

Abstract
The question as to why some hosts can eradicate their tumors whilst others succumb to tumor-progression remains unanswered. Here, I propose a provocative concept that intrinsic differences in the T cell receptor (TCR) repertoire of individuals may influence the outcome of anti-tumor immunity by affecting the frequency and/or variety of tumor-reactive CD8 and/or CD4 tumor-infiltrating lymphocytes. This idea implicates that the TCR repertoire in a given patient might not provide sufficiently different TCR clones that can recognize tumor antigens, namely, “a hole in the TCR repertoire” might exist. This idea may provide a novel perspective to further dissect the mechanisms underlying heterogeneous anti-tumor immune responses in different hosts. Besides tumor-intrinsic heterogeneity and host microbiome, I also discuss the various factors that may constantly shape the dynamic TCR repertoire. Elucidating mechanistic differences in different individuals’ immune systems will allow us to better harness immune system to design new personalized cancer immunotherapy.
Keywords: heterogeneity of anti-tumor immunity, cancer immunology, TCR repertoire, tumor immunogenicity
Graphical Abstract:
Underlying mechanisms that dictate the immunological heterogeneity in cancers remain elusive. Besides tumor-intrinsic heterogeneity and environmental factors, intrinsic differences in the T-cell receptor (TCR) repertoire may play a role. “A hole in the TCR repertoire” hypothesis is proposed to explain heterogeneity of anti-tumor responses that may significantly impact cancer immunotherapy.
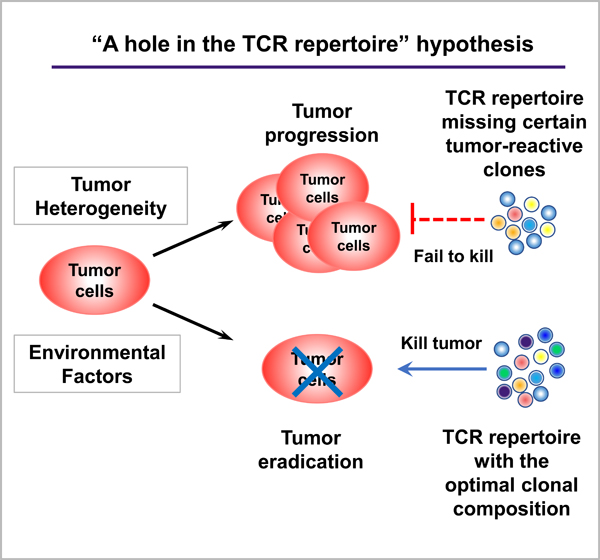
Introduction
Tumor heterogeneity presents a huge challenge when it comes to treating cancer patients. Intertumoral heterogeneity is frequently observed between patients who often do not respond to the same treatments. Even within the same tumor and the same patient, subclones with different mutations exist [1], so called intratumoral heterogeneity, which can mean that certain cells can resist treatments, and the cancer can relapse. Such flexibility displayed by tumors is probably the reason why cancer immunotherapy has become so popular – the adpative immune system can fight tumors’ flexibility with equal adaptability and specificity. Cancer immunotherapy works by enhancing the powers of the host’s own immune system to fight cancers [2]. Such an approach is advantageous, because immune cells can attack cancer cells systemically throughout the body, which is particularly relevant in metastatic cancers that can no longer be targeted by surgical intervention. Also, immune cells have great specificity – they can be trained to recognize and target cancer cells, which means fewer and less serious side effects against other cells in the body. Lastly, immune cells can develop memory responses against cancer cells, which means long-term, durable protection. In the case of melanoma treatment, some patients are in remission for more than 5 to 10 years [3–5]. Thus, if successful, immunotherapy can potentially eliminate cancers for a lifetime.
Our adaptive immune system includes T and B cells. T cells can be divided into two types: CD8 and CD4 T cells. CD8 T cells can be activated and differentiate into cytotoxic T lymphocytes (CTLs) that can directly kill cancer cells. CD4 T cells are also called helper T lymphocytes, and these cells have many accessory functions (e.g., secreting cytokines or helping B cells to produce antibodies). To prevent uncontrolled immune responses and collateral tissue damage, our immune system develops mechanisms to intrinsically shut down immune responses, which include the expression of co-inhibitory receptors on activated T cells, such as programmed death 1 (PD-1) [6–8]. Immune checkpoint inhibitors (ICIs), such as anti-PD-1 (a monoclonal antibody against PD-1), can block co-inhibitory receptors on activated T cells and unleash T cells that have become dysfunctional in the suppressive tumor microenvironment (TME) [6,9,10]. In turn, these “re-activated” T cells themselves can kill the cancer cells (Figure 1). While ICIs have become popular, unfortunately, they do not work in all patients; it remains poorly understood why some patients respond while others do not. One of the predictive indicators of anti-PD-1 therapy involves how infiltrated the tumor was in the first place [11]. Cancer patients without sufficient T cell infiltration before treatment were much less likely to respond to anti-PD-1 [11]. Thus, it is important to better understand what factors determine whether and how our T cells recognize cancer cells.
Figure 1. Mechanisms of action of immune checkpoint inhibitors.

(A) PD-1/PD-L1 interaction leads to exhaustion of CD4 and CD8 TILs. MHC class II or class I presents antigens to CD4 or CD8 T cells, respectively. PD-1 is expressed on activated CD4 or CD8 TILs, which can interact with PD-L1 expressed on cancer cells or other immune cells in the TME (e.g., myeloid cells). PD-1 engagement results in CD4 or CD8 TIL dysfunction. (B) Immune checkpoint inhibitors rescue CD4 or CD8 TILs. Anti-PD-1 or anti-PD-L1 blocks the interaction between PD-1/PD-L1, thereby leading to the reactivation of CD4 or CD8 TILs.
Anti-tumor immune responses are highly heterogeneous in different individuals.
The presence of tumor infiltrating lymphocytes (TILs) indicates an on-going anti-tumor immune response in that T cells have already recognized tumor-associated or tumor-specific antigens, which appears to be necessary, but not always sufficient, for clinical benefit of anti-PD-1 [11]. It has been increasingly recognized that a higher level of CD8 TIL infiltration correlates with better clinical parameters [12,13]; however, the infiltration pattern of TILs in human cancers is highly variable [14,15]. It remains poorly understood why anti-tumor immune responses vary so much in different individuals. Basically, anti-tumor immunity appears to be heterogeneous and individual-dependent. This poses a huge challenge for us to dissect the underlying mechanisms.
Many mouse tumor models that attempt to replicate human tumors do not adequately demonstrate tumor heterogeneity [16–18]. Other groups attempt to look into tumor heterogeneity by making a collection of different models, with different origins and different mutations, then comparing those models to each other [19]. However, such approaches examine too many variables at once (e.g., different oncogenic mutations); thus, many aspects of the tumor and TME can vary. With such confounding variables, it becomes difficult to study what the immune system might be doing differently in each situation. Ideally, we need a model system in which the tumor cells are derived from the same primary tumors driven by the same oncogenic mutations, yet these tumor cells could behave differently when transplanted into recipient mice with identicial genetic background. By controlling the tumors, we could largely minimize the effects of tumor-intrinsic factors, thus, be able to understand the mechanisms that immune cells use to respond to tumors, namely, whether host-intrinsic differences in immune surveillance make a difference in anti-tumor immune responses.
Several ICIs (e.g., anti-PD1) have been approved for various cancers including melanoma, lung cancers, and head and neck cancers but efficacy varies considerably in different patients [3,20–23]. In addition, effective rate remains low in patients of certain cancer types such as prostate or pancreatic cancers that usually exhibit low tumor immunogenicity [24]. Variable responses to therapies in cancer patients may be partially attributed to the fact that anti-tumor immune responses are heterogeneous in patients, evidenced by a highly variable level of T cell infiltration before treatment. However, it is very difficult to model such heterogeneity due to too many uncontrollable variables in human patients. Ideally, we need to establish a model system in which the same tumor cells can be transplanted into recipient mice with identical genetic background that exhibit different levels of T cell infiltration and respond differentially to anti-PD-1 treatment. A model as such may allow us to identify the fundamental principles shared between human and mouse that govern the heterogeneous anti-tumor immune responses in different individuals.
What tumor-intrinsic or tumor-extrinsic mechanisms can dictate heterogeneous anti-tumor immune responses?
An obvious question is whether heterogeneous anti-tumor immune responses depend on tumor-intrinsic (i.e. differences in tumor cells) or host-intrinsic factors. While active research is still on-going to address how different types of tumors influence the immune profile within the TME, many recent studies have focused on the tumor-intrinsic mechanisms, such as differentiation and different oncogenic drivers, that dictate heterogeneity [19]. Furthermore, human carcinogenesis is a highly complex process, even within the same type of cancers, there are many different subtypes caused by different mechanisms. For instance, head and neck squamous cell carcinomas (HNSCCs) can be associated with mutagens (e.g., smoking or alcohol) or human papilloma virus (HPV) [25]. Studies suggest that HPV− and HPV+ HNSCCs exhibit different levels of immune infiltration or respond to ICI treatment differentially [14,26]. Additionally, it is important to consider tumor heterogeneity in the context of immunological responses since tumor cells express different classes of tumor antigens. Neoantigen (NeoAg) is a distinct class of tumor antigens that are derived from somatic mutations in the cancer genome [27,28]. The critical role of neoAgs in ICI-based immunotherapy has been extensively reviewed elsewhere [29–31]. Individual tumor cells may harbor different genetic mutations that can lead to distinct expression pattern of different neoAgs; furthermore, tumor cells may evolve during metastasis or treatment and lose or acquire different neoAgs, which may in turn affect T cell-mediated anti-tumor immune responses [32]. Hence, tumor-intrinsic heterogeneity probably contributes to heterogeneous anti-tumor immune responses.
It remains possible that differences in environmental factors such as the host microbiome also affect tumor growth or tumor responses to ICI treatment. Prior studies in mouse models showed that host microbiome may influence their responses to ICI treatment [33]. Recent clinical studies using fecal samples collected from responders vs. non-responders also showed that patient microbiome correlated to their responses to ICI treatment [34–36]. However, it is unclear what composition of human microbiome favors clinical response to ICI treatment since different studies identified different groups of bacteria that were associated with responders or non-responders. Nevertheless, it is possible that differences in human microbiome likely contribute to heterogeneous anti-tumor immune responses.
However, there is a potentially very important factor that has received little attention previously, namely, the intrinsic differences in the TCR repertoire of an individual host. I propose a novel idea that intrinsic differences in the TCR repertoire of individuals may influence the outcome of anti-tumor immunity by affecting the frequency and/or variety of tumor antigen-reactive CD8 and/or CD4 TILs. This is due to the stochastic nature of TCR generation, which leads to a scenario in that some patients’ repertoires may be intrinsically deficient in harboring TCRs suitable for mounting efficient responses against a given tumor antigen. While antigen-specific T cells are known to influence the outcome of anti-tumor immunity or ICI treatment efficacy, this is a novel and provocative idea that does not have any support from experimental data yet. In addition, it may be difficult to test this idea because it can always be argued that tumors are different instead of TCR repertoire. There are technological and economic restrictions for testing whether the tumors are indeed “identical” in terms of their transcriptional or mutational profile in each individual clone; furthermore, it is possible that tumor cells diverge in their antigen profile as a function of time and in vivo outgrowth in different individuals. This idea does not exclude the contribution of tumor-intrinsic factors to heterogeneous anti-tumor responses; however, it may provide a novel perspective to test whether intrinsic differences in TCR repertoire contribute to heterogeneous anti-tumor responses in different hosts.
TCR repertoire is generated stochastically via random V(D)J recombination.
T and B cell development requires V(D)J recombination, a somatic DNA recombination process that generates antigen receptors for T and B cells [37,38] (Figure 2). V(D)J recombination occurs in a random and stochastic manner in different individuals (Figure 2). The TCR of most conventional T cells consists of two different protein chains, an alpha (α) chain and a beta (β) chain (encoded by TRA and TRB, respectively), linked via disulfide bonds. During T cell development, which normally occurs in the thymus [39] (Figure 2), progenitor T cells start as a double negative (DN) (CD4−CD8−) population that undergoes V(DJ) recombination of the TCRβ locus. The DN population then progresses to double positive (DP) (CD4+CD8+) population that undergoes V(D)J recombination of the TCRα locus. Successfully rearranged DP T cells will undergo thymic selection (positive and negative selection) and differentiate into single positive (SP) CD4+ or CD8+ T cells [39]. A TCR clonotype consists of unique TCRα and TCRβ chains that have unique VDJ usage and complementarity-determining region 3 (CDR3). CDR3 encompasses the highly divergent junction of V(D)J recombination that determines TCR specificity; its unique nucleotide sequences can act as a barcode for individual TCRs.
Figure 2. Overview of T cell development and V(D)J recombination.

(A) V(D)J recombination during T cell development. TCRβ gene rearrangement occurs in double negative (DN) (CD4−CD8−) progenitor T cell (Pro T) population. TCRα gene rearrangement occurs in double positive (DP) (CD4+CD8+) pre-T cell population. CD4 or CD8 SP immature T cells undergo thymic selection before their emigration of thymus. In peripheral lymphoid organs, 95% of human T cells are αβ T cells, whereas about 5% of human T cells are γδ T cells. (B) Schematics of V(D)J recombination in TCRβ locus. Top: germline configuration of human TCRβ locus. RAGs (recombination activating genes) initiate V(D)J recombination by generating DNA breaks at the V, D or J gene segments. Non-homologous end-joining (NHEJ) DNA repair pathway joins the broken V, D, J gene segments and completes V(D)J recombination.
However, our knowledge of formation of the human TCR repertoire is rather limited because it is difficult to access human thymus samples and impossible to manipulate the variables in vivo. Recent studies used the humanized mouse model to study formation of the human TCR repertoire that was generated by transplanting immunodeficient mice with human thymus and hematopoietic stem cells (HSCs) from the same or different donors [40]. The study concluded that formation of the human TCR repertoire is largely stochastic and the TCR repertoires can be totally divergent in thymi of animals with identical HSCs, thymus, genetic background, and environment [40]. Basically, this means that each individual has an almost completely different TCR repertoire – even in the case of identical twins − a fact that may explain the incomplete penetrance of genetically controlled autoimmune diseases in monozygotic twins [41].
Like the human TCR repertoires, I predict that each individual mouse will have an almost completely different thymic TCR repertoire from another mouse even if they have identical genomes. The initially formed TCR repertoire is continuously shaped by additional factors. For instance, certain clones of peripheral T cells will encounter antigens and undergo clonal expansion; thus, the number of such T cells that share the same TCR clonotype will increase compared with their thymic CD4 or CD8 SP counterparts. Nonetheless, the peripheral TCR repertoire will still be divergent in each individual mouse. Taken together, it is likely that the intrinsic differences in diverse TCR repertoire may also contribute to heterogeneous anti-tumor immune responses in different hosts, which may offer a new explanation for why some hosts would harbor T cells that can eradicate tumors, while others would not.
T cells recognize antigens in the context of major histocompatibility complex (MHC). MHC class I or class II molecules present peptides derived from self- or foreign-antigens to CD8 or CD4 T cells, respectively. Hence, there is a tripartite interaction among MHC/peptide and individual TCRs. Of note, the aforementioned model system investigated formation of the TCR repertoire in identical genetic background of the same donors [40], which means that the MHC alleles are identical in the thymic environment. However, cancer patients harbor vastly different alleles of human leukocyte antigen (HLA), the human version of MHC genes. The diversity of HLA constitutes another important aspect of formation of the TCR repertoire in humans. Different HLA types between individuals may influence the TCR repertoire through positive and negative selection during T cell development in the thymus. Prior studies reported a strong association between genetic variation in the MHC locus and usage of the TCR V-genes in a large human cohort, suggesting that MHC genotypes may influence the profiles of V-gene usage in each individual’s TCR repertoire [42]. However, the precise nature of this evolutionarily ancient MHC-TCR interaction remains controversial, and the functional significance of different MHC alleles and their influence on TCR diversity remain poorly understood [42–46]. Besides influencing formation of the TCR repertoire, different HLA alleles can present different epitopes, derived from either mutated or non‐mutated tumor antigens, to T cells, thereby leading to the differences in clonal expansion of T cells and affecting the TCR repertoire of CD8 or CD4 TILs in the TME. Consistently, it has been shown that HLA alleles were associated with clinical outcomes of ICI treatment [47,48]. In conclusion, the highly divergent TCR repertoires in different cancer patients likely contribute to heterogeneous anti-tumor immune responses.
Anti-tumor immune responses can occur in an antigen-specific manner.
Antigen-specific anti-tumor immunity plays an essential role in eradicating cancers. Apart from the well-studied model antigens, recent studies also highlight the potential application of neoAgs. NeoAgs are tumor-specific antigens that are absent in the normal host tissues and can be generated by somatic mutations in cancers [27,49–51]. NeoAgs are capable of inducing cancer-specific immune responses that not only eradicate cancers but also spare normal tissues, leading to less severe side-effects. Despite this potential, neoAgs are not always useful for anti-tumor immune surveillance. While some neoAgs are useful and induce robust T cell responses, other neoAgs resemble normal proteins too closely, and T cells do not bind them with an appropriate affinity and/or do not get activated properly. It is well-known that a fraction of human cancer samples lack immune cell infiltration, so-called “immune-excluded or immune desert tumors” [52,53]. However, it remains less well-understood whether this is because tumor cells lack any antigenicity, for example, they do not express tumor-specific neoAgs, or because the host lacks the proper TCRs that can recognize such tumor-specific antigens. Overall, these are fundamental questions in the field of cancer immunology that remain to be addressed.
It remains unknown how TCRs with different affinities to neoAgs dictate T cell responses in an in vivo tumor model. Moreover, enhancing affinity excessively impaired T cell function and diminished their killing activity [54–56]. Overall, successful anti-tumor immunity may require optimal-affinity TCRs, which cannot be too high or too low. Using a diabetes/ovalbumin (OVA) mouse model, TCR affinity was shown to dictate not only the magnitude of T cell activation but also the differentiation status of responding T cells [57]. Similar phenotypes were observed for pathogen-specific TCRs [58,59]. However, it remains to be determined how TCRs with different affinities dictate T cell activation, proliferation, and differentiation status in the context of successful vs. failed anti-tumor responses in vivo. I propose that a key determinant of successful anti-tumor immunity is the ability of a host to develop a “selected diverse” anti-tumor immune response, which means that robust anti-tumor responses may require a “right” combination of multiple T cell clones that are selected with optimal affinity of antigen/TCR interaction.
New ideas to explain the heterogeneous responses of anti-tumor immunity
While the “starting point” of T cells for each individual is highly different as a result of the stochastic nature of V(D)J recombination in TCRα and TCRβ loci, the TCR repertoire in mice and humans are also dynamic [60]. Because naïve T cells could be produced by the thymus continuously and lead to accumulative production of a variety of T cells [60]. However, in aged human patients, thymic atrophy has already occurred and a majority of their CD8 T cells do not exhibit naïve T cell phenotypes anymore due to constant antigen stimulation in periphery of a lifetime [61]. Therefore, the number of TCR clonotypes in an aged individual presumably would be much smaller. This means that the TCR repertoire in a given patient might not provide sufficiently different TCR clones that can recognize tumor antigens, namely, “a hole in the TCR repertoire” might exist. Of course, there are technological and economic restrictions for comprehensively studying the TCR repertoire: repertoire sequencing is a subsampling, especially for the naïve T cell population, whose clonal frequency is very low. Currently, there is no data to support the possibility of “any hole” in the TCR repertoire when this repertoire is considered to be dynamic and constantly shaped by various factors.
Another factor that needs to be considered is the characteristics of tumor-specific antigens in that the tumor-specific antigens are usually not that different from endogenous proteins, which make them more difficult to distinguish from endogenous self-peptides. In contrast, foreign antigens are very different from endogenous proteins in general. While it may be true that there is hardly “any hole in the TCR repertoire” of an immunocompetent mouse or human in the context of recognizing foreign peptides, it is likely that the TCR repertoire of aged human patients run out of their choices at recognizing tumor-specific antigens that have only one amino acid difference from their endogenous counterparts. This is especially relevant to tumorigenesis, which takes time to develop, and the tumor clones that can be recognized easily by T cells will presumably be eliminated during the early stage of tumor development.
One additional factor that could potentially shape the TCR repertoire in cancer patients is the different therapeutic regimens applied to the patients. Apart from immunotherapy, cancer patients may receive chemotherapy or radiation therapy. It is well-known that chemotherapy has detrimental effects on the adaptive immune system and causes T-cell immunodeficiency [62,63]. Radiation therapy is also highly toxic to radiosensitive T cells that are killed by ionizing radiation (IR) [64]. For instance, prior studies showed that the numbers of lymphocytes (CD4 and CD8 T cells) and leukocytes were reduced in the blood and spleen of C57BL/6 mice treated with IR with doses up to 3 Gy [65]. Hence, the T cell compartment has to be reconstituted after such therapies; as a result, the TCR repertoire in cancer patients may change, depending on the type, dose, and duration of different therapies. Furthermore, high doses of IR led to a loss of spleen and thymus mass in mice [65]. High-dose or low-dose IR can also damage hematopoietic progenitor cells [64]. Given that T cell development occurs in thymus and requires hematopoietic progenitor cells, IR may also negatively impact the reconstitution phase of T cell compartment. After chemotherapy, the numbers of both CD4 and CD8 T cells can recover in patients; however, recovery mechanisms differ greatly for CD4 and CD8 T cells [63,66,67]. Thymic-dependent pathways are required for an efficient recovery of CD4 T cells; however, these pathways undergo an age-dependent decline, thereby leading to prolonged CD4 T cell depletion in aged adults [63]. While the number of total CD8 T cells can recover relatively quickly post-chemotherapy, the disruption of different CD8 subsets often persists for a long time [63]. In particular, the subpopulation of CD8 T cells with reconstitution capacity exhibited elusive phenotypes (CD8+CD28−CD57−) that persisted for several months after chemotherapy [68,69]. The recovery of CD8 T cells may be mediated by a process termed “peripheral expansion” involving the proliferation of mature T cells [63]. While this process can increase CD8 T cell number, it may not restore the diversity of the TCR repertoire. As suggested by prior studies, while peripheral homeostatic process appears to be intact, the naïve TCR repertoire is not completely restored in aged patients [63,67]. Thus, I propose that such therapeutic interventions may cause contraction and reduce diversity of the TCR repertoire, further supporting the possibility of “a hole in the TCR repertoire” in the context of anti-tumor immunity.
I propose that intrinsic differences in the TCR repertoire of individual hosts may influence the outcome of anti-tumor immune responses by affecting the number and/or composition of TCR clones against tumor antigens. While heterogeneity of responses to ICI is a general phenomenon observed in many cancer types, the underlying mechanisms remain elusive. While T cell-inflamed tumors likely contain a selected diverse TIL repertoire that can successfully eliminate tumors upon ICI treatment, I speculate that the most effective and tumor-reactive T cell clones may pre-exist in hosts at higher frequencies by a random chance in the central or peripheral TCR repertoire before tumor development. In contrast, tumors that present as an “immune desert” lack the necessary diversity of TIL TCR repertoire to recognize tumor antigens; furthermore, these hosts may also lack the most useful clones in their central and peripheral TCR repertoires, namely, these hosts do not have the needed T cells generated in the first place or the frequency of such T cell clones is too low, and thus cannot respond effectively to ICI immunotherapy.
Conclusions and outlook
While formation of the human TCR repertoire is largely stochastic and can be totally divergent in individuals that have identical genetic background [40], it remains unknown how the stochastically generated TCR repertoire and the selection of TME cooperatively shape the outcome of anti-tumor immunity. It is critical to better understand whether and how differences in the TCR repertoire of individual hosts contribute to TIL responses because this may provide a completely new explanation for why certain hosts can eradicate tumors while others fail to do so. In addition, such studies may fundamentally influence overall strategies of cancer immunotherapy by increasing focus on either enhancing tumor antigenicity or diversifying TCR repertoire of the host. Further understanding the mechanistic basis of heterogeneous anti- tumor immune responses may profoundly impact the development of more effective personalized cancer immunotherapy.
Acknowledgments
I apologize to those whose work was not cited due to length restrictions. I thank Rachel A. Woolaver for her suggestions and proofreading of the manuscript. This work was supported by University of Colorado School of Medicine and Cancer Center startup funds and a THI pilot grant to J.H.W., NIH R01-DE027329, R01-DE028420, R21-CA184707, R21-AI110777, R01-CA166325, R01-CA229174, R01-CA249940 and a fund from Cancer League of Colorado to J.H.W.
Footnotes
Conflict of interests: I have declared that no conflict of interest exists.
References cited
- 1.Chen Z, Elos MT, Viboolsittiseri SS, Gowan K, et al. 2016. Combined deletion of Xrcc4 and Trp53 in mouse germinal center B cells leads to novel B cell lymphomas with clonal heterogeneity. J Hematol Oncol 9: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll DM. 2012. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer 12: 252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Postow MA, Callahan MK, Wolchok JD. 2015. Immune Checkpoint Blockade in Cancer Therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 33: 1974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, et al. 2019. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. The New England journal of medicine 381: 1535–46. [DOI] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, et al. 2019. Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamoto K, Hatae R, Honjo T. 2020. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int J Clin Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishimura H, Nose M, Hiai H, Minato N, et al. 1999. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11: 141–51. [DOI] [PubMed] [Google Scholar]
- 8.Ishida Y, Agata Y, Shibahara K, Honjo T. 1992. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO journal 11: 3887–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Han X. 2015. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. The Journal of clinical investigation 125: 3384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou W, Wolchok JD, Chen L. 2016. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8: 328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ayers M, Lunceford J, Nebozhyn M, Murphy E, et al. 2017. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. The Journal of clinical investigation 127: 2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Ruiter EJ, Ooft ML, Devriese LA, Willems SM. 2017. The prognostic role of tumor infiltrating T-lymphocytes in squamous cell carcinoma of the head and neck: A systematic review and meta-analysis. Oncoimmunology 6: e1356148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen N, Bellile E, Thomas D, McHugh J, et al. 2016. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canning M, Guo G, Yu M, Myint C, et al. 2019. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front Cell Dev Biol 7: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen SMY, Krinsky AL, Woolaver RA, Wang X, et al. 2020. Tumor immune microenvironment in head and neck cancers. Mol Carcinog. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosely SI, Prime JE, Sainson RC, Koopmann JO, et al. 2017. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer immunology research 5: 29–41. [DOI] [PubMed] [Google Scholar]
- 17.Vigneswaran N, Wu J, Song A, Annapragada A, et al. 2011. Hypoxia-induced autophagic response is associated with aggressive phenotype and elevated incidence of metastasis in orthotopic immunocompetent murine models of head and neck squamous cell carcinomas (HNSCC). Exp Mol Pathol 90: 215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Highfill SL, Cui Y, Giles AJ, Smith JP, et al. 2014. Disruption of CXCR2-Mediated MDSC Tumor Trafficking Enhances Anti-PD1 Efficacy. Sci Transl Med 6: 237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Byrne KT, Yan F, Yamazoe T, et al. 2018. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 49: 178–93.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santini FC, Hellmann MD. 2018. PD-1/PD-L1 Axis in Lung Cancer. Cancer J 24: 15–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, et al. 2016. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. The New England journal of medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seiwert TY, Burtness B, Mehra R, Weiss J, et al. 2016. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. The Lancet Oncology 17: 956–65. [DOI] [PubMed] [Google Scholar]
- 23.Munhoz RR, Postow MA. 2018. Clinical Development of PD-1 in Advanced Melanoma. Cancer J 24: 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macherla S, Laks S, Naqash AR, Bulumulle A, et al. 2018. Emerging Role of Immune Checkpoint Blockade in Pancreatic Cancer. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leemans CR, Braakhuis BJ, Brakenhoff RH. 2011. The molecular biology of head and neck cancer. Nature reviews Cancer 11: 9–22. [DOI] [PubMed] [Google Scholar]
- 26.Perri F, Ionna F, Longo F, Della Vittoria Scarpati G, et al. 2019. Immune Response Against Head and Neck Cancer: Biological Mechanisms and Implication on Therapy. Transl Oncol 13: 262–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. 2015. Tumor neoantigens: building a framework for personalized cancer immunotherapy. The Journal of clinical investigation 125: 3413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gubin MM, Zhang X, Schuster H, Caron E, et al. 2014. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515: 577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schumacher TN, Schreiber RD. 2015. Neoantigens in cancer immunotherapy. Science 348: 69–74. [DOI] [PubMed] [Google Scholar]
- 30.Desrichard A, Snyder A, Chan TA. 2016. Cancer Neoantigens and Applications for Immunotherapy. Clin Cancer Res 22: 807–12. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava RM, Purohit TA, Chan TA. 2020. Diverse Neoantigens and the Development of Cancer Therapies. Semin Radiat Oncol 30: 113–28. [DOI] [PubMed] [Google Scholar]
- 32.Riaz N, Havel JJ, Makarov V, Desrichard A, et al. 2017. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171: 934–49 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fessler JL, Gajewski TF. 2017. The Microbiota: A New Variable Impacting Cancer Treatment Outcomes. Clin Cancer Res 23: 3229–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, et al. 2018. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359: 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Routy B, Le Chatelier E, Derosa L, Duong CPM, et al. 2018. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359: 91–7. [DOI] [PubMed] [Google Scholar]
- 36.Matson V, Fessler J, Bao R, Chongsuwat T, et al. 2018. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359: 104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bassing CH, Swat W, Alt FW. 2002. The mechanism and regulation of chromosomal V(D) J recombination. Cell 109 Suppl: S45–55. [DOI] [PubMed] [Google Scholar]
- 38.Jung D, Alt FW. 2004. Unraveling V(D)J recombination; insights into gene regulation. Cell 116: 299–311. [DOI] [PubMed] [Google Scholar]
- 39.Germain RN. 2002. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol 2: 309–22. [DOI] [PubMed] [Google Scholar]
- 40.Khosravi-Maharlooei M, Obradovic A, Misra A, Motwani K, et al. 2019. Crossreactive public TCR sequences undergo positive selection in the human thymic repertoire. The Journal of clinical investigation 130: 2446–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Redondo MJ, Yu L, Hawa M, Mackenzie T, et al. 2001. Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 44: 354–62. [DOI] [PubMed] [Google Scholar]
- 42.Sharon E, Sibener LV, Battle A, Fraser HB, et al. 2016. Genetic variation in MHC proteins is associated with T cell receptor expression biases. Nat Genet 48: 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott-Browne JP, White J, Kappler JW, Gapin L, et al. 2009. Germline-encoded amino acids in the alphabeta T-cell receptor control thymic selection. Nature 458: 1043–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holland SJ, Bartok I, Attaf M, Genolet R, et al. 2012. The T-cell receptor is not hardwired to engage MHC ligands. Proceedings of the National Academy of Sciences of the United States of America 109: E3111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vrisekoop N, Monteiro JP, Mandl JN, Germain RN. 2014. Revisiting thymic positive selection and the mature T cell repertoire for antigen. Immunity 41: 181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.La Gruta NL, Gras S, Daley SR, Thomas PG, et al. 2018. Understanding the drivers of MHC restriction of T cell receptors. Nat Rev Immunol 18: 467–78. [DOI] [PubMed] [Google Scholar]
- 47.Chowell D, Morris LGT, Grigg CM, Weber JK, et al. 2018. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 359: 582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Havel JJ, Chowell D, Chan TA. 2019. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nature reviews Cancer 19: 133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. 2014. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nature reviews Cancer 14: 135–46. [DOI] [PubMed] [Google Scholar]
- 50.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, et al. 2005. Cancer/testis antigens, gametogenesis and cancer. Nature reviews Cancer 5: 615–25. [DOI] [PubMed] [Google Scholar]
- 51.Heemskerk B, Kvistborg P, Schumacher TN. 2013. The cancer antigenome. The EMBO journal 32: 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gajewski TF, Schreiber H, Fu YX. 2013. Innate and adaptive immune cells in the tumor microenvironment. Nature immunology 14: 1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hegde PS, Chen DS. 2020. Top 10 Challenges in Cancer Immunotherapy. Immunity 52: 17–35. [DOI] [PubMed] [Google Scholar]
- 54.Zhong S, Malecek K, Johnson LA, Yu Z, et al. 2013. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proceedings of the National Academy of Sciences of the United States of America 110: 6973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chervin AS, Stone JD, Soto CM, Engels B, et al. 2013. Design of T-cell receptor libraries with diverse binding properties to examine adoptive T-cell responses. Gene Ther 20: 634–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soto CM, Stone JD, Chervin AS, Engels B, et al. 2013. MHC-class I-restricted CD4 T cells: a nanomolar affinity TCR has improved anti-tumor efficacy in vivo compared to the micromolar wild-type TCR. Cancer immunology, immunotherapy : CII 62: 359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.King CG, Koehli S, Hausmann B, Schmaler M, et al. 2012. T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity 37: 709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ueno T, Tomiyama H, Fujiwara M, Oka S, et al. 2004. Functionally impaired HIV-specific CD8 T cells show high affinity TCR-ligand interactions. Journal of immunology 173: 5451–7. [DOI] [PubMed] [Google Scholar]
- 59.Thomas S, Xue SA, Bangham CR, Jakobsen BK, et al. 2011. Human T cells expressing affinity-matured TCR display accelerated responses but fail to recognize low density of MHC-peptide antigen. Blood 118: 319–29. [DOI] [PubMed] [Google Scholar]
- 60.Kumar BV, Connors TJ, Farber DL. 2018. Human T Cell Development, Localization, and Function throughout Life. Immunity 48: 202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thomas R, Wang W, Su DM. 2020. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun Ageing 17: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kotsakis A, Sarra E, Peraki M, Koukourakis M, et al. 2000. Docetaxel-induced lymphopenia in patients with solid tumors: a prospective phenotypic analysis. Cancer 89: 1380–6. [DOI] [PubMed] [Google Scholar]
- 63.Mackall CL. 2000. T-cell immunodeficiency following cytotoxic antineoplastic therapy: a review. Stem Cells 18: 10–8. [DOI] [PubMed] [Google Scholar]
- 64.Manda K, Glasow A, Paape D, Hildebrandt G. 2012. Effects of ionizing radiation on the immune system with special emphasis on the interaction of dendritic and T cells. Front Oncol 2: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pecaut MJ, Nelson GA, Gridley DS. 2001. Dose and dose rate effects of whole-body gamma-irradiation: I. Lymphocytes and lymphoid organs. In Vivo 15: 195–208. [PubMed] [Google Scholar]
- 66.Mackall CL, Fleisher TA, Brown MR, Andrich MP, et al. 1997. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood 89: 3700–7. [PubMed] [Google Scholar]
- 67.Sfikakis PP, Gourgoulis GM, Moulopoulos LA, Kouvatseas G, et al. 2005. Age-related thymic activity in adults following chemotherapy-induced lymphopenia. Eur J Clin Invest 35: 380–7. [DOI] [PubMed] [Google Scholar]
- 68.Fagnoni FF, Lozza L, Zibera C, Zambelli A, et al. 2002. T-cell dynamics after high-dose chemotherapy in adults: elucidation of the elusive CD8+ subset reveals multiple homeostatic T-cell compartments with distinct implications for immune competence. Immunology 106: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Onyema OO, Decoster L, Njemini R, Forti LN, et al. 2015. Chemotherapy-induced changes and immunosenescence of CD8+ T-cells in patients with breast cancer. Anticancer Res 35: 1481–9. [PubMed] [Google Scholar]