

Abstract
Clinical application of drug cocktails for cancer therapy is limited by their severe systemic toxicity. To solve a catch-22 dilemma between safety and efficacy for drug cocktails, a hetero-targeted nano-cocktail (PPPDMA) with traceless linkers has been developed. In the PPPDMA nanogel, a hetero-targeting strategy is employed to improve its tumor selective targeting efficacy by overcoming the cancer cell mono-ligand density limitation. Benefit from its glutathione and reactive oxygen species responsiveness, the loaded paclitaxel and doxorubicin can be quickly and tracelessly released into the cytoplasm in their original form, which bestows PPPDMA nanogels the capability to overwhelm the processing capacity of cancer cell’s P-glycoprotein efflux pump allows, and ultimately kill them without inducing side effects. The PPPDMA treatment reduced its tumor burden over 99% (in tumor weight) and 96% (in tumor number). Most importantly, no detectable tumor in more than half of the PPPDMA treated mice. We conclude that traceless linker and hetero-targeted nano-cocktail strategy could be a safe and effective approach for cancer treatment.
Keywords: Nano-cocktail, traceless linker, hetero-targeted, dual responsive, cancer
Graphical Abstract
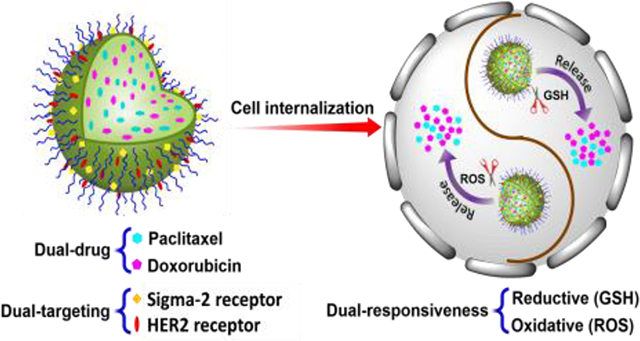
Hetero-targeted Tracelly-releasing Nano-cocktail, PPPDMA, is the integration of hetero-targeting, cancer cell environment responsive release, traceless linking, and drug cocktail concepts. PPPDMA synergistically enhances the anticancer efficacy of the nanogel while eliminating the systemic toxicity of the combination of doxorubicin and paclitaxel. Consequently, PPPDMA effectively eradicate the tumor in a metastatic ovarian cancer model.
1. Introduction
Due to the heterogeneous nature of tumors and the limitation of anticancer drugs, a mono-compound treatment usually can only kill a fraction of cancer cells while sparing non-responsive ones, which can continue proliferating and result in the recurrence of cancer.[1] To address the inadequacy of a single drug, a drug combination, or so-called “drug cocktail”, comprising multiple anticancer drugs of different anticancer mechanisms has been proposed and explored for the treatment of cancer.[2] In spite of many promising in vitro and in vivo results achieved with drug cocktails in cancer animal models, translation of the drug cocktail concept into a successful cancer treatment remains a challenge due to the lack of a safe and effective delivery system.[3]
Our previous research designed a two-drug cocktail nanogel system by encapsulating drugs through their hydrophobic interaction with a pH and redox potential dual responsive poly[(2-(pyridin-2-yldisulfanyl)ethyl acrylate)-co-[poly(ethylene glycol)]] (PDA-PEG) polymer.[4] It was found that the release of the encapsulated drugs during circulation can be reduced while significantly boosting its discharge in acidic pH or reducing environments. In the meantime, the nano-cocktail exhibits a synergistic effect in killing cancer cells. Although the nanogel based system could attenuate drug burst release, there were still about 20% of drug discharging before it reaches its targeted tissue,[4] which could potentially deteriorate the health condition of an already compromised cancer patient. Besides that, the ratio between the two compounds in the nano-cocktail could not be freely tuned according to their required potencies due to the constraint of their relative hydrophobicity.
To further minimize premature-release induced side effects, polymer-drug conjugate based nanoparticles, in which drug molecules are linked with a polymer carrier via covalent bonds, including hydrazone bonds,[5] ester bonds,[6] amide bonds,[7] and disulfide bonds,[8] have attracted a lot of attention. Since the loaded drug can only be released when the linkage is cleaved, the systemic toxicity of the drug can be significantly reduced. Before its conjugation onto a polymer, most drugs have to be modified first to introduce a functional group. Consequently, after the break of the linkage, the liberated molecules from the polymer are not in their original therapeutic form, which results in significantly diminished potency.[8a, 8b] Thus, the theoretical advantages of drug cocktails are locked in a catch-22 dilemma between safety and efficacy.
Herein, we designed an ErbB2 receptor and sigma-2 receptor hetero-targeted dual-responsive nano-cocktail (PPPDMA) to deliver both paclitaxel (PTX) and doxorubicin (DOX) into tumor tissue with a cancer cell responsive, traceless linking strategy. Once PPPDMA enters cancer cells through the multivalent effect of its hetero-ligands, the conjugated PTX and DOX will be tracelessly released upon the high intracellular redox and reactive oxygen species (ROS) levels, and subsequently eradicate the cancer cells (Figure 1a,b).
Figure 1.

The scheme for the fabrication of the nanogels and property characterization. (a) Fabrication scheme of PPP, PPPM, PPD, PPPD, and PPPDM nanogels, and AEB-conjugated PPDA, PPPDA, and PPPDMA nanogels. A polymer or polymer mixture self-assembles in aqueous media and further crosslinks through disulfide bonds to form polymer nanogels, which could be further decorated with AEB. (b) Schematic illustration of the dual-stimuli-responsive drug release in cancer cells. After the nanogels are delivered into cancer cells through the sigma 2 and/or HER-2 facilitated self-delivery, on-demand drug release will be triggered by the elevated GSH and ROS within cancer cells. (c) TEM image and the size distribution spectrum acquired by DLS (insert) of PPPDMA nanogels. Scale bar is 100 nm. (d) Drug release profiles of PTX from PPP nanogels. (E) Drug release profiles of DOX from PPD nanogels. Schematic illustration of the bioactivatable self-quenched nanogel for targeted photodynamic therapy.
2. Results and Discussion
2.1. Nanogel preparation and characterization
PTX and DOX were conjugated onto PDA-PEG polymer through the traceless linkers as described in Scheme S1–2 with the help of disulfide bonds. Our pilot study found that PTX:DOX at the ratio of 1:19 produces a synergistic effect in killing cancer cells. Thus, the nano-cocktail containing both PTX and DOX (PPPD) at that ratio was fabricated by dissolving PDA-PEG-PTX and PDA-PEG-DOX polymers in dimethyl oxide (DMSO) and dialyzing against water. The formation of nanogel structure was triggered by the addition of tris(2-carboxyethyl)phosphine (TCEP) as reported.[4] TEM revealed that the size of the PPPDMA is about 100 nm with a spherical shape, which matches well with the size of 114.9 nm achieved through dynamic light scattering (DLS) (Figure 1c). The loading content for PTX and DOX were 2.3% and 27.6%, respectively. It was found that the surface charge of the nanogels is −18.5 mV, suggesting good stability during blood circulation.[9]
Since most anti-cancer drugs show tempered therapeutic efficacy post modification, it is crucial to ensure that the conjugated drug can be released in its original form at the site of action. To achieve that, we introduced the traceless release design and expected the conjugated PTX and DOX to be released through the scheme as described in (Scheme S3). The abundance of ROS and glutathione (GSH) are two hallmarks of cancer cells.[10] An elevated level of ROS and GSH promotes tumor progression and metastasis.[10–11] HPLC confirmed that both free PTX and the released compound from the nanogel under reducing or elevated ROS level environments have the same elution time (Figure S8). Furthermore, the signal peak of PTX was also confirmed by LC-MS (Figure S9) in the released product. Similar results were also observed for the released DOX under reducing or elevated ROS level environments (Figure S10 and S11). Thus, we validated the concept that the drug loaded nanogel can tracelessly release its payloads in a cancer cell specific environment.
2.2. Nanogel size stability and drug release kinetics
To evaluate the physical stability of the nanogels, the size of the nanogel was monitored in different buffers. Figure S12 shows that the size of the PPPDMA nanogel remained nearly constant over eight days in PBS or a serum-containing medium, suggesting the excellent biocompatibility of PPPD with blood components during circulation, which may be attributed to its negative surface charge.
To investigate the stability of the drug-loaded nanogel during blood circulation and inside cancer cells, a drug release kinetics study was employed. Figure 1d shows that only trace amounts (< 5% ) of PTX was released in pH 5.0, pH 7.4, or pH 7.4 environment supplemented with 10% serum protein over 36 h. Interestingly, the addition of 10 mM of GSH or H2O2 triggered more than 70% and 55% of drug release within 12 h, respectively. A very similar release pattern was also observed for DOX in Figure 1e. Furthermore, after 24 h of incubation, only less than 5% and 10% of loaded PTX (Figure S13) and DOX (Figure S14), respectively, were released in 0.1 mM GSH supplemented media, a condition mimicking the circulating blood environment. The outstanding stability of the nanogel ensures that it will not release its payloads prematurely during blood circulation, while its responsiveness to GSH and elevated ROS bestows the capability of tracelessly dumping more PTX and DOX inside cancer cells than the processing capacity of the cell’s P-glycoprotein efflux pump allows, ultimately killing them.
2.3. Hetero-ligand effect on the cellular uptake and cell-killing efficacy of the nanogel
It has been reported that HER2, or so called ErbB2 receptors, and sigma 2 receptors are overexpressed in many cancers, including ovarian cancer, breast cancer, and head and neck cancer.[12] To facilitate the nanogels entering ovarian cancer cells, 4-methoxybenzamide (MBA) which targets sigma 2 receptors and Anti-ErbB2 affibody (AEB) which targets ErbB2 receptors were conjugated onto the surface of the nanogel separately. Cell proliferation assay found that the initial increase of ligand density could boost the cell-killing effect of the nanogel in SKOV-3 ovarian cancer cells. However, once the ligand density reached a certain extent, further increasing the ligand density could not yield additional potency, both for MBA and AEB ligands (Figure S18 and S19). We postulate that, when the ligand density of the nanogels is greater than or equals to the density of its corresponding receptor, the cellular uptake rate of the nanogel is limited by the receptors expressed on the cell surface. Thus, the further addition of a targeting ligand cannot yield enhanced efficacy. To break the receptor density limitation of a mono-ligand, a hetero-dual targeting strategy was adopted by conjugating both MBA and AEB onto the surface of the PPPD nanogel simultaneously.[13] The mass contents of MBA and AEB ligands in the PPPDMA nanogel are 1.8% and 0.6%, respectively. Confocal microscopy revealed that both MBA and Anti-ErbB2 affibody could boost the cellular uptake of PPPD nanogel. As expected, the dual-targeted nanogel, PPPDMA, yields higher DOX fluorescence signals inside treated cells (Figure 2a and Figure S20) than those treated with its mono-targeted counterparts. In contrast, neither MBA nor Anti-ErbB2 affibody showed a boosting effect for the cellular uptake of PPPD nanogel in normal cells, NIH-3T3 cells (Figure S21). Flow cytometry further confirmed that PPPDMA treatment results in the highest cellular uptake efficiency for PPPD nanogels (Figure 2b).
Figure 2.

The in vitro targeting effect of the PPPDMA nanogel. (a) Representative CLSM images of SKOV-3 cells after various treatments for 3h. Cell nuclei were stained with Hoechst 33342. The red fluorescence is from DOX, and the blue fluorescence is from Hoechst 33342. The scale bars are 20 μm. (b) Flow cytometry analysis of SKOV-3 cells after various treatments for 3h. (c) Cell viability of SKOV-3 cells after various treatments with free PTX, free DOX, a combination of PTX and DOX, and PPPD nanogels for 72 h. (d) Cell viability of SKOV-3 cells after various treatments with the combination of free PTX and DOX, PPPD, PPPDM, PPPDA, and PPPDMA nanogels for 72 h. Data represent the means ± SD, n=4.
To investigate whether the enhanced cellular uptake of PPPDMA could yield better cell-killing effect, a cell proliferation assay was employed. Figure 2c confirmed that the combination of free PTX and DOX is more potent than either PTX or DOX alone in killing SKOV-3 cells. Since non-targeted PPPD nanogel is less efficient in entering cancer cells (Figure 2b), the cell-killing efficacy is lower than that of the free drug combination (Figure 2c). With the help of targeting ligands, both mono-targeted nanogels, PPPDM and PPPDA, exhibited better cell-killing effect than non-targeted PPPD (Figure 2d). As expected, the PPPDMA displayed the highest potency in killing cancer cells (IC50 of 6.5 nM for PTX and 123.5 nM for DOX). Based on the results shown in Figure 2, we concluded that the hetero-ligand strategy facilitates PPPDMA entering cancer cells and boosts its potency in killing cancer cells.
2.4. Biodistribution and systemic toxicity of the PPPDMA nanogel
Encouraged by the exciting in vitro results above, we further investigated the tumor targeting and tumor growth inhibitory effect of PPPDMA in vivo according to the treatment schedule shown in Figure 3a. A mouse peritoneal metastatic ovarian tumor model was established following our previously reported method by inoculating luciferase-expressing SKOV-3 (SKOV-3/Luc) cells intraperitoneally to female nude mice.[14] Since the bioluminescence intensity is proportional to the population of SKOV-3/Luc cells in the tumor,[15] the size and distribution of tumors could be monitored by recording the intensity of bioluminescence emitted from the mice after the injection of luciferin. Besides serving as an active pharmaceutical ingredient, DOX in the nanogel can also be used as a fluorescent probe. Thus, the biodistribution of nanogel can be tracked with the help of a non-invasive IVIS imaging system. It was found that the fluorescence emission of DOX was detected mainly in livers, kidneys, and tumors of nanogel treated mice (Figure 3b), which suggests that the nanogels could take advantage of the so-called enhanced permeability and retention (EPR) effect to target tumor tissue selectively.[16] As expected, nanogels with MBA or AEB ligands were more effective in targeting tumor tissue as evidenced by stronger fluorescence signals. More importantly, the strongest fluorescence signal was detected in the tumor of the dual-targeted nanogel, PPPDMA, treated mice (Figure S22), indicating the success of our hetero-targeted strategy. Since PPPDMA nanogel is made of PDA-PEG polymer, the PEG segments will form a hydrophilic corona around the nanogel to prevent the detection of the immune system. The high tumor targeting efficiency of the PPPDMA nanogel in Figure 3b validated its protective effect.
Figure 3.

Treatment schedule, biodistribution, and systemic toxicity of the nanogels in a peritoneal metastatic ovarian tumor mouse model. (a) Schedule of treatment of the nanogels. (b) Ex vivo fluorescence images of mice organs and tumors after treatments with various nanogels. (c) Body weight changes over the duration of treatment. Mice in the PTX and DOX group died after receiving 2 weeks of treatment. (d) Representative histological features of the tissues after 6 weeks of nanogel treatment. Mice in the PTX and DOX group only received 2 weeks of treatment.
In the in vivo anticancer efficacy assay, tumor-bearing nude mice were intraperitoneally administrated with one of the following groups: control (PBS), PTX+DOX (the free drug combination of DOX and PTX in the formulation of Taxol), PPPD, PPPDM, PPPDA, and PPPDMA nanogels, at a combined dose of 1.0 mg/kg equivalent to PTX and 12 mg/kg equivalent to DOX, corresponding to a 1:19 molar ratio between PTX and DOX. Such a dose is higher than the reported combined maximum tolerated dose (MTD) of the two drugs (1.0 mg/kg PTX plus 1.5 mg/kg DOX) for nude mice.[17] It has been reported that a lower combined dosage of 5.0 mg/kg (PTX+DOX in total) could cause over 30% body weight loss to mice in 10 days because of the elevated toxic side effects of the drug cocktail.[18] As a result, mice in the free PTX+DOX treatment group exhibited drastic body weight loss (about 25%) in one week as shown in Figure 3c, and died within two weeks. On the contrary, no obvious body weight loss was observed in mice in other groups even after six weeks of treatment, which indicates that the formulation of nanogels could effectively circumvent the severe systemic toxicity of the free drug combination, attributed to the targeted delivery of the drug cocktail.
To further evaluate the potential systemic toxicity of the nano-cocktail, histological analysis of the liver, kidney, heart, and spleen were carried out. Compared with the control group, no noticeable structural difference was detected in the livers, kidneys, and spleens from other treatment groups (Figure 3d). However, the heart tissue from the animals treated with free drug combination (PTX + DOX) contains lots of empty space and disoriented structure, indicating the severe cardiotoxicity of the treatment.[19] On the contrary, none of the nano-cocktail treatments resulted in such effect, which confirmed that targeted, traceless release technology effectively attenuated the DOX-associated cardiotoxicity.
2.5. Tumor growth inhibitory effect of the PPPDMA nanogel
Aside from reducing the toxic side effects of the combined drugs, the drug-loaded nanogels also presented plausible tumor growth inhibition to varying degrees. As shown in Figure 4a, for mice treated with PBS in the control group, strong luminescence signals covered nearly the entire abdominal area of the mice after 6 weeks of treatment, indicating tumors had spread all over the abdomen of the mice. The rapid tumor growth was also quantitatively reflected by the dramatic increase in the luminescence intensity of mice shown in Figure 4b. However, contrary to the control group, the non-targeted PPPD nanogels treatment considerably delayed the tumor growth because the luminescence signals were detected only in about a quarter of the abdomen area of mice (Figure 4a), and the luminescence intensity was much weaker (Figure 4b), by which the enhanced anticancer effectiveness of the nano-cocktail was confirmed. The almost constant luminescence intensity throughout the whole treatment process in the PPPDM treated group proved its efficacy in controlling the growth of the tumor (Figures 4a and 4b). Furthermore, it was revealed that PPPDA was even more efficient in inhibiting tumor growth, as evidenced by the progressively reduced luminescence signals (Figure 4a and 4b). Most importantly, among all groups, the PPPDMA exhibited the highest potency in inhibiting the progression of tumors. In this group, after 6 weeks of treatment, mice emitted little to no luminescence signals. As presented in Figure 4a, a very faint bioluminescence signal was observed in 2 of the 5 PPPDMA treated mice, while no detectable bioluminescence signal was recorded in the other 3 mice, indicating the eradication of the disease. To quantify the efficacies of different treatments in more detail, tumors were harvested for analysis after the mice were sacrificed. As shown in Figure 4d, more than 10 solid tumors were collected from the abdominal cavity of mice in the control group. In contrast, much fewer tumors were found in mice treated with the PPPD nano-cocktail, and the tumor sizes were smaller than those in the control group, which were also quantitatively evidenced by the dramatically reduced tumor weight (Figure 4c) and tumor numbers (Figure 4d). As expected, all targeted nano-cocktail treatments further diminished the tumor burden both in tumor number and in tumor size. Specifically, compared with the control group, the hetero-targeted PPPDMA treatment reduced its tumor burden over 99% (in tumor weight) and 96% (in tumor number). Most importantly, only one tiny solid tumor was collected in 2 mice in the PPPDMA treatment group, while no detectable tumor in the other 3, suggesting that the original tumors were totally eradicated in those mice. These results are in good agreement with those obtained from bioluminescence analysis (Figure 4a), which collectively confirm the success of our dual-drug, dual-responsiveness, tracelless linkage, and hetero-targeting strategy in eradicating advanced ovarian cancer.
Figure 4.

Tumor growth inhibitory effect of nanogels. (a) Whole body bioluminescence images of mice before and after various treatments for 6 weeks. (b) The bioluminescent intensity of mice at predetermined weekly time points. The inset: enlargement corresponding to the first three weeks. (c) Tumor weight of mice after receiving different treatments for 6 weeks. (d) Tumor number of mice after receiving different treatments for 6 weeks. The insert: representative images of tumors harvested from sacrificed mice after receiving different treatments for 6 weeks. Data represent the means ± SD, n=5. *P < 0.05; **P < 0.01; and ***P < 0.001.
3. Conclusion
In summary, we developed a hetero-targeted, dual-responsive nano-cocktail system, PPPDMA nanogel, which is safe and effective in eradicating cancer in a metastatic ovarian tumor model. Due to the existence of covalent linkages between the polymer and the drugs, PPPDMA nanogel is free of premature release during bloodstream circulation. With the help of MBA and anti-ErbB2 affibody, PPPDMA can selectively enter tumor tissue and cancer cells. Upon the trigger of elevated redox potential and ROS in the cytoplasm of cancer cells, PPPDMA tracelessly releases it payloads, PTX and DOX. Consequently, the integration of the hetero-targeting, cancer cell environment responsive release, traceless linkage, and drug cocktail concepts synergistically enhances the anticancer efficacy of PPPDMA and cures ovarian cancer while eliminating the systemic toxicity of the drug cocktail. More importantly, due to the interchangeable nature of the payload and targeting ligands, our results pave the way for developing a safe and effective delivery platform for eradicating other cancers as well.
4. Experimental Section
Materials
2-Mercaptoethanol (BME), glacial acetic acid, acryloyl chloride, 3,3′-dithiodipropionic acid, 4-(dimethylamino)pyridine (DMAP), 4-methoxybenzoic acid, cystamine dihydrochloride, N-hydroxysuccinimide (NHS), N,N-diisopropylethylamine (DIPEA), poly(ethylene glycol) methacrylate (PEGMA, average Mn = 360 Da), poly(ethylene glycol) methyl ether methacrylate (PEGMMA, average Mn = 480 Da), 2,2′-azobis(2-methylpropionitrile) (AIBN), L-glutathione (GSH), hydrogen peroxide (30 wt.% in H2O), TWEEN®80, and phosphate buffered saline (PBS) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). 2,2’-Dipyridyl disulfide, triethylamine (TEA), N,N′-dicyclohexylcarbodiimide (DCC), DL-dithiothreitol (DTT), tris(2-carboxyethyl)phosphine (TCEP), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N,N′-disuccinimidyl carbonate (DSC), (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and silica gel (spherical, 100 μm) were purchased from Tokyo Chemical Industry Co., Ltd (Portland, OR, USA). Paclitaxel (PTX) and doxorubicin hydrochloride (DOX) were purchased from LC Laboratories (Woburn, MA, USA). Anti-ErbB2 affibody (AEB) was purchased from Abcam plc. (Cambridge, MA, USA). Cremophor EL® was purchased from EMD Millipore Corp. (Billerica, MA, USA). XenoLightTM D-luciferin potassium salt was purchased from PerkinElmer, Inc. (Waltham, MA, USA). Gibco™ Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin (PS), trypsin-EDTA, and Invitrogen™ Hoechst 33342 were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Deuterated solvents chloroform and dimethyl sulfoxide were purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA). All the other solvents used in this research were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA) and directly used without further purification. Thiolated paclitaxel, PTX-SH, was synthesized with commercial PTX as the starting material following a reported two-step procedure.[20] The PDSA monomer, 2-(pyridin-2-yldisulfanyl) ethyl acrylate (PDSA), was prepared for the polymerization reactions as previously described in literature.[4]
Synthesis of MBA-SS-MBA
In a 25 mL round-bottom flask equipped with a magnetic stirring bar, 4-methoxybenzoic acid (456 mg, 3.0 mmol) was dissolved in 10 mL methanol, followed by the addition of DIPEA (1.74 mL, 10 mmol), NHS (230 mg, 2.0 mmol), and EDC (575 mg, 3.0 mmol) in order. After stirring at room temperature for 1 h, cystamine dihydrochloride (225 mg, 1.0 mmol) was added. The reaction mixture was kept stirring at room temperature for 24 h and then the solvent was removed under reduced pressure. The resulted residue was extracted with dichloromethane (DCM) and H2O and washed three times with brine. The organic layer was dried over Na2SO4, then filtered and condensed in vacuum. The crude product was separated by flash column chromatography with silica gel (100 μm) using the gradient elution solvents of hexane and ethyl acetate (70/30) to yield MBA-SS-MBA as a white solid (320 mg, 76%). The molecular structure of the product was confirmed by mass spectrometry (MS) and nuclear magnetic resonance spectroscopy (NMR).
Synthesis of MBA-SH
A mixture of MBA-SS-MBA (210 mg, 0.5 mmol) and DTT (154 mg, 1.0 mmol) was dissolved in 10 mL DMF in a 25 mL round-bottom flask equipped with a magnetic stirring bar. After stirring at room temperature for 1 h, TEA (0.7 mL, 5 mmol) was added to the reaction mixture and stirred at room temperature overnight. The reaction mixture was condensed under vacuum, and then extracted with DCM/H2O and washed three times with brine. The organic phase was dried by anhydrous Na2SO4, filtered, and then evaporated in vacuo to yield the targeted compound as a white solid (171 mg, 81%). The chemical structure of MBA-SH was confirmed by LC-MS and 1H NMR results.
Synthesis of PDA-PEG1 and PDA-PEG2
The polymers, PDA-PEG1 and PDA-PEG2, were synthesized through free radical polymerization as reported previously.2 In brief, 482 mg (2.0 mmol) PDSA and 2.0 mmol PEG derivatives (720 mg PEGMA for PDA-PEG1, 960 mg PEGMMA for PDA-PEG2) were dissolved into 10 mL anisole in a 50 mL round-bottom flask and degassed with nitrogen for 30 min at room temperature. After that, a degassed solution of AIBN (32.8 mg, 0.2 mmol) in 1 mL anisole was added dropwise into the reaction mixture. Then the flask was immersed in an oil bath maintained at 65 °C and stirred for 24 h in the dark. Following the reactions, the resulted polymers were collected by precipitation with ice-cold diethyl ether. For further purification, the collected polymers were dissolved in DCM and then precipitated with ice-cold diethyl ether for three times. The purified polymers were dried under vacuum in the dark until the solvents were completely removed. The structural composition of PDA-PEG1 and PDA-PEG2 was analyzed by NMR using CDCl3 as the solvent. The yields of PDA-PEG1 and PDA-PEG2 are 81% and 78%, respectively. The average molecular weight of the polymers was determined by gel permeation chromatography (GPC).
Synthesis of PDA-PEG-PTX
The polymer PDA-PEG-PTX was synthesized through thiol-disulfide exchange reaction between the thiol group of PTX-SH and the disulfide bond of PDA-PEG1. Typically, 18 mg PDA-PEG1 was dissolved in 500 μL DMSO, followed by dropwise addition of 14 mg PTX-SH in 200 μL DMSO. The reaction mixture was stirred at room temperature overnight in the dark and then dialyzed towards DMSO using Spectra/Por® dialysis tube (MWCO: 8 kDa) to get rid of unreacted PTX-SH. The final product was obtained from precipitation with ice-cold diethyl ether. Then the polymer was dissolved into DCM, precipitated with ice-cold diethyl ether twice to eliminate DMSO residue, and dried under vacuum in the dark for 48 h (30 mg, 94%). The chemical structure of PDA-PEG-PTX was confirmed by 1H NMR and the content of PTX in PDA-PEG-PTX was measured with high performance liquid chromatography (HPLC).
Synthesis of PDA-PEG-MBA
PDA-PEG-MBA was synthesized in the same way as the synthesis of PDA-PEG-PTX. 3.2 mg MBA-SH in 200 μL DMSO was added to 500 μL DMSO solution of 18 mg PDA-PEG1 drop by drop. The resulted solution was kept at room temperature in the dark overnight, followed by dialysis against DMSO with Spectra/Por® dialysis tube (MWCO: 1 kDa) to remove unreacted MBA-SH. The desired polymer was collected from precipitation with ice-cold diethyl ether and further purified by precipitating from DCM solution with ice-cold diethyl ether twice. After being dried in vacuo for 48 h in the dark (19.5 mg, 92%), the polymer PDA-PEG-MBA was analyzed with 1H NMR to confirm its chemical structure, and its content of MBA was determined by HPLC.
Synthesis of PDA-PEG-BME
The intermediate polymer PDA-PEG-BME was prepared via a thiol-disulfide exchange reaction between 2-mercaptoethanol (BME) and polymer PDA-PEG2. PDA-PEG2 (22 mg) was dissolved in 500 μL DCM with a catalytic amount of glacial acetic acid. While vigorously stirring, 1.6 mg BME in 200 μL DCM was added dropwise. The reaction was kept stirring overnight in the dark at room temperature. Then the targeting product was precipitated with ice-cold diethyl ether and further purified through precipitation with DCM and ice-cold diethyl ether for two more times. The polymer PDA-PEG-BME was dried under vacuum in the dark for 48 h (22 mg, 93%), and its structural composition was confirmed by 1H NMR.
Synthesis of PDA-PEG-DSC
The free hydroxyl group of PDA-PEG-BME was replaced by N-succinimidyl carbonate group to produce PDA-PEG-DSC. PDA-PEG-BME (20 mg) and 7.7 mg N,N′-disuccinimidyl carbonate (DSC) were dissolved in 500 μL DMSO. Then 8.4 μL TEA was added, and the reaction solution was stirred overnight at room temperature in the dark. The unconjugated DSC was removed by dialyzing the reaction mixture towards DMSO using Spectra/Por® dialysis tube (MWCO: 8 kDa). The desired product PDA-PEG-DSC was gathered by precipitation with ice-cold diethyl ether. Further removal of DMSO residue was performed twice via DCM/ice-cold diethyl ether precipitation. After in vacuo dryness in the dark for 48 h (20 mg, 83%), the polymer was analyzed by 1H NMR to verify its chemical structure.
Synthesis of PDA-PEG-DOX
The anticancer drug DOX was conjugated to the polymer through a reaction between the newly introduced succinimidyl group of PDA-PEG-DSC and the amino group of DOX. To a solution of 20 mg PDA-PEG-DSC in 500 μL DMSO, 16.3 mg DOX was added, followed by the addition of 10 μL TEA. The resulting reaction mixture was kept in the dark, stirring at room temperature for 24 h. Spectra/Por® dialysis tube (MWCO: 8 kDa) was utilized to remove unreacted free DOX through dialysis against DMSO. Then the purified polymer solution was dialyzed towards deionized water to exchange DMSO into water. The final polymer PDA-PEG-DOX was collected through lyophilization in the dark (25 mg, 96%). The molecular structure of PDA-PEG-DOX was confirmed by 1H NMR and the content of DOX in PDA-PEG-DOX was determined with HPLC.
Fabrication of nanogels
Polymer nanogels were prepared via TCEP-induced crosslinking method according to our previous study.[21] Generally, 0.3 mg TCEP in 20 μL DMSO was added to a solution of 10 mg polymer (or a mixture of different polymers) in 0.5 mL DMSO upon vigorous stirring. After equilibration for 15 min, the resulted mixture was dropped into 5 mL deionized water under robust stirring and maintained stirring overnight. Following that, the polymer nanogels were dialyzed in Spectra/Por® dialysis tube (MWCO: 8 kDa) against PBS (pH 7.4) for 24 h to eliminate small molecules. At last, the nanogels were filtered through 0.45 μm syringe filters and stored at 4 °C. The particle size, size distribution (polydispersity index, PDI), and surface charge of the nanogels were determined by dynamic light scattering (DLS) and zeta potential measurement, recorded on Zetasizer (Zetasizer Nano ZS, Malvern Instruments Ltd, Malvern, UK). The morphology of polymer nanogels was observed using Hitachi HT7800 transmission electron microscopy (TEM, Hitachi High-Technologies Corporation, Tokyo, Japan). The amount of drugs contained in the nanogels was analyzed with HPLC. For the preparation of a series of MBA functionalized PPP nanogel, PPPM (PPPM1, PPPM2, PPPM3, and PPPM4), mixtures of different ratios of PDA-PEG-PTX:PDA-PEG-MBA (w/w) at 9:1, 8:2, 6:4, and 5:5 were used, respectively. The size distribution (Dp and PDI), and zeta potential data of all the PPPM series of polymer nanogels are summarized in Table S1.
Conjugation of AEB
The tumor-targeting Anti-ErbB2 affibody (AEB), a thiol-containing affibody, was conjugated to polymer nanogels by thiol-disulfide exchange reaction. The above-prepared PPD or PPPD nanogels dispersed in PBS (pH 7.4) were mixed with different amount of AEB dissolved in PBS (pH 7.4) to prepare AEB-conjugated PPDA or PPPDA nanogels, respectively. The resulted reaction mixtures were stirred at 4 °C overnight. The conjugation of AEB was monitored by measuring the release of 2-pyridinethione with UV-Vis spectrophotometer (DU 650 Spectrophotometer, Beckman Coulter, USA) at 375 nm (molar absorption coefficient: 8080 M−1 cm−1). The unconjugated free AEB, which has a molecular weight of 6 kDa, was removed from the nanogel system by centrifuging with Amicon® ultra centrifugal filter (MWCO: 30 kDa). The AEB-decorated nanogels were collected and dispersed into PBS (pH 7.4) and then stored at 4 °C for future use. For the preparation of a series of AEB affibody functionalized PPD nanogel, PPDA (PPDA1, PPDA2, PPDA3, and PPDA4), mixtures of different ratios of PPD:AEB (w/w) at 640:1, 320:1, 160:1, and 100:1 were used, respectively. The size distribution (Dp and PDI), and zeta potential data of all the PPDA series of polymer nanogels are summarized in Table S3. The ratio between PDA-PEG-PTX plus PDA-PEG-DOX and PDA-PEG-MBA was 8:2 by weight in PPPDM and PPPDMA, and the ratios (w/w) of PPPD and PPPDM to AEB were 160:1 in PPPDA and PPPDMA, respectively.
Drug release
The release profiles of PTX and DOX from PPP and PPD nanogels were investigated at 37 °C with different buffer solutions (containing 1% TWEEN®80, m/v) as release media, including acetate buffer (pH 5.0), PBS buffer (pH 7.4), PBS with 10% FBS (pH 7.4), PBS with 10 mM GSH (pH 7.4), and PBS with 10 mM H2O2 (pH 7.4). Nanogels (equivalent to 200 nmol PTX/DOX) suspended in certain release medium (without TWEEN®80) were instantly sealed into Spectra/Por® dialysis tube (MWCO: 8 kDa) and immediately immersed into 100 mL pre-warmed respective release medium. At predetermined time points, 10 mL release medium outside of the dialysis tube was sampled and same amount of fresh release medium was replaced to make the total volume of the release system constant. The sample was lyophilized and then dissolved into acetonitrile for HPLC analysis.
Serum stability
The hydrodynamic stability of the nanogels were monitored by DLS. Nanogels were first dispersed in PBS buffer (pH 7.4) and PBS buffer with 10% FBS (pH 7.4), and then incubated at 37 °C for up to 8 days. The size of the nanogels were determined with DLS at predetermined time points.
Cell culture
Human ovarian cancer cells, SKOV-3 and SKOV-3/Luc cells, were cultured in Gibco™ DMEM supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37 °C in 75 mL culture flasks under a humidified atmosphere of 5% CO2. Cells were sub-cultured when the cell confluence reached ~80%.
Cytotoxicity assay
The anticancer activities of polymer nanogels against SKOV-3 cells were evaluated by MTT assay. Cells were seeded in 96-well plates at a density of 5,000 cells/well for 24 h prior to the test at 37 °C with 5% CO2. Then cells were treated with varying concentrations of PTX/DOX or polymer nanogels in fresh medium and further incubated for 72 h. In the control group, cells were allowed to grow without any treatment. After that, the medium was replaced with 100 μL fresh medium containing MTT reagent (final concentration 1 mg/mL) and cells were further incubated for 4 h. The purple MTT crystal was dissolved with 100 μL MTT stop solution and the optical density at 595 nm was recorded on a microplate reader (ELX808, Bio-Tech Instrument, Inc.).
Confocal microscopy
The cellular uptake of poly-nano-prodrugs was qualitatively examined by confocal laser scanning microscopy (CLSM) with SKOV-3 cells. Cells were seeded in 35 mm2 Petri dish with a glass window at a density of 200,000 cells/dish for 24 h at 37 °C with 5% CO2. Then cells were washed with PBS (pH 7.4) and incubated with free DOX or DOX-contained prodrugs with equivalent concentration of 2 μM DOX for 3h. Cells without any incubation were utilized as control. All cells were subsequently washed three times with PBS and fixed with paraformaldehyde (4% in PBS) for 10 min at room temperature. Cells were washed with PBS again for three times after the removal of paraformaldehyde, and the nuclei of cells were stained with Hoechst 33342 (final concentration 1μg/mL) for 10 min. At last, cells were washed three times with PBS and then imaged under a confocal microscope (LSM 700, Carl-Zeiss Inc.).
Flow Cytometry
The uptake of prodrugs by SKOV-3 cells was further quantitatively determined by flow cytometry. Cells were seeded in 6-well plates at a density of 300,000 cells/well for 24 h at 37 °C with 5% CO2. Then cells were washed with PBS (pH 7.4) and incubated with free DOX or DOX-contained nanogel with an equivalent concentration of 1 μM DOX for 3h. Cells with no incubation were utilized as a control. After that, cells were washed with PBS, trypsinized with trypsin-EDTA, and collected through centrifuging (2000 rpm, 3 min). Cells were suspended into PBS and then centrifuged for two more times. Finally, collected cells were re-suspended into PBS for analysis. Intracellular fluorescence intensity was quantified by flow cytometer (BD Accuri C6, BD Biosciences) at λex 488 and λem 560 nm.
Animal model
All animal experiments were conducted in accordance with NIH regulations and approved by the Institutional Animal Care and Use Committee of the University of South Carolina. A mouse model of intraperitoneal (IP) ovarian metastatic tumor was established as described in our previous study.[22] In brief, SKOV-3/Luc cells, a luciferase-expressing cell line derived from SKOV-3, were cultured in DMEM culture medium. 2,000,000 cells were suspended in 200 μL DMEM and intraperitoneally injected to a female nude mouse (8–10 week old, ~20 g, Jackson Laboratories). The tumor burdens were monitored with a whole body imaging system weekly and the animals were observed for body weight change and signs of pain every other day throughout the duration of experiments.
Whole-body imaging
The tumor growth of mice was monitored using a noninvasive IVIS Lumina III whole-body imaging system (PerkinElmer Inc., Waltham, USA). The tumor-bearing nude mice were first anesthetized by 2% isoflurane and then injected intraperitoneally with 200 μL D-luciferin (15 mg/mL in PBS). After approximately 15 min, the whole body of mice was imaged to record the bioluminescence emitted from SKOV-3/Luc tumors. The imaging time was optimized and all images were collected under identical system settings. The images were analyzed by Living Image® software and the intensity of recorded bioluminescence signals was quantified in radiance.
In vivo biodistribution
Three weeks after the inoculation of SKOV-3/Luc cells, the tumor-bearing mice were administered with various nanogels by intraperitoneal injection at a dose of 1 mg/kg equivalent to DOX. PBS (pH 7.4) was used as control. Mice were sacrificed after 6 h post-injection, and the organs and tumors were collected for imaging. The fluorescence was recorded ex vivo with the IVIS Lumina III whole body imaging system.
Anti-tumor efficacy
After inoculation with SKOV-3/Luc cells for two weeks, the tumor-bearing mice were randomly assigned into six groups (n=5 for each group) and were intraperitoneally administrated with PTX+DOX or nanogels at a dose of 1 mg/kg equivalent to PTX and 12 mg/kg equivalent to DOX. Free PTX was administrated in the formulation of Taxol. PBS (pH 7.4) was used as control. Mice were marked and weighed prior to treatment. The treatments were given to mice once per week. After six weeks, all mice were sacrificed to harvest the organs and tumors for further analysis.
Histological analysis
The collected organs (heart, liver, spleen, and kidney) were fixed in 4% paraformaldehyde solution. Then the fixed organs and tumor tissues were embedded in optimal cutting temperature (OCT) gel, sectioned into ~10 μm, stained with hematoxylin and eosin (H&E), and analyzed under a light microscope. The histology was performed in a blinded fashion by professional personnel at the University of South Carolina.
Statistical analysis
All data were processed and demonstrated as means with standard deviations (mean ± SD). Student’s t-test was utilized to analyze statistical difference between parallel groups. P < 0.05 from a two-tailed test was considered statistically significant.
Supplementary Material
Acknowledgments
The authors want to thank National Institutes of Health (1R15CA188847-01A1 and 1R01AG054839-01A1) for financial support of the research. We also want to thank the University of South Carolina Center for Targeted Therapeutics Microscopy and Flow Cytometry Core Facility (National Institutes of Health 5P20GM109091) for technical support.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Masui K, Gini B, Wykosky J, Zanca C, Mischel PS, Furnari FB, Cavenee WK, Carcinogenesis 2013, 34, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Al-Eisawi Z, Beale P, Chan C, Yu J, Huq F, J. Ovarian Res. 2013, 6, 78; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Caraglia M, Marra M, Budillon A, Meo G, Ricciardiello F, Bismuto E, Brachelente G, Francini G, Giordano A, Correale P, Abbruzzese A, Cancer Biol. Ther. 2005, 4, 1159–1167; [DOI] [PubMed] [Google Scholar]; c) Grunberg SM, Dugan M, Muss H, Wood M, Burdette-Radoux S, Weisberg T, Siebel M, Support. Care Cancer 2009, 17, 589–594; [DOI] [PubMed] [Google Scholar]; d) Sato S, Itamochi H, Kigawa J, Oishi T, Shimada M, Naniwa J, Uegaki K, Nonaka M, Terakawa N, Cancer Sci. 2009, 100, 546–551; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wang H, Zhao Y, Wu Y, Hu Y.-l., Nan K, Nie G, Chen H, Biomaterials 2011, 32, 8281–8290. [DOI] [PubMed] [Google Scholar]
- [3].a) Tas F, Yildiz I, Kilic L, Ciftci R, Keskin S, Sen F, Am. J. Ther. 2013; [Google Scholar]; b) Qi WX, Fu S, Zhang Q, Guo XM, J. Chemother. 2015, 27, 181–187; [DOI] [PubMed] [Google Scholar]; c) Bayo J, Avino V, Toscano F, Jimenez F, Breast J. 2018, 24, 462–467; [DOI] [PubMed] [Google Scholar]; d) da Silva WC, de Araujo VE, Lima E, Dos Santos JBR, Silva M, Almeida P, de Assis Acurcio F, Godman B, Kurdi A, Cherchiglia ML, Andrade EIG, BioDrugs 2018, 32, 585–606; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xu R, Xu C, Liu C, Cui C, Zhu J, Onco. Targets Ther. 2018, 11, 8605–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bahadur K. C R, Xu P, Adv. Mater. 2012, 24, 6479–6483. [DOI] [PubMed] [Google Scholar]
- [5].Duan XP, Xiao JS, Yin Q, Zhang ZW, Yu HJ, Mao SR, Li YP, ACS Nano 2013, 7, 5858–5869. [DOI] [PubMed] [Google Scholar]
- [6].Cho JK, Chun C, Kuh HJ, Song SC, Eur. J. Pharm. Biopharm. 2012, 81, 582–590. [DOI] [PubMed] [Google Scholar]
- [7].Pinhassi RI, Assaraf YG, Farber S, Stark M, Ickowicz D, Drori S, Domb AJ, Livney YD, Biomacromolecules 2010, 11, 294–303. [DOI] [PubMed] [Google Scholar]
- [8].a) Remant BK, Chandrashekaran V, Cheng B, Chen H, Pena MM, Zhang J, Montgomery J, Xu P, Mol. Pharm. 2014, 11, 1897–1905; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou Z, Shen Y, Tang J, Fan M, Van Kirk EA, Murdoch WJ, Radosz M, Adv. Funct. Mater. 2009, 19, 3580–3589; [Google Scholar]; c) Meng FH, Hennink WE, Zhong Z, Biomaterials 2009, 30, 2180–2198. [DOI] [PubMed] [Google Scholar]
- [9].Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS, Biomaterials 2011, 32, 3435–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bansal A, Simon MC, J. Cell Biol. 2018, 217, 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Panieri E, Santoro MM, Cell Death Dis. 2016, 7, e2253; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gill JG, Piskounova E, Morrison SJ, Cold Spring Harb. Sym. 2016, 81, 163–175. [DOI] [PubMed] [Google Scholar]
- [12].a) Zeng C, Vangveravong S, McDunn JE, Hawkins WG, Mach RH, Br. J. Cancer 2013, 109, 2368–2377; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Peluso JJ, Steroids 2011, 76, 903–909; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Slamon D, Godolphin W, Jones L, Holt J, Wong S, Keith D, Levin W, Stuart S, Udove J, Ullrich A, a. et, Science 1989, 244, 707–712; [DOI] [PubMed] [Google Scholar]; d) Huang Y-S, Lu H-L, Zhang L-J, Wu Z, Med. Res. Rev. 2014, 34, 532–566. [DOI] [PubMed] [Google Scholar]
- [13].a) Yu B, Mao Y, Yuan Y, Yue C, Wang X, Mo X, Jarjoura D, Paulaitis ME, Lee RJ, Byrd JC, Lee LJ, Muthusamy N, Biomaterials 2013, 34, 6185–6193; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tang J, Zhang L, Liu Y, Zhang Q, Qin Y, Yin Y, Yuan W, Yang Y, Xie Y, Zhang Z, He Q, Int. J. Pharm. 2013, 454, 31–40; [DOI] [PubMed] [Google Scholar]; c) Kibria G, Hatakeyama H, Ohga N, Hida K, Harashima H, J. Control. Release 2011, 153, 141–148; [DOI] [PubMed] [Google Scholar]; d) Chen WH, Xu XD, Luo GF, Jia HZ, Lei Q, Cheng SX, Zhuo RX, Zhang XZ, Sci. Rep. 2013, 3, 3468; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Saul JM, Annapragada AV, Bellamkonda RV, J. Control. Release 2006, 114, 277–287. [DOI] [PubMed] [Google Scholar]
- [14].Cho EJ, Sun B, Doh K-O, Wilson EM, Torregrosa-Allen S, Elzey BD, Yeo Y, Biomaterials 2015, 37, 312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].He H, Markoutsa E, Li J, Xu P, Acta Biomater. 2018, 68, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Maeda H, Matsumura Y, Adv. Drug Deliv. Rev. 2010; [DOI] [PubMed] [Google Scholar]; b) Fang J, Nakamura H, Maeda H, Adv. Drug Deliv. Rev. 2010. [Google Scholar]
- [17].Ahmed F, Pakunlu RI, Brannan A, Bates F, Minko T, Discher DE, Control J Release 2006, 116, 150–158. [DOI] [PubMed] [Google Scholar]
- [18].Chen Y, Zhang W, Huang Y, Gao F, Sha X, Fang X, Int. J. Pharm. 2015, 488, 44–58. [DOI] [PubMed] [Google Scholar]
- [19].Cheng B, Gao F, Maissy E, Xu P, Acta Biomater. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zou J, Zhang FW, Zhang SY, Pollack SF, Elsabahy M, Fan JW, Wooley KL, Adv. Healthc. Mater. 2014, 3, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].He H, Cattran AW, Nguyen T, Nieminen A-L, Xu P, Biomaterials 2014, 35, 9546–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].He H, Markoutsa E, Li J, Xu P, Acta Biomater. 2018, 68, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.