

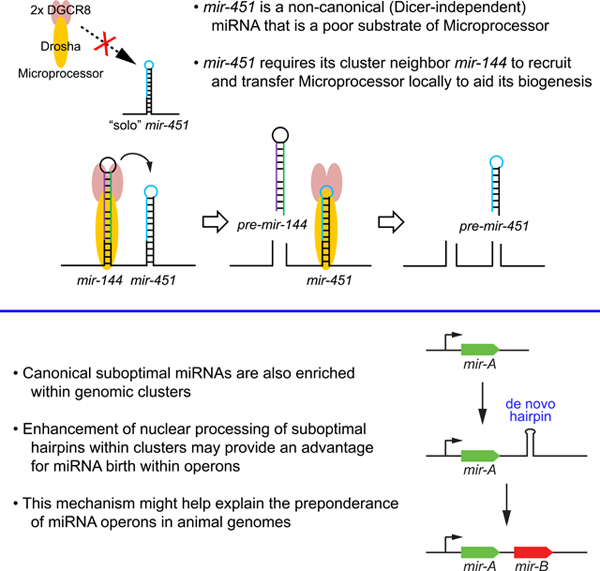
Summary
Nuclear processing of most miRNAs is mediated by Microprocessor, comprised of RNase III enzyme Drosha and its cofactor DGCR8. Here, we uncover a hidden layer of Microprocessor regulation via studies of Dicer-independent mir-451, which is clustered with canonical mir-144. Although mir-451 is fully dependent on Drosha/DGCR8, its short stem and small terminal loop render it an intrinsically weak Microprocessor substrate. Thus, it must reside within a cluster for normal biogenesis, although the identity and orientation of its neighbor are flexible. We use DGCR8 tethering assays and operon structure-function assays to demonstrate that local recruitment and transfer of Microprocessor enhances suboptimal substrate processing. This principle applies more broadly since genomic analysis indicates suboptimal canonical miRNAs are enriched in operons, and we validate several of these experimentally. Proximity-based enhancement of suboptimal hairpin processing provides a rationale for genomic retention of certain miRNA operons, and may explain preferential evolutionary emergence of miRNA operons.
eTOC blurb
Shang et al. investigate the biogenesis of clustered miRNAs, which were largely considered to be processed independently. They document multiple mammalian miRNA operons bearing suboptimal substrates of the Drosha/DGCR8 (Microprocessor) complex, which require close proximity to canonical miRNA neighbors to recruit and locally transfer Microprocessor for their optimal nuclear biogenesis.
Graphical Abstract
Introduction
microRNAs (miRNAs) comprise an abundant family of ~22 nucleotide (nt) RNAs, derived from inverted repeat transcripts, that mediate extensive gene regulatory networks. In the canonical pathway, a primary miRNA (pri-miRNA) hairpin is cleaved by the nuclear RNase III enzyme Drosha and its double-stranded RNA binding (dsRBD) partner DGCR8 (“Microprocessor”) to release the pre-miRNA hairpin, which is cleaved by the cytoplasmic RNase III enzyme Dicer to yield a miRNA/star duplex. This is loaded into an Argonaute effector, and matured to a single-stranded complex that seeks complementary targets for regulation. In addition, a variety of non-canonical miRNA substrates are known. For example, a variety of Drosha-independent and Dicer-independent miRNA biogenesis pathways have been documented (Yang and Lai, 2011), which made it possible to design synthetic, RNase III-independent miRNA biogenesis strategies in mammalian cells (Maurin et al., 2012). Improved mechanistic knowledge of miRNA biogenesis is important not only to understand this endogenous regulatory system, but also to exploit these pathways for experimental gene silencing.
Although many canonical miRNAs are “solo” loci, ~1/3 of vertebrate miRNAs are expressed as operons, in which two or more miRNA hairpins are hewn from a single primary transcript (Altuvia et al., 2005). The biological imperatives that drive miRNA clustering are not fully known. Where tested, miRNAs derived from operons are generally functional when expressed as individual pri-miRNA constructs (Bejarano et al., 2012; He et al., 2005; Mavrakis et al., 2010; Silver et al., 2007). This argues against the widespread existence of dispersed cis-elements that are essential for miRNA processing within clusters. Nevertheless, idiosyncrasies of miRNA cluster biogenesis have been reported, such as stepwise processing of inner miRNAs within the mir-17~92 cluster (Donayo et al., 2019; Du et al., 2015), or dependencies in the maturation of select miRNAs on their neighbors. The latter has been observed in certain Drosophila (Truscott et al., 2016), mammalian (Hutter et al., 2019; Lataniotis et al., 2017) and even viral (Haar et al., 2016) miRNA clusters. Overall, the mechanistic reasons are not clearly defined, but such dependencies were suggested to correlate with suboptimal processing. Alternatively, the frequent arrangement of miRNA operons might have to do with the nature of preferential evolutionary emergence within clusters (Marco et al., 2013; Mohammed et al., 2014), or with selection to maintain precise cellular co-expression of miRNAs or perhaps co-targeting by operonic miRNAs (Bushati et al., 2008; Han et al., 2015; Wang et al., 2016).
Many features of optimal, canonical miRNA biogenesis were elucidated from detailed analyses of individual mutagenized miRNA precursors, along with largescale sequencing-based assays using randomized miRNA model backbones. These approaches reveal that a double-stranded stem of ~35 basepairs, flanking single-stranded regions, a terminal loop of >10 nts, and specific sequence motifs within the terminal loop and flanking regions, all contribute positively to miRNA biogenesis (Auyeung et al., 2013; Fang and Bartel, 2015; Han et al., 2006; Kwon et al., 2019; Kwon et al., 2016; Ma et al., 2013; Nguyen et al., 2015; Zeng and Cullen, 2005; Zeng et al., 2005; Zhang and Zeng, 2011). A majority of conserved miRNAs conform to these general structural features and contain one or more recognizable cis-motifs, suggesting that such miRNAs are under selection for molecular mechanisms that enable efficient biogenesis. On the other hand, miRNA loci that are evolutionary young tend to conform less strictly to this menu of features, exhibit more heterogeneous processing, and generate low-expressed mature miRNAs.
Studies of the conserved vertebrate operon mir-144/451 (Figure S1) showed that mir-451, in contrast to canonical mir-144, is matured by an unusual strategy (Yang and Lai, 2010). While it has a typical “lower stem” that mediates hairpin cleavage by Drosha/DGCR8, the resulting pre-miRNA is only 42 nt in length (Cheloufi et al., 2010; Yang et al., 2010). Since its stem is only 17 bp in length, it is too short to be cleaved by Dicer; instead, pre-mir-451 hairpins load directly into Ago proteins (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). If pre-mir-451 binds “Slicer” Ago2, the sole vertebrate Ago-class factor with efficient catalytic activity, it is cleaved on the 3’ hairpin arm and subsequently further trimmed at its 3’ end by PARN (Yoda et al., 2013) to yield mature, functional miR-451. Although miR-451 is the only conserved vertebrate miRNA matured by this strategy, its backbone can be readily reprogrammed to produce synthetic Dicer-independent miRNAs (Cheloufi et al., 2010; Shang et al., 2015; Yang et al., 2012; Yang et al., 2010). This strategy does not produce a miRNA* sequence, which is a substantial source of off-targeting effects.
In this study, we reveal unexpected dependency of mir-451 cropping on proximity to mir-144. This requirement can be substituted by other canonical miRNAs, and the underlying mechanism involves suboptimal features of the mir-451 terminal loop that render it a poor Microprocessor substrate in vivo. This makes mir-451 processing subservient to that of mir-144 and explains why these miRNAs remain tightly clustered across evolution. We use tethering assays and structural variants of the mir-451 locus to provide evidence that Microprocessor is recruited to its vicinity and transferred locally to facilitate the nuclear biogenesis of this suboptimal miRNA hairpin. Moreover, we extend this principle by showing that suboptimal canonical miRNAs are also enriched within clusters. This strategy by which the nuclear biogenesis of a suboptimal miRNA hairpin is enhanced within a cluster may contribute to the high frequency of miRNA operons in present day metazoan genomes.
Results
Biogenesis of non-canonical mir-451 is dependent on its neighbor mir-144
Although miR-451 is Dicer-independent, its biogenesis requires the canonical nuclear miRNA processing machinery (Yang et al., 2010). In particular, miR-451 cannot be matured upon knockout of Drosha (Cheloufi et al., 2010) or DGCR8 (Jee et al., 2018). Of note, prior ectopic expression studies used constructs containing genomic DNA covering both mir-144 and mir-451 (Figure 1A) (Cheloufi et al., 2010; Yang et al., 2010), or synthetic pre-mir-451 that bypasses Drosha cleavage (Cheloufi et al., 2010; Cifuentes et al., 2010).
Figure 1. Biogenesis and function of conserved vertebrate mir-451 requires its operon neighbor mir-144.

(A) Schematics of wild-type and variant mir-144/451 constructs. 144 and 451 are smaller expression constructs that still contain extensive flanking genomic segments; Δ144–451 refers to the starting pri-mir-144/451 expression construct bearing a deletion in the pre-mir-144 hairpin, while 144LD-451 contains only a deletion in the terminal hairpin loop of mir-144. The order of the miRNAs is reversed in 451–144. (B) Processing of different wild-type and variant mir-144/451 constructs by Northern blotting in HEK293T cells. Mir-144/451 and mir-375 expression constructs were cotransfected and blotted sequentially; endogenous let-7a and U6 snRNA were also assayed as further loading controls. RNA size markers (nt) are shown on the left. Mature miR-144 is processed regardless of miR-451 status, while normal maturation of miR-451 requires the transcription of mir-144 in cis. Note that the loop-deletion form of mir-144 accumulates a modest amount of hairpin precursor (pre-mir-144LD) but no mature miRNA. (C) Activity of wild-type and variant mir-144/451 constructs on luciferase sensors. (D) Replacement of pri-mir-144 sequences by other miRNAs. (E) Processing and (F) activity of mir-451 from constructs with different neighbors. mir-7a and mir-545 can effectively substitute for mir-144, but another copy of mir-451 cannot.
Surprisingly, when we tested mir-144 and mir-451 solo expression constructs (Figure 1A), we observed normal maturation of miR-144 but the latter were extremely poor at generating pre-mir-451 and mature miR-451 (Figure 1B). Co-expression of primir-144 in trans did not rescue activity or maturation of solo pri-mir-451, implying a cis-requirement for mir-144 during mir-451 biogenesis (Figure 1B). This notion was further supported by testing pri-mir-144/451 variants. Not only did deletion of pre-mir-144 from this longer primary transcript block maturation of miR-451 (Δ144–451), so did a small deletion of the terminal loop of pre-mir-144 (144LD-451, Figure 1A). The latter construct barely expressed the truncated pre-mir-144LD hairpin and failed to yield miR-144, and was similarly inhibited for miR-451 maturation (Figure 1B). Finally, the relative position of miRNAs within the cluster was flexible, as mir-144 supported effective miR-451 maturation from a downstream position (Figure 1A–B). Consistent with these processing data, luciferase sensor assays of all these constructs showed that mir-451 yielded substantial repression only when expressed from the same primary transcript as mir-144, whereas mir-144 activity was independent of mir-451 (Figure 1C).
To explore if the integrity of this specific miRNA cluster is needed to generate miR-451, we swapped other miRNAs in place of mir-144 (Figure 1D). We found that mir-7a and mir-545 successfully promoted effective miR-451 maturation and function, while a second copy of mir-451 had little effect on either readout (Figure 1E–F). Thus, miR-451 has a generic biogenesis requirement for proximity to a canonical miRNA, but mir-451 itself is inherently suboptimal and additional copies do not help.
In vitro assays have been powerful to dissect substrate preference and cleavage site selection by Drosha/DGCR8 complex. At face value, these data with transfected constructs appeared at odds with prior observations that pri-mir-451 could be cropped in vitro by Drosha/DGCR8 (Cheloufi et al., 2010). We compared the processing of pri-mir-144/451 and pri-(Δ144)mir-451 using Drosha/DGCR8-IP material, using internally labeled substrates or unlabeled substrates followed by Northern blotting (Figure S2A). The appearance of pre-mir-451 hairpin in these reactions from the solo context was slightly delayed relative to the operon context (Figure S2B). Nevertheless, pri-mir-451 was cropped in vitro, as previously reported (Cheloufi et al., 2010), and was not markedly delayed relative to mir-144 when processed from the operon. We attempted to sensitize substrate availability and reaction kinetics, but did not obtain further differential processing (Figure S2C). Thus, our in vitro assay conditions did not fully model strong in vivo dependence of mir-451 biogenesis on a neighboring canonical miRNA.
Biogenesis of endogenous miR-451 requires neighboring mir-144
Because of discrepancies between transfected constructs and in vitro tests, we sought definitive assays of the endogenous locus. To do so, we used CRISPR/Cas9 to engineer K562 cells, which express mir-144/451 abundantly. As summarized in Figure 2A, we isolated clones with biallelic deletions encompassing both mir-144 and mir-451 hairpins, biallelic deletion of mature miR-451, and biallelic or monoallelic deletion of the mir-144 hairpin (Figure S3). Northern blotting yielded clear results, in that single deletions of the cognate miRNAs eliminated production of the expected miRNAs (Figure 2B), as expected. However, biallelic deletion of pre-mir-144 also resulted in complete loss of pre-mir-451 and mature miR-451. Moreover, monoallelic loss of pre-mir-144 correspondingly reduced the hairpin and mature forms of both miR-144 and miR-451 by ~50% (Figure 2B). These data provide stringent evidence that cropping and maturation of endogenous miR-451 indeed depends on the neighboring mir-144 hairpin in cis.
Figure 2. K562 knockout cells recapitulate dependency of mir-451 on mir-144.

(A) Schematics for endogenous mir-144/451 knockout by CRISPR-Cas9 in K562 cells. (B) Biogenesis of endogenous mir-144 and mir-451 in KO cell lines by Northern blotting. The ratio of (pre + mature) miRNA to U6 snRNA is indicated for each lane. Note that miR-451 is lost in 144KO homozygous cells, and both miRNAs are reduced by ~50% in 144het cells. (C) Expression of pri-mir-144/451 transcripts in KO cell lines by RT-qPCR. GAPDH mRNA was used as a reference control. pri-mir-144/451 is increased in 144KO and 144/451-dKO cells to comparable levels, but is not increased in 451KO cells. (D) Model to interpret mir-144/451 processing. The nuclear biogenesis of suboptimal mir-451 is enhanced by its clustered neighbor mir-144. Based on qPCR data from mutants, there appears to be sequential processing of the hairpins, where the recruitment of Microprocessor to mir-451 is facilitated by local transfer from neighboring mir-144.
We tested for reciprocal changes in pri-mir-144/451 using a qPCR amplicon located upstream of the miRNAs. Deletion of the mir-144 hairpin increased the level of the primary transcript to the same extent as did the double miRNA deletion, while deletion of mir-451 did not markedly affect pri-mir-144/451 (Figure 2C). This further supports the notion that mir-451 is not effectively cropped in the absence of mir-144. A corollary implication of these pri-mir-144/451 data is that there is sequential cleavage of the miRNA hairpins (Figure 2D). We sought to test the tenets of this implied mechanism, in which mir-451 is a suboptimal Microprocessor substrate whose effective biogenesis relies upon increased local availability of Drosha/DGCR8 due to proximity to a neighboring canonical miRNA.
Suboptimal features of mir-451 render it a poor Microprocessor substrate
We first addressed why mir-451 might be a poor substrate for Microprocessor. Current knowledge invokes that optimal substrates involve recognition of hairpins of sufficient length, with the cleavage site measured from the basal junction of the single stranded and duplex region (Fang and Bartel, 2015; Han et al., 2006; Kwon et al., 2019). In addition, specific motifs help position Microprocessor, such as the mGHG motif within the lower stem that is bound by the Drosha dsRBD, UG at the basal junction that is recognized by Drosha, and UGU near the apical junction that is bound by DGCR8. Mir-144 and mir-451 have basal UG sequences but lack apical UGU and lower stem mGHG motifs (Figure S1). Moreover, although little studied until recently, effective miRNA biogenesis requires a sufficiently sized terminal loop (Zeng et al., 2005; Zhang and Zeng, 2011). The mir-451 hairpin not only has a short duplex, but also exhibits a notably small terminal loop of 4 nt (Figure S1).
We constructed mir-451 variants where we lengthened its stem into a typical pre-miRNA and/or enlarged its terminal loop (Figure 3A). These were prepared in the context of mir-144LD/451 bearing a non-functional loop deletion variant of mir-144, and in the context of solo mir-451. Indeed, enlarging the terminal loop of mir-451 enhanced its maturation in both Dicer-independent and Dicer-dependent formats, with the combination of these yielding optimal miRNA biogenesis and activity that is fully independent of mir-144 (Figure 3B–D). Thus, the short terminal loop intrinsically renders both canonical and non-canonical hairpins as suboptimal Microprocessor substrates. Interestingly, although pri-mir-144LD is not effectively diced, it remains a Drosha substrate. We observed that optimized mir-451 bearing both longer stem and enlarged loop (mir-451LSM) enhanced the accumulation of pre-mir-144LD (Figure 3B). Thus, we can uncouple the capacity of a clustered miRNA to enhance Drosha processing of a neighboring substrate, from the fate of the resulting hairpin in miRNA maturation.
Figure 3. Mechanistic basis of suboptimal nuclear processing of mir-451.

(A) Schematics of mir-451 variants with a larger loop (LM), longer stem (SM) or both alterations (LSM). These variants were induced within the mir-144/451 operon bearing an inhibitory loop-deletion of mir-144 (144LD), as well as in a solo mir-451 context. (B) Processing of variant mir-144/451 precursors or solo mir-451 constructs. These were cotransfected with mir-375 expression construct and blotted sequentially; endogenous let-7a and U6 snRNA were also assayed as further loading controls. Enlarging the mir-451 loop stimulated miR-451 biogenesis, in both 144LD and solo expression vectors. This effect of increasing loop size was synergistic with increasing stem length. (C, D) Activity of mir-451 from different mutants in mir-144 loop-deleted (C) or solo (D) mir-451 constructs. (E) Overexpression of Microprocessor (Drosha/DGCR8) selectively enhanced the biogenesis of miR-451 from the suboptimal solo context, but not from the mir-144/451 operon. (F) Schematics of Pol III>mir-451 variants under control of the H1 promoter. (G) Northern blotting shows that miR-451 was still enhanced by a neighboring canonical miRNA when transcribed by Pol III, but was not appreciably matured from the other constructs. Maturation of cotransfected miR-375 serves as a control. (H) Comparison of λN-DGCR8 overexpression on naive and tethered miRNA hairpins. (I) Schematics of wild-type and BoxB-substituted pri-mir-451. Note that the distal BoxB sequence is mostly G:C pairs, which inhibit mir-451 trimming following Ago2 cleavage. (J, K) Processing of wt mir-451 (J) or mir-451-BoxB chimera (K) precursors under expression of control GFP, DGCR8, or λN-DGCR8. The ratio of (pre + mature) miR-451 to U6 snRNA is indicated. λN-DGCR8 selectively enhanced the biogenesis of 451BoxB.
Since mir-451 is an obligate Microprocessor substrate, albeit a suboptimal one, we asked if we could improve its biogenesis by global elevation of Microprocessor. Compared to control EGFP transfection, introduction of Drosha/DGCR8 expression constructs did not substantially affect the amounts of miR-144 or miR-451 produced from an optimal mir-144/451 backbone, nor did it affect endogenous let-7a (Figure 3E). By contrast, the latter condition dramatically increased the production of pre-mir-451 and mature miR-451 species from a solo mir-451 construct (Figure 3E). Thus, unlike overexpression of Ago proteins, which stabilize all co-expressed miRNAs (Diederichs and Haber, 2007), elevated Microprocessor selectively enhances suboptimal mir-451 only from the solo context.
The cropping reaction is co-transcriptional and localized on chromatin (Ballarino et al., 2009; Liu et al., 2016; Morlando et al., 2008; Pawlicki and Steitz, 2008). Recently, evidence was reported that DGCR8 binds CTD-phosphorylated RNA Pol II, coupling of transcription to cropping (Church et al., 2017). This mechanism preferentially promotes biogenesis of a class of suboptimal miRNAs (lacking apical UGU). As mentioned, neither mir-144 nor mir-451 bears apical UGU motifs, and the small terminal loop of mir-451 appears to be a poor DGCR8 binding site in particular (Figure S1).
To test possible impact of Pol II coupling on the mir-144/451 locus, we expressed a panel of constructs from an RNA Pol III promoter (Figure 3F). We observed meager biogenesis of miR-451 from a solo H1>mir-451 or from H1>mir-144LD-451, containing a deletion in the mir-144 hairpin loop (Figure 3G). By contrast, H1>mir-144/451 efficiently produced mature miR-144 and miR-451 (Figure 3G). Thus, Pol II coupling to Microprocessor is not required for enhancement of miR-451 biogenesis within the cluster, even though Microprocessor appears to be limiting for nuclear cleavage of mir-451.
Direct recruitment of DGCR8 can selectively promote miR-451 biogenesis
We found that co-expression of Drosha/DGCR8 was far more potent at inducing miR-451 biogenesis than either individual factor (Figure S4). This might be expected since these factors work as a complex and are known to stabilize/solubilize each other (Han et al., 2009). We hypothesized that DGCR8 expression might represent a sensitized situation for tethering assays. If the small terminal loop of mir-451 impedes effective recruitment of DGCR8, then direct tethering of DGCR8 to its binding site may further improve biogenesis above general overexpression (Figure 3H). We therefore replaced the distal portion of the mir-451 hairpin with a BoxB sequence (Figure 3I).
A caveat of this design is that, by replacing the distal mir-451 duplex with BoxB, we convert this normally A:U-rich region to nearly complete G:C pairing (Figure 3I). We previously showed such a configuration inhibits 3’ trimming of Ago2-cleaved mir-451-like hairpins (Yang et al., 2012). In spite of this, we indeed observed specific biogenesis enhancement by λN-DGCR8. As mentioned, DGCR8 only modestly enhanced maturation of miR-451 from solo-mir-451, compared to co-expressed Drosha+DGCR8 (Figure 3E vs. 3J). However, λN-DGCR8 selectively yielded additional enhancement of miR-451 biogenesis only from solo mir-451-BoxB, particularly at the level of pre-mir-451BoxB, while its effect on native mir-451 was similar to untethered DGCR8 (Figure 3J–K). Therefore, direct recruitment of DGCR8 to pri-mir-451 can bypass the need for a neighboring canonical miRNA.
Evidence for local Microprocessor recruitment and transfer to promote miR-451 biogenesis
We next sought evidence to support the model that Microprocessor is transferred from the local vicinity to mir-451 to promote its biogenesis (Figure 2D). We substituted the mir-144 hairpin with 1 or 5 BoxB sites, allowing us to separate neighboring miRNA biogenesis from local increase of Microprocessor (Figure 4A). DGCR8 overexpression only mildly promoted biogenesis of miR-451 from either construct, similar to λN-DGCR8 on untethered mir-451 (Figure 4B). By contrast, λN-DGCR8 elicited 2-fold greater miR-451 maturation than did DGCR8 via BoxB-mir-451, and was nearly 20-fold more effective than DGCR8 on 5xBoxB-mir-451. These specific, dose-sensitive data provide evidence for a local effect on mir-451 biogenesis.
Figure 4. Local recruitment and transfer of Microprocessor promotes nuclear mir-451 biogenesis.

(A) Schematics of constructs tested. Using Δ144–451 as a starting construct, we inserted 1 or 5x BoxB elements at the site of the mir-144 hairpin. We also made a version in which BoxB was introduced at the terminal loop of mir-144 (144BoxB-451). (B) Northern blotting shows that λN-DGCR8 is slightly better than DGCR8 at promoting biogenesis of BoxB-451, but is far better on the 5xBoxB-451 substrate (almost 20-fold). Notably, the single BoxB in 144BoxB-451 yields much better enhancement of miR-451 biogenesis with λN-DGCR8 compared to DGCR8 (10 fold). (C) Varying the spacer length between mir-144 and mir-451. Note that these distances correspond to the nts between Drosha cleavages and therefore the “0” nt spacing actually deletes all the lower sequences between these miRNAs while the “30” nt spacing removes one side of the lower single-stranded flanking sequences for both miRNAs. (D) mir-144/451 and mir-375 expression constructs were cotransfected and blotted sequentially; endogenous let-7a and U6 snRNA were also assayed as further loading controls. The biogenesis of miR-144 is relatively stable across these length variants (excepting “0”, which is expected to be non-functional for both miRNAs), whereas biogenesis of miR-451 was optimal at its normal spacing and gradually declined with greater inter-miRNA distance. Little miR-451 was produced at a 2kb spacing. No miR-451 was produced at the 30-nt spacing, which would leave insufficient flanking single-stranded sequence for Microprocessor to recognize pri-mir-451 following mir-144 cropping. (E) Sensor assays of the mir-144-(Xnt)-451 length variants. The activity of miR-451 declines with increasing distance from mir-144. (F) Enhancement at a longer distance. Although tempered, mir-144 still enhances mir-451 biogenesis across a range of longer spacer distances. These are all due specifically to mir-144, since deletion of the mir-144 loop (144LD) within all of these constructs abolishes both miR-144 and miR-451 function.
In principle, tethered DGCR8, presumably in association with endogenous Drosha, should not affect mir-451 biogenesis until it was released. However, recruitment to BoxB sites might be relatively stable, since the short BoxB hairpin is not a Drosha cleavage substrate. We therefore prepared another variant in which a single BoxB site replaced the terminal region of mir-144 (Figure 4A). In this setup, we hypothesized that locally recruited DGCR8 may have greater availability to transfer to pri-mir-451 following release of pre-mir-144-BoxB. Indeed, recruitment of λN-DGCR8 to a single site on mir-144-BoxB boosted miR-451 biogenesis ~5-fold higher than did a single BoxB site per se, and was only ~2-fold less than 5xBoxB sites (Figure 4B). This experiment strongly supports the concept that local recruitment and release of Microprocessor is responsible for enhancement of mir-451 biogenesis in vivo. Notably, pre-mir-144-BoxB proved to be a poor Dicer substrate, as it accumulated mostly as a hairpin and yielded little mature miRNA (Figure 4B). Thus, biogenesis interactions that promote nuclear pri-miRNA cleavages within clusters can be fully uncoupled from subsequent Dicer cleavages that yield mature miRNAs.
Another implication of our model for local Microprocessor transfer is that proximity-based enhancement of cropping might be constrained by inter-miRNA distance. Indeed, the close pairing of mir-144/451 is relatively conserved across vertebrates (Cheloufi et al., 2010; Cifuentes et al., 2010; Yang et al., 2010). We inserted spacers of varying lengths between these miRNAs, up to ~2kb (Figure 4C and Figure S5). Northern blotting showed that while biogenesis miR-144 was similar across these constructs, the maturation of miR-451 was maximal from the wild-type construct, and declined progressively as the inter-miRNA distance increased (Figure 4D). The maturation of miR-451 was severely impaired at ~2kb separation from mir-144.
We also tested the functionality of miR-144 and miR-451 across these length variants using luciferase sensors. Consistent with blotting, miR-144 activity was comparable across all constructs, but repression by miR-451 was progressively reduced with increasing distance from mir-144 (Figure 4E). We could confirm that all alterations of mir-451 activity in these length variants were specifically due to mir-144. When we deleted the terminal loop of mir-144, which we showed strongly inhibits its cropping (Figure 1A–B), we observed concomitant loss of miR-144 and miR-451 activity from the entire panel of length variants (mir-144LD-[Xnt]-451 constructs, Figure 4F). Thus, the pri-mir-144 hairpin preferentially enhances pri-mir-451 processing when they are close. We note that the nucleotide lengths of these spacers should not be taken as linear measurements of physical distance between these miRNA hairpins within cells. Nevertheless, the fact that increasing inter-miRNA distance correlates with loss of biogenesis capacity and function of suboptimal mir-451 provides robust support for spatially localized transfer of Microprocessor between hairpin substrates.
We also tested a few constructs in which we moved the miRNAs closer (Figure 4C and Figure S5). The spacing noted refers to the distance between the pre-miRNAs, such that 0-nt actually removes all the lower sequences between the pre-miRNA hairpins, and 30-nt removes one side of the lower single-stranded flanking sequences for both miRNAs. Unsurprisingly, neither miRNA was produced nor functional from the shortest construct (Figure 4D–E). Although less miR-144 was generated from the 30-nt spacer construct, no miR-451 emerged. In this case, once mir-144 is released from mir-144-[30]-451, the single-stranded region 5’ to pri-mir-451 is too short to be recognized by Microprocessor substrate (Figure S5).
Overall, these tests support our model that the basis of biogenesis enhancement of suboptimal mir-451 involves local recruitment and transfer of Microprocessor from a neighboring canonical miRNA that resides within suitable proximity.
Suboptimal canonical miRNAs are enriched within genomic clusters
Although mir-451 is the only well-expressed Dicer-independent mammalian miRNA, we were curious if this principle could be generalized, since many canonical miRNAs reside in clusters. Although all miRNAs adopt hairpin structures that can computationally predicted, the details of experimentally-determined miRNA hairpin structures often differ from predictions (Starega-Roslan et al., 2011). In particular, RNA folding methods tend to over-predict base-pairing in silico, which in reference to our interests, would create a systematic bias in calling suboptimal small terminal loops. Moreover, quantitative data are lacking to assess the relative efficiency with which different miRNAs are cleaved by Drosha/DGCR8. To overcome this, we exploited two newly-generated largescale datasets: (1) SHAPE-MaP structural probing data for >400 pri-miRNAs (B. Kim, S. Baek, and V.N. Kim, unpublished data), and (2) systematic in vitro Microprocessor data reflecting the relative processing efficiency of these miRNAs (K. Kim, S. Baek, and V.N. Kim, unpublished data).
As shown in Figure 5A–B, there is a directional tendency for experimentally derived structures to exhibit less pairing within the terminal loop, compared to RNAstructure predictions. We emphasize the value of the empirical data with selected pri-miRNA hairpins whose structures are substantially discrepant between the two methods (Figure 5C and Figure S6). In summary, the distal loop regions tend to be less structured in genuine miRNA hairpins than predicted by RNA folding algorithms (Figure 5D). With this in mind, it was relevant to consider how to classify pri-miRNAs with suboptimal loop properties. A simple analysis of the very terminal hairpin region (“simple loop”) revealed dozens of loci with 3–4 nt loops, potentially analogous to mir-451 (Figure 5E). However, as exemplified by let-7a-1 (Figure 5F), many of these loci still harbor long segments above the apical junction that might contribute to DGCR8 binding in vivo. Therefore, we classified the length of the apical segment above the ds-ssRNA junction as comprising the “terminal loop”. With this criterion, we observed a relatively few pri-miRNAs with very small terminal loops, but their numbers increased substantially between 7-nt to 8-nt loops (Figure 5E). In total, there were 45 pri-miRNAs with short (≤7nt) terminal loop segments (Table S1).
Figure 5. Genomewide analyses of suboptimal canonical miRNAs.

(A, B) Global analyses of predicted and SHAPE-MaP data show substantially less base-pairing of nucleotides distal to the miRNA duplex in experimentally-derived structures. (C) Example miRNA hairpin that illustrates how computational data tend to over-predict base-pairing in the terminal loop of pri-miRNAs. (D) Schematic of the structural discrepancy between SHAPE-MaP data and computationally predicted hairpins. (E) Defining an appropriate criterion of suboptimal loops. The “simple loop” records the very apical loop of the hairpin, whereas a more inclusive “terminal loop” describes the region apical to the ds-ss junctions distal to miRNA annotation sites (i.e., above the apical junction, “AJ”). Many miRNAs are recorded as having very small simple loops, but small terminal loops are much rarer. (F) Illustration of hairpin loop calculations. For pri-let-7a-1, the simple loop is only 4 nt, but the terminal loop is calculated as 27 nt. We subsequently used the terminal loop calculation as a more accurate reflection of a suboptimal feature. (G) Processing efficiency of miRNAs with short (≤ 7nt) vs longer (> 7nt) terminal loops; only miRNA hairpins lacking apical “UGU” were considered. Two-tailed Mann-Whitney U test is used to show the significance. (H) Intra-cluster distance distribution between non-UGU miRNAs and their neighbors. The suboptimal miRNAs exhibit significantly higher residence within clusters, as emphasized by replotting only the data up to 2 kb neighbors (H’). (I) Processing efficiency of small loop (≤ 7nt) miRNAs, with or without neighbors within 2 kb, relative to other miRNAs; only miRNA hairpins lacking apical “UGU” were considered. Only the suboptimal miRNAs that live in clusters exhibit decreased processing efficiency relative to the control group.
We analyzed whether miRNAs with short loops were differentially processed in vitro from those with longer loops, using processing data generated with individual pri-miRNAs. We observed a trend for poorer processing of short loop miRNAs, but the difference was not significant (Figure S7A–B). However, when we narrowed our scope to pri-miRNAs lacking UGU motif, which is expected to sensitize for DGCR8 recruitment, the difference was greater and now statistically significant (Figure 5G, two-tailed Mann-Whitney U test). Therefore, even though the in vitro Microprocessor assay is readily saturable (Figure S2), quantitative comparisons reveal that smaller terminal loops weaken miRNA cropping efficiency.
Next, we examined the tendency of suboptimal miRNAs to reside in operons. Here, we measured the distance between each pre-miRNA and its closest pre-miRNA. On account of our experimental data (Figure 4C–F), we set a cutoff of ±2000 nts as reflecting genomic clustering relevant to Microprocessor enhancement. We observed that pri-miRNAs with short terminal loops were indeed more likely to reside in operons (Table S1). This is easily visualized in CDF plots of closest neighbor miRNA distances (Figure S7C–D), especially when restricting the comparison to those miRNAs lacking UGU (Figure 5H, miRNAs with <2 kb neighbors shown in 5H’). This emphasizes that the strong majority of miRNAs with combined suboptimal features have close genomic neighbors.
Finally, we integrated these analyses to test whether the in vitro processing efficiency of suboptimal, operonic pri-miRNAs is different from others. We find that these miRNAs indeed have lower efficiency than solo pri-miRNAs with large terminal loops, particularly if they lack apical UGU (Figure 5I and Figure S7E–F). Together, these genomic analyses support and expand the notion that processing interactions with clustered neighbors can facilitate the biogenesis of suboptimal canonical miRNAs. Interestingly, we note that several solo pri-miRNAs (lacking a neighbor within 2 kb) that bear short terminal loops had similar overall processing efficiency to the control group (Figure 5I). This suggests that these seemingly suboptimal miRNAs that lack neighbors may harbor other features that enhance their biogenesis in the solo context.
Validation that canonical miRNAs with small terminal loops are enhanced by residence in operons
We selected several suboptimal loci for experimental validation. We initially took note of suboptimal mir-374b and mir-374a (Table S1), which also happen to be the two shortest canonical pre-miRNA hairpins determined from Drosha fCLIP-seq, which precisely mapped pri-miRNA cleavage sites in cells (Kim et al., 2017). Both harbor very short (4 nt) terminal loops based on SHAPE-MaP data and both are clustered (Figure 6A), with inter-hairpin distances similar to mir-144/451 (hsa-mir-374b/421, 108nt; has-mir-374a/mir-545, 109nt). Note that mir-421 and mir-545 are distinct miRNAs, so these clusters have diverged from each other and thus are not identical duplicate clusters.
Figure 6. Validation of suboptimal canonical miRNAs whose biogenesis is enhanced within operons.

(A) Schematics of mir-374a/545 and mir-374b/421 clusters, for which mir-374a/b both have 4 nt terminal loops. Also shown are mir-374a/b variants with enlarged terminal loops (LM), which were engineered into cluster constructs in which their partner miRNA carried a loop deletion to inhibit processing (545LD or 421LD). (B, C) Processing of wt and variant mir-374a/545 constructs (B) and mir-374b/421 constructs (C) in HEK293T cells. Note these cells lack miR-374a/miR-545, but do express miR-374b/miR-421; asterisk indicates blots indicates background band hybridized to miR-374a probe. (D, E) Activity of mir-374a (D) or mir-374b (E) from wt and mutated constructs. These assays demonstrate that biogenesis and activity of miR-374a and miR-374b require their miRNA neighbors, but can be compensated by enlarging their terminal loops. (F) Schematic of mir-449c/b/a cluster, which yields three similar mature miRNAs; only mir-449c bears a small (5 nt) terminal loop. (G) To avoid cross-hybridization, we compared the properties of solo mir-449c with an artificial cluster with mir-545. (H) Biogenesis and (I) activity of mir-449c is enhanced when transcribed from an operon relative to a solo context.
We prepared constructs to test whether biogenesis or function of mir-374a or mir-374b are influenced by their neighboring miRNAs. Northern blotting indicates that optimal biogenesis of both miR-374a and miR-374b depends on their clustered context (Figure 6B–C). Moreover, deletion of either the partner miRNA hairpin or even just the terminal loop (LD) of the partner miRNA within the respective operon constructs was sufficient to abrogate the enhancing effects of the neighboring miRNA (Figure 6B–C). The relative capacity to mature miR-374a and miR-374b from these various constructs correlated well with activity sensor measurements (Figure 6D–E).
Finally, we tested if the small terminal loops of these loci were causal to their suboptimality. To do so, we re-engineered mir-374a and mir-374b to have larger terminal loops (“LM” variants, Figure 6A), within the context of “LD” operon constructs where their neighbors were inactivated by loop deletions. For both miRNAs, this restored normal biogenesis (Figure 6B–C), and activity on sensors (Figure 6D–E), providing direct evidence that their small terminal loops are the relevant suboptimal feature.
We tested additional loci with small terminal loops for possible suboptimal biogenesis. We were curious about mir-449c, since it is a member of the well-studied miR-34/449 miRNA family that has overlapping activities in cilia development and cancer (Lv et al., 2019). Three mir-449 members are located in a cluster, with mir-449c located ~1.5 kb away from the closely paired mir-449b/a (Figure 6F). This case is somewhat hidden, since the operonic miRNAs are highly similar (Figure 6F). To distinguish the role of miRNA neighbors in its processing, we excised mir-449c as a solo locus and compared to an operon version with mir-545 (Figure 6G). With these reagents, we could clearly see that both biogenesis and activity of miR-449c were enhanced by residence near a normal canonical miRNA (Figure 6H–I).
Competition amongst suboptimal miRNAs within an operon further supports the Microprocessor transfer model
The availability of additional validated suboptimal miRNAs allowed us to perform a final set of critical tests of our model for Microprocessor transfer during operon biogenesis. Recall that diverse miRNAs are able to substitute for mir-144 to promote mir-451 biogenesis, and mir-451-solo biogenesis can be enhanced by generally elevating Microprocessor or by direct recruitment of DGCR8. However, can we locally block this enhancing effect? If so, that would further support the model for local transfer.
We set a scheme by placing suboptimal mir-374a (Figure 6A) in various locations within a mir-144/451 construct (Figure 7A). Unlike mir-451 itself (Figure 1E), mir-374a mildly enhanced miR-451 biogenesis and repression activity, although this was modest compared to mir-144 (Figure 7B–C). Although both mir-374a and mir-451 are demonstrably suboptimal for Microprocessor recruitment, mir-374a has a conventional pri-miRNA stem length.
Figure 7. Location-dependent competition amongst suboptimal clustered miRNAs.

(A) Schematics of miRNA constructs. In addition to Δ144–451 solo and mir-144/451 constructs used earlier, we replaced mir-144 with suboptimal mir-374a (374a-451), inserted mir-374a in between mir-144 and 451 (144–374a-451), or placed it upstream in this operon (374a-144–451). (B) Northern blotting shows that mir-374a only modestly enhances mir-451 biogenesis. However, miR-374a can be enhanced by mir-144, but in so doing, it blocks biogenesis of downstream mir-451. Placing mir-144 in between these suboptimal miRNAs allows both to experience some biogenesis enhancement. (C) Sensor assays of these constructs correlate well with the Northern results, indicating location-dependent competition between mir-374a and mir-451 for benefitting from proximity to mir-144. Reciprocally, miR-144 activity is little affected by the presence or number of suboptimal neighbors. (D) In vivo test of competition for locally released Microprocessor by suboptimal primary miRNA hairpins. λN-DGCR8 is tethered to the terminal loop of mir-144 and its capacity to locally promote the biogenesis of neighboring suboptimal miRNAs is assayed; untethered DGCR8 is used as a control. (E) Northern blotting shows that local recruitment and release of λN-DGCR8 from neighboring pre-mir-144BoxB can strongly potentiate mir-451 biogenesis, but this is blocked by proximal location of suboptimal mir-374a.
We then asked whether mir-374a could compete with mir-451 for biogenesis and/or functional enhancement by mir-144. When we placed mir-374a in between mir-144 and mir-451, we observed increased miR-374a levels and sensor repression at the expense of miR-451 (Figure 7B–C). However, when mir-144 was in between mir-374 and mir-451, we observed intermediate enhancement of both suboptimal miRNAs (Figure 7B–C). To summarize these tests, proximity of the suboptimal miRNA to the optimal miRNA provides a competitive advantage for biogenesis enhancement.
In a final test of the competition model, we used the mir-144BoxB system to assess the biogenesis enhancement of linked mir-374a/451 miRNAs by λN-DGCR8 (Figure 7D). As shown previously (Figure 4A–B), λN-DGCR8 promotes efficient maturation of miR-451 by mir-144BoxB. However, insertion of mir-374a between these miRNAs (144BoxB-374a-451) strongly inhibits maturation of miR-451, concomitant with the appearance of miR-374a (Figure 7D–E). Thus, there is competition between suboptimal miRNAs to take advantage of a fixed amount of Microprocessor recruited to a neighboring pri-miRNA hairpin, and these tests further imply that a limited amount of serial transfer between hairpins is possible.
Discussion
Regulated biogenesis of miRNAs
Although the miRNA pathway is often perceived as linear and unfettered, the reality is that miRNA biogenesis does not proceed exclusively via canonical or optimal routes (Treiber et al., 2019). In the case of the former, a diversity of non-canonical miRNA biogenesis strategies have been documented that bypass either Drosha or Dicer (Yang and Lai, 2011), or in principle can bypass both (Maurin et al., 2012). In the case of the latter, there are numerous miRNAs whose processing is inhibited at the Drosha step or requires activation by a trans-acting factor, and analogous regulation has been found for Dicer cleavage as well as for control of mature miRNA levels and/or activity (Treiber et al., 2019).
Previous studies have defined sequence and structure requirements for optimal Microprocessor substrates (Fang and Bartel, 2015; Kwon et al., 2019), but individual miRNA often lack one or more of these features. In this study, we studied a hidden layer for miRNA biogenesis regulation in which the processing efficiency of a suboptimal miRNA can be enhanced by a neighboring canonical miRNA within the same primary transcript. This phenomenon was documented for certain miRNA clusters in diverse species. For example, the biogenesis of Drosophila mir-998 is dependent on its neighbor mir-11 in a position-independent manner (Truscott et al., 2016), accumulation of mammalian miR-497a is dependent on its neighbor mir-195a (Lataniotis et al., 2017), and the expression of Epstein-Barr virus (EBV) miR-BHRF1–3 is dependent on its cluster neighbor mir-BHRF1–2 (Haar et al., 2016).
Although a mechanism for these cluster dependencies was not established in these studies, miRNAs that require cluster residency exhibit unusual secondary structures that in some cases were shown as poor Microprocessor substrates, as with pri-mir-BHRF1–3 (Haar et al., 2016). We provide evidence for a similar rationale for suboptimality of nuclear processing of Dicer-independent mir-451, for which proximity of a nearby strong Microprocessor substrate presumably increases the local concentration of this enzymatic activity, thereby facilitating recognition and processing of the suboptimal hairpin (Figure 2D). Beyond known elements sought by Microprocessor during the challenging process of correctly identifying miRNA substrates (Auyeung et al., 2013; Fang and Bartel, 2015), we show a key role for terminal loop size in DGCR8 recruitment. We extend this by providing explicit evidence that local Microprocessor recruitment and release are the key determinants for promoting the nuclear biogenesis of clustered suboptimal miRNAs.
Many, but not all, suboptimal Drosha/DGCR8 substrates are located within clusters. For other miRNAs with small apical loops but lacking close neighbors (Figure 5I), perhaps some are regulated by RNA binding proteins (RBPs) that promote their biogenesis. It is conceivable that regulators of suboptimal miRNA biogenesis have already been revealed in the course of largescale RBP-miRNA interaction studies (Nussbacher and Yeo, 2018; Treiber et al., 2017). A preprint by Herzog and colleagues also investigates how the clustered neighbor mir-16 promotes the nuclear biogenesis of suboptimal mir-15a (Hutter et al., 2019). Notably, they identify SAFB2 as a trans-acting factor that promotes biogenesis of miR-15a from the cluster by interacting with Drosha. As SAFB2 is required for optimal biogenesis of certain other clustered miRNAs (Hutter et al., 2019), it will be interesting to elucidate if suboptimal miRNAs generally exhibit common or distinct mechanistic involvement for this factor.
Evolution of miRNA clusters
Our findings may have implications for miRNA evolution. This hidden layer of regulation may help to explain the preference of some miRNAs within operons, perhaps within the local vicinity of miRNA genomic loci that concentrates Microprocessor activity. In this model, the biogenesis of fortuitous, evolutionarily emergent, suboptimal hairpins lacking the full menu of miRNA features will be aided by proximity to pre-existing canonical miRNAs, compared to a “solo” location. Since miRNAs operate as concentration-dependent molecules, establishing sufficient maturation is a key step in their capacity to enter into and influence endogenous regulatory networks, and to have such activities be shaped by natural selection. As it may be challenging for completely de novo miRNAs to emerge with fully optimal features for biogenesis, this mechanism should privilege the birth of functional miRNA loci within operons. Such preferred biogenesis may help to explain the frequent arrangement of miRNAs in operons seen in present-day metazoan genomes (Altuvia et al., 2005; Marco et al., 2013; Mohammed et al., 2014).
We envision that genomic retention of some of these clusters may have to do with the utility of their co-expression, which may have gone hand-in-hand with the gradual acquisition of beneficial targets in the shared domain of such miRNAs. However, in some cases, as with mir-144/451, the non-canonical nature miR-451 biogenesis demands retention of the cluster for normal processing. As we have shown (Figure 3), we can relieve the dependency of mir-451 on its neighbor by increasing its terminal loop or its stem length. Since neither of these alterations has occurred during vertebrate evolution, we imagine a biological imperative to keep miR-451 biogenesis subservient to miR-144 during the dynamics of erythropoiesis. As other miRNAs have been retained as suboptimally processed loci within clusters (Figures 5–6), this may reflect other biological rationales for regulated miRNA biogenesis that remain to be elucidated.
STAR★Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric C. Lai (laie@mskcc.org), or by Renfu Shang (shangr@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293T cells were grown in DME-high glucose media containing 10% FBS, 1% non-essential amino acids, 1% sodium pyruvate, penicillin/streptomycin, L-glutamate, and 0.1% 2-mercaptoethanol. K562 cells were grown in RPMI1640 media containing 10% FBS and 1% penicillin/streptomycin. Mycoplasma contaminations were regularly tested for the cell lines. The K562 cell lines for knockout were authenticated via genotyping.
METHOD DETAILS
Constructs
Plasmids for expression of all the miRNAs used in this study were constructed by inserting amplified DNA fragments containing the miRNA precursors from genomic DNA of HEK293T cells between Bgl II and Xho I sites downstream of a CMV promoter. The luciferase plasmids containing bulge or perfect miRNA sensors were constructed by inserting annealed DNA oligonucleotides containing miRNA sensor sequences between Nhe I and Xba I (for bulge sensors) or Xho I and Xba I (for perfect sensors) sites in the 3’ UTR of the firefly luciferase gene (Wu and Belasco, 2005). The CRISPR-sgRNA plasmids were constructed by inserting annealed DNA oligonucleotides containing guide RNA sequences in the BsmB I site in lentiCRISPRv2 vectors containing a GFP or puromycin-resistant gene as selection marker (Shalem et al., 2014). All the details and oligonucleotide sequences used to clone these constructs are listed in Table S2.
Generation of mir-144/451 knockout K562 cell lines using CRISPR/Cas9
Lentiviral particles containing CRISPR-Cas9 were produced by transfecting HEK293T cells with pMD2.G (400 ng/well), psPAX2 (800 ng/well) and the lentiviral sgRNA plasmids (800 ng/well) using Lipofectamine 2000 (Thermo Fisher) in 6-well plates. Cell culture supernatants were collected 48 hr after transfection and filtered through a 0.45 μm filtration membrane. K562 cells growing in 6-well plates were infected using paired lentiviral sgRNAs particles (one containing GFP and the other containing puromycin-resistant gene) and selected by puromycin (4 μg/mL) for 7 days. We collected cells and performed cell sorting to split the GFP-positive cells into each well containing 100 μl of RPMI1640 media into 96-well plates containing 1 cell/well. After two weeks of culturing, the cells were collected for genotyping to identify colonies with the desired mir-144 or mir-451 deletion alleles. Oligo sequences for sgRNAs and genotyping are listed in Table S2.
Sensor assays
Transient transfection of the HEK293T cells with miRNA expressing plasmids (150~200 ng/well) and luciferase plasmids containing miRNA sensors (15 ng/well) was performed in 24-well cell culture plates using Lipofectamine2000 (Thermo Fisher) according to the manufacturer’s protocol. Cells were harvested 24 hr post-transfection and then Firefly and Renilla luciferase (co-transfected as reference gene) activities were measured using the Dual-Glo luciferase assay system (Promega).
Northern blotting
Total RNAs from cultured cells were prepared using Trizol reagent. Equal amounts of total RNAs (10~15 μg) were denatured at 95 °C and fractionated by electrophoresis on a 20% urea polyacrylamide gel. Then the gel was transferred to GeneScreen Plus membrane (Perkin Elmer), UV-crosslinked and baked at 80 °C for 30 min and then hybridized with γ−32P-labeled probes at 42 °C overnight. Probe sequence s are listed in Table S2.
Quantitative RT-PCR (qRT-PCR)
Total RNAs (1 μg) were used for cDNA preparation by DNase I treatment and reverse transcription using SuperScript III Reverse Transcriptase (Invitrogen). qPCR reactions were performed using SYBR Select master mix (Life Technologies). Data were normalized to GAPDH amplification. Primer sequences for qPCR are listed in Table S2.
Immunoprecipitation of Drosha/DGCR8 complex
HEK293T cells grown in 6-well plate were transiently co-transfected with plasmids (1 μg for each) encoding for Drosha-Flag and HA-DGCR8. After 48 hr, the cells were washed with phosphate-buffered saline (PBS) and harvested in lysis buffer containing 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 and protease inhibitor cocktail. After rotation at 4 °C for 20 min, each l ysate was clarified by centrifugation at 14,000 × g at 4 °C for 15 min. A total of 500 μl supernatant was mixed with 20 μl of EZview Red Anti-FLAG Affinity Gel (Sigma-Aldrich) at room temperature for 2 hr. The beads were washed four times with Tris-buffered saline (TBS) for the following in vitro processing assays.
In vitro Microprocessor assays
Pri-mir-144/451 and variants are generated and internally labeled with α−32P-UTP by in vitro transcription using HiScribe™ T7 High Yield RNA Synthesis Kit (NEB) and purified from 6% urea polyacrylamide gel. In vitro processing of pri-miRNAs was carried out in 30 μl reaction containing 6.4 mM MgCl2, 1 U/μl RNase Inhibitor, 400 ng of pri-miRNA transcripts and 5 μl of immunoprecipitated Drosha-Flag/HA-DGCR8 complex from HEK293T cells. The reaction mixture was incubated at 37 °C for different time points. The RNA was purified from the reaction mixture and analyzed on 20% urea polyacrylamide gel. Primer sequences for T7 template amplification are listed in Table S2.
High throughput in vitro Microprocessor assay
We first prepared DNA templates of pri-miRNAs by taking advantage of massively parallel synthesis technology (Cellemics). To minimize cost and maximize synthetic accuracy, we confined the essential parts of 125 nt centered at pre-miRNA. This 125 nt region encompasses all known primary sequence motifs (UG, UGU, mGHG and CNNC) and shows higher phyloP conservation scores compared to the surrounding regions. The templates were synthesized with two adapter sequences (18 nt each) at both ends for amplification. After synthesis and amplification, pri-miRNA substrates were prepared by T7 in vitro transcription and gel purification. To purify Microprocessor enzyme, DROSHA-FLAG and DGCR8-HA constructs were transfected into HEK293E cells. The cells were harvested and sonicated, and then Microprocessor was purified from the supernatant through FLAG-IP followed by 3XFLAG-peptide elution. For in vitro processing, pri-miRNA substrates were incubated with the recombinant Microprocessor for 1 hour at 37°C. Input, processed, and unprocess ed RNAs were separately gel-purified and sequenced.
Calculation of processing efficiency
Two libraries were generated: one from input RNA before processing and the other after in vitro processing reaction. Processing efficiency for pri-mir-X was defined as below, where Xi and Xu are pri-mir-X read counts from the input library and the unprocessed reads from the processing library, respectively.
Due to the depth imbalance between input and unprocessed libraries, read count in unprocessed library was adjusted by dividing by Nu / Ni, where N refers to negative control RNA.
SHAPE-MaP
RNA folding and SHAPE probing were performed as described in (Siegfried et al., 2014). RNA mixture of pri-miRNAs was folded in 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM MgCl2 at 37°C for 20 min. After folding, RNAs were modified in the presence of 10 mM 1M7 and incubated at 37°C for 75 sec. The no-reagent control sample, containing neat DMSO instead of SHAPE reagent, was performed in parallel. For the denatured sample, RNAs were modified using 1M7 under 50 mM HEPES pH 8.0, 4 mM EDTA and 50% formamide at 95°C for 1 min. After the modification, RNAs were precipitated by ethanol. RNAs were then subjected to reverse transcription for 3 h at 42°C using SuperScript II (Invitrogen) in 0.5 mM premixed dNTPs, 50 mM Tris-HCl pH 8.0, 75 mM KCl, 6 mM MnCl2 and 10 mM DTT buffer. After reverse transcription, cDNAs were purified by Agencourt RNAClean XP (Beckman). Sequencing libraries were generated using one-step PCR approach. The one-step PCR was performed for 3 cycles to tag cDNAs and generated the final library for sequencing. The library was sequenced by BGI platform. Empirical structures of pri-miRNAs were obtained by Shape-Mapper (version 1.0, http://chem.unc.edu/rna/software.html) with sequencing data as inputs.
Computational analysis of pri-miRNA structures
Terminal loop of pri-miRNAs was defined as the RNA segment above the apical ds-ssRNA junction. The position of apical ds-ssRNA junction was determined as the junction that satisfies the following criteria: the distance from the basal ds-ssRNA junction (>31 and <39), the distance from the 5′ end of 5p miRNA (>17) and the maximal number of unpaired bases. To sort pri-miRNAs with the UGU motif, the UGU sequence was scanned at +20~+24 bp from the 5′ end of 5p miRNA. In silico prediction of RNA secondary structure and pairing probability was performed using the RNAstructure package v5.8 (Reuter and Mathews, 2010).
Genomic analysis of pri-miRNA clusters
To measure the genomic distance to the nearest neighbor miRNA locus, we retrieved the coordinates of human miRNAs from miRBase v21 (Kozomara and Griffiths-Jones, 2014). We assigned two adjacent pri-miRNAs as “neighbors” if the two locate within 1000 kb in the same strand of the same chromosome. If a pri-miRNA has neighbor(s) within 2 kb, then it was considered as “clustered” for further analysis.
DATA AND CODE AVAILABILITY
A summary of pri-miRNA processing efficiencies and terminal loop sizes determined by SHAPE-MaP are provided in Table S1. ShapeMapper execution files and codes are available from the Kevin Weeks lab website (http://weeks.chem.unc.edu/software.html). The miRNA processing datasets were generated as part of another study and will be reported separately, but are available from Narry Kim (narrykim@snu.ac.kr) upon request. Codes that analyzed processing efficiency, secondary structures, genomic information of pri-miRNAs were deposited at https://github.com/schanbaek/mirclustering.
Supplementary Material
Table S1, Related to Figure 5. This table summarizes properties of miRNAs with small terminal loops, including data on their relative in vitro processing efficiency (PE), loop size determined from SHAPE-MaP analysis, presence of UGU motif, and distance to nearest miRNA neighbor
Table S2. Related to Figures 1–4, 6–7. This table summarizes oligonucleotide sequences used to clone constructs, to measure gene expression, and for Northern blotting.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| TOP10 Chemically Competent E. coli | Our lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ATP, [γ-P32] | PerkinElmer | Cat#BLU502Z250UC |
| UTP, [α-P32] | PerkinElmer | Cat#BLU007H250UC |
| Puromycin Dihydrochloride | Thermo Fisher | Cat#A1113803 |
| Penicillin-Streptomycin | Life Technologies | Cat#15070063 |
| Trizol reagent | Life Technologies | Cat#15596018 |
| Protease Inhibitor Cocktail tablets, EDTA-free | Roche | Cat#11836170001 |
| Lipofectamine 2000 | Thermo Fisher | Cat#11668030 |
| T4 Polynucleotide Kinase | NEB | Cat#M0201L |
| T4 DNA Ligase | NEB | Cat#M0202L |
| RNaseOUT Ribonuclease Inhibitor | Thermo Fisher | Cat#10777019 |
| Gel Loading Buffer II | Invitrogen | Cat#AM8547 |
| TURBO DNase | Thermo Fisher | Cat#AM2238 |
| Opti-MEM Reduced Serum Medium | Thermo Fisher | Cat#51985034 |
| Hexadimethrine bromide | Sigma-Aldrich | Cat#107689 |
| Critical Commercial Assays | ||
| Dual Glo luciferase assay system | Promega | Cat#E2940 |
| HiScribe™ T7 High Yield RNA Synthesis Kit | NEB | Cat#E2040S |
| SYBR select master mix | Life Technologies | Cat#4472942 |
| Superscript III RT kit | Invitrogen | Cat#18080044 |
| Decade marker system | Thermo Fisher | Cat#AM7778 |
| AccuPrime pfx DNA Polymerase | Thermo Fisher | Cat#12344024 |
| Q5 Hot Start High-Fidelity DNA Polymerase | NEB | Cat#M0493L |
| ANTI-FLAG M2 Affinity Gel | Sigma-Aldrich | Cat#A2220 |
| SequaGel UreaGel System | National Diagnostics | Cat#EC-833 |
| Experimental Models: Cell Lines | ||
| HEK293T | Our lab | N/A |
| K562 | Michael G. Kharas | N/A |
| miR-144 knockout K562 | This study | N/A |
| miR-451 knockout K562 | This study | N/A |
| miR-144 and miR-451 double knockout K562 | This study | N/A |
| Oligonucleotides | ||
| Oligonucleotides for plasmid construction, see Table S2 | This study | N/A |
| Oligonucleotides for in vitro transcription, see Table S2 | This study | N/A |
| Oligonucleotides for sgRNA and genotyping, see Table S2 | This study | N/A |
| Oligonucleotides for Northern blotting probes, see Table S2 | This study | N/A |
| Oligonucleotides for qPCR, see Table S2 | This study | N/A |
| Recombinant DNA | ||
| All the miRNA expression plasmids, see Table S2 | This study | N/A |
| All the miRNA sensor plasmids, see Table S2 | This study | N/A |
| pCK-Drosha-Flag | V. Narry Kim | N/A |
| CMV-HA-DGCR8 | V. Narry Kim | N/A |
| pCMV-λN-DGCR8 | This study | N/A |
| pCMV-λN-EGFP | This study | N/A |
| pcDNA6.2-miR-375 | Our lab | N/A |
| LentiCRISPRv2 GFP | Addgene | Cat#82416 |
| LentiCRISPRv2 PuroR | Addgene | Cat#52961 |
| Software and Algorithms | ||
| Multi Gauge V3.0 software | FUJIFILM | N/A |
| RNAstructure package v5.8 | Reuter and Mathews, 2010 | N/A |
| miRBase v21 | Kozomara and Griffiths-Jones, 2014 | N/A |
| Other | ||
| Illustra MicroSpin G-25 columns | GE Healthcare | Cat#27532501 |
| GeneScreen Plus hybridization transfer membrane | PerkinElmer | Cat#NEF1017001PK |
| Stericup-GP Sterile Vacuum Filtration System | Millipore | Cat#SCGPU05RE |
| Millex-HV Syringe Filter Unit, 0.45 μm | Millipore | Cat#SLHV033RB |
Highlights:
mir-451 is a Dicer-independent miRNA whose biogenesis requires its neighbor mir-144
Both small loop and short stem render nuclear processing of mir-451 suboptimal
mir-451 processing involves local recruitment and transfer of Microprocessor
Genomic analyses show suboptimal canonical miRNAs preferentially reside in operons
Acknowledgements
We thank Dr. Ligang Wu (SIBCB, CAS) for providing the backbone plasmids for constructing miRNA expression and sensor vectors. Work in the V.N.K lab was supported by the Institute for Basic Science from the Ministry of Science and ICT of Korea (IBS-R008-D1). Work in the E.C.L. lab was supported by the Susan and Peter Solomon Divisional Genomics Program, NIH grants R01-GM083300, R01-NS083833 and R01-HL135564, and by the MSK Core Grant P30-CA008748.
Footnotes
Declaration of Interests.
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T, and Margalit H. (2005). Clustering and conservation patterns of human microRNAs. Nucleic acids research 33, 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auyeung VC, Ulitsky I, McGeary SE, and Bartel DP (2013). Beyond Secondary Structure: Primary-Sequence Determinants License Pri-miRNA Hairpins for Processing. Cell 152, 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballarino M, Pagano F, Girardi E, Morlando M, Cacchiarelli D, Marchioni M, Proudfoot NJ, and Bozzoni I. (2009). Coupled RNA processing and transcription of intergenic primary microRNAs. Molecular and cellular biology 29, 5632–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejarano F, Bortolamiol-Becet D, Dai Q, Sun K, Saj A, Chou YT, Raleigh DR, Kim K, Ni JQ, Duan H, et al. (2012). A genome-wide transgenic resource for conditional expression of Drosophila microRNAs. Development 139, 2821–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Stark A, Brennecke J, and Cohen SM (2008). Temporal Reciprocity of miRNAs and Their Targets during the Maternal-to-Zygotic Transition in Drosophila. Curr Biol 18, 501–506. [DOI] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM, and Hannon GJ (2010). A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465, 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church VA, Pressman S, Isaji M, Truscott M, Cizmecioglu NT, Buratowski S, Frolov MV, and Carthew RW (2017). Microprocessor Recruitment to Elongating RNA Polymerase II Is Required for Differential Expression of MicroRNAs. Cell reports 20, 3123–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson N, et al. (2010). A Novel miRNA Processing Pathway Independent of Dicer Requires Argonaute2 Catalytic Activity. Science 328, 1694–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs S, and Haber DA (2007). Dual Role for Argonautes in MicroRNA Processing and Posttranscriptional Regulation of MicroRNA Expression. Cell 131, 1097–1108. [DOI] [PubMed] [Google Scholar]
- Donayo AO, Johnson RM, Tseng HW, Izreig S, Gariepy A, Mayya VK, Wu E, Alam R, Lussier C, Jones RG, et al. (2019). Oncogenic Biogenesis of pri-miR-17 approximately 92 Reveals Hierarchy and Competition among Polycistronic MicroRNAs. Molecular cell. [DOI] [PubMed] [Google Scholar]
- Du P, Wang L, Sliz P, and Gregory RI (2015). A Biogenesis Step Upstream of Microprocessor Controls miR-17 approximately 92 Expression. Cell 162, 885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W, and Bartel DP (2015). The Menu of Features that Define Primary MicroRNAs and Enable De Novo Design of MicroRNA Genes. Molecular cell 60, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haar J, Contrant M, Bernhardt K, Feederle R, Diederichs S, Pfeffer S, and Delecluse HJ (2016). The expression of a viral microRNA is regulated by clustering to allow optimal B cell transformation. Nucleic acids research 44, 1326–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, and Kim VN (2006). Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125, 887–901. [DOI] [PubMed] [Google Scholar]
- Han J, Pedersen JS, Kwon SC, Belair CD, Kim YK, Yeom KH, Yang WY, Haussler D, Blelloch R, and Kim VN (2009). Posttranscriptional crossregulation between Drosha and DGCR8. Cell 136, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YC, Vidigal JA, Mu P, Yao E, Singh I, Gonzalez AJ, Concepcion CP, Bonetti C, Ogrodowski P, Carver B, et al. (2015). An allelic series of miR-17 approximately 92-mutant mice uncovers functional specialization and cooperation among members of a microRNA polycistron. Nature genetics 47, 766–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. (2005). A microRNA polycistron as a potential human oncogene. Nature 435, 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter K, Lohmuller M, Jukic A, Eichin F, Avci S, Labi V, Hoser SM, Huttenhofer A, Villunger A, and Herzog S. (2019). SAFB2 enables the processing of suboptimal stem-loop structures in clustered primary miRNA transcripts. bioRxiv doi: 10.1101/858647. [DOI] [PubMed] [Google Scholar]
- Jee D, Yang JS, Park SM, Farmer DT, Wen J, Chou T, Chow A, McManus MT, Kharas MG, and Lai EC (2018). Dual Strategies for Argonaute2-Mediated Biogenesis of Erythroid miRNAs Underlie Conserved Requirements for Slicing in Mammals. Molecular cell 69, 265–278 e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Jeong K, and Kim VN (2017). Genome-wide Mapping of DROSHA Cleavage Sites on Primary MicroRNAs and Noncanonical Substrates. Molecular cell 66, 258–269 e255. [DOI] [PubMed] [Google Scholar]
- Kozomara A, and Griffiths-Jones S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research 42, D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SC, Baek SC, Choi YG, Yang J, Lee YS, Woo JS, and Kim VN (2019). Molecular Basis for the Single-Nucleotide Precision of Primary microRNA Processing. Molecular cell 73, 505–518 e505. [DOI] [PubMed] [Google Scholar]
- Kwon SC, Nguyen TA, Choi YG, Jo MH, Hohng S, Kim VN, and Woo JS (2016). Structure of Human DROSHA. Cell 164, 81–90. [DOI] [PubMed] [Google Scholar]
- Lataniotis L, Albrecht A, Kok FO, Monfries CAL, Benedetti L, Lawson ND, Hughes SM, Steinhofel K, Mayr M, and Zampetaki A. (2017). CRISPR/Cas9 editing reveals novel mechanisms of clustered microRNA regulation and function. Scientific reports 7, 8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liang C, Kollipara RK, Matsui M, Ke X, Jeong BC, Wang Z, Yoo KS, Yadav GP, Kinch LN, et al. (2016). HP1BP3, a Chromatin Retention Factor for Co-transcriptional MicroRNA Processing. Molecular cell 63, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J, Zhang Z, Pan L, and Zhang Y. (2019). MicroRNA-34/449 family and viral infections. Virus research 260, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Wu Y, Choi JG, and Wu H. (2013). Lower and upper stem-single-stranded RNA junctions together determine the Drosha cleavage site. Proceedings of the National Academy of Sciences of the United States of America 110, 20687–20692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Ninova M, Ronshaugen M, and Griffiths-Jones S. (2013). Clusters of microRNAs emerge by new hairpins in existing transcripts. Nucleic acids research 41, 7745–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin T, Cazalla D, Yang JS, Bortolamiol-Becet D, and Lai EC (2012). RNase III-independent microRNA biogenesis in mammalian cells. RNA 18, 2166–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Khan AA, Leslie CS, et al. (2010). Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nature cell biology 12, 372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J, Siepel A, and Lai EC (2014). Diverse modes of evolutionary emergence and flux of conserved microRNA clusters. RNA 20, 1850–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, and Proudfoot NJ (2008). Primary microRNA transcripts are processed co-transcriptionally. Nature structural & molecular biology 15, 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TA, Jo MH, Choi YG, Park J, Kwon SC, Hohng S, Kim VN, and Woo JS (2015). Functional Anatomy of the Human Microprocessor. Cell 161, 1374–1387. [DOI] [PubMed] [Google Scholar]
- Nussbacher JK, and Yeo GW (2018). Systematic Discovery of RNA Binding Proteins that Regulate MicroRNA Levels. Molecular cell 69, 1005–1016 e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlicki JM, and Steitz JA (2008). Primary microRNA transcript retention at sites of transcription leads to enhanced microRNA production. The Journal of cell biology 182, 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, and Mathews DH (2010). RNAstructure: software for RNA secondary structure prediction and analysis. BMC bioinformatics 11, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang R, Zhang F, Xu B, Xi H, Zhang X, Wang W, and Wu L. (2015). Ribozyme-enhanced single-stranded Ago2-processed interfering RNA triggers efficient gene silencing with fewer off-target effects. Nature communications 6, 8430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried NA, Busan S, Rice GM, Nelson JA, and Weeks KM (2014). RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nature methods 11, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver SJ, Hagen JW, Okamura K, Perrimon N, and Lai EC (2007). Functional screening identifies miR-315 as a potent activator of Wingless signaling. Proceedings of the National Academy of Sciences of the United States of America 104, 18151–18156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starega-Roslan J, Krol J, Koscianska E, Kozlowski P, Szlachcic WJ, Sobczak K, and Krzyzosiak WJ (2011). Structural basis of microRNA length variety. Nucleic acids research 39, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber T, Treiber N, and Meister G. (2019). Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nature reviews. Molecular cell biology 20, 5–20. [DOI] [PubMed] [Google Scholar]
- Treiber T, Treiber N, Plessmann U, Harlander S, Daiss JL, Eichner N, Lehmann G, Schall K, Urlaub H, and Meister G. (2017). A Compendium of RNA-Binding Proteins that Regulate MicroRNA Biogenesis. Molecular cell 66, 270–284 e213. [DOI] [PubMed] [Google Scholar]
- Truscott M, Islam AB, and Frolov MV (2016). Novel regulation and functional interaction of polycistronic miRNAs. RNA 22, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Luo J, Zhang H, and Lu J. (2016). microRNAs in the Same Clusters Evolve to Coordinately Regulate Functionally Related Genes. Molecular biology and evolution 33, 2232–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, and Belasco JG (2005). Micro-RNA regulation of the mammalian lin-28 gene during neuronal differentiation of embryonal carcinoma cells. Molecular and cellular biology 25, 9198–9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, and Lai EC (2010). Dicer-independent, Ago2-mediated microRNA biogenesis in vertebrates. Cell cycle 9, 4455–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, and Lai EC (2011). Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Molecular cell 43, 892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Maurin T, and Lai EC (2012). Functional parameters of Dicer-independent microRNA biogenesis. RNA 18, 945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, and Lai EC (2010). Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proceedings of the National Academy of Sciences of the United States of America 107, 15163–15168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda M, Cifuentes D, Izumi N, Sakaguchi Y, Suzuki T, Giraldez AJ, and Tomari Y. (2013). Poly(A)-specific ribonuclease mediates 3’-end trimming of Argonaute2-cleaved precursor microRNAs. Cell reports 5, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, and Cullen BR (2005). Efficient processing of primary microRNA hairpins by Drosha requires flanking non-structured RNA sequences. The Journal of biological chemistry. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Yi R, and Cullen BR (2005). Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. The EMBO journal 24, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Zeng Y. (2011). The terminal loop region controls microRNA processing by Drosha and Dicer. Nucleic acids research 38, 7689–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Related to Figure 5. This table summarizes properties of miRNAs with small terminal loops, including data on their relative in vitro processing efficiency (PE), loop size determined from SHAPE-MaP analysis, presence of UGU motif, and distance to nearest miRNA neighbor
Table S2. Related to Figures 1–4, 6–7. This table summarizes oligonucleotide sequences used to clone constructs, to measure gene expression, and for Northern blotting.
Data Availability Statement
A summary of pri-miRNA processing efficiencies and terminal loop sizes determined by SHAPE-MaP are provided in Table S1. ShapeMapper execution files and codes are available from the Kevin Weeks lab website (http://weeks.chem.unc.edu/software.html). The miRNA processing datasets were generated as part of another study and will be reported separately, but are available from Narry Kim (narrykim@snu.ac.kr) upon request. Codes that analyzed processing efficiency, secondary structures, genomic information of pri-miRNAs were deposited at https://github.com/schanbaek/mirclustering.