

Summary
Tumor organoids are promising tools for cancer biology investigations and pre-clinical drug screenings because they are often representative of the histology and drug responses of patients. Here, we introduce a facile protocol to overcome technical limitations by generating patient-derived tumor organoids using a simplified ring-like geometry. This facilitates media exchange and drug treatment for histopathology characterization and automated high-throughput drug screenings.
For complete details on the use and execution of this protocol, please refer to Phan et al. (2019).
Graphical Abstract
Highlights
-
•
Mini-ring format permits facile and automated highthroughput drug screening
-
•
Maxi-ring setup for immunohistochemistry and downstream biochemical assays
-
•
Empty center of the wells allows for easy media exchange and drug treatment
-
•
Adaptability of screening protocol for biology and translational applications
Tumor organoids are promising tools for cancer biology investigations and pre-clinical drug screenings because they are often representative of the histology and drug responses of patients. Here, we introduce a facile protocol to overcome technical limitations by generating patient-derived tumor organoids using a simplified ring-like geometry. This facilitates media exchange and drug treatment for histopathology characterization and automated high-throughput drug screenings.
Before You Begin
CRITICAL: All the procedures described are performed in a Class II biological hood following standard aseptic techniques. Tumor organoids are cultured in a humidified 37°C incubator at 5% CO2.
Organoid Culture Medium
Timing: 30 min
-
1.
Prepare the complete serum-free medium using for organoid culture by adding all supplements to the base medium following the volumes indicated in the table below:
| Mammocult | 450 mL |
|---|---|
| Mammocult supplement | 50 mL |
| Hydrocortisone | 2.5 mL |
| Heparin | 1 mL |
Matrigel
Note: We recommend preparing 500 μL aliquots of Matrigel ahead of time. To prepare aliquots, thaw a bottle of Matrigel, gently vortex and place in a beaker containing ice in the biosafety cabinet. Rapidly pipette 500 μL of Matrigel to pre-chilled Eppendorf tubes. Aliquots can be stored in a -20°C freezer.
-
2.
Place aliquots of Matrigel on ice for 1–2 h to thaw completely
-
3.
Gently vortex for 5 s at intermediate speed to mix completely
-
4.
Leave on ice until needed
Collagenase Stock Solution
Note: We recommend storing Collagenase IV as a Stock Solution at 10,000 U/mL in a –20°C freezer. Different batches of Collagenase IV have different U/mg; make sure to adjust your calculations accordingly (see below).
-
5.
Prepare a Stock Solution of collagenase (10,000 U/mL) by weighing X mg of collagenase powder of known Y U/mg in a 50 mL Falcon tube. We recommend preparing 30–50 mL per tissue sample (6–10 biopsy cores or 1–3 cm3 from tissue resection)
-
6.
Dissolve the powder in Z mL of cold HBSS, where Z = X∗Y/10,000. To help fully dissolving the powder, invert the tube and gently vortex for few seconds at low rpm
-
7.
Once fully dissolved, filter using a 50 mL, 0.22 μm pore size Millipore Steriflip
-
8.
Prepare 1 mL aliquots and store in a –20°C freezer
Collagenase Working Solution
Note: The Working Solution (200 U/mL) is prepared fresh just prior to tissue digestion.
-
9.
Thaw sufficient Stock solution aliquots ahead of time by placing them on ice
-
10.
To prepare 50 mL of Working Solution, add a single 1 mL stock aliquot to 49 mL of PBS
-
11.
Vortex to mix
-
12.
Filter using a 50 mL, 0.22 μm pore size Millipore Steriflip
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Human tumor samples | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MammoCult™ Human Medium Kit | Stemcell Technologies | Ca#05620 |
| Hydrocortisone Stock Solution | Stemcell Technologies | Ca#07925 |
| Heparin Solution | Stemcell Technologies | Ca#07980 |
| Matrigel Basement Membrane Matrix | Corning | Ca#354234 |
| Collagenase type IV | Thermo Fisher | Ca#17-104-019 |
| Dispase | Life Technologies | Ca#17105-041 |
| Histogel | Thermo Fisher | Ca#R904012 |
| Ammonium Chloride Solution | Stem Cell Technology | Ca#07850 |
| Hanks' Balanced Salt Solution (HBSS) | Stem Cell Technology | Ca#37150 |
| Cryostor®CS10 | Sigma Aldrich | Ca#C2874 |
| Recovery™ Cell Culture Freezing Medium | Thermo Fisher | Ca#12648010 |
| PBS | Thermo Fisher | Ca#14190-144 |
| 10% Neutral Buffered Formalin | VWR | Ca#89370-094 |
| Critical Commercial Assays | ||
| CellTiter-Glo 3D | Promega | Ca#G968B |
| AO/PI Staining Solution | Nexcelom | Ca#S2-0106-5mL |
| Software and Algorithms | ||
| Celigo S Software | Nexcelom | N/A |
| Other | ||
| Corning 96-well assay plate | Corning | Ca#CLS3610-48EA |
| Masterblock plate | Greiner bio-one | Ca#786261 |
| MicroAmp Optical Adhesive Film | Thermo Fisher | Ca#4311971 |
| Tissue Path Microcassette 6-Compartiment | Thermo Fisher | Ca#15182705C |
| Millipore Steriflip 50ml | Thermo Fisher | Ca#SE1M179M6 |
| Cellometer Auto 2000 Cell Viability Counter | Nexcelom | N/A |
| Cellometer Disposable Imaging Chambers | Nexcelom | Ca#CHT4-SD100-002 |
| Celigo S Image Cytometer | Nexcelom | Ca#200-BFFL-S |
| epMotion 96 | Eppendorf | Ca#5069000004 |
| epT.I.P.S.® Motion as Reload System 50ul | Eppendorf | Ca#0030014421 |
| epT.I.P.S.® Motion as Reload System 300ul | Eppendorf | Ca#0030014464 |
| 10μl Biotix™ uTIP™ Low-Retention Filter Pipette Tips | Biotix | Ca#M-0011-9FC |
| 20μl Biotix™ uTIP™ Low-Retention Filter Pipette Tips | Biotix | Ca# M-0020-9FC |
| 100μl Biotix™ uTIP™ Low-Retention Filter Pipette Tips | Biotix | Ca#M-0100-9FC |
| 1000μl Biotix™ uTIP™ Low-Retention Filter Pipette Tips | Biotix | Ca#M-1000-9FC |
Resource Availability
Lead Contact
Further information requests should be directed to and will be fulfilled by the Lead Contact, Alice Soragni (alices@mednet.ucla.edu)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code.
Materials and Equipment
Alternatives: Matrigel can be substituted with other substrates such as Cultrex BME (Trevigen 3423-010-01).
Alternatives: Masterblock plates can be substituted with Axygen plates (Axygen P-96-450V-C-S).
Step-By-Step Method Details
Human Tumor Tissue Processing
The following steps are critical to obtain a homogeneous cell suspension to minimize variability for downstream experiments and high-throughput drug screenings in particular.
-
1.
Transfer the tumor sample to a sterile petri dish in a biological hood
-
2.
Add 5 mL of Collagenase Working Solution to keep the tissue moist and use a sterile single-use bistoury to process into 1–3 mm3 fragments
-
3.
Collect the fragments in a 50 mL Falcon tube using the bistoury and wash the dish with an additional 5 mL of Collagenase Working Solution to ensure that all fragments are collected
-
4.
Incubate the tissue in Collagenase Working Solution at 37°C in the cell incubator for 2 h, vortexing every ∼10 min to facilitate tissue digestion
Note: For larger specimens, 10 mL of Working Solution may not be sufficient. In that case, increase the volume accordingly.
Note: After approximately 2 h, you should notice that the solution became increasingly turbid. Larger tissue fragments may still be present and will require iterative digestion and collection steps.
-
5.
Place the Falcon tube in the biological hood and wait 2–3 min for the larger undigested fragments to deposit on the bottom of the tube
-
6.
Gently remove the supernatant containing the cell suspension and place it in a new 50 mL Falcon tube
-
7.
Spin the cell suspension at 400–600 × g for 5 min, immediately followed by red blood cells lysis (#9–18)
-
8.
Resuspend the remaining tissue fragments in 10 mL of Collagenase Working Solution and place back in the incubator for further digestion
Note: Spinning speed may vary depending on tissue type. If unsure, start at the slowest recommended speed (400 × g) and spin a second time at higher speed if the supernatant is still turbid.
Note: Repeat steps 4–8 until the tissue is completely dissolved. We have observed variability in incubation times not only for different types of tissue but also for individual tumors within the same tumor type. We recommend visual inspection of the sample to verify tissue digestion. Digestions of >24 h can result in reduced cell viability.
Red Blood Cell Lysis
Most tumor cell pellets will appear red due to the presence of contaminating red blood cells. While the amount of blood cells can vary significantly from sample to sample, we recommend performing a minimum of one lysis for all samples. For some specimens, excessive red blood cell contaminations will require multiple iterations of the lysis protocol or a larger amount of lysis buffer. Adjust the protocol accordingly.
-
9.
At the end of the spin, gently aspirate the supernatant without disrupting the cell pellet
-
10.
Gently resuspend the pellet in 1 mL of cold, sterile and serum-free RPMI medium
-
11.
Add 9 mL of cold Ammonium Chloride solution drop-by-drop and invert the tube to mix
-
12.
Incubate on ice for 10 min, inverting the tube every 2–3 min
-
13.
Spin at 600 × g for 5 min
-
14.
Gently aspirate all supernatant
Note: If the pellet appears red, repeat steps 9–14.
-
15.
Resuspend cell pellet in 1 mL cold, sterile and serum-free RPMI medium, pipetting up and down several times to obtain a homogenous cell suspension
-
16.
Place a 100 μm strainer on a new 50 mL Falcon and pipette the cell suspension through the strainer
-
17.
Wash the tube with an additional 2–9 mL of cold, sterile and serum-free RPMI medium and pass through the strainer to obtain a final cell suspension of 2–10 mL
-
18.
Leave the tube on ice
Note: The volume of RPMI depends on the size of the cell pellet and method used to count the cells. Cells can be diluted by adding more medium or concentrated by spinning and resuspending in a smaller volume if needed
Note: From step 18 onward, leave the cells on ice unless stated otherwise
Note: Cells from sequential digestions can be pooled at this step. Store the cells on ice while waiting for further digestions.
Viable Cell Counting
Here, we determine the number of viable cells/mL prior to seeding. If multiple cell harvesting steps are performed, pool all cells and count at that point.
-
19.
Invert the Falcon containing the cell suspension in RPMI multiple times
-
20.
Remove a 25 μL aliquot and place in an Eppendorf tube
-
21.
Add 25 μL of AO/PI Staining Solution
-
22.
Mix by pipetting up and down 8–10 times paying attention not to introduce bubbles
-
23.
Add 20 μL to each of the two counting chambers in a Cellometer Disposable Imaging slide
-
24.
Image and count both chambers with a Cellometer Auto 2000 Cell Viability Counter using the “AO/PI Primary Cell/Cell lines” program to distinguish debris from nucleated, viable cells
-
25.
Average the counts to obtain the number of viable cells/mL
Note: Cell counting can be performed in a variety of ways. Regardless of the approach chosen, we recommend selecting a method that allows to use a small volume of cells and can discriminate debris from viable nucleated cells.
Pause Point: Cells can be frozen at this step, if desired. We recommend freezing cells in Cryostor®CS10 or Recovery™ Cell Culture Freezing Medium following manufacturer's instructions.
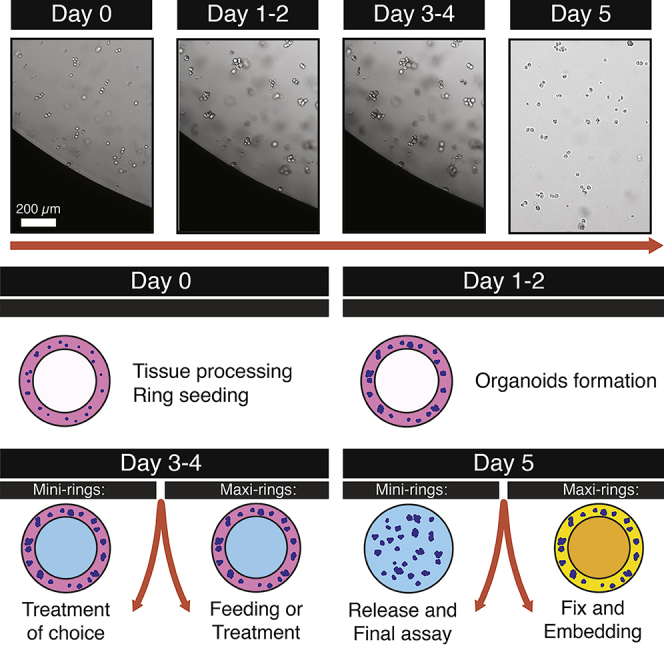
Seeding Mini-Rings for High-Throughput Screenings and Drug Discovery Studies
We establish mini-rings of Matrigel and tumor cells in 96-well plates for screening. For typical primary samples, we use 5,000 cells/well (range: 5,000–10,000). The same procedure can be used with cell lines as well. For cell lines, we recommend seeding 1,000–5,000 cells/well. Refer to Video S1 for a visual example.
-
26.
Firstly, determine how many wells you wish to generate. For each well, you need 10 μL of cells in Matrigel at a ratio of 1:1.33 (cell suspension to Matrigel). For example, if you want to generate 100 rings, you will need 500,000 cells to be resuspended in 430 μL of cold complete Mammocult medium and 570 μL of Matrigel (see below) for a total volume of 1 mL (10 μL/well).
Note: Always calculate in excess so as to generate about 30% more rings than the number you desire to plate. For example, if you want to generate 100 rings, prepare enough cell suspension in Matrigel to obtain 130.
-
27.
Prepare an aliquot of cells according to the calculation above
-
28.
Spin at 600 × g for 5 min
-
29.
Gently aspirate the supernatant without disturbing the pellet
-
30.
Add the calculated volume of cold Mammocult and thoroughly resuspend the cells by pipetting
-
31.
Add the calculated volume of Matrigel to the cells directly on ice and pipette multiple times to ensure homogenous mixing
-
32.
Vortex 3 times for 5 s at low rpm, placing the tube back on ice after each vortexing step
-
33.
Keep the mixture in a small beaker containing ice inside the biosafety cabinet and immediately proceed plating the mini-rings
-
34.
Using a P20 pipette with a low attachment 20 μL tip, pipette up and down a few times and aspirate 10 μL of the cell solution
-
35.
Keeping the tip at a 45 degree angle, place at the bottom of a 96-well plate and eject the mixture slowly while moving the tip in a circular motion around the rim. It should take ∼2 s to complete one ring.
Note: Mini-rings can be generated in any 96-well plate and do not require any specialized or functionalized support (Phan et al, 2019). Choose the 96-well plate on the basis of the assay that you wish to perform. For ATP release luminescence assay (see below), use white plates. Fluorescent-based assays or specific imaging approaches may be require black plates or glass-bottom plates.
-
36.
Once all rings are plated, quickly move the plate to a humidified 37°C incubator at 5% CO2 and incubate for a minimum of 30 min
-
37.
While the rings are solidifying, prepare a Masterblock media plate. Each well will need to be fed with 100 μL pre-warmed Mammocult. Thus, prepare a Masterblock by adding Mammocult to each well using a multi-well pipette or a microplate dispenser
Note: Add a 20% excess to ensure equal volume will be added to each well. For instance, if you have a single 96 mini-ring well plate, add 120 μL of Mammocult to each well.
Note: If mini-rings are only present in a subset of wells, add pre-warmed PBS to all empty wells.
-
38.
Cover the Masterblock media plate and leave in the incubator for a minimum of 30 min
-
39.
Once the mini-rings are solidified, add 100 μL of Mammocult from the Masterblock media plate to the center of each well
-
40.
Incubate in a humidified 37°C incubator at 5% CO2 for 3 days prior to media exchange or drug treatment (see below)
Note: To minimize uneven evaporation, add warm PBS in the space between wells.
Note: We use a 3 day incubation followed by two sequential drug treatments as described in Phan et al. (2019). However, duration of each step can be adjusted depending on tissue type, experiment type, etc.
Note: We strongly recommend using a 96-well automatic pipettor such as an Eppendorf EpMotion or an automated Beckman Biomek to add media as well as for all subsequent steps. If using an EpMotion, set the media dispensing speed to “1” to avoid disrupting the rings. Refer to Video S1 for an example.
Note: We recommend imaging each plate daily to visualize organoid establishment and growth. We use a Celigo S Image Cytometer in brightfield mode that allows for even illumination of the wells.
High-Throughput Drug Screening
Here, we present two alternatives for high-throughput screening. Steps 41–47 describe the procedure we follow using an EpMotion or equivalent liquid handler. Steps 48–52 include the procedure for using an automated liquid handler such as a Beckman Coulter Biomek.
After 3 days, proceed with drug treatment or medium exchange if you wish to maintain the organoids for longer times. In both cases, proceed to fully remove the medium and replace it as indicated below.
-
41.
At least 30 min prior to media exchange, prepare a Masterblock plate containing either fresh pre-warmed Mammocult or drugs of choice and incubate at 37°C
-
42.
Prepare a set of tips (300 μL, filtered) for each plate requiring a media exchange
-
43.
Retrieve the mini-ring plate and aspirate all medium by placing the tips at the bottom of the wells, in the empty center. Parameters: up “2”, volume “200 μL”
-
44.
Dispense the aspirated medium in a “waste plate”
-
45.
Repeat 43–44 until no medium is left in any of the wells and mini-rings are visible by visual inspection of the plate
-
46.
Aspirate 100 μL from the new Masterblock plate and dispense at the center of the mini-rings. Recommended parameters: up “2”, down “1”, volume “100 μL”
-
47.
Immediately place the mini-ring plate back in the incubator
Note: Repeat all steps 24 h after the first treatment for drug studies. For media exchange, replace media every 2–3 days.
Note: When performing high-throughput drug treatments, we recommend scattering negative control wells (vehicle) across the plate. Positive controls (in our case, Staurosporine at 1 μM and 10 μM) are placed in the corner wells.
Alternatives: If using an automated liquid hander such as a Beckman Coulter Biomek instrument, follow steps 48–52. The steps below are an alternative to steps 41–47.
For this type of automated liquid handler, we prepare compound library plates containing the desired drug stocks at 100x concentration in DMSO.
-
48.
At least 30 min prior to media exchange, prepare a dilution Masterblock plate containing Mammocult (120 μL/well) and incubate at 37°C
-
49.
Using filtered Beckman Coulter Biomek p50 tips, program the robot to first transfer 1.2 μL of drug at 100x concentration in DMSO from the library compound plate to the dilution Masterblock plate and thoroughly mix
-
50.
The robot then removes 100 μL of media from the mini-ring plate in two steps and disposes of it in a “waste plate”
-
51.
Lastly, program the robot to add 100 μL from the dilution Masterblock plate directly to the mini-ring plate in two steps
-
52.
Immediately place the mini-ring plate back in the incubator
Note: Repeat all steps 24 h after the first treatment for drug studies. For media exchange, replace media every 2–3 days.
Note: When performing high-throughput drug treatments, we recommend to scatter negative control wells (vehicle) and positive controls (in our case, Staurosporine at 1 μM and 10 μM) across the plate.
ATP-Release Assay
Several different assays can be performed to evaluate organoid growth and drug treatment effect (Phan et al, 2019). Here, we focus on the assay we employ more frequently, the ATP-release assay.
Note: Dispase will be added to each well of the mini-ring plate to help release the organoids from Matrigel prior to performing any assay. Before starting, calculate how much powder is needed taking into account that, while 50 μL/well of a 5 mg/mL solution are ultimately added to each mini-ring, a 50%–70% excess is needed to account for both the volume lost at the filtration step and the extra volume added to the dispase Masterblock plate. Once dissolved, keep the dispase tube or plate on ice at all times and use within 30 min. The enzyme can very rapidly precipitate.
Note: For this assay, you will need to prepare a pre-warmed PBS Masterblock plate, a cold dispase Masterblock plate, 2 sets of 300 μL epTIPS and one set of 50 μL epTIPS.
-
53.
Prepare a Masterblock plate by adding PBS (120 μL) to each well and warm up at 37°C for a minimum of 30 min before starting
-
54.
Prepare a fresh dispase solution immediately prior to the assay by dissolving dispase powder in cold, serum-free RPMI to a final concentration of 5 mg/mL
-
55.
Invert the tube or gently vortex to dissolve the powder without forming bubbles
-
56.
Once the powder is dissolved, filter using a 50 mL Millipore Steriflip
-
57.
Using a multi-well pipette or a microplate dispenser, add 75 μL to each well of a Masterblock plate
-
58.
Cover the plate and store on ice as you proceed to retrieve the mini-ring plate and PBS Masterblock plate from the incubator
-
59.
Using one set of 300 μL epTIPS, remove all media from the mini-ring plate by placing the tips at the bottom of the wells, in the empty center. Parameters: up “2”, down “5”, volume “200 μL”
-
60.
Dispense the aspirated medium in a “waste plate”
-
61.
Repeat 59–60 until no medium is left in any of the wells and mini-rings are visible by visual inspection of the plate
-
62.
Using a second set of 300 μL epTIPS, aspirate 100 μL from the PBS Masterblock plate and add it to the mini-ring plate to wash. Parameters are as follows: up “2”, down “1”, volume “100 μL”
-
63.
Using the same tips, aspirate all PBS following the steps in 59–61
-
64.
Lastly, using a set of 50 μL epTIPS, remove 50 μL of dispase from the dispase Masterblock plate and add it to the empty mini-ring plate. Parameters are as follows: up “2”, down “1”, volume “50 μL”
-
65.
Incubate at 37°C for 25 min
-
66.
After the incubation, shake for 5 min at 80 rpm using a plate shaker
-
67.
Using a microscope, confirm that the organoids are now in solution and released from the rings. If not, incubate for an additional 5 min, followed by shaking for 5 min as in #66
-
68.
Add 75 μL of CellTiter-Glo 3D Reagent to each well using a multi-well pipette and without generating any air bubbles
-
69.
Cover the plate with a MicroAmp Optical Adhesive film
-
70.
Shake for 5 min at 80rpm using a plate shaker in the dark
-
71.
Incubate for 20 min at room temperature in the dark
-
72.
Measure luminescence using a SpectraMax iD3. Parameters: shake for 5 min prior to reading, read all wavelengths, 500 ms integration time.
Note: While we report instructions on how to perform this protocol using an EpMotion instrument, the same process can be programmed on a Biomek or similar liquid handler.
Seeding Maxi-Rings for Histologic Characterization or Biochemical Analysis
We establish maxi-rings of Matrigel and tumor cells in 24 well plates for histologic characterization or downstream biochemical analysis. The procedure is similar to the mini-ring protocol, with minor modifications such as cells/well (100,000 vs 5,000 for mini-rings). For a visual guide, refer to Video S2 (generating and feeding maxi-rings).
-
73.
Firstly, determine how many wells you wish to generate and how much Mammocult and Matrigel is required. For each well, you need 70 μL of cells in Matrigel at a ratio of 1:1.33 (cell suspension to Matrigel). For example, if you want to generate 10 rings, you will need 1,000,000 cells to be resuspended in 300 μL of cold Mammocult medium and 400 μL of Matrigel (see below) for a total volume of 0.7 mL or 70 μL/well).
Note: Always calculate in excess so to generate about 30% more rings than the number you desire to plate.
-
74.
Prepare an aliquot of cells according to the calculation above
-
75.
Spin at 600 × g for 5 min
-
76.
Gently aspirate the supernatant without disturbing the pellet
-
77.
Add the calculated volume of cold Mammocult and thoroughly resuspend the cells by pipetting
-
78.
Add the calculated volume of Matrigel to the cells directly on ice and pipette multiple times to ensure homogenous mixing
-
79.
Vortex 3 times for 5 s at low rpm, placing the tube back on ice after each vortexing step
-
80.
Keep the mixture in a small beaker containing ice inside the biosafety cabinet and immediately proceed plating the maxi-rings
-
81.
Using a P200 pipette with a low attachment 300 μL tip, pipette up and down a few times and aspirate 70 μL of the cell solution
-
82.
Keeping the tip at a 45 degree angle, place at the bottom of a single well in a 24 well plate and eject the mixture slowly while moving the tip in a circular motion around the rim
-
83.
Once all rings are plated, quickly move the plate to a humidified 37°C incubator at 5% CO2 and incubate for a minimum of 45 min
-
84.
While the rings are solidifying, prepare a Falcon tube containing 1 mL of Mammocult for each of the maxi-rings seeded and incubate at 37°C for a minimum of 30 min
-
85.
After the rings are visibly solidified, add 1 mL of the pre-warmed Mammocult drop-by-drop directly to the center of the wells using a P1000
-
86.
Incubate the plate in a humidified 37°C incubator with 5% CO2, imaging every day using a Celigo S Image Cytometer
Note: Make sure to add the media to the center of the well and not to the side as when working with 2D cells or Matrigel drops in order to avoid disrupting the maxi-rings.
Media Exchange for Maxi-Rings
Organoids in maxi-rings should be fed after 3 days, and subsequently every 1–3 days for maintenance. For a visual guide, refer to Video S2 (generating and feeding maxi-rings).
-
87.
Make an aliquot of Mammocult (1 mL for each well) and put in the incubator at least 30 min to pre-warm
-
88.
Aspirate all the medium from the center of the well, paying attention not to aspirate or touch the ring
-
89.
Add 1 mL of the pre-warmed Mammocult drop-by-drop directly to the center of the wells using a P1000
-
90.
Incubate the plate in a humidified 37°C incubator with 5% CO2
Note: To apply a treatment, follow the same protocol and replace Mammocult with Mammocult + drug of choice
Sample Preparation for Histologic Analysis
Here, we focus on generating samples for histopathology. For a visual guide, refer to Video S3 (preparation of sample for embedding).
-
91.
First, pre-warm an aliquot of PBS (1 mL/well)
-
92.
Remove the maxi-ring plate from the incubator, and aspirate all the medium from the center of the well, paying attention not to aspirate or touch the ring
-
93.
Wash in PBS once, adding it to the center of the well and removing it without breaking the rings
-
94.
Add 500 μL of 10% buffered formalin to each well
-
95.
Incubate at 37°C for 5 min
-
96.
Incubate on ice for 30 min and then leave at 4°C at least 24 h
-
97.
The following day, transfer all organoids to a Falcon tube. Use a pipette tip to dislodge the rings and wash the wells in PBS to ensure that all cells are aspirated and transferred to the tube
-
98.
Spin the organoids for 5 min at 2,000 × g
-
99.
In the meantime, microwave Histogel for ∼5 s on low settings
-
100.
Aspirate as much liquid as possible without disturbing the pellet and discard formalin according to your institution’s regulations
-
101.
Spin the organoids for 5 min at 2,000 × g a second time and remove all remaining buffer
-
102.
Label a histology cassette using a pencil
-
103.
Add 4 μL of Histogel to the pellet, vortex and spin for a few seconds
-
104.
Let it solidify on ice for a few min
-
105.
Add 5 μL of Histogel to the cassette and let it solidify
-
106.
Use a spatula to place the mixture of cell and histogel on top of the Histogel bed already solidified in the cassette
-
107.
Pipet an additional 5 μL of Histogel on top of the cells to stabilize the small pellet
-
108.
Close the cassette, wrap it in parafilm and leave it on ice for 2–3 min
-
109.
Unwrap the parafilm and place the cassette in a beaker containing 70% EtOH
-
110.
Proceed with standard embedding, sectioning and staining
Expected Outcomes
-
•
Tissue processing. Tumors are typically fully dissolved within 2–24 h and yield viable cells. After red blood cell lysis, the cell pellet should appear white and not red or pink color. Viability tends to correlate with pathology findings; i.e. tumors showing 60%–99% necrosis will result in low number of viable cells to be used for establishing rings.
-
•
Maxi- and mini-rings. Rings should appear homogenous in terms of volume, shape, and number of cells. Neither broken rings nor bubbles should be present. Any defective mini-ring identified by imaging QC should be marked and omitted for downstream analysis. ATP results should fall within the dynamic range of the assay and correlate with cell viability. Replicates across the plates are expected to result in similar readout values.
Limitations
The quality of mini-rings is dependent on the operator preparing the plate. A skilled operator can learn to prepare good quality, homogenous mini-rings in a few days. We recommend practicing ahead of time to master the technique before starting the experiment.
Troubleshooting
Problem 1
Liquid remaining in mini-ring wells (steps 43–45, 59–61)
During drugs treatment or ATP assay steps, it is necessary to aspirate all media from each well, which is important to obtain consistent and accurate quantitative results. We noticed that, due to the plate construction and unevenness, wells in the center of 96-well plates tend to have liquid remaining when using an automated pipettor.
Potential Solution
When using an EpMotion, try to apply more pressure to make sure that the tips can reach the center wells, then gently release the plate holder. Visually inspect the plate and repeat until all liquid is gone.
Problem 2
Open/partial rings (steps 35 and 82)
It is possible, particularly when learning the technique, that mini- or maxi-rings are left open, resulting in a nick or gap in the ring.
Potential Solution
When plating a mini or maxi-ring, always make sure to close the ring by completing a full circle around the well while releasing the cell/Matrigel mixture. If all the mixture is released before you complete the circle, hold the pipette piston on the first resistance and complete the ring. That is sufficient to close a ring and the mixture will redistribute more evenly.
Problem 3
Air bubbles in rings (steps 35 and 82)
Bubbles in rings can make the rings inhomogeneous and unstable and even result in catastrophic breaks of maxi-rings.
Potential Solution
While preparing the cell/Matrigel mixture, follow the instructions paying attention not to introduce air bubbles. Similarly, always stop pipetting when encountering the first resistance of the pipet, even if not all Matrigel is released. For mini-rings: mark and discard mini-rings with air bubbles. For maxi-rings: try to aspirate the bubble to remove it. If not possible, one can aspirate the whole ring and attempt to re-plate it into clean well.
Acknowledgments
We acknowledge all members of the Soragni lab for their contributions to the optimization of these protocols and for helpful comments. This work was supported by the NIH (R01CA240718 to A.S.), Worldwide Cancer Research (#16-0253 to A.S.), and an Iris Cantor-UCLA Executive Advisory Board/CTSI Pilot Award (to A.S.).
Author Contributions
H.T.L.N. optimized the protocol and wrote the manuscript. A.S. conceived the protocol and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100056.
References
- Phan N., Hong J.J., Tofig B., Mapua M., Elashoff D., Moatamed N., Huang J., Memarzadeh S., Damoiseaux R., Soragni A. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun. Biol. 2019;2:78. doi: 10.1038/s42003-019-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.