

Abstract
Biological production of chemicals often requires the use of cellular cofactors, such as nicotinamide adenine dinucleotide phosphate (NADP+). These cofactors are expensive to use in vitro and difficult to control in vivo. We demonstrate the development of a noncanonical redox cofactor system based on nicotinamide mononucleotide (NMN+). The key enzyme in the system is a computationally designed glucose dehydrogenase with a 107-fold cofactor specificity switch toward NMN+ over NADP+ based on apparent enzymatic activity. We demonstrate that this system can be used to support diverse redox chemistries in vitro with high total turnover number (~39,000), to channel reducing power in Escherichia coli whole cells specifically from glucose to a pharmaceutical intermediate, levodione, and to sustain the high metabolic flux required for the central carbon metabolism to support growth. Overall, this work demonstrates efficient use of a noncanonical cofactor in biocatalysis and metabolic pathway design.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Biomanufacturing is the synthesis of chemicals from renewable resources by engineered microbes. Although numerous fuels, pharmaceuticals, and commodities have been biomanufactured, most of these processes failed to proceed beyond laboratory scale because the productivity, titer, and yield are still low1. This problem highlights the existence of a critical knowledge gap in our understanding of cell metabolism. This knowledge gap exists largely due to the extraordinary complexity of metabolic systems2,3.
To overcome the complexity problem, one solution is to insulate the much simpler, engineered pathways in an orthogonal metabolic system that operates in parallel to the hosts’ complex native metabolism2,4. Catabolism and anabolism are the most universal orthogonal metabolic systems in nature. These two seemingly opposing processes coexist without interference largely because they each have a designated redox cofactor, nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+), respectively. Therefore, it has been hypothesized that a third, orthogonal metabolic system could be established if one could introduce a noncanonical redox cofactor inside the cells5.
In addition to their applications in vivo, noncanonical redox cofactors have also been explored as alternatives to NADP+ during in vitro biotransformation, where purified enzymes are used to manufacture chemicals. The majority of industrial biotransformation processes developed so far involve oxidoreductase enzymes6,7, which require redox cofactors. Analogs of NADP+ with simpler structures are easier to synthesize and have faster mass transfer rates, which may greatly reduce the cost of in vitro biotransformation8–10.
However, with the exception of some flavoenzymes9,10, most natural enzymes have low activity with noncanonical redox cofactors. Moreover, shifting enzymes’ cofactor preference toward noncanonical redox cofactors remains a challenging task8,10–12. For example, Sulfolobus solfataricus glucose dehydrogenase (Ss GDH) has been engineered to use simpler NAD+ analogs10. However, the catalytic efficiency of the engineered enzyme was still low (kcat/Km ≈ 5.17 × 10−3 mM−1 s−1, for the cofactor 3-carbamoyl-1-phenethylpyridin-1-ium chloride, where kcat is turnover number and Km is the Michaelis constant for the cofactor). Furthermore, most engineered enzymes still retain substantial levels of activity with the natural redox cofactor NADP+, which lowers their orthogonality in vivo. For example, a metabolic circuit based on the synthetic cofactor nicotinamide cytosine dinucleotide (NCD+) has been constructed in E. coli5. However, the engineered phosphite dehydrogenase enzyme responsible for reducing NCD+ only had a ~four-fold preference of NCD+ over NAD+, which resulted in cross-talk between the metabolic circuit and the NAD+-dependent natural metabolism.
To overcome this challenge, we used computational approaches to design new interactions between the enzyme and the noncanonical redox cofactor nicotinamide mononucleotide (NMN+). In this work, glucose dehydrogenase from Bacillus subtilis (Bs GDH) is targeted. The best variant has an overall specificity switch of ~2 × 107-fold and ~1 × 107-fold toward NMN+ from NAD+ and NADP+, respectively, based on apparent enzymatic activity. By coupling the engineered Bs GDH with various enzymes, we first demonstrated that NMN+ can support diverse chemistries during in vitro biotransformation processes, which include the reduction of C=C double bonds, C≡C triple bonds, and nitro groups, as well as supply electrons to cytochrome P450. In particular, C=C double bonds were reduced with industrially relevant activity and robustness (total turnover number (TTN) ≈ 39,000)13. Next, we demonstrated in E. coli whole cells that NMN+ can mediate orthogonal reducing power delivery from glucose to the production of a pharmaceutical intermediate, levodione, while keeping other NAD(P)H-dependent competing reactions inactive. Finally, we demonstrate the proof of concept that a noncanonical cofactor, NMN+, can be utilized in vivo with sufficiently high flux to sustain life-essential central metabolism.
Results
Engineering Bs GDH to use NMN+.
NMN+ is very similar to NADP+ in redox chemistry. With no modification on the nicotinamide, NMN+ is suggested to have very similar redox potential to that of NADP+ (ref.14) (standard biochemical redox potential, E° = −320 mV; ref.15), which makes NMN+ potentially compatible with all NADP+-mediated chemistries in metabolism. More than 2,000 types of redox reaction directly use NADP+ as their cofactor, which comprise one-quarter of all metabolic reaction types known so far (according to the KEGG database).
On the other hand, NMN+ is structurally distinct from NADP+ because it lacks the adenosine monophosphate (AMP) moiety (Fig. 1a). We hypothesized that with the AMP ‘recognition handle’ missing, enzymes would require stronger interaction with the remaining NMN+ moiety to use the cofactor efficiently. Specifically, we sought to design new interactions with the ribose-phosphate group of NMN+ based on the principle of increasing electrostatic complementarity (Fig. 1a). We targeted glucose dehydrogenase because it is widely used to generate reducing power in biomanufacturing10. The glucose dehydrogenase from B. subtilis (Bs GDH, UniProt ID: A0A1B2ATD9_BACIU) was chosen based on its high expression level in E. coli.
Fig. 1 |. Engineering Bs GDH to use NMN+ as redox cofactor efficiently in biotransformation in vitro.
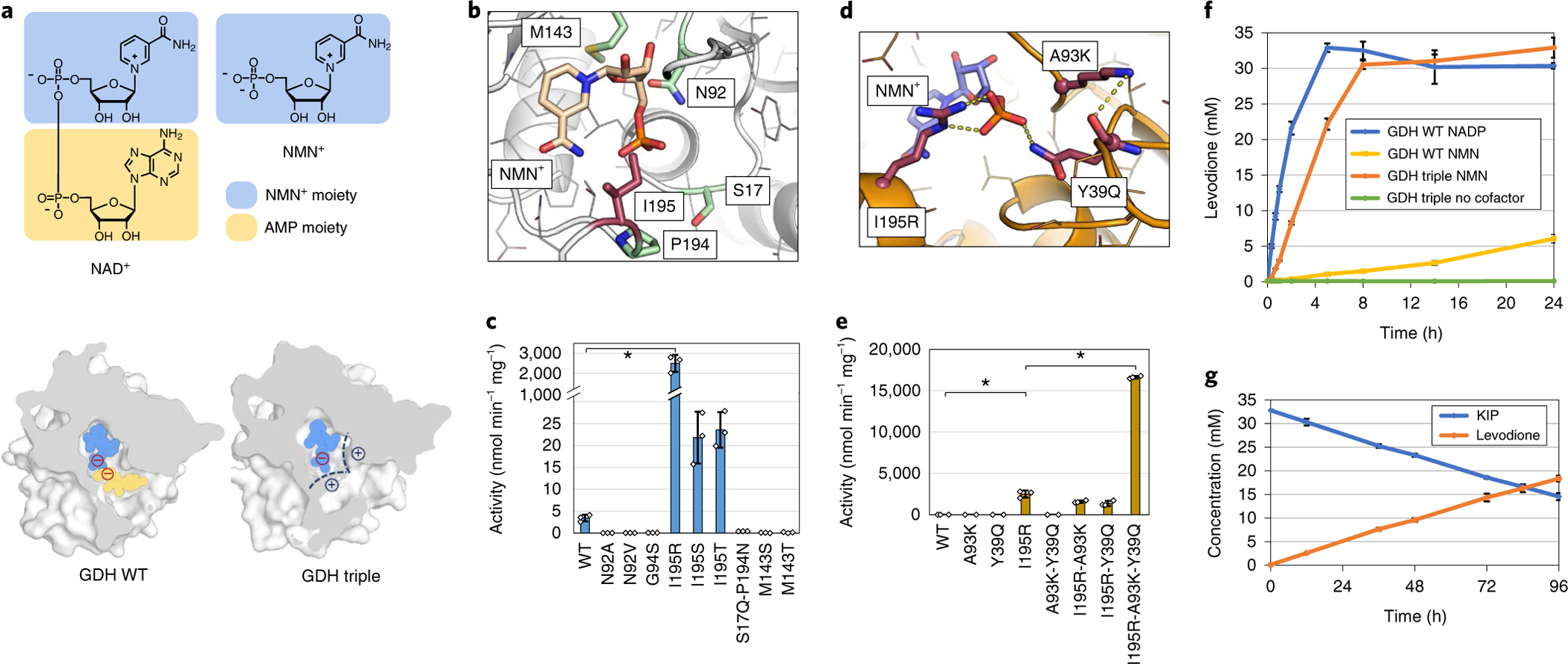
a, Structures of the natural redox cofactor NAD+, and the noncanonical redox cofactor NMN+, and the sliced representation of the NAD+/NMN+ binding pocket. Wild-type Bs GDH (GDH WT) interacts with both the NMN+ moiety (blue) and the AMP moiety (yellow) of NAD+. The Bs GDH Triple (I195R-A93K-Y39Q) has an engineered positively charged region (black dashed lines) for the monophosphate of NMN+ to anchor the ligand in a catalytically relevant conformation. b, The homology model of Bs GDH with NMN+ docked in the active site. The residues where mutations resulted in solubly expressed variants in the first round of engineering are shown in sticks. The mutations on position Ile195 (highlighted in red) resulted in three variants with improved activity. c, Specific activities of first-round variants toward NMN+. d, The model of Bs GDH Triple (I195R-A93K-Y39Q) with NMN+ docked in the active site. The mutations I195R and Y39Q are predicted to form hydrogen bonds with the phosphate on NMN+. A93K is predicted to facilitate and support the loop of Y39Q to be in the proper conformation to interact with NMN+. e, Individual mutations’ contribution to the increased NMN+-dependent activity of Bs GDH Triple. Omitting any of the three mutations abolishes the activity. f, Bs GDH Triple (I195R-A93K-Y39Q), in combination with Pseudomonas putida XenA, supported NMN(H)-dependent levodione generation from KIP in vitro. g, Bs GDH Triple was stable for at least 96 h, as suggested by the steadily increasing levodione and decreasing KIP conversions over 96 h. reactions were performed in 200 mM potassium phosphate buffer (pH 7.5), 1 M NaCl, 300 mM d-glucose, 6 mM NMN+ and 33 mM ketoisophorone at 30 °C while mixing. Bs GDH variants were supplied at 0.33 mg ml−1 (f) or 0.0132 mg ml−1 (g). Values are an average of at least three biological replicates with error bars representing one standard deviation. Two-tailed t-tests were used to determine statistical significance (*P < 0.05).
In the first round of design, a total of nine variants of Bs GDH were tested (Fig. 1b,c). Three variants displayed improvement in activity toward NMN+, which all contained a polar amino acid substitution of Ile195 (Fig. 1c). The best variant from this set, I195R, improves the specific activity of Bs GDH by 545-fold toward NMN+. This arginine residue is predicted to make two new interactions with the negatively charged phosphate on the NMN+ ligand. Protein sequence conservation analysis of Bs GDH against its enzyme family (Pfam ID: PF13561.5) reveals that the Ile195 position is highly variable, where all 20 amino acids are present at varying frequencies (Supplementary Table 1). This suggests that Ile195 is a position with high tolerance to mutational changes, primed for tuning the cofactor binding functionality.
Using I195R as the starting chassis, a second round of design was carried out to introduce more electrostatic interactions with NMN+ from a second site in the binding pocket (Fig. 1a). From this round of design simulation, A93K was predicted to form a new hydrogen bond with the backbone carbonyl oxygen of residue Tyr39 (Fig. 1d). This new interaction was predicted to stabilize a backbone geometry that brings Tyr39 within 6 Å of the phosphate on NMN+, opening new possibilities for engineering (Fig. 1d). Amino acids that could potentially form hydrogen bonds with NMN+ (Q, N, R, K and H) at Tyr39 were systematically introduced with their Rosetta energies evaluated. These simulations predicted that Y39Q could form a new hydrogen bond with the phosphate of NMN+ (Fig. 1d). Consistent with the simulation, I195R-A93K-Y39Q has further improved activity over I195R alone (Fig. 1e). Based on the apparent kinetic parameters (Table 1), this triple mutant (I195R-A93K-Y39Q, Bs GDH Triple) exhibits a kcat/Km of ~0.51 mM−1 s−1, which corresponds to a 1,000-fold increase in catalytic efficiency over the wild-type enzyme toward NMN+. To the best of our knowledge, this represents the largest specificity switch toward noncanonical nicotinamide cofactors achieved until now. The strength of computational design is highlighted by the discovery of the three mutations that function with high cooperativity (Fig. 1e), in that omitting any one mutation diminishes the activity.
Table 1 |.
Apparent kinetics parameters of Bs GDH variants
| Enzyme | Cofactor | Km (mM) | kcat (s−1) | kcat/Km (mM−1s−1) |
|---|---|---|---|---|
| Bs GDH WT (wild type) | NAD+ | (3.0 ± 0.1) × 10−2 | 5.5 ± 1.1 | (1.8 ± 0.3) × 102 |
| NADP+ | (1.5 ± 0.1) × 10−2 | 4.3 ± 0.1 | (2.8 ± 0.1) × 102 | |
| NMN+ | NDa | NDa | (4.7 ± 0.5) × 10−4 | |
| Bs GDH Triple (I195R-A93K-Y39Q) | NAD+ | 3.7 ± 0.9 | (4.1 ± 0.3) × 10−1 | (1.1 ± 0.2) × 10−1 |
| NADP+ | (6.1 ± 1.5) × 10−1 | 4.4 ± 0.1 | 7.5 ± 1.8 | |
| NMN+ | 6.4 ± 0.8 | 3.1 ± 0.1 | (5.1 ± 0.6) × 10−1 | |
| Bs GDH Ortho (I195R-A93K-Y39Q-S17E) | NAD+ | 6.5 ± 1.3 | (2.5 ± 0.2) × 10−2 | (3.8 ± 0.5) × 10−3 |
| NADP+ | 2.0 ± 0.1 | (2.2 ± 1.3) × 10−2 | (1.1 ± 0.8) × 10−2 | |
| NMN+ | 5.9 ± 1.0 | 1.2 ± 0.1 | (2.1 ± 0.2) × 10−1 |
Reactions were performed in 35 mM Tris-HCl buffer (pH 8.0) at 25 °C using a constant glucose concentration of 140 mM. Detailed rate equations can be found in the Methods. Errors shown are the standard deviation of three independent measurements. ND, not determined.
Enzyme could not be saturated with the cofactor concentrations tested.
In vitro NMN(H) cycling supports efficient biocatalysis.
We next sought to determine whether NMN+ and Bs GDH Triple could efficiently sustain enzymatic biotransformation in vitro. Previous studies using enoate reductases revealed true opportunities for industrial use of biomimetic cofactors at preparative scale16. The model reaction we chose was the asymmetric reduction of the activated C=C double bond in ketoisophorone (KIP) catalyzed by the enoate reductase XenA from P. putida9, producing the chiral pharmaceutical intermediate, levodione17. XenA has been shown to be promiscuous for a range of biomimetic redox cofactors9. Using kinetic assays, we first showed that the reduced NMN+ (NMNH) was also well accepted by XenA with comparable activity to NADH and reduced NADP+ (NADPH) (Supplementary Fig. 1). Subsequently, KIP biotransformation was performed using Bs GDH variants to generate NMNH in situ. When using 6 mM NADP+ as the cycling cofactor, the wild-type Bs GDH supported an initial productivity of ~3.00 μM s−1 (Fig. 1f). However, when NMN+ was used in place of NADP+, the initial productivity dropped to ~0.05 μM s−1. The engineered Bs GDH Triple improved the NMN+-dependent productivity by 22-fold, reaching ~1.15 μM s−1. Given an enzyme loading of Bs GDH Triple at 11.7 μM, the initial turn over frequency of the triple mutant reached ~0.10 s−1, which is within the range of industrial catalysts (10−2–102 s−1)13. With no redox cofactors added, the system showed virtually no conversion, which ruled out the possibility that the observed conversion in NMN(H) cycling reaction was conferred by the NADP+ contamination in recombinant proteins10.
The stability of an enzymatic catalyst is critical for its practicality. Using a lower enzyme loading of 0.47 μM for Bs GDH Triple, longer-term KIP biotransformation was performed. The results showed robust conversion over 96 h (Fig. 1g). The TTN of Bs GDH Triple was calculated to be ~39,000, which is substantially higher than those of previously reported biomimetic cofactor recycling methods involving an artificial metalloenzyme (TTN ≈ 2,000)18 or an engineered S. solfataricus GDH (TTN ≈ 1,183)10.
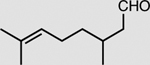
XenA is active toward a broad range of substrates containing activated C=C double bonds19. In addition to a >99% conversion of 33 mM KIP to levodione in 24 h (Table 2), the coupled XenA–Bs GDH Triple system also achieved ~76% conversion of 10 mM citral and ~49% conversion of 50 mM trans-2-hexen-1-al in 24 h, using NMN+ as the cycling cofactor (Table 2).
Table 2 |.
Conversion of a range of substrates using NMN+ as the cycling cofactor
| Substrate | Product | Enzyme | Substrate concentrationa | Conversion (%) |
|---|---|---|---|---|
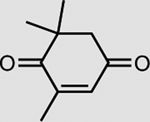 |
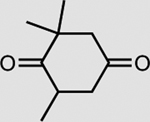 |
XenA | 33 mM | >99 |
 |
 |
XenA | 10 mM | 76 ± 2 |
| XenA | 50 mM | 49 ± 3 | ||
 |
 |
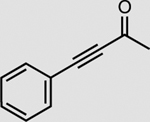
OYE3 | 5 mM | >99 |
 |
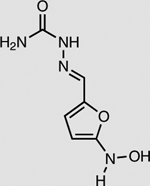 |
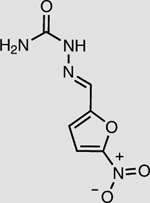
NfsB | 2 mM | 92 ± 1 |
| Cytochrome c (oxidized) | Cytochrome c (reduced) | P450 BM3 W1046S | 50 μM | >99 |
Reactions were preformed in 200 mM potassium phosphate buffer (pH 7.5), 1 M NaCl, 300 mM d-glucose, 6 mM NMN+, and substrates at 30 °C while mixing for 24 h. Bs GDH was supplied at 0.33 mg ml−1. XenA, OYE3, NsfB, and P450 BM3 W1046S were supplied at 0.75 mg ml−1. Values are an average of at least three replicates with values after ± represent one standard deviation.
Substrate concentration was limited by substrate solubility in the buffer.
In vitro NMN(H) cycling supports diverse chemistries.
XenA’s promiscuity for noncanonical redox cofactors may be attributed to its ping-pong mechanism of catalysis involving the flavin prosthetic group. Specifically, the hydride transfer from cofactors to flavin might be less sensitive to the differences in binding modes of various cofactors9. Several other classes of flavoenzymes have been shown to accept biomimetic redox cofactors20–22. Given the versatility of flavoenzymes, there might be opportunities to extend the application of the NMN(H) cycling systems to other chemistries.
The NMN(H) cycling process was coupled with three enzymes other than XenA (Table 2). In the presence of Bs GDH Triple and NMN+, the enoate reductase OYE3 from Saccharomyces cerevisiae23 reduced the activated C≡C triple bond in 4-phenyl-3-butyn-2-one with >99% conversion and the nitro reductase NfsB from E. coli21,24 reduced the nitro group in nitrofurazone with ~92% conversion (Table 2). Cytochrome P450 BM3 from Bacillus megaterium natively has low activity with NMNH. By mutating the highly conserved ‘shielding’ tryptophan at the cofactor binding site to serine22, we first improved the NMNH-dependent activity of BM3 by ~46-fold (Supplementary Fig. 2). Subsequently, we showed that Bs GDH Triple generated NMNH in situ to supply electrons to the engineered BM3 W1046S, allowing the latter to reduce cytochrome c with >99% conversion (Table 2). A control experiment performed without BM3 enzyme showed very low conversion (Supplementary Fig. 3).
Engineering Bs GDH to exclude NADP+.
To achieve orthogonality in vivo, the Bs GDH enzyme must be selective toward NMN+ over NAD+ and NADP+. Therefore, additional mutations that could further disrupt the binding of NAD+ and NADP+ without substantial effects on NMN+ were explored. Specifically, mutations to negatively charged residues that exploited the differential electrostatic repulsion of phosphates between NMN+ and NADP+ were evaluated through docking simulations using Rosetta.
Notable differences in binding modes of these ligands were observed on the introduction of S17E (Fig. 2a,b). The predicted binding mode of NMN+ with the quadruple mutant I195R-A93K-Y39Q-S17E (Bs GDH Ortho) remains similar to that of the Bs GDH Triple, with the hydrogen bonds between I195R and NMN+ unaltered (Figs. 1d and 2a). On the other hand, due to electrostatic repulsion and steric hindrance, S17E is predicted to repel NAD+ into a binding mode that disrupts all favorable interactions with I195R and Y39Q (Fig. 2b). More importantly, the S17E mutation is predicted to force the AMP moiety of NAD+ to further bend outward toward solvents (Fig. 2c), making this ‘recognition handle’ no longer available for the enzyme.
Fig. 2 |. Engineering Bs GDH to exclude NADP+.
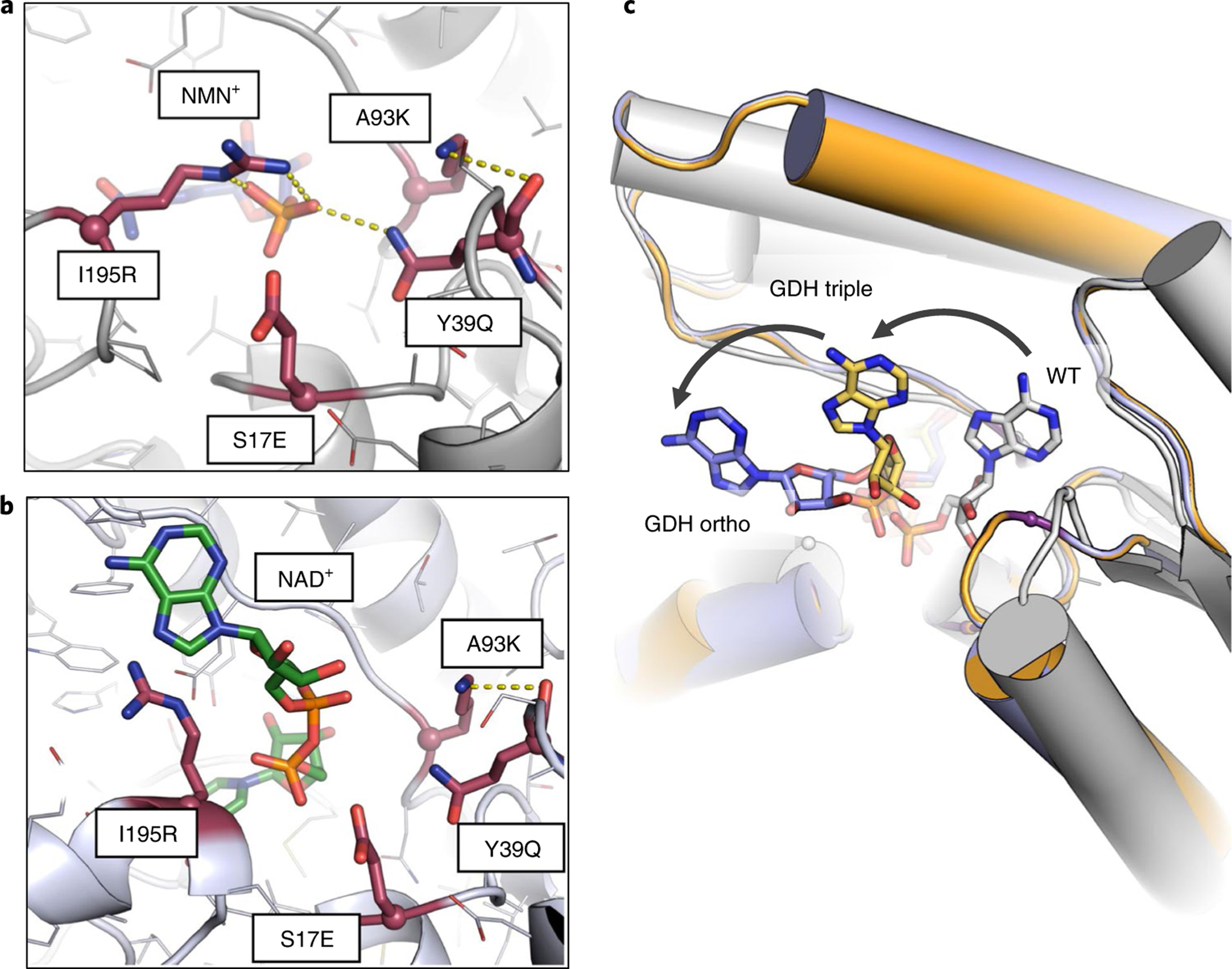
a,b, Comparison of the predicted binding sites of Bs GDH Ortho (I195R-A93K-Y39Q-S17E) with NMN+ (a) and with NAD+ (b). Bs GDH Ortho binds NMN+ in a similar binding mode as observed in Bs GDH Triple (I195R-A93K-Y39Q). In contrast, when NAD+ was docked into this quadruple mutant, S17E repells the diphosphate on NAD+ and leads to a binding mode where the beneficial interactions with I195R and Y39Q are disrupted. c, The predicted conformational changes of NAD+ bound in the wild-type Bs GDH, Bs GDH Triple (I195R-A93K-Y39Q) and Bs GDH Ortho (I195R-A93K-Y39Q-S17E). The introduced mutations in Bs GDH Triple and Bs GDH Ortho move the NAD+ from its native binding mode toward solvent through occluding of the AMP moiety. This causes the ‘recognition handle’ to no longer be available for the enzyme, which switches the specificity toward NMN+.
These predictions are supported by the apparent catalytic efficiencies observed. Comparing Bs GDH Ortho with Bs GDH Triple, the catalytic efficiencies, based on apparent kinetic parameters, dropped 220- and 890-fold for NAD+ and NADP+ on the introduction of the S17E mutation (Table 1), while the activity for NMN+ was only modestly reduced. Compared to the wild type, Bs GDH Ortho has an overall specificity switch of ~2 × 107- and ~1 × 107- fold toward NMN+ from NAD+ and NADP+, respectively, based on apparent kinetic parameters.
Kinetic analysis of the engineered enzymes.
Wild-type GDH follows an ordered bi–bi mechanism in catalysis25 (Supplementary Fig. 4). As indicated by the product inhibition study, the engineered GDH follows the same mechanism using NMN+ as the cofactor (Supplementary Fig. 4). According to the rate equation of the ordered bi–bi mechanism with cofactor binding first in catalysis26, we characterized the full steady-state kinetic parameters of Bs GDH WT, Bs GDH Triple, and Bs GDH Ortho, as well as the microscopic rate constants (Supplementary Table 2). GDH Triple and GDH Ortho have an increased on-rate of cofactor (k1) for NMN+ and decreased on-rate for NADP+ compared to wild-type Bs GDH. The on-rate of glucose (k3) after binding of the natural cofactor NADP+ to the enzyme also drastically decreased for GDH Triple and GDH Ortho. This is consistent with the computational prediction that the natural cofactor may bind to the engineered enzymes with an altered conformation, which may be far less optimal for subsequent glucose binding. The composite microscopic rate constants k1k3/k2 (where k2 is the off-rate of cofactor) has been shown as a useful parameter to compare the performance of ordered bi–bi enzymes26. Compared to Bs GDH WT, the catalytic performance (k1k3/k2) for NMN+ increased by ~36-fold and ~10-fold for Bs GDH Triple and Bs GDH Ortho, respectively. On the other hand, the catalytic performance (k1k3/k2) for NADP+ decreased by 106 to 105-fold for engineered enzymes compared to wild type (Supplementary Table 2). Overall, Bs GDH Ortho has ~1 × 108- and ~3 × 106-fold switch toward NMN+ from NAD+ and NADP+, respectively, in catalytic performance, based on full steady-state kinetic analysis.
In vivo NMN(H) cycling supports E. coli growth.
To investigate Bs GDH Ortho’s NMN+ cycling function in vivo, we sought to link its activity to cell growth and use the latter as an easy readout. First, we disrupted the Embden–Meyerhof–Parnas pathway and the pentose phosphate pathway by disrupting zwf, gnd and pgi genes (Fig. 3a). This resulted in a strain that cannot grow with glucose as the sole carbon source. Next, we built the entry way of glucose to the Entner–Doudoroff (ED) pathway by over-expressing the genes encoding glucose facilitator27 (glf from Zymomonas mobilis), gluconate kinase28 (gntK from Ralstonia eutropha) and wild-type or engineered Bs GDH (Fig. 3a). In this system, cell growth on glucose is specifically linked to the function of GDH because the conversion of glucose to gluconate is an essential step in the single glycolytic pathway available, the ED pathway.
Fig. 3 |. in vivo NMN+ cycling supports E. coli growth.
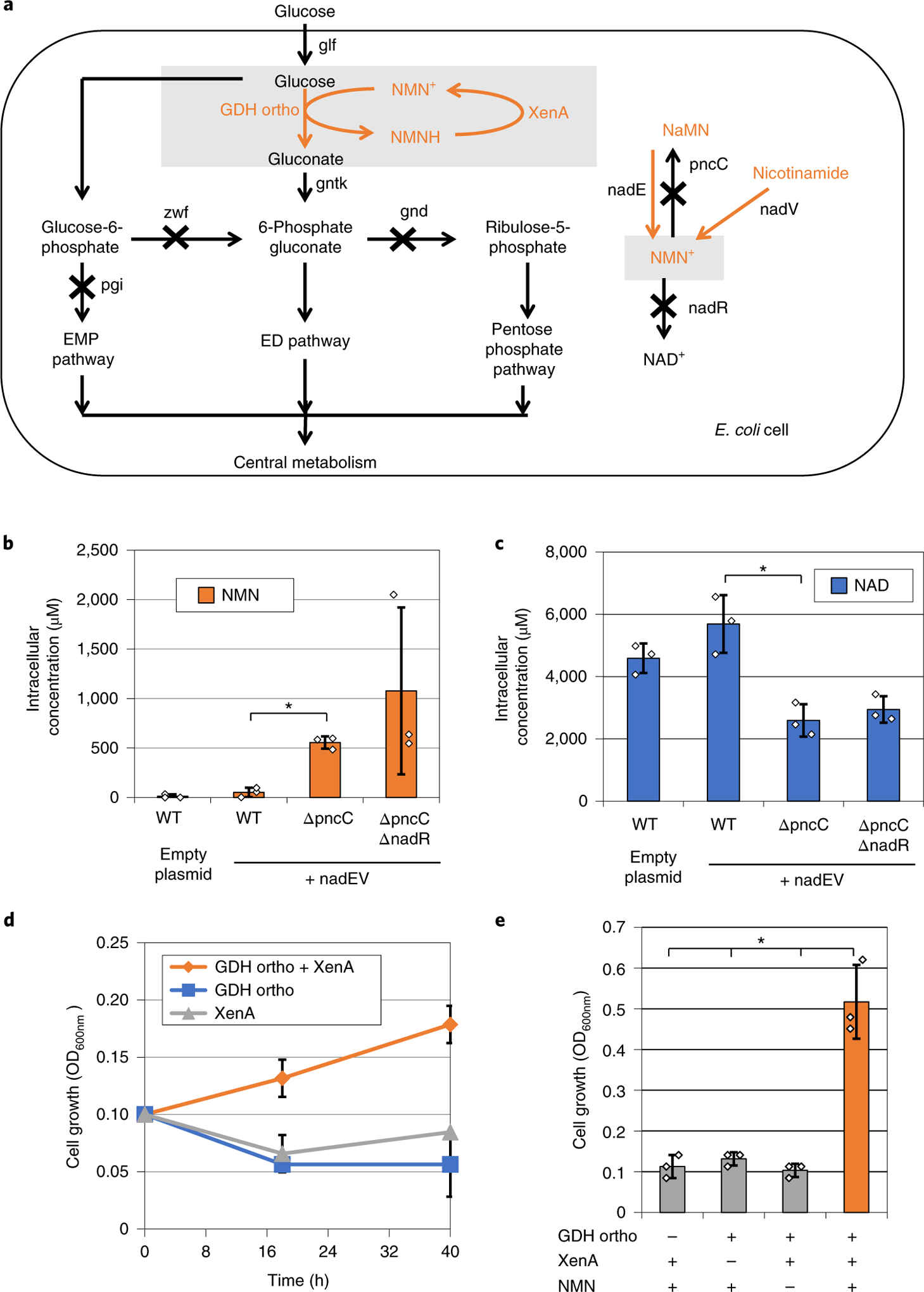
a, An engineered ED pathway that couples GDH activity to E. coli growth on glucose. b, Disruption of NMN+ degrading genes (pncC and nadR) and over-expression of NMN+ producing genes (Ft nadE and Ft nadV) enabled elevated intracellular levels of NMN+. c, Intracellular levels of NAD+ were slightly lowered in the ΔpncC and ΔnadR cells. d, Using the synthetic pathway to supply NMN+ intracellularly, Bs GDH Ortho supported E. coli growth on glucose when XenA was present to recycle NMNH. e, Addition of 5 mM NMN+ extracellularly supported higher cell growth when both Bs GDH Ortho and XenA were present. Values are an average of at least three biological replicates with error bars representing one standard deviation. Two-tailed t-tests were used to determine statistical significance (*P < 0.05). XenA from P. putida. Ft, Francisella tularensis.
Although NMN+ is naturally produced by DNA ligases in E. coli in small amounts29, we hypothesized that this low level may not be sufficient to support effective redox cycling. Other microorganisms possess NMN+ biosynthetic pathways as part of the salvage pathway of NAD+ (ref.29). To build up the intracellular NMN+ pool in E. coli, we over-expressed the genes encoding nicotinamide phosphoribosyltransferase (nadV) and NMN synthetase (nadE) from Francisella tularensis30 in the above-mentioned strain (Fig. 3a). Furthermore, we disrupted potential NMN+-degradation pathways in E. coli by knocking out genes pncC and nadR29 (Fig. 3a). These manipulations together resulted in a ~1,000-fold increase in intracellular NMN+ concentration compared to that of wild-type E. coli (from ~1 to ~1,077 μM, Fig. 3b). In the highest NMN+-producing strain, the level of the NMN+ reached around 30% that of NAD+ (Fig. 3c).
Subsequently, we introduced the NMN(H)-cycling enzymes (Fig. 3a,d). In minimal medium with glucose as the sole carbon source, only the cells harboring both Bs GDH Ortho and XenA were able to grow, suggesting that these two enzymes form a closed redox cycle in vivo that sustained the engineered glycolytic pathway. Consistently, cells with only one-half of the redox pair did not grow (Fig. 3d). Although we did not supply XenA substrates, such as KIP, in the medium, previous studies have shown that oxygen, quinones and intracellular metabolites with α,β-unsaturated aldehydes, and ketones could be used by enoate reductases as electron acceptors31. Overall, these results support that NMN+ cycles in vivo, which can provide sufficiently high flux to support the life-essential, central carbon metabolism. Moreover, NMNH is an orthogonal reducing power in E. coli because native enzymes in the host could not consume NMNH efficiently enough to replace the need for a heterologous, NMNH-consuming enzyme (for example, XenA).
Although the built-in NMN+ pool could support effective redox cycling, we found that supplementing NMN+ in the medium afforded an increased growth rate (Fig. 3 and Supplementary Fig. 5). Again, we showed that cells with all three components of the redox cycle (Bs GDH Ortho, XenA and NMN+ supplementation) reached a much higher cell density than those with any one of the three components omitted (Fig. 3e). The dependence of growth on NMN+ concentration again indicates that Bs GDH Ortho uses this noncanonical redox cofactor in vivo. To the contrary, wild-type Bs GDH was able to use the natural cofactors NADP+ in vivo and rescued growth without XenA over-expression or NMN+ supplementation (Supplementary Fig. 5). E. coli cells have been shown to both uptake and excrete NMN+ (ref.32,33). As NMN+-transporting systems continue to be discovered in various organisms34,35, genomics information may aid in the elucidation of transport mechanism of NMN+ in E. coli.
NMN(H) enables specific electron delivery in E. coli.
Compared to in vitro biotransformation, whole cell-based processes are considered more robust and inexpensive. However, one drawback is that the host’s natural metabolism often interferes with the desired biotransformation reaction in vivo. For example, aldehydes are often reduced by the numerous, nonspecific, C=O bond reducing-enzymes native to the hosts36–38. As such, a tool is needed to deliver reducing power only to the desired reaction in vivo. We sought to test if the Bs GDH Ortho-mediated NMN(H) cycling system can serve as such a tool.
We chose the levodione production reaction (Fig. 1f,g) as the model system. In the substrate KIP, three sites are susceptible to enzymatic reduction (Fig. 4a). While reduction of the C=C double bond by XenA yields the desired product levodione, reduction of the two C=O groups by the enzymes levodione reductase39 (LVR from Corynebacterium aquaticum) and alcohol dehydrogenase40 (ADH, from Ralstonia sp.) will result in the side products phorenol and 4-hydroxyisophorone (HIP), respectively.
Fig. 4 |. Bs gDH ortho selectively provides reducing power for levodione production in E. coli whole cells.
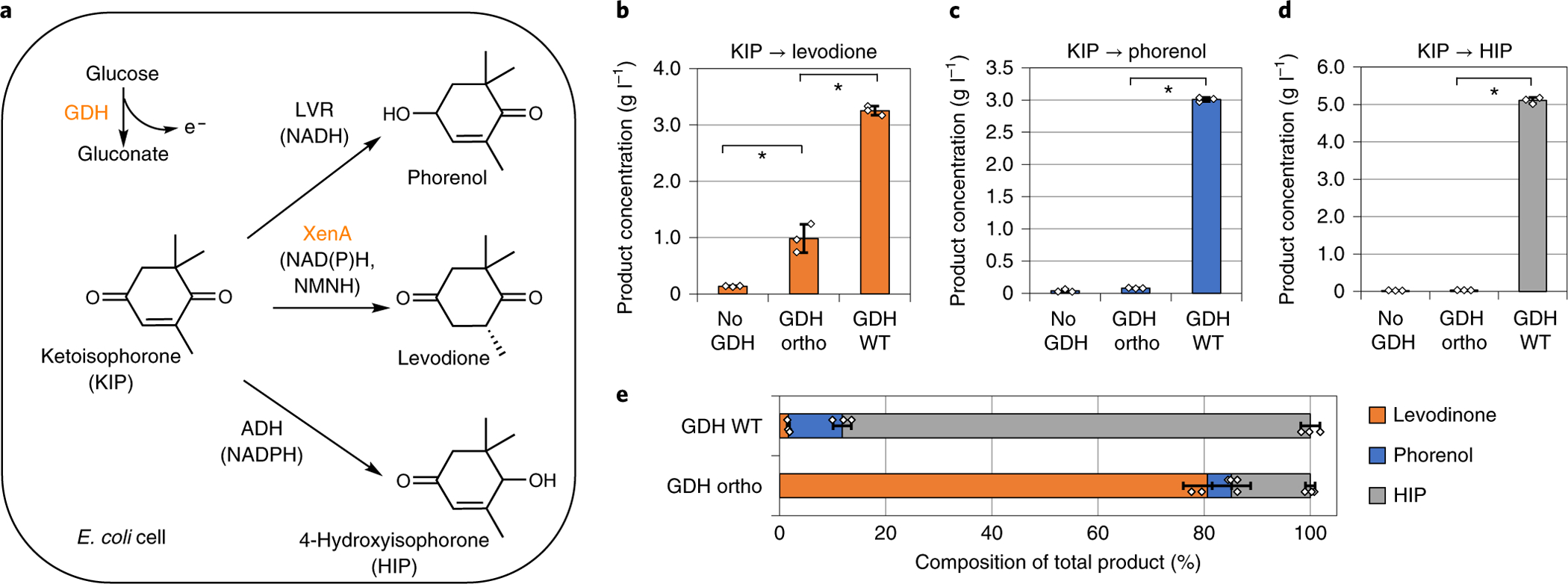
a, Bs GDH derives reducing power from glucose, which can support the biotransformation enzymes LVR, XenA and ADH to convert ketoisophorone (KIP) into phorenol, levodione and HIP, respectively. b–d, KIP conversion using resting E. coli cells expressing Bs GDH variants and individual conversion enzymes XenA (b), LVR (c) or ADH (d). Wild-type Bs GDH supported all three conversion reactions, while Bs GDH Ortho specifically facilitated the conversion of KIP to levodione, which is catalyzed by an NMNH-using enzyme, XenA. e, KIP conversion using resting E. coli cells expressing Bs GDH variants and all three conversion enzymes (LVR, XenA and ADH) simultaneously. Total product composition switched toward levodione production when Bs GDH Ortho was expressed instead of Bs GDH WT. Whole-cell biotransformation was performed using resting E. coli cells of OD600nm of ~2, in buffer containing 100 mM potassium phosphate, 2 mM NMN+, 200 mM glucose, and 5 g l−1 KIP, at 30 °C for 48 h. Values are an average of at least three biological replicates with error bars representing one standard deviation. Two-tailed t-tests were used to determine statistical significance (*P < 0.05). XenA from P. putida; LVR from C. aquaticum; ADH from Ralstonia sp.
We first built the whole-cell biotransformation chassis by disrupting pgi, zwf and gntK genes in E. coli (Supplementary Fig. 6), leaving Bs GDH as the only enzyme that can generate reducing power from glucose. Next, we paired Bs GDH wild-type or Bs GDH Ortho with XenA, LVR or ADH, individually (Fig. 4b–d). In resting cells with XenA and wild-type Bs GDH, the whole cells produced ~3 g l−1 levodione from 5 g l−1 KIP, using glucose as the co-substrate. Bs GDH Ortho was also able to power the XenA-catalyzed reaction with NMN+ supplementation in the buffer (~1 g l−1 levodione was formed under the same conditions) (Fig. 4b). In contrast, LVR and ADH-catalyzed reactions were only active when wild-type Bs GDH was used (Fig. 4c,d). When Bs GDH Ortho was used, minimal levels of phorenol or HIP was produced (Fig. 4c,d), indicating that LVR and ADH, which catalyze the competing reactions in our model system, were not supplied with reducing power.
Last, we over-expressed XenA, LVR and ADH simultaneously in resting cells, which resulted in a mixture of products being formed from KIP. By using different Bs GDH variants to generate reducing power, the composition of the mixtures can shift greatly (Fig. 4e). When using wild-type Bs GDH to generate NADH and NADPH, all three enzymes were active. However, XenA could not compete favorably with LVR and ADH for substrate conversion, which led to low product-to-byproduct ratio (levodione only constituted ~2% of total products by mass, Fig. 4e). On the other hand, when using Bs GDH Ortho, the fraction of levodione in the product mixture increased to ~80%. Analysis of the levodione, phorenol, and HIP concentrations suggested that the improved ratio was largely due to the substantially decreased byproduct formation (Supplementary Fig. 7). These results support that NMNH can be established as an alternative reducing power in E. coli to increase selectivity in whole-cell biotransformation. E. coli’s endogenous enoate reductase, NemA, might also contribute to levodione production. Based on a previous report, this contribution may be minor due to the relatively low background activity of NemA without over-expression41.
Discussion
In summary, we have developed a noncanonical redox cofactor system based on NMN+. We explored its applications in redox cycling in vitro and specific electron delivery in vivo. This was enabled by a computationally designed Bs GDH that possesses the largest specificity switch toward a noncanonical nicotinamide cofactor achieved so far. Given the catalytic roles of the nicotinamide ring and its surrounding residues, most mutational candidates will likely lie in residues that could form new interactions with the negatively charged phosphate when NMN+ is the desired cofactor. Our future work will aim to mine a diverse ensemble of oxidoreductase scaffolds42 primed for the incorporation of hydrogen bond-donating amino acids to bind NMN+, which could potentially result in a large panel of enzymes capable of harboring NMN+ for catalysis. More than 150,000 sequences have been annotated as or are predicted to be NADPH-dependent dehydrogenases43. Given that Bs GDH binds cofactors with a conserved Rossmann fold, the design principles discovered here may be applicable to other Rossmann fold enzymes. Future work is needed to extend this work to targeting other cofactor binding folds, such as the TIM-barrel and the dihydroquinoate synthase-like folds.
We have engineered E. coli cells to require NMN+-based redox balance to grow. This growth phenotype may enable high-throughput selection, which can open opportunities for engineering NMN+-dependent enzymes through directed evolution, or optimizing NMN+-dependent pathways in vivo in a combinatorial manner. Similar redox balance-based, high-throughput selection platforms have been established for the two natural redox cofactors NAD+ and NADP+ (refs.44–46).
To increase the reaction rate of NMN+-dependent whole-cell biotransformation, future work may focus on further improving the catalytic efficiencies of the NMN+-dependent enzymes, potentially via directed evolution as mentioned above. Furthermore, recent efforts demonstrated pathways for efficient NMN+ biosynthesis47, which could be used to further increase intracellular NMN+ concentration toward ultimately eliminating the need for NMN+ supplementation. We did not observe substantial growth deficiency in NMN+-accumulating cells. Future work is needed to characterize the physiological impacts of NMN+ accumulation, such as potentially identifying host enzymes that are regulated by NMN(H) and studying transcriptomic changes caused by varying NMN(H) level.
This work demonstrated that cofactor specificity switch between NMN+ and NADP+ can be achieved by introducing step-wise point mutations, which indicates that this switch may be readily accessible during natural evolution. Especially, since NMN+ has simpler structure, one hypothesis is that it may be an evolutionary precursor of NADP+. However, given that ancient biochemistries remain imprinted in modern metabolism, this hypothesis is not supported by the fact that, to our knowledge, no existing metabolic pathways use NMN+ as a redox cofactor. In fact, it has been suggested that NADP+ predated NMN+ in evolution, and the AMP moiety in NADP+ represents a relic of the RNA-based catalysts14, rather than an additional recognition handle gained later. This view is consistent with the fact that the AMP moiety is common in many universal coenzymes such as coenzyme A and adenosine triphosphate.
Methods
Bs GDH design strategy.
Our design process focused on building and introducing new interactions to NMN+ while keeping the nicotinamide ring binding and catalytic residues unaltered. Since no crystal structure of Bs GDH was available, a HMMER48 search was performed to identify orthologous protein structures to determine essential versus designable residues of Bs GDH. Glucose dehydrogenase from B. megaterium (Bm GDH, PDB 1GCO) with NAD+ bound was found from this search. Pairwise sequence alignment shows that Bs GDH and Bm GDH share a pairwise sequence identity of 83.5%. The NAD+ in this crystal structure forms hydrogen bonds with Thr193 and Asn196 through the amide group on the catalytically essential nicotinamide moiety. In addition, Tyr158 and Lys162 of the catalytic triad hydrogen bond with both hydroxyl groups on the ribose of the nicotinamide half of NAD+ (Supplementary Fig. 8)49. Therefore, the equivalent residues were kept constant during all subsequent design simulations and geometric constraints enforcing the chemical interactions observed in Bm GDH were used throughout subsequent simulations to prevent the interactions critical to the enzyme’s function from being changed. Based on the Bm GDH structure, a molecular model of Bs GDH was produced using RosettaCM50 to obtain its three-dimensional structure and design simulations were carried out with RosettaDesign51 and Foldit52, as detailed below.
Homology modeling and design of Bs GDH.
A homology model of Bs GDH was produced using the RosettaCM50 protocol. The protein sequence of Bs GDH was used to search against the Protein Data Bank using HMMER to identify homologous crystal structures for modeling generation. 2,500 simulations were run to generate the models. The lowest scoring model based on total system energy was chosen as the starting point for design. An example run of the homology modeling simulation containing all inputs, command lines, and simulation variables can be found in the Supplementary Information.
The three-dimensional representation of NMN+ was built based on the nicotinamide adenine dinucleotide molecule found from the crystal structure 1GCO (a closely related enzyme with a pairwise sequence identity of 83.5% relative to Bs GDH). This output was then optimized and its conformer library generated using Spartan53. The resulting conformer library was used for a docking and design simulation with the homology model of Bs GDH using RosettaDesign with distance and angle constraints present (Supplementary Information). A total of 1,000 simulations were run, and the top 20 best scoring outputs sorted based on protein-ligand interface energy and total system energy were selected for further optimization and design using Foldit52. During the design simulations, all side chains within 6 Å of the NMN+ ligand were allowed to be designed, and any residues within 8 Å of the ligand were relaxed with backbone movements enabled. For the docking of NAD+, the docking protocol was the same, but the conformer library was generated in a different manner. Due to the flexibility of NAD+, the NAD+ conformer library was generated by sampling the representative ensembles of NAD+ conformers listed in CoFactor database. Overall, 31 conformers of NAD+ were included in the conformer library for the docking simulation. An example run of the design simulation containing all inputs, command lines, and simulation variables can be found in the Supplementary Information.
Plasmid and strain construction.
All plasmids and strains used in this study are summarized in Supplementary Table 3. All plasmids were constructed using Gibson isothermal DNA assembly method54. PCR reactions were performed using PrimeSTAR Max DNA Polymerase (TaKaRa). Cloning was done in E. coli XL1-Blue (Stratagene). The detailed methods for plasmid construction are described below.
The Bs gdh gene was amplified by PCR from chromosomal DNA of B. subtilis. The resulting PCR product was gel-purified and assembled with a vector backbone that contains a 6× His-tag at the N terminus (ColE1 ori, AmpR) by the Gibson isothermal DNA assembly method. Site-directed mutagenesis was performed by introducing point mutations on the primers, followed by the assembly of the mutation-containing PCR fragments. R. eutropha gntK, E. coli nfsB and S. cerevisiae OYE3 genes were amplified by PCR from corresponding chromosomal DNA. C. aquaticum lvr, R. sp adh, F. tularensis nadE and nadV, P. putida xenA and Z. mobilis glf were amplified from synthesized DNA templates. P450 BM3 was amplified from the plasmid pBsaBM3, a gift from T. Nagamune (Addgene plasmid no. 85102). All genes were also inserted in their respective vectors similarly.
For multi-gene plasmids, the genes were inserted sequentially. For example, pLZ219 (Supplementary Table 3) was constructed by using a forward primer that binds immediately downstream of the Bs gdh gene on pSM107 together with a reverse primer that binds at the 3′-end of the Bs gdh gene, yielding a backbone DNA fragment containing the Bs gdh gene and the vector. Subsequently, the backbone and amplified PCR fragment of P. putida xenA were assembled by Gibson assembly. Empty plasmids pSM105 and pSM108 were constructed by eliminating the Bs gdh from pEK101 and pSM107, respectively.
The multi-gene deletions in strains MX101, MX102 and MX103, were created by P1 phage transduction followed by flippase recombinase-mediated excision of the corresponding kanamycin resistance cassette as described previously55. Derivatives of E. coli K-12 strain BW25113 carrying single gene deletions were used as the donors, which carry a flippase recognition target-flanked kanamycin resistance cassette in place of the target gene. Donors were obtained from the Yale E. coli Genetic Stock Center.
Protein expression and purification.
Proteins were purified as 6× His-tag fusion at the N terminus. E. coli BL21 (DE3) with plasmids were inoculated into LB medium with 200 mg l−1 ampicillin. Cells were induced with 0.5 mM IPTG, and the proteins were expressed for 24 h at 30 °C while shaking at 250 r.p.m. BM3 cytochrome P450 was extracted with the BugBuster Plus Lysonase Kit (EMD Millipore) and purified using the HisPur Ni-NTA Superflow Purification System (Thermo Fisher) according to the manufacturer’s instructions. Others proteins were purified using His-Spin Protein Miniprep kit (Zymo Research Corporation). The purified proteins were quantified by Bradford assay.
GDH enzymatic assays and kinetics study.
The GDH activity was measured as described previously25. The reactions were started by adding the purified protein to the assay mixture containing 35 mM Tris-HCl buffer (pH 8.0), cofactors, and glucose. The absorbance variation at 340 nm was detected by a spectrophotometer at 25 °C. For specific activity, 3 mM NAD+, NADP+ or NMN+ and 140 mM glucose were used. For apparent kinetics parameter determination, cofactor concentrations were varied with a constant 140 mM glucose. For full steady-state kinetics parameter determination, both cofactor and glucose concentrations were varied.
For apparent kinetic parameter analysis, the results were fitted to the standard Michaelis–Menten equation.
| (1) |
where v0 is the initial reaction rate, Et is the total enzyme concentration and S is the concentration of cofactor. In the case of Bs GDH wild type with NMN+, The reaction rate is linearly proportional to the cofactor concentration, and the enzyme could not be saturated with the cofactor concentrations tested. Therefore, the data was fitted to equation (2), which is a simplified form of equation (1) under Km ≫ S:
| (2) |
For full steady-state kinetic parameter analysis, the results were fitted to the following ordered bi–bi rate equation26:
| (3) |
where A and B are the concentrations of cofactor and glucose, respectively, KA and KB are Michaelis constants for cofactor and substrate, respectively, and kia is the dissociation constant for cofactor binding.
In the case of wild-type Bs GDH with NMN+, and Bs GDH Ortho with NAD+, the data fitted poorly to equation (3) due to a very large KA. The reaction rate is linearly proportional to the cofactor concentration, and the enzyme could not be saturated with the cofactor concentrations tested. Therefore, equation (4) was used, which is a simplified form of equation (3) when KA ≫ A:
| (4) |
Similarly, in the case of Bs GDH Triple with NAD+, the reaction rate is linearly proportional to the glucose concentration and the enzyme could not be saturated with the glucose concentrations tested. Therefore, equation (5) was used, which is a simplified form of equation (3) when KB ≫ B:
| (5) |
The microscopic reaction rates were calculated from the relationships26:
| (6) |
where k1 is the on-rate of cofactor, k2 is the off-rate of cofactor and k3 is the on-rate of glucose.
For product inhibition studies, the assay mixture contains 35 mM Tris-HCl buffer (pH 8.0), 50 mM d-glucose, various concentrations of oxidized cofactor, and reduced cofactor. NMNH was generated enzymatically as detailed below. The conversion was performed in 50 mM Tris-Cl (pH 8.5), 0.2 mM EDTA, 50 mM Na2HPO4, 5 mM d-glyceraldehyde-3-phosphate, and 4 mM NMN+. The conversion was started by spiking 28 μM of purified E. coli glyceraldehyde-3-phosphate dehydrogenase. The mixture was incubated at 30 °C for 8 h. The mixture was then filtered through an Amicon Ultra 3 K filter (Millipore) to remove the protein. The filtrate was concentrated by Speedvac and resuspended in 150 μl of water. The NMNH concentration was measured by comparing absorbance at 340 nm to a standard curve of NADPH.
XenA activity assays.
The enzymatic buffer contained 200 mM phosphate buffer (pH 7.5), 5 mM ketoisophorone, 0.2 mM NADPH, NADH or NMNH, and 8 μg ml−1 purified XenA protein. The initial reaction rate was quantified by absorbance variation at 340 nm using a spectrophotometer at 37 °C. Reactions with no ketoisophorone added were performed to quantify the nonspecific reaction rate. The net reaction rate was calculated by subtracting the nonspecific reaction rate from the total reaction rate. For generating NMNH, a microcentrifuge tube (2 ml) containing 35 mM Tris-HCl (pH 8.0), 4 mM NMN+, 1 M NaCl and 140 mM glucose, was incubated for ~3 h at 30 °C with purified Bs GDH I195R. An aliquot of this reaction system was taken to measure the accumulation of NMNH by reading absorbance at 340 nm. The mixture was then filtered through an Amicon Ultra 3K filter (Millipore) to remove the protein. The NMNH concentration was measured by comparing absorbance at 340 nm to a standard curve of NADPH. The solvent was evaporated, and the residue was collected to give NMNH as a yellowish oil.
P450 BM3 assay conditions.
Reaction mixtures (0.2 ml) contained 0.2 M potassium phosphate buffer (pH 7.5), 0.3 M glucose, 1 M NaCl, 2 mM NMNH with 50 μM cytochrome c (C2506, Sigma). Reactions were initiated by the addition of 0.75 mg ml−1 purified BM3 variants. Reduction of cytochrome c was monitored spectroscopically at 550 nm. Quantification was performed using an extinction coefficient ε550 of 21.1 mM−1 cm−1 (ref.22). NMNH was generated as described above for XenA activity assays.
Coupled enzymatic biotransformation.
All biotransformation reactions were performed in buffer A at 30 °C for 24 h. Buffer A, modified from a previous system19, contained 200 mM potassium phosphate buffer (pH 7.5), 1 M NaCl, 300 mM d-glucose, 6 mM NMN+ (or NADP+ as the positive control), and substrates. All assays were performed in triplicate, with no proteins or no cofactors added as negative controls. The protein loading for GDH variants was kept at 0.33 mg ml−1 or 11.7 μM. The various enzymes for biotransformation reactions were added at the concentration of 0.75 mg ml−1.
For XenA–Bs GDH coupled cycling assays, the substrates ketoisophorone, citral, or trans-2-hexen-1-al were added at 33, 10 or 50 mM, respectively. For the OYE3-GDH coupled cycling assays, the substrate 4-phenyl-3-butyn-2-one23 was added at 5 mM. At various time points over 24 h, 100 μl samples were taken and extracted with 100 μl ethyl acetate. Conversion was determined via gas chromatography-flame ionization detection (GC–FID) with octanol as an internal standard (see below). For NfsB-Bs GDH coupled cycling assays, 2 mM nitrofurazone was used as the substrate24. Nitrofurazone conversion was measured spectroscopically at 400 nm and quantification was performed using a standard curve24. The initial levels added of the above-mentioned substrates were mainly determined by their solubility in the assay buffer. For P450 BM3-Bs GDH coupled cycling assays, 50 μM cytochrome c (C2506, Sigma) was used as the substrate. The reduction of cytochrome c was measured spectroscopically at 550 nm, and the quantification was performed using extinction coefficient ε550 of 21.1 mM−1 cm−1 (ref.22).
NMN+-dependent whole-cell biotransformation.
One biotransformation plasmid expressing XenA, LVR or ADH with a GDH (selected from pLZ217-pLZ225) and pSM104 containing the glucose transport facilitator were transformed into strain MX102 by electroporation. Then, 4 ml seed cultures of 2xYT media with appropriate antibiotics, 0.1 mM IPTG and 0.2% (w/v) glucose were cultured at 30 °C while shaking at 250 r.p.m. for 16 h. Next, 0.5% (v/v) seed cultures were used to inoculate 150 ml of 2xYT media with appropriate antibiotics, A5 trace metals with cobalt and 0.5 mM IPTG in a 250 ml baffled shake flask and cultured at 30 °C at 250 r.p.m. When an optical density (OD600) of ~0.4 was reached, protein expression was induced with 0.1% (w/v) arabinose, and the cells were cultured for an additional 8 h at 30 °C while shaking at 250 r.p.m. Cells were collected by centrifugation for 15 min at 20 °C at 3,750 r.p.m. The supernatant was discarded. Cells were washed three times with 50 ml of 100 mM potassium phosphate (pH 7.5), followed by being resuspended to an OD600 of 100 in assay buffer consisting of 100 mM potassium phosphate buffer (pH 7.5), 200 mM d-glucose, 0.5% arabinose, and 0.5 mM IPTG. Then 1 ml of resuspended cells were added to 20 ml of identical assay buffer in a 250 ml unbaffled, screw-cap shake flask. KIP was spiked into the flask to 5 g l−1 to initiate the reaction. Flask caps were secured tightly to prevent evaporative loss of substrate or products. Flasks were incubated at 30 °C while shaking at 250 r.p.m for 48 h. After 48 h, 1 ml of culture was pelleted, and the supernatant was used for analysis. 200 μl of supernatant was extracted with an equal volume of ethyl acetate containing 200 mg l−1 octanol as an internal standard, and the samples were analyzed by GC (see detailed method below). For samples expressing all three conversion enzymes (XenA, LVR and ADH) on the same vector (pLZ226), the Bs GDH was expressed individually on a separate vector (pSM106, pSM107, or pSM108). The Zm glf gene was also expressed in this system (pSM110).
GC–FID analytical methods.
All GC analysis was performed on an Agilent 6850 (Agilent Technologies) equipped with an FID. An Agilent DB-WAXetr capillary column (30 m × 0.56 mm × 1 μm) was used for separation. The inlet and detector were held at 250 and 260 °C, respectively. The GC was operated in constant pressure mode with a pressure of 3.66 psi. Helium was used as the carrier gas. Air and hydrogen were supplied to the FID at 350 and 40 ml min−1, respectively. All gasses were purchased from Airgas and 5 μl of sample was injected with a split ratio of 2:1.
For analysis of citral and its reduction product citronellal, the oven was initially held at 150 °C for 10 min and then ramped at a rate of 45 °C min−1 to 240 °C. Citral and citronellal eluted at 9.32 and 4.50 min, respectively. Octanol was used as an internal standard.
For analysis of trans-2-hexen-1-al and its reduction product trans-2-hexan-1-al, the oven was initially held at 50 °C for 1 min, the oven was ramped at 15 °C min−1 to 120 °C, then ramped at 20 °C min−1 to 230 °C and held for 3 min. Trans-2-hexen-1-al and trans-2-hexan-1-al eluted at 6.41 and 4.78 min, respectively. Octanol was used as an internal standard.
For analysis of 4-phenyl-3-butyn-2-one (containing C≡C triple bond) and its fully reduced product 4-phenyl-2-butanone, as well as the intermediate 4-phenyl-2-butene-2-one (containing a C=C double bond), the oven was initially held at 200 °C for 1 min, then ramped at 5 °C min−1 to 230 °C and held for 1 min. Octanol was used as an internal standard. In vitro ketoisophorone reduction to levodione was analyzed using the same method. Elution times are as follows: 4-phenyl-3-butyne-2-one (5.53 min), 4-phenyl-2-butene-2-one (6.76 min), 4-phenyl-2-butanone (4.55 min), ketoisophorone (3.65 min), levodione (4.08 min) and octanol (2.80 min).
For analysis of in vivo ketoisophorone biostransformation, the oven was held at 200 °C for 15 min. Elution times are as follows: octanol (2.80 min), ketoisophorone (3.76 min), levodione (7.25 min), phorenol (8.35 min) and 4-hydroxyisophorone (12.90 min).
GDH TTN determination.
TTN (Fig. 1g) was determined by the number of moles of product formed divided by the moles of purified GDH added. The assays were performed in reaction buffer A, as shown above, at 30 °C and 33 mM ketoisophorone was used as the substrate. The reaction was started by spiking purified proteins (0.0132 mg ml−1 or 0.47 μM for GDH, 0.75 mg ml−1 for XenA). Samples were taken every 12 h for 96 h. The extraction and GC–FID analysis were performed using a similar method to that mentioned above.
Quantification of intracellular NMN+ and NAD+ levels.
A plasmid containing Ft nadE and nadV (pWB203) was transformed into E. coli strains BW25113, JW2670–1 and MX101 to examine their effects on NMN+ generation. Overnight cultures were grown at 30 °C while shaking at 250 r.p.m. in 2xYT media containing 0.1 mM IPTG, 0.2% d-glucose and appropriate antibiotics for 12 h. To cultivate cells for nucleotide analysis, 10 ml of 2xYT media containing 0.5 mM IPTG, 1 mM nicotinamide and appropriate antibiotics in a 50 ml conical tube was inoculated with 1% v/v overnight culture. Tubes were incubated at 30 °C at 250 r.p.m. for 4 h.
Before gathering cells, cell density was measured at 600 nm and 1 ml of culture was pelleted in a 1.5 ml microcentrifuge tube. Supernatant was removed by pipetting. The cell pellet was washed once with 1 ml of room temperature deionized water, repelleted, and the supernatant was removed by pipetting. Cells were lysed with 1 ml of 95 °C 1% formic acid with 1 μM 1-methylnicotinamide as an internal standard. Cells were incubated at 95 °C for 2 min while intermittently vortexing to ensure complete lysis. Lysates were quenched in an ice water bath before pelleting cell debris. Supernatant was run on an ultrahigh-performance-tandem mass spectrometry (UPLC-MS/MS) system for analysis. The UPLC-MS/MS method is detailed in the Supplementary Information. Values from liquid chromatography (LC)–MS/MS were correlated back to intracellular concentration using the number of cells per OD600 of 1 in 1 ml of culture = 1 × 109 and the intracellular volume of an E. coli cell as 1 × 10−15 l per cell56.
Liquid chromatography was performed on a Waters ACQUITY UPLC with a Waters ACQUITY UPLC CSH C18 column (1.7 μm × 2.1 mm × 50 mm). Mobile phases used in the separation were (A) water with 2% acetonitrile and 0.2% acetic acid and (B) acetonitrile with 0.2% acetic acid. The compounds were separated with a linear gradient from 10 to 90% buffer B over 1 min, held at 90% buffer B for 1 min, then returned to 10% buffer B and held at 10% buffer B for 1 min. Flow rate was held constant at 0.3 ml min−1 and 10 μl of sample was injected for analysis.
MS/MS detection was performed by a Waters Micromass Quattro Premier XE Mass Spectrometer operating in positive ion, MRM mode. Capillary voltage was set to 3.3 kV. Desolvation gas flow rate was 800 l h−1 at 300 °C. Cone gas flow rate was 50 l h−1. The source was maintained at 120 °C. Primary mass, fragment mass, cone voltages and collision energies for each compound are listed in Supplementary Table 5.
Supporting E. coli growth with NMN+-dependent glycolysis.
E. coli strain with ΔpgiΔzwfΔgnd knockout (strain MX103, Supplementary Table 3) cannot metabolize glucose. The strain also has ΔnadRΔpncC knockouts to potentially preserve intracellular NMN+ (ref.29). The strain was transformed with a plasmid (pSM103) containing F. tularens nadE and nadV, Z. mobilis glf and R. eutropha gntK in a synthetic operon. In addition, the strain was also transformed with one of the three plasmids: pLZ214 (XenA alone), pSM106 (Bs GDH Ortho alone) or pLZ215 (Bs GDH Ortho with XenA).
Colonies were picked in triplicate and grown overnight (15 h) at 30 °C in 4 ml of 2xYT media. Overnight cultures were used to inoculate (0.5%, v/v) 4 ml of expression media (2xYT, 20 g l−1 glucose, 1 mM nicotinamide). Cells were grown at 30 °C with shaking for 3 h before induction with 0.5% (w/v) arabinose. Cultures were allowed to express for 3 h at 30 °C before being collected and washed with M9 minimal media (1× M9 salts, 0.1 mM CaCl2, 1 mM MgSO4, 1× A5 trace metals with cobalt) three times. Washed cultures were used to inoculate 4 ml of growth media to an initial OD600 of 0.1. Growth media consisted of the previously mentioned M9 minimal media with the addition of 20 g l−1 glucose, 0.05% (w/v) arabinose and 1 mM nicotinamide. Cultures were supplemented with 0, 1, 2, or 5 mM NMN+ when examining the effects of extracellular NMN+ on growth rate. Cultures were grown while shaking at 30 °C. All media contained 200 mg l−1 ampicillin (except overnight cultures that had 100 mg l−1), 50 mg l−1 spectinomycin, 50 mg l−1 kanamycin, and 0.1 mM IPTG. Cell growth was monitored by measuring optical density at 600 nm.
Rosetta runs files.
Files used for Rosetta docking and design runs include Docking.xml file, Flags file, GluDH.enzdes.cst file (specify chemical constraints), GluDH_model.pdb file, X00.params file (describe properties of the NMN+ ligand), and submit.sh file (commandline job submission). Files used for homology modeling run include alignment.fasta file, Bacil_thread.fasta file, setup_cm.sh file, template.pdb file, flags file, rosetta_cm.xml file, and hybridize.sh file (commandline submission file). For further information and description on these files please refer to Rosetta documentation on RosettaCM and RosettaDesign at: https://www.rosettacommons.org/docs/latest/application_documentation/structure_prediction/RosettaCM and https://www.rosettacommons.org/docs/latest/application_documentation/design/enzyme-design
Examples of these run files can be found at: https://github.com/siegel-lab-ucd/Engineering-a-nicotinamidemononucleotide-redox-cofactor-system-for-biocatalysis
Due to the flexibility of NAD+, the NAD+ conformer’s library was generated by sampling the representative ensembles of NAD+ conformer listed in CoFactor database57. Overall, 31 conformers of NAD+ were used for the calculation.
Supplementary Material
Acknowledgements
H.L. acknowledges support from University of California, Irvine, the National Science Foundation (NSF) (award no. 1847705), and the National Institutes of Health (NIH) (award no. DP2 GM137427). S.M. acknowledges support from the NSF Graduate Research Fellowship Program (grant no. DGE-1839285). W.B.B. acknowledges support from Graduate Assistance in Areas of National Need fellowship funded by the U.S. Department of Education. J.B.S., W.S.M. and Y.C. acknowledge support from the University of California, Davis, by the NSF (award nos. 1827246, 1805510, 1627539), the National Institute of Environmental Health Sciences of the NIH (award no. P42ES004699) and the NIH (award no. R01 GM 076324-11). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the NSF. We thank the University of California, Irvine Mass Spectrometry Facility and F. Grun for help with LC-MS analysis.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-019-0402-7.
Data availability
The authors declare that all relevant data supporting the findings of this study are available within the manuscript and its Supplementary Information.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-019-0402-7.
References
- 1.Lee SY & Kim HU Systems strategies for developing industrial microbial strains. Nat. Biotechnol 33, 1061–1072 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Mampel J, Buescher JM, Meurer G & Eck J Coping with complexity in metabolic engineering. Trends Biotechnol 31, 52–60 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Paddon CJ & Keasling JD Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol 12, 355 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Pandit AV, Srinivasan S & Mahadevan R Redesigning metabolism based on orthogonality principles. Nat. Commun 8, 15188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L et al. Synthetic cofactor-linked metabolic circuits for selective energy transfer. ACS Catal 7, 1977–1983 (2017). [Google Scholar]
- 6.Martínez AT et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv 35, 815–831 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Wildeman SMAD, Sonke T, Schoemaker HE & May O Biocatalytic reductions: from lab curiosity to ‘first choice’. Acc. Chem. Res 40, 1260–1266 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Campbell E, Meredith M, Minteer SD & Banta S Enzymatic biofuel cells utilizing a biomimetic cofactor. Chem. Commun 48, 1898–1900 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Knaus T et al. Better than Nature: nicotinamide biomimetics that outperform natural coenzymes. J. Am. Chem. Soc 138, 1033–1039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nowak C, Pick A, Lommes P & Sieber V Enzymatic reduction of nicotinamide biomimetic cofactors using an engineered glucose dehydrogenase: providing a regeneration system for artificial cofactors. ACS Catal 7, 5202–5208 (2017). [Google Scholar]
- 11.Flores H & Ellington AD A modified consensus approach to mutagenesis inverts the cofactor specificity of Bacillus stearothermophilus lactate dehydrogenase. Protein Eng. Des. Sel 18, 369–377 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Lo HC & Fish RH Biomimetic NAD(+) models for tandem cofactor regeneration, horse liver alcohol dehydrogenase recognition of 1,4-NADH derivatives, and chiral synthesis. Angew. Chem 41, 478–481 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Hagen J Industrial Catalysis: A Practical Approach 2nd edn (Wiley-VCH, 2006). [Google Scholar]
- 14.Everse J, Anderson B & You K-S The Pyridine Nucleotide Coenzymes (Academic Press, 1982). [Google Scholar]
- 15.Paul CE, Arends IWCE & Hollmann F Is simpler better? Synthetic nicotinamide cofactor analogues for redox chemistry. ACS Catal 4, 788–797 (2014). [Google Scholar]
- 16.Paul CE et al. Mimicking nature: synthetic nicotinamide cofactors for C=C bioreduction using enoate reductases. Org. Lett 15, 180–183 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Kataoka M et al. Old Yellow Enzyme from Candida macedoniensis catalyzes the stereospecific reduction of the C=C bond of ketoisophorone. Biosci. Biotechnol. Biochem 66, 2651–2657 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Okamoto Y, Köhler V, Paul CE, Hollmann F & Ward TR Efficient in situ regeneration of NADH mimics by an artificial metalloenzyme. ACS Catal 6, 3553–3557 (2016). [Google Scholar]
- 19.Chaparro-Riggers JF, Rogers TA, Vazquez-Figueroa E, Polizzi KM & Bommarius AS Comparison of three enoate reductases and their potential use for biotransformations. Adv. Synth. Catal 349, 1521–1531 (2007). [Google Scholar]
- 20.Paul CE et al. Nonenzymatic regeneration of styrene monooxygenase for catalysis. ACS Catal 5, 2961–2965 (2015). [Google Scholar]
- 21.Knox RJ et al. Virtual cofactors for an Escherichia coli nitroreductase enzyme: relevance to reductively activated prodrugs in antibody directed enzyme prodrug therapy (ADEPT). Biochem. Pharmacol 49, 1641–1647 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Ryan JD, Fish RH & Clark DS Engineering cytochrome P450 enzymes for improved activity towards biomimetic 1,4-NADH cofactors. Chem. Bio. Chem 9, 2579–2582 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Müller A, Stürmer R, Hauer B & Rosche B Stereospecific alkynereduction: novel activity of old yellow enzymes. Angew. Chem. Int. Ed 46, 3316–3318 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Race PR et al. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone. Reversed binding orientations in different redox states of the enzyme. J. Biol. Chem 280, 13256–13264 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Hilt W, Pfleiderer G & Fortnagel P Glucose dehydrogenase from Bacillus subtilis expressed in Escherichia coli. I: Purification, characterization and comparison with glucose dehydrogenase from Bacillus megaterium. Biochim. Biophys. Acta 1076, 298–304 (1991). [DOI] [PubMed] [Google Scholar]
- 26.Solanki K, Abdallah W & Banta S Engineering the cofactor specificity of an alcohol dehydrogenase via single mutations or insertions distal to the 2′-phosphate group of NADP(H). Protein Eng. Des. Sel 30, 373–380 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Schewe H, Kaup BA & Schrader J Improvement of P450(BM-3) whole-cell biocatalysis by integrating heterologous cofactor regeneration combining glucose facilitator and dehydrogenase in E. coli. Appl. Microbiol. Biotechnol 78, 55–65 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Pohlmann A et al. Genome sequence of the bioplastic-producing ‘Knallgas’ bacterium Ralstonia eutropha H16. Nat. Biotechnol 24, 1257–1262 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Gazzaniga F, Stebbins R, Chang SZ, McPeek MA & Brenner C Microbial NAD metabolism: lessons from comparative genomics. Microbiol. Mol. Biol. Rev 73, 529–541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorci L et al. Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. Proc. Natl Acad. Sci. USA 106, 3083–3088 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stott K, Saito K, Thiele DJ & Massey V Old Yellow Enzyme. The discovery of multiple isozymes and a family of related proteins. J. Biol. Chem 268, 6097–6106 (1993). [PubMed] [Google Scholar]
- 32.Wang X et al. Engineering Escherichia coli nicotinic acid mononucleotide adenylyltransferase for fully active amidated NAD biosynthesis. Appli. Environ. Microbiol 83, e00692–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witholt B Method for isolating mutants overproducing nicotinamide adenine dinucleotide and its precursors. J. Bacteriol 109, 350–364 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grozio A et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab 1, 47–57 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grose JH et al. Assimilation of nicotinamide mononucleotide requires periplasmic AphA phosphatase in Salmonella enterica. J. Bacteriol 187, 4521–4530 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunjapur AM & Prather KL Microbial engineering for aldehyde synthesis. Appl. Environ. Microbiol 81, 1892–1901 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez GM & Atsumi S Toward aldehyde and alkane production by removing aldehyde reductase activity in Escherichia coli. Metab. Eng 25, 227–237 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hall M, Hauer B, Stuermer R, Kroutil W & Faber K Asymmetric whole-cell bioreduction of an α,β-unsaturated aldehyde (citral): competing prim-alcohol dehydrogenase and C–C lyase activities. Tetrahedron Asymmetry 17, 3058–3062 (2006). [Google Scholar]
- 39.Yoshisumi A, Wada M, Takagi H, Shimizu S & Nakamori S Cloning, sequence analysis, and expression in Escherichia coli of the gene encoding monovalent cation-activated levodione reductase from Corynebacterium aquaticum M-13. Biosci. Biotechnol. Biochem 65, 830–836 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Kulig J, Frese A, Kroutil W, Pohl M & Rother D Biochemical characterization of an alcohol dehydrogenase from Ralstonia sp. Biotechnol. Bioeng 110, 1838–1848 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Oberleitner N, Peters C, Rudroff F, Bornscheuer UT & Mihovilovic MD In vitro characterization of an enzymatic redox cascade composed of an alcohol dehydrogenase, an enoate reductases and a Baeyer–Villiger monooxygenase. J. Biotechnol 192 Pt B, 393–399 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mak WS et al. Integrative genomic mining for enzyme function to enable engineering of a non-natural biosynthetic pathway. Nat. Commun 6, 10005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart JD in Future Directions in Biocatalysis (ed. Matsuda T) Ch. 12 (Elsevier Science, 2007). [Google Scholar]
- 44.Liang K & Shen CR Selection of an endogenous 2,3-butanediol pathway in Escherichia coli by fermentative redox balance. Metab. Eng 39, 181–191 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Machado HB, Dekishima Y, Luo H, Lan EI & Liao JC A selection platform for carbon chain elongation using the CoA-dependent pathway to produce linear higher alcohols. Metab. Eng 14, 504–511 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, King E, Luo R & Li H Development of a high-throughput, in vivo selection platform for NADPH-dependent reactions based on redox balance principles. ACS Synth. Biol 7, 1715–1721 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marinescu GC, Popescu RG, Stoian G & Dinischiotu A Beta-nicotinamide mononucleotide (NMN) production in Escherichia coli. Sci. Rep 8, 12278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finn RD et al. HMMER web server: 2015 update. Nucleic Acids Res 43, W30–W38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto K et al. Crystal structure of glucose dehydrogenase from Bacillus megaterium IWG3 at 1.7 A resolution. J. Biochem 129, 303–312 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Song Y et al. High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richter F, Leaver-Fay A, Khare SD, Bjelic S & Baker D De novo enzyme design using Rosetta3. PloS One 6, e19230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kleffner R et al. Foldit Standalone: a video game-derived protein structure manipulation interface using Rosetta. Bioinformatics 33, 2765–2767 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shao Y et al. Spartan’08, Wavefunction, Inc. Irvine, CA. Phys. Chem. Chem. Phys 8, 3172–3191 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Datsenko KA & Wanner BL One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Findik BT & Randall LL Determination of the intracellular concentration of the export chaperone SecB in Escherichia coli. PLoS One 12, e0183231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fischer J, Holliday G & Thornton J The CoFactor database: organic cofactors in enzyme catalysis. Bioinformatics 26, 2496–2497 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.