pair in place. The inlet and outlet channels of the bioprinted blood/lymphatic vessels were connected with steel connectors (27G, 0.4 mm in diameter) and these connectors further connected with PVC tubes to aid flow of the drugs into the vessels or to drain the interstitial liquid. After perfusable vessel was bioprinted in the bioreactor, 0.2 ml of 5% w/v GelMA having 0.5% w/v PI was added in the chamber embedding perfusable bioprinted vessel, and crosslinked under 500 mW·cm−2 of UV light for 30 s. FITC and FITC-BSA (50 μg·mL−1) were introduced through the perfusable tube by peristaltic pump at 1 μL·s−1. Fluorescence images were taken at different times in the bioreactor at 37 °C using a Ti-U microscope (Japan). Effect diffusional permeability coefficients (Pd) were calculated by the following equation:
| Eq. 1 |
, where ΔI is the difference in averaged intensities of the 60 min and background images, δI is the difference between 55 min and 60 min images, δt is 5 min, and d is the diameter of the tube[82]. Standard curves verified that the fluorescence signal was proportional to fluorophore concentration in the range of 0.2–50 μg·mL−1.
Fabrication of one end-blinded vessels using bioprinting
To mimic the structure of lymphatic vessels, CaCl2 flow was controlled and discontinued through the inner layer of coaxial nozzle device to form channels with one end closed and crosslinked with lymphatic vessel bioink to form hollow or solid tubes.
Construction of TOC-BBL
The coaxial nozzle was attached to bioprinter to control the precise assembly of the vessels. The bioreactor was partly modified from the bioreactor used for measurement of permeability. The main chamber contained two pairs of inlet and outlet channels for connecting the bioprinted blood and lymphatic vessel. For biomolecule/drug delivery within the blood vessel, both inlet and outlet were connected, while in the case of lymphatic vessel, only the open end was connected for applying the draining pressure to ensure the removal of the drugs. Then, 5% w/v GelMA containing different initial concentrations of MCF-7 cells (1 × 106, 5 × 106, 1 × 107, 5× 107 cells·mL−1) was added into the main chamber (to perform different biological assays) after the tubes were bioprinted and connected.
Calculation of drug transport metrics
The transport of FITC-BSA (50 μg·mL−1), FITC-dextran (50 μg·mL−1), and DOX (0.5 µM) in TOC-BBL was calculated by Fick’s Law:
| Eq. 2 |
, where Mt is the concentration of related solution at time, M∞ is the concentration of solution at infinite time, Mt/M∞ is the fractional release, De is the effective diffusion coefficient, t is time, and δ is half of the hydrogel thickness. From Fick’s equation, it follows that Mt/M∞ is directly proportional to t½ and a plot of Mt/M∞ versus t½ would give De [83]. A mass balance was performed to find Mt, which can be can be calculated from the intensity of fluorescence according to the standard curve[84–89]. All the experiments were refined until the diffusion of three sequentially refined models differed by <1%.
To create biologically relevant vessels, HUVECs and HLECs were seed in the interior surfaces of the bioprinted blood and lymphatic vessels, respectively, at 1× 107 cells·mL−1. After 4 days of culture, both of the vessels were stained with anti-CD-31 (ab24590, 1:200) and incubated with Alexa Fluor-588 goat anti-mouse IgG (1:500), and nuclei was counter-stained with DAPI (1:500). Images were obtained using a confocal laser scanning microscope (LSM880, Zeiss). Both of the endothelialized vessels were assembled as in the case without the cells to form the TOC-BBL, where the transport of DOX (0.5 µM) was calculated.
Post-treatment viability assay and dose-responses of DOX
A Live/Dead Cell Viability Kit was used to assess cell viability at different time points (Days 1, 3, 5, and 7), by following the manufacturer instructions. For F-actin staining, the samples were fixed on different days (Day 1, 3, 5, and 7) with 4% w/v PFA for 1 h at room temperature, and soaked in 0.1% w/v Triton X-100 in PBS for 30 min, while non-specific binding was inhibited using 10% w/v bovine serum albumin (BSA) for 1 h at room temperature. Samples were then incubated with F-actin at 1:40 dilution in 10% w/v BSA and 1× PBS solution, overnight at 4 °C. Nuclei of the cells were stained by DAPI, and images were observed using a fluorescence microscope[90].
For 2D monolayer culture, 1000 cells per well were seeded in to 96-well microplates. Following a 24-h incubation, the cells were exposed to DOX and incubated for 6 days. A dose-response curve for DOX was obtained with final concentrations per well between 0.2 nM and 200 μM (12-point dose-response curve). At the conclusions of both 2D/3D assays, a final concentration of 600-μM PrestoBlue cell viability reagent (Thermofisher) was added to each well and incubated for 4–6 h at 37°C. The intensity was detected using a microplate reader (570 nm, INFINITE M PLEX, Tecan). In addition, 3D cell cultures were also measured for viability at the conclusions of the assays with the addition of 2 μM calcein AM dye (Life Technologies).
Statistical analysis
T-test was used to compare between two groups. Statistical significance was determined at P<0.05.
Supplementary Material
Acknowledgments
X. Cao and R. Ashfaq contributed equally to this work. This work was supported by the National Institutes of Health (K99CA201603, R00CA201603, R21EB025270, R21EB026175) and the New England Anti-Vivisection Society. X. Cao acknowledges the support from the National Natural Science Foundation of China (81503025).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Xia Cao, Division of Engineering in Medicine, Brigham and Women’s Hospital; Department of Medicine, Harvard Medical School Cambridge, MA, 02139; Department of Pharmaceutics and Tissue Engineering, School of Pharmacy, Jiangsu University, Zhenjiang 212013, P.R. China.
Ramla Ashfaq, Division of Engineering in Medicine, Brigham and Women’s Hospital; Department of Medicine, Harvard Medical School Cambridge, MA, 02139; National Center of Excellence in Molecular Biology, University of the Punjab, 87 West Canal Bank Rd, Thokar Niaz Baig, Lahore 53700, Pakistan.
Feng Cheng, Division of Engineering in Medicine, Brigham and Women’s Hospital; Department of Medicine, Harvard Medical School Cambridge, MA, 02139.
Sushila Maharjan, Division of Engineering in Medicine, Brigham and Women’s Hospital; Department of Medicine, Harvard Medical School Cambridge, MA, 02139.
Jun Li, Division of Engineering in Medicine, Brigham and Women’s Hospital; Department of Medicine, Harvard Medical School Cambridge, MA, 02139.
Guoliang Ying, Division of Engineering in Medicine, Brigham and Women’s Hospital; Department of Medicine, Harvard Medical School Cambridge, MA, 02139.
Shabir Hassan, Division of Engineering in Medicine, Brigham and Women’s Hospital; Department of Medicine, Harvard Medical School Cambridge, MA, 02139.
Haiyan Xiao, South China Advanced Institute for Soft Matter Science and Technology, School of Molecular Science and Engineering, South China University of Technology, Guangzhou 510640, P.R. China State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, P.R. China.
Kan Yue, South China Advanced Institute for Soft Matter Science and Technology, School of Molecular Science and Engineering, South China University of Technology, Guangzhou 510640, P.R. China State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, P.R. China.
Yu Shrike Zhang, Division of Engineering in Medicine, Brigham and Women’s Hospital; Department of Medicine, Harvard Medical School Cambridge, MA, 02139.
References
- [1].Pereira PMR, Berisha N, Bhupathiraju N, Fernandes R, Tome JPC, Drain CM, Plos One 2017, 12, e0177737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hirst J, Pathak HB, Hyter S, Pessetto ZY, Ly T, Graw S, Koestler DC, Krieg AJ, Roby KF, Godwin AK, Cancer Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mosaad EO, Chambers KF, Futrega K, Clements JA, Doran MR, Sci Rep-Uk 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chung M, Ahn J, Son K, Kim S, Jeon NL, Adv Healthc Mater 2017, 6. [DOI] [PubMed] [Google Scholar]
- [5].Anari F, Ramamurthy C, Zibelman M, Future Oncol 2018, 14, 1409. [DOI] [PubMed] [Google Scholar]
- [6].Mebarki M, Bennaceur A, Bonhomme-Faivre L, Drug Discov Today 2018, 23, 857. [DOI] [PubMed] [Google Scholar]
- [7].Albanese A, Lam AK, Sykes EA, Rocheleau JV, Chan WC, Nat Commun 2013, 4, 2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Quartararo CE, Reznik E, Decarvalho AC, Mikkelsen T, Stockwell BR, ACS Med Chem Lett 2015, 6, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].He XT, Wu RX, Xu XY, Wang J, Yin Y, Chen FM, Acta Biomater 2018, 71, 132. [DOI] [PubMed] [Google Scholar]
- [10].Puliafito A, De Simone A, Seano G, Gagliardi PA, Di Blasio L, Chianale F, Gamba A, Primo L, Celani A, Sci Rep 2015, 5, 15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jovanovic B, Beeler JS, Pickup MW, Chytil A, Gorska AE, Ashby WJ, Lehmann BD, Zijlstra A, Pietenpol JA, Moses HL, Breast Cancer Res 2014, 16, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Betriu N, Recha-Sancho L, Semino CE, J Vis Exp 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang Y, Tong L, Yi L, Zhang C, Hai L, Li T, Yu S, Wang W, Tao Z, Ma H, Liu P, Xie Y, Yang X, Mol Med Rep 2018, 17, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rajangam T, Park MH, Kim SH, Tissue Eng Part C Methods 2016, 22, 679. [DOI] [PubMed] [Google Scholar]
- [15].Anjum F, Lienemann PS, Metzger S, Biernaskie J, Kallos MS, Ehrbar M, Biomaterials 2016, 87, 104. [DOI] [PubMed] [Google Scholar]
- [16].Mi SL, Yi XM, Du ZC, Xu YY, Sun W, Biofabrication 2018, 10. [Google Scholar]
- [17].Luo Y, Zhang XL, Li YJ, Deng J, Li XR, Qu YY, Lu Y, Liu TJ, Gao ZG, Lin BC, Rsc Adv 2018, 8, 25409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Beissner N, Albero AB, Fuller J, Kellner T, Lauterboeck L, Liang JH, Bol M, Glasmacher B, Muller-Goymann CC, Reichl S, Eur J Pharm Biopharm 2018, 126, 57. [DOI] [PubMed] [Google Scholar]
- [19].Martin JD, Fukumura D, Duda DG, Boucher Y, Jain RK, Cold Spring Harb Perspect Med 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Willyard C, Nat Med 2017, 23, 138. [DOI] [PubMed] [Google Scholar]
- [21].Lee H, Kim S, Chung M, Kim JH, Jeon NL, Microvasc Res 2014, 91, 90. [DOI] [PubMed] [Google Scholar]
- [22].Kappings V, Grun C, Ivannikov D, Hebeiss I, Kattge S, Wendland I, Rapp BE, Hettel M, Deutschmann O, Schepers U, Adv Mater Technol-Us 2018, 3. [Google Scholar]
- [23].Kim S, Lee H, Chung M, Jeon NL, Lab on a Chip 2013, 13, 1489. [DOI] [PubMed] [Google Scholar]
- [24].Lee H, Kim S, Chung M, Kim JH, Jeon NL, Microvascular Research 2014, 91, 90. [DOI] [PubMed] [Google Scholar]
- [25].Morgan JP, Delnero PF, Zheng Y, Verbridge SS, Chen JM, Craven M, Choi NW, Diaz-Santana A, Kermani P, Hempstead B, Lopez JA, Corso TN, Fischbach C, Stroock AD, Nature Protocols 2013, 8, 1820. [DOI] [PubMed] [Google Scholar]
- [26].Rockson SG, Am J Med 2001, 110, 288. [DOI] [PubMed] [Google Scholar]
- [27].Sato M, Sasaki N, Ato M, Hirakawa S, Sato K, Sato K, PLoS One 2015, 10, e0137301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Harris AR, Perez MJ, Munson JM, Bmc Cancer 2018, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Obinu A, Gavini E, Rassu G, Maestri M, Bonferoni MC, Giunchedi P, Expert Opin Drug Del 2018, 15, 459. [DOI] [PubMed] [Google Scholar]
- [30].Pisano M, Triacca V, Barbee KA, Swartz MA, Integr Biol (Camb) 2015, 7, 525. [DOI] [PubMed] [Google Scholar]
- [31].Bitu CC, Kauppila JH, Bufalino A, Nurmenniemi S, Teppo S, Keinanen M, Vilen ST, Lehenkari P, Nyberg P, Coletta RD, Salo T, PLoS One 2013, 8, e70925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fujiwara H, Rinsho Ketsueki 2018, 59, 1895. [DOI] [PubMed] [Google Scholar]
- [33].Chaudary N, Pintilie M, Schwock J, Dhani N, Clarke B, Milosevic M, Fyles A, Hill RP, Cancers (Basel) 2012, 4, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim S, Chung M, Jeon NL, Biomaterials 2016, 78, 115. [DOI] [PubMed] [Google Scholar]
- [35].Osaki T, Serrano JC, Kamm RD, Regen Eng Transl Med 2018, 4, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yin J, Yan M, Wang Y, Fu J, Suo H, ACS Appl Mater Interfaces 2018, 10, 6849. [DOI] [PubMed] [Google Scholar]
- [37].Gohl J, Markstedt K, Mark A, Hakansson K, Gatenholm P, Edelvik F, Biofabrication 2018, 10. [DOI] [PubMed] [Google Scholar]
- [38].Truby RL, Lewis JA, Nature 2016, 540, 371. [DOI] [PubMed] [Google Scholar]
- [39].Van Engeland NCA, Pollet A, Den Toonder JMJ, Bouten CVC, Stassen O, Sahlgren CM, Lab Chip 2018, 18, 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang H, Xiao L, Li Q, Qi X, Zhou A, Biomicrofluidics 2018, 12, 024119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hussein KH, Park KM, Teotia PK, Yang JW, Kim HM, Hong SH, Yang SR, Park IC, Park SM, Woo HM, Transpl P 2013, 45, 3092. [DOI] [PubMed] [Google Scholar]
- [42].Kulkarni V, Bodas D, Paknikar K, Acs Biomater Sci Eng 2018, 4, 1407. [DOI] [PubMed] [Google Scholar]
- [43].Verjans ET, Doijen J, Luyten W, Landuyt B, Schoofs L, J Cell Physiol 2018, 233, 2993. [DOI] [PubMed] [Google Scholar]
- [44].Chung M, Lee S, Lee BJ, Son K, Jeon NL, Kim JH, Adv Healthc Mater 2018, 7. [DOI] [PubMed] [Google Scholar]
- [45].Jusoh N, Oh S, Kim S, Kim J, Jeon NL, Lab Chip 2015, 15, 3984. [DOI] [PubMed] [Google Scholar]
- [46].Kang M, Park W, Na S, Paik SM, Lee H, Park JW, Kim HY, Jeon NL, Small 2015, 11, 2789. [DOI] [PubMed] [Google Scholar]
- [47].Guo Y, Li L, Li F, Zhou H, Song Y, Lab Chip 2015, 15, 1759. [DOI] [PubMed] [Google Scholar]
- [48].Jang MJ, Nam Y, Macromol Biosci 2015, 15, 613. [DOI] [PubMed] [Google Scholar]
- [49].Wang ZH, Yu Y, Ma J, Zhang HR, Zhang H, Wang XQ, Wang JC, Zhang X, Zhang Q, Mol Pharmaceut 2012, 9, 2646. [DOI] [PubMed] [Google Scholar]
- [50].Tada A, Horie S, Mori S, Kodama T, Cancer Sci 2017, 108, 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pajoumshariati SR, Azizi M, Wesner D, Miller PG, Shuler ML, Abbaspourrad A, Acs Appl Mater Inter 2018, 10, 9235. [DOI] [PubMed] [Google Scholar]
- [52].Zhang Y, Sun T, Jiang C, Acta Pharm Sin B 2018, 8, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Matsunaga T, Matsunaga N, Kusunose N, Ikeda E, Okazaki H, Kakimoto K, Hamamura K, Koyanagi S, Ohdo S, Biochem Biophys Res Commun 2018, 498, 86. [DOI] [PubMed] [Google Scholar]
- [54].Marungruang N, Arevalo Sureda E, Lefrancoise A, Westrom B, Nyman M, Prykhodko O, Fak Hallenius F, Neurogastroenterol Motil 2018, 30, e13285. [DOI] [PubMed] [Google Scholar]
- [55].Nasr SH, Kouyoumdjian H, Mallett C, Ramadan S, Zhu DC, Shapiro EM, Huang X, Small 2018, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Baker A, Kim H, Semple JL, Dumont D, Shoichet M, Tobbia D, Johnston M, Breast Cancer Res 2010, 12, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ostensson K, Lun S, Acta Vet Scand 2008, 50, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Meijer EF, Baish JW, Padera TP, Fukumura D, Methods Mol Biol 2016, 1458, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jia W, Gungor-Ozkerim PS, Zhang YS, Yue K, Zhu K, Liu W, Pi Q, Byambaa B, Dokmeci MR, Shin SR, Khademhosseini A, Biomaterials 2016, 106, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang YS, Arneri A, Bersini S, Shin SR, Zhu K, Goli-Malekabadi Z, Aleman J, Colosi C, Busignani F, Dell’erba V, Bishop C, Shupe T, Demarchi D, Moretti M, Rasponi M, Dokmeci MR, Atala A, Khademhosseini A, Biomaterials 2016, 110, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Liu W, Zhong Z, Hu N, Zhou Y, Maggio L, Miri AK, Fragasso A, Jin X, Khademhosseini A, Zhang YS, Biofabrication 2018, 10, 024102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wei W, Li Y, Yang H, Nassab R, Shahriyari F, Akpek A, Guan X, Liu Y, Taranejoo S, Tamayol A, Zhang YS, Khademhosseini A, Jang HL, Macromol Biosci 2017, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Massa S, Sakr MA, Seo J, Bandaru P, Arneri A, Bersini S, Zare-Eelanjegh E, Jalilian E, Cha BH, Antona S, Enrico A, Gao Y, Hassan S, Acevedo JP, Dokmeci MR, Zhang YS, Khademhosseini A, Shin SR, Biomicrofluidics 2017, 11, 044109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhang W, Zhang YS, Bakht SM, Aleman J, Shin SR, Yue K, Sica M, Ribas J, Duchamp M, Ju J, Sadeghian RB, Kim D, Dokmeci MR, Atala A, Khademhosseini A, Lab Chip 2016, 16, 1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fu WX, Wang Q, Zhang YS, Li Y, Xu T, He S, Ren H, Sun T, Eur Rev Med Pharmacol Sci 2015, 19, 602. [PubMed] [Google Scholar]
- [66].Zhang YS, Wang Y, Wang L, Wang Y, Cai X, Zhang C, Wang LV, Xia Y, Theranostics 2013, 3, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Son YJ, Kim HS, Mao W, Park JB, Lee D, Lee H, Yoo HS, Nanoscale 2018, 10, 6051. [DOI] [PubMed] [Google Scholar]
- [68].Jia WT, Gungor-Ozkerim PS, Zhang YS, Yue K, Zhu K, Liu WJ, Pi Q, Byambaa B, Dokmeci MR, Shin SR, Khademhosseini A, Biomaterials 2016, 106, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Di Giuseppe M, Law N, Webb B, Macrae RA, Liew LJ, Sercombe TB, Dilley RJ, Doyle BJ, J Mech Behav Biomed 2018, 79, 150. [DOI] [PubMed] [Google Scholar]
- [70].Ozcelikkale A, Moon HR, Linnes M, Han B, Wires Nanomed Nanobi 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li LM, Wang XY, Hu LS, Chen RS, Huang Y, Chen SJ, Huang WH, Huo KF, Chu PK, Lab on a Chip 2012, 12, 4249. [DOI] [PubMed] [Google Scholar]
- [72].Morikis VA, Radecke C, Jiang YY, Heinrich V, Curry FR, Simon SI, Biorheology 2015, 52, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Miri AK, Hosseinabadi HG, Cecen B, Hassan S, Zhang YS, Acta Biomater 2018, 77, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Valente KP, Khetani S, Kolahchi AR, Sanati-Nezhad A, Suleman A, Akbari M, Drug Discov Today 2017, 22, 1654. [DOI] [PubMed] [Google Scholar]
- [75].Brunsteiner M, Khinast J, Paudel A, Pharmaceutics 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang X, Luo J, He L, Cheng X, Yan G, Wang J, Tang R, J Colloid Interface Sci 2018, 525, 269. [DOI] [PubMed] [Google Scholar]
- [77].Jhawat V, Gupta S, Saini V, Drug Deliv 2016, 23, 3573. [DOI] [PubMed] [Google Scholar]
- [78].Miri AK, Hosseinabadi HG, Cecen B, Hassan S, Zhang YS, Acta Biomaterialia 2018, 77, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zhu K, Chen N, Liu X, Mu X, Zhang W, Wang C, Zhang YS, Macromol Biosci 2018, 18, e1800127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hausmann MK, Ruhs PA, Siqueira G, Lauger J, Libanori R, Zimmermann T, Studart AR, ACS Nano 2018, 12, 6926. [DOI] [PubMed] [Google Scholar]
- [81].Duan H, Shao Z, Zhao M, Zhou Z, Carbohydr Polym 2016, 137, 92. [DOI] [PubMed] [Google Scholar]
- [82].Huxley VH, Curry FE, Am J Physiol 1987, 252, H395. [DOI] [PubMed] [Google Scholar]
- [83].Zustiak SP, Leach JB, Biotechnology and Bioengineering 2011, 108, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Tripathi D, Jhorar R, Beg OA, Shaw S, Meccanica 2018, 53, 2079. [Google Scholar]
- [85].Cao QL, Han XT, Li L, J Phys D Appl Phys 2012, 45. [Google Scholar]
- [86].Prakash J, Tripathi D, J Mol Liq 2018, 256, 352. [Google Scholar]
- [87].Khan AA, Masood F, Ellahi R, Bhatti MM, J Mol Liq 2018, 258, 186. [Google Scholar]
- [88].Mondal S, De S, Electrophoresis 2013, 34, 668. [DOI] [PubMed] [Google Scholar]
- [89].Yew AG, Pinero D, Hsieh AH, Atencia J, Appl Phys Lett 2013, 102. [Google Scholar]
- [90].Loessner D, Meinert C, Kaemmerer E, Martine LC, Yue K, Levett PA, Klein TJ, Melchels FP, Khademhosseini A, Hutmacher DW, Nat Protoc 2016, 11, 727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.