

Abstract
Metabolic flux analysis (MFA) is highly relevant to understanding metabolic mechanisms of various biological processes. While the pace of methodology development in MFA has been rapid, a major challenge the field continues to witness is limited metabolite coverage, often restricted to a small to moderate number of well-known compounds. In addition, isotopic peaks from an enriched metabolite tend to have low abundances, which makes liquid chromatography tandem mass spectrometry (LC-MS/MS) highly useful in MFA due to its high sensitivity and specificity. Previously we have built large-scale LC-MS/MS approaches that can be routinely used for measurement of up to ~1,900 metabolite/feature levels [Gu et al. Anal. Chem. 2015, 87, 12355–12362. Shi et al. Anal. Chem. 2019, 91, 13737–13745.]. In this study, we aim to expand our previous studies focused on metabolite level measurements to flux analysis and establish a novel comprehensive isotopic targeted mass spectrometry (CIT-MS) method for reliable MFA analysis with broad coverage. As a proof-of-principle, we have applied CIT-MS to compare the steady-state enrichment of metabolites between Myc(oncogene)-On and Myc-Off Tet21N human neuroblastoma cells cultured with U-13C6-glucose medium. CIT-MS is operationalized using multiple reaction monitoring (MRM) mode and is able to perform MFA of 310 identified metabolites (142 reliably detected, 46 kinetically profiled) selected from >35 metabolic pathways of strong biological significance. Further, we developed a novel concept of relative flux, which eliminates the requirement of absolute quantitation in traditional MFA and thus enables comparative MFA under the pseudosteady state. As a result, CIT-MS was shown to possess the advantages of broad coverage, easy implementation, fast throughput, and more importantly, high fidelity and accuracy in MFA. In principle, CIT-MS can be easily adapted to track the flux of other labeled tracers (such as 15N-tracers) in any metabolite detectable by LC-MS/MS and in various biological models (such as mice). Therefore, CIT-MS has great potential to bring new insights to both basic and clinical metabolism research.
Graphical Abstract

Metabolism is ubiquitous in almost every aspect of biology, and abnormal metabolism often signals underlying pathology or is itself the etiology of serious morbidities, such as inborn errors of metabolism, obesity, diabetes, cardiovascular disease, Alzheimer's disease, and cancer.1–10 Currently, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database has indexed more than 8,700 metabolic reactions and annotated 16,000 metabolites. Defining these metabolic pathways and characterizing their physiological roles have resulted in many significant findings. For example, carcinogenesis and cell proliferation may cause aberrant concentration and flux changes in numerous metabolites and metabolic pathways; the Warburg effect stipulates that many cancer cells switch from producing most of their energy via the TCA cycle in the mitochondria, to lactic acid production in the cytosol.4,11,12 Thus, in addition to gene- and protein-level studies, understanding the dynamic role of metabolites and metabolic pathways is a critically important and novel area of research in both basic and clinical settings.
Metabolic flux analysis (MFA) using stable isotope labeled tracers enables a dynamic description of the cellular metabolome in a living system.8,13–22 The whole metabolome can be conceptualized as a vast metabolic reaction network, comprised of metabolites (nodes) and metabolic pathways (edges). Analysis of metabolite levels provides cross-sectional information on biochemical abundances; however, cellular metabolism in a living system is not static. In addition, many metabolites are involved in multiple pathways; therefore, concentration changes of a particular metabolite can result from perturbations in several pathways, often confounding the analysis of metabolite level data. In contrast, MFA investigates the rate of metabolite turnover in biochemical reactions derived from stable isotope labeled tracers, through tracking isotopic conversions taking place in the metabolic network.8,16–21 Furthermore, MFA can be easily linked to other important areas in systems biology, such as proteomics and genomics. Therefore, pathway-specific information from MFA is often required to fully understand a biological process.
MFA places a high requirement for analytical measurements and computational modeling, since flux rates are not directly measurable. For metabolite level measurements, mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy are often used to detect and quantify the isotopic peaks of product compounds of interest. While NMR is highly useful to examine the position of isotopes (such as 13C) within isotopomers, MS is significantly more sensitive and has a much wider metabolite coverage.23,24 MS is a preferred analytical method to detect low-abundance peaks, which is of great significance in MFA as isotopic peaks (not M + 0 peaks) tend to have low intensities in many cases.
Mathematical models are applied to translate the measurement data into metabolic flux, and kinetic flux profiling (KFP) is a practical and efficient modeling approach for obtaining metabolic flux in live cells under the pseudosteady state.25–28 Briefly, the enrichment of tracers into various metabolites is determined. Mass isotopomer distribution (MID) and mean enrichment (ME) are calculated based on the isotopic intensities for each identified metabolite (X). The time-course ME data (MEt) are then used to fit an exponential function, and the apparent first-order rate constant of decay (kx) is derived from eq 1
| (1) |
where XT is the normalized total pool size of the metabolite of interest (constant in pseudosteady MFA), which requires absolute quantitation in traditional MFA. XU,t denotes unlabeled X at time t (), and α is a constant. Finally, the flux of metabolite X, fX, can be calculated using eq 2:
| (2) |
While the pace of methodology development in MFA has been rapid and MFA has been successfully applied in many studies,13,15,26,27,29–40 a number of technical bottlenecks have prevented realization of the full potential of MFA. A major challenge the field continues to witness is limited metabolite coverage. Currently, MFA is severely restricted to a small to moderate number of compounds from main pathways. For example, central carbon metabolism metabolites have been heavily studied in MFA.9,20,41 Although these metabolites do play important roles in biological processes, the whole metabolome consists of numerous other metabolites and metabolic pathways of different purposes and functions. Furthermore, isotopic peaks from the same metabolite (M + n) often have low signals; therefore, isotopic peaks should be detected with the best sensitivity offered to ensure optimal MFA accuracy. It is of great significance to develop state-of-the-art methods and platforms to enable broad MFA with high reliability.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) is widely used in analytical measurements, and it is often operated in multiple reaction monitoring (MRM) mode.42–45 The combination of precursor ions and product ions makes MRM highly sensitive and specific, especially for detecting exponentially decaying isotopic signals. In addition, LC-MS/MS generally has excellent quantitative performance, providing a wide linear dynamic range and good reproducibility. Notably, large-scale LC-MS/MS approaches have been established and successfully used in a growing number of metabolomics studies.45–49 For example, we developed novel LC-MS/MS approaches that have the analytical advantages of both traditional untargeted profiling and targeted detection.50,51 In this study, we aim to expand our previous studies focused on metabolite level measurements to flux analysis and establish a novel comprehensive isotopic targeted mass spectrometry (CIT-MS) method for reliable MFA analysis with broad coverage. As a proof of principle, we examined the performance of CIT-MS to study cellular metabolic reprogramming as a result of differential Myc-oncogene expression in Tet21N human neuroblastoma cells.
EXPERIMENTAL METHODS
Development of the overall analytical strategy is shown in Figure 1. This large-scale, LC-MS/MS-based MFA approach can be conceptualized as follows:
Figure 1.
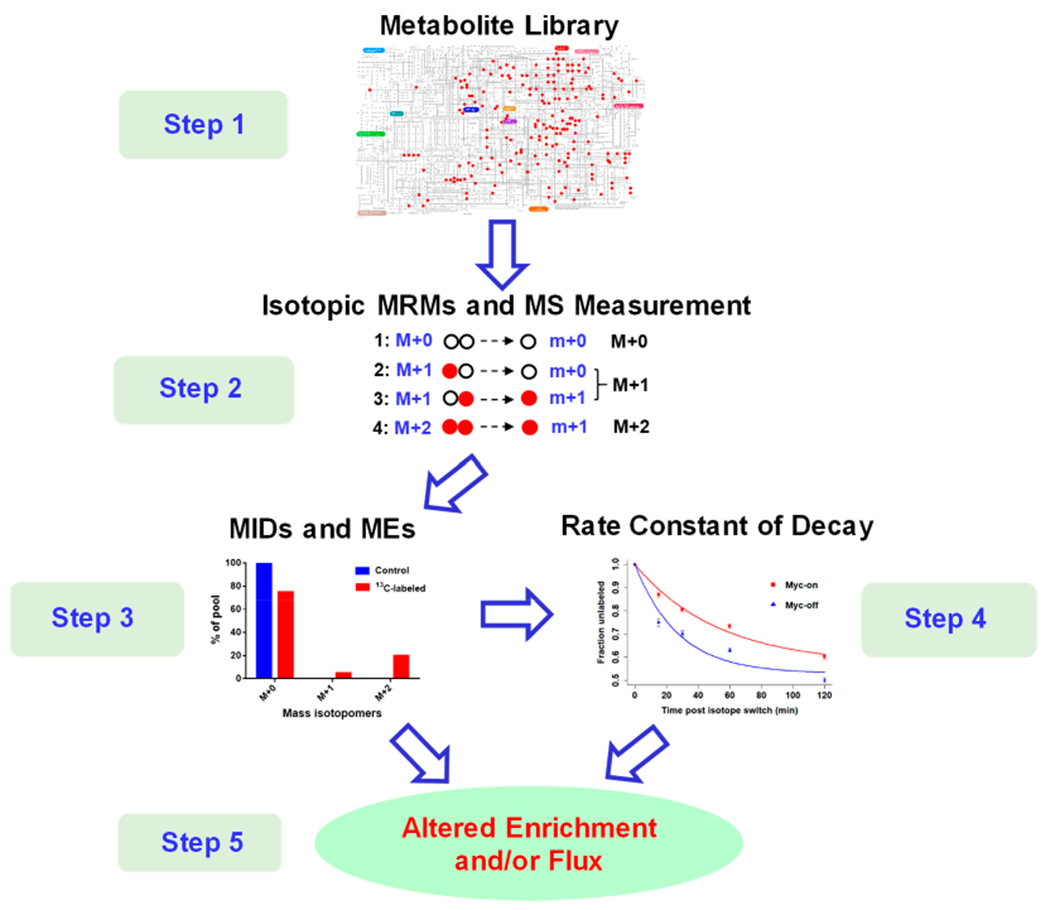
Overall analytical strategy for CIT-MS in MFA.
Step 1. An in-house database was constructed comprising 310 metabolites selected from >35 metabolic pathways of strong biological significance, and this in-house database was assembled from a large-scale LC-MS/MS assay that has been widely used in a growing number of studies.45,48,49,51–57 Notably, unlabeled chemical standards were available for all these metabolites, enabling both the optimization of elution and MRM parameters of the LC-QQQ-MS system as well as identification of detected metabolites.
Step 2. The isotopic MRM library was generated including all MRM transitions for each metabolite isotopologue. Since the chemical formula is known for each selected metabolite (i.e., the number of each element is known), it is easy to determine the precursor ion m/z value (M + n) of the isotopologue when n (n = 0 – the number of C in this metabolite) carbon atoms are enriched, using tracking 13C as an example. For the product ion, we examined the chemical formula in the HMDB and METLIN databases58,59 to determine the number of carbon atoms. These isotopic MRMs were then used in LC-MS/MS measurements, and the isotopic intensity of this isotopologue was the sum of MRMs of M + n → all possible product ions when n 12C atoms were replaced by 13C.
Step 3. Using IsoCor software,32 the MID and ME values for each metabolite were calculated.
Step 4. In case of a time-course study under the pseudosteady state,25–28 the rate constant of decay (k) of a metabolite was determined by fitting an exponential equation (eq 1), similar to KFP. Notably, we also developed a novel concept of relative flux (fR) for comparative MFA, without requiring absolute quantitation as with traditional MFA.
Step 5. Depending on the experimental design, altered metabolite enrichment and/or relative flux were examined, thereby allowing inspection of the mechanisms underlying metabolic reprogramming.
Chemicals and Reagents.
HPLC-grade acetonitrile (ACN) and methanol (MeOH) were purchased from Sigma-Aldrich (St. Louis, MO). Ammonium acetate and acetic acid were obtained from Fisher Scientific (Pittsburgh, PA). Deionized water was provided in-house by a Millipore Milli-Q Academic Water Purification System (Billerica, MA). Pure metabolite standards in the Arizona Metabolomics Laboratory (AML) were purchased from Sigma-Aldrich and Fisher Scientific. Stable isotope-labeled standards were purchased from Cambridge Isotope Laboratories (Tewksbury, MA).
Cell Culture and Preparation.
For the unlabeled culture, Tet21N human neuroblastoma cells (Myc-On cells) were cultured in DMEM with 25 mM glucose and 2 mM glutamine (Gibco, Grand Island, NY), supplemented with 10% FBS (HyClone, South Logan, UT) and 1% penicillin/streptomycin (Gibco). Doxycycline (1 μg/mL, Clontech Laboratories, Inc., Mountain View, CA) was used to repress N-Myc expression, thereby enabling comparison of Myc-On and Myc-Off Tet21N cells. For the labeled culture, the experimental conditions were the same, except that the cells received an isotopically labeled tracer (U-13C6-glucose; Cambridge Isotope Laboratories, Tewksbury, MA). When cells were 80% confluent in a 2 cm diameter well (~1 × 106 cells), the culture medium was quickly switched to a 2-mL isotope-containing medium with glucose/glutamine-free DMEM (Gibco) supplemented with 10% dialyzed FBS (Gibco), 5.5 mM U-13C6-glucose, and 2 mM l-glutamine (Sigma-Aldrich, St. Louis, MO). For steady-state growth, cell samples were cultured for 0, 24, 48, and 96 h after introduction of U-13C6-glucose to the culture medium, and cell passage was carried out every 24 h. For pseudosteady state growth, the time points of sample collection were 0, 0.5, 1, 2, 4, 8, 12, and 27 h (for metabolites that can be quickly enriched, only the first few time points were used).
The cell samples were processed using standard operating procedures (SOPs) we established in previous studies.36,60–62 Briefly, the cells were rinsed using PBS (prechilled to 4 °C). Afterward, 0.8 mL of methanol/H2O (v:v = 80:20, prechilled to −80 °C) was added to quench cellular metabolism and perform metabolic extraction on dry ice. Cells were then scraped into a new tube. Then, 0.3 mL of prechilled methanol/H2O (v:v = 80:20) was added to the plate, and cells were scraped again. The two extraction solutions were combined and sonicated for 30 min in an ice bath. The samples were then centrifuged at 14000 rpm for 10 min at 4 °C. Of the supernatant, 1,000 μL was transferred to a new tube and dried using the speed vacuum concentrator (Labconco, Kansas City, MO). The resulting sample pellet was used to assay total protein concentration for normalization, using the BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL).
For MFA using gas chromatography (GC)-mass spectrometry, extracted samples were incubated with 30 μL of O-methylhydroxylamine hydrochloride solution in pyridine (Sigma-Aldrich, St. Louis, MO) at 60 °C for 90 min. Next, 70 μL of N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA, Sigma-Aldrich, St. Louis, MO) was added and placed at 60 °C for 30 min.
instrumentation.
All LC-MS/MS experiments were performed on an Agilent 1290 UPLC-6490 QQQ-MS system (Santa Clara, CA). The flow rate was 0.3 mL/min, autosampler temperature was set at 4 °C, and the column compartment was set to 40 °C. The separations were performed on a Waters XBridge BEH Amide column (150 × 2.1 mm, 2.5 μm particle size; Waters Corporation, Milford, MA). The mobile phase was composed of Solvents A (10 mM ammonium acetate, 10 mM ammonium hydroxide in 95% H2O/5% ACN, pH = 9) and B (10 mM ammonium acetate, 10 mM ammonium hydroxide in 5% H2O/95% ACN, pH = 7). After an initial 1 min isocratic elution of 90% B, the percentage of Solvent B decreased to 40% at t = 11 min. The composition of solvent B was maintained at 40% for 4 min (t = 15 min), after which the percentage of solvent B gradually returned to 90%, in preparation for the next injection. The QQQ-MS system was operated with a capillary voltage of 3,500 V. The nebulizer gas (N2) pressure was set at 30 psi with a drying gas (N2) flow rate of 15 L/min and a temperature of 175 °C. The flow rate of sheath gas (N2) was set at 11 L/min with a temperature of 225 °C. After data collection, the extracted MRM peaks were integrated using Agilent Mass Hunter Quantitative Data Analysis software (Santa Clara, CA).
Briefly, GC-MS spectral acquisition was performed on an Agilent 7820A GC system coupled to an Agilent 5977B MSD system (Agilent Technologies, Inc., Santa Clara, CA). For analysis, 1 μL of derivatized sample was injected using the splitless mode, and helium (purity >99.999%) was used as the carrier gas. The flow rate of carrier gas was 0.5 mL/min. The temperature of injection port and transfer line was 250 and 290 °C, respectively. Chemical derivatives in the samples were separated using an HP-5 MS capillary column coated with 5% phenyl–95% methylpolysiloxane (30 m × 250 μm i.d. × 0.25 μm film thickness, Agilent Technologies). The column temperature was maintained at 60 °C for 1 min, then increased at a rate of 10 °C/min to 325 °C, and held at this temperature for 10 min. The solvent delay was 9.69 min. Mass spectral signals were recorded in full scan mode using electron ionization (EI, 70 eV), ion source temperature was 230 °C, EM voltage was 1,225 V, and mass range was 50–600 m/z with a scan frequency of 2.7 scans/s.
Data Analysis.
For each metabolite, MID and ME were calculated after correction for natural abundance using IsoCor software.32 The time-course data were used to determine flux, and exponential fitting was carried out using R (version 4.0.0). Studenťs t-test was performed using Microsoft Excel (Redmond, WA). For analysis of Myc samples, principal component analysis (PCA) was performed using MetaboAnalyst (https://www.metaboanalyst.ca/).63 Data were mean-centered prior to model construction. The metabolic map was generated using IPath tools (https://pathways.embl.de/).64
RESULTS
CIT-MS Development.
Large-scale LC-MS/MS has been routinely used in metabolomics, mainly due to its high sensitivity and reliable quantitative performance.25–27,30–32,44–46 As shown in Table S1, we have compiled an in-house metabolite library that contains 310 unlabeled chemical standards and their optimized MS parameters, such as precursor and product ions. The retention times (RTs) of metabolites were acquired through hydrophilic interaction chromatography (HILIC). Further, we used IPath tools to annotate these metabolites in the KEGG global pathway map; however, although belonging to known pathways, only 179 (red dots) of these 310 metabolites could be indicated in Figure S1 given software constraints and consideration of optimal pathway view.
To determine isotopic MRM transitions, we first used the large-scale LC-MS/MS method to target these 310 metabolites in a quality control (QC) sample that was pooled from unlabeled Myc-On and Myc-Off Tet21N cells. Following reliable detection of 142 metabolites, precursor ions and corresponding product ions were structurally annotated by searching spectral libraries using the HMDB58 and METLIN59 databases. For an identified metabolite, the MRM parameters can be easily generated from the number of carbon atoms (for 13C-tracers) in the chemical formula.
Table S2 shows all isotopic MRM transitions in CIT-MS. Using alanine as an example, Table 1 shows the MRM transition editing process. Alanine has a precursor m/z of 90.1 and generates a two-carbon fragment with m/z of 44.0 in positive ion mode. To detect 13C2-labeled alanine (M + 2), 2 Da must be added to the precursor ion to form 92.1. However, for product ions, two possibilities exist: 13C-labeled fragment or 13C2-labeled fragment. Therefore, two transitions should be used to monitor 13C2-labeled alanine: 92.1 → 45 and 92.1 → 46, and the sum of them is the intensity for the M + 2 peak. The LC-MS/MS parameters for these MRMs were the same for each metabolite.
Table 1.
MRM Transitions of Alanine Isotopomers in CIT-MS
| MRM | precursor ion | product ion | collision energy | polarity |
|---|---|---|---|---|
| alanine-M+0-0 | 90.1 (C3H8NO2) | 44.0 (C2H6N) | 9 | positive |
| alanine-M+1-0 | 91.1 | 44 | 9 | positive |
| alanine-M+1-1 | 91.1 | 45 | 9 | positive |
| alanine-M+2-1 | 92.1 | 45 | 9 | positive |
| alanine-M+2-2 | 92.1 | 46 | 9 | positive |
| alanine-M+3-2 | 93.1 | 46 | 9 | positive |
Then, identification and deconvolution of isotopologues were conducted through searching both the labeled and unlabeled samples. We examined ~30 metabolite standards and their isotopic standards in various metabolic pathways, and the following criteria were used to determine isotopologue groups (only peaks satisfying all criteria were regarded as isotopic peaks generated from the same metabolite):
For the unlabeled sample, the base peak is the M + 0 peak, and other M + n peaks should continuously decrease as m/z increases;
For the labeled sample, if the base peak is the M + 0 peak, there will be at least one other isotopic peak higher than the corresponding peak from the unlabeled sample after base peak normalization;
For the labeled sample, the base peak can be the M + n peak (n being an integer between 1 and the number of carbon atoms in the metabolite);
The RT difference between isotopic peaks should be small (δRT; e.g., <0.1 min).
The collected data were then subjected to enrichment analysis (MID and ME) and/or relative flux (fR) calculations. Under the pseudosteady state, fR can be obtained for probing cellular metabolic flux based on the kinetics of assimilation of isotope-labeled nutrients into a myriad of downstream metabolites.25–28 Fitted from eq 1, kX is the apparent first-order rate constant for disappearance of the unlabeled metabolite. Notably, traditional MFA requires absolute quantitation of the intracellular pool size of a metabolite to calculate its flux (eq 2), which is able to compare flux of different metabolites. However, in many cases, we only need to compare flux through the same metabolite but in different biological conditions. Therefore, we used the relative intracellular pool size of the metabolites, normalized intensities by total protein, to calculate fR.
Validation Using Glucose Isotopologues.
For the initial validation, we prepared 8 standard mixture solutions, with different mass ratios of glucose (G), 1-13C-G, 1,6-13C2-G, and U-13C6-G. The ratios were 0:100:0:0, 0:0:100:0, 0:0:0:100, 91:9:0:0, 71:9:10:10, 21:28:31:20, 0:38:32:30, and 0:0:11:89, respectively. Figure 2 compares the percentage of these glucose isotopologues derived from CIT-MS to theoretical values. It is evident that CIT-MS results fit well with expected values. In Table S3, the linear regression analysis of the observed% against theoretical% showed all coefficient of determination (R2) values to be more than 0.998. Therefore, these results provide solid initial evidence that CIT-MS is able to perform reliable flux analysis.
Figure 2.
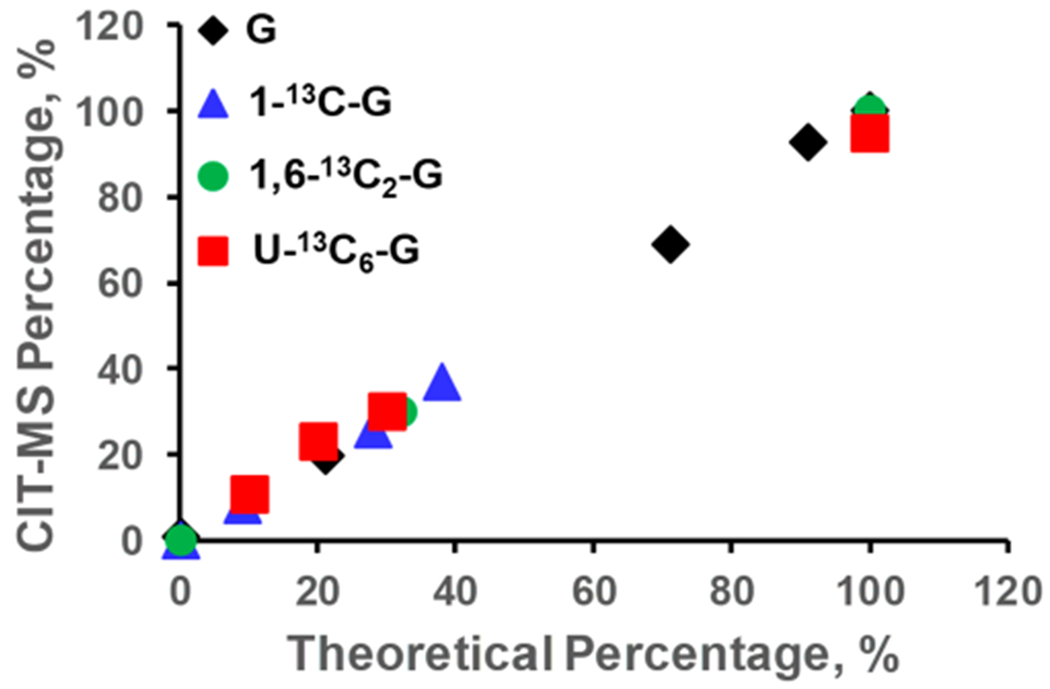
Comparison of the theoretical percentage values of glucose isotopologues to those derived from CIT-MS.
Steady State Enrichment Analysis.
Myc is a well-studied oncogene that causes significant alterations in global gene and protein expression and metabolic reprogramming.40,61,65,66 Using Myc-On and Myc-Off Tet21N cells, we applied CIT-MS for a long-term steady state enrichment analysis. The cells were cultured up to 96 h with U-13C6-glucose, and cell samples were collected at 0, 24, 48, and 96 h. Ultimately, 80 metabolites were able to be enriched by U-13C6-glucose, of which 63 were annotated by IPath (https://pathways.embl.de/). As shown in Figure 3, the enriched metabolites were distributed in diverse pathways such as glycolysis, the TCA cycle, alanine/aspartate/glutamate metabolism, purine metabolism, pyrimidine metabolism, pentose and glucuronate interconversions, arginine biosynthesis, glutathione metabolism, and the hexose monophosphate shunt, many of which are currently rarely studied.
Figure 3.
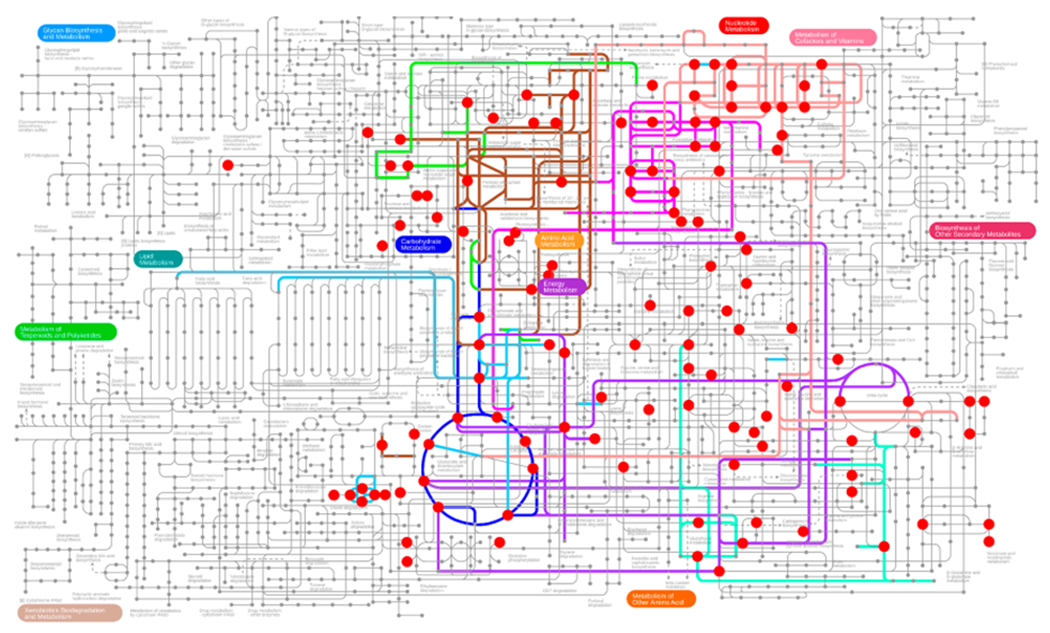
Pathway view of enriched metabolites from U-13C6-glucose that were detected by CIT-MS in a steady state study using Tet21N cells.
To investigate long-term U-13C6-glucose labeling, Figures 4 and S2–4 show labeled enrichment of various metabolites at different time points. It was found that enrichment of many nucleotides and amino acid metabolites stabilized after 48 h for both Myc-On and Myc-Off cells. Consistent with previous studies,37,67–69 most TCA cycle metabolites and other metabolites of carbohydrate metabolism quickly achieved steady state (~24 h). Therefore, 96 h after labeling with U-13C6-glucose was selected in this study to compare differences between labeled enrichment of Myc-On and Myc-Off cell lines.
Figure 4.
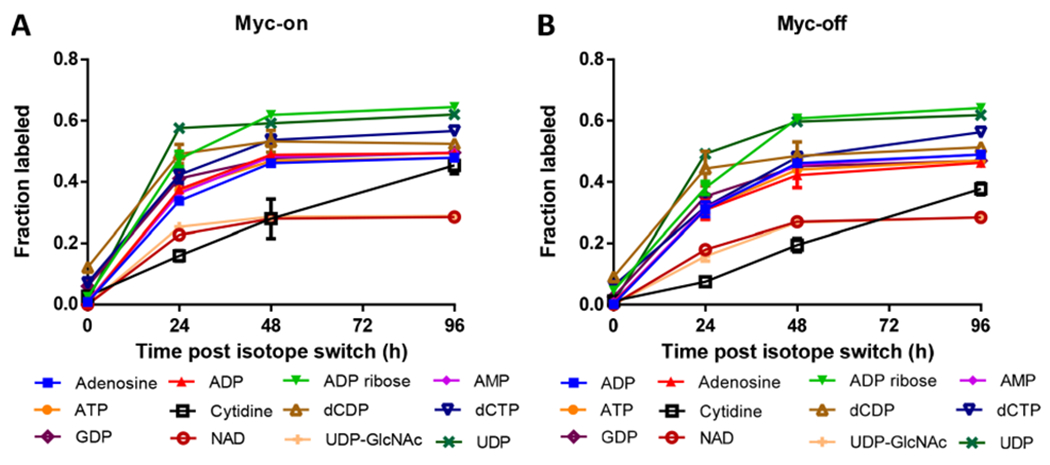
Enriched nucleotide metabolites from U-13C6-glucose that were detected by CIT-MS in a steady state study using (A) Myc-On and (B) Myc-Off Tet21N cells.
Based on the enrichment data at 96 h, Figure 5A showed that Myc-On and Myc-Off cells could be clearly separated in the PCA score plot. The total variance explained by PC1 and PC2 was more than 87%. In addition, as shown in Figure 5B, comparison of Myc-On and Myc-Off variants of the human Tet21N cells revealed differential enrichment of nine detected metabolites with a p value < 0.05 and fold change (FC) > 2. These metabolites were γ-aminobutyric acid (GABA), methylmalonic acid, 2-methylglutaric acid, glutamine, asparagine, α-ketoglutaric acid, hydroxyproline, aspartate, and glutamic acid.
Figure 5.
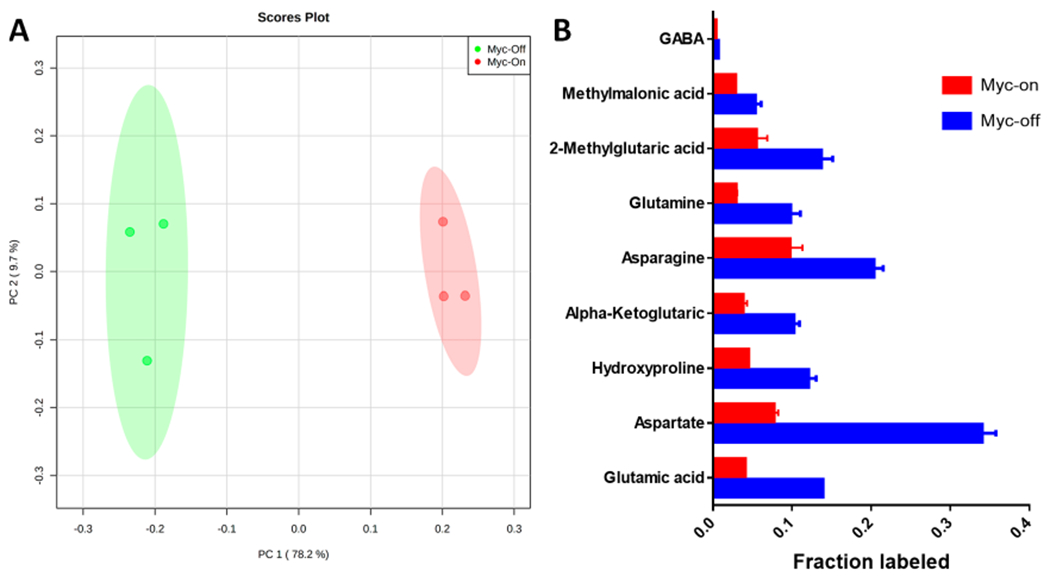
A) The PCA score plot and B) metabolites with p < 0.05 and FC > 2 for the comparison of Myc-On and Myc-Off Tet21N cell lines at 96 h post U-13C6-glucose labeling.
Pseudosteady State and Relative Flux.
Myc-On and Myc-Off samples were collected during pseudosteady state cell growth. KFP was used to calculate the rate constant of decay (k) following U-13C6-glucose labeling using eq 1, and curve fitting was estimated by assessing the correlation coefficient (r). An r value > 0.75 was regarded as good fitness. Relative flux (fR) of metabolites was calculated using eq 2. Here, XT denotes the peak area for each metabolite after normalization with total protein. Using citrate as an example, Figure 6A shows the disappearance of unlabeled citrate to be an exponential decay function. The k values of Myc-On and Myc-Off cells were calculated as 0.576 and 0.786, respectively. The r values were 0.986 and 0.979, indicating excellent fit to the exponential function. In addition, fR was calculated to be 1168.26 ± 65.56 and 7487.99 ± 1385.49 (n = 4) in Myc-On and Myc-Off cells, respectively.
Figure 6.
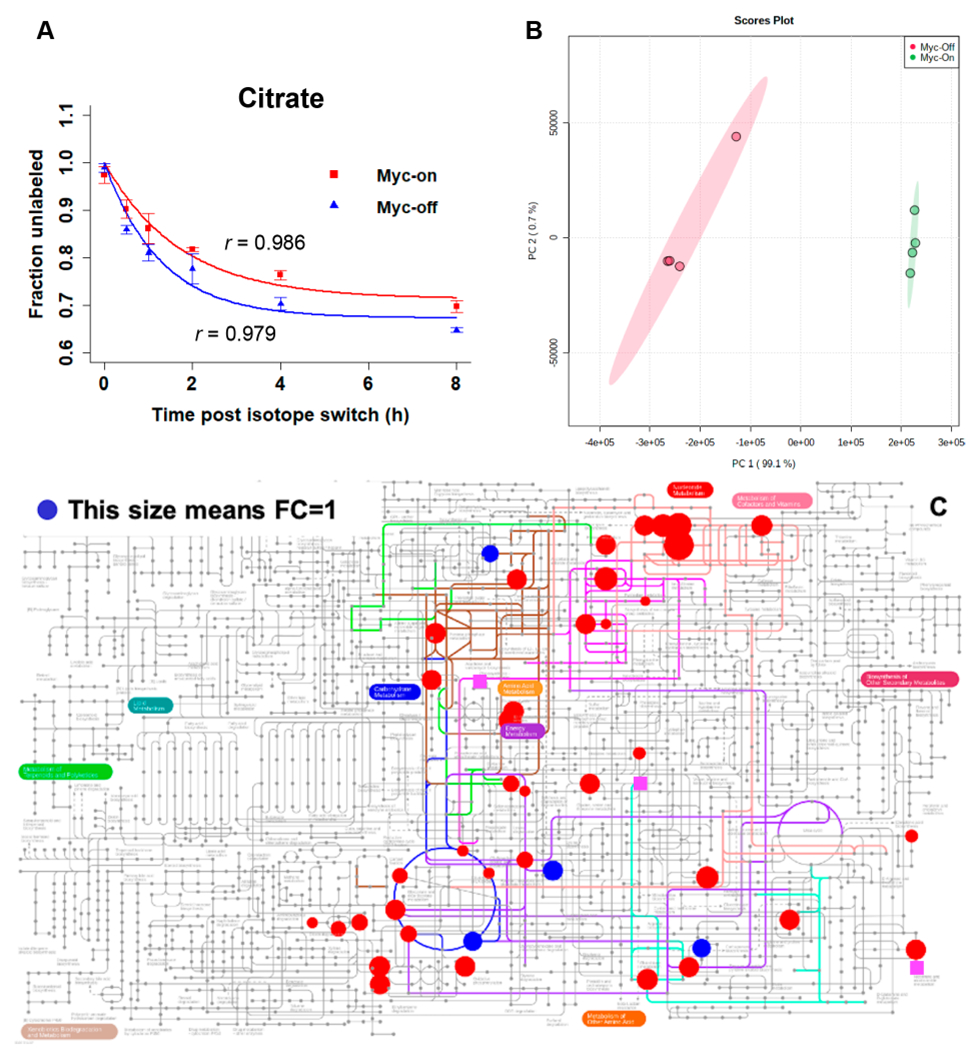
A) Exponential fitting of the time-course data of citrate, B) the PCA score plot, and C) pathway view of metabolites with relative flux derived, for the comparison of Myc-On and Myc-Off Tet21N cells under pseudosteady state following U-13C6-glucose labeling: red circles have p values < 0.05, blue circles have p values > 0.05, and pink squares indicate metabolites present only in the Myc-Off group.
In total, 46 metabolites were fitted for k and fR under the pseudosteady state. Figures S5–S8 show the kinetics of disappearance for unlabeled nucleotide, carbohydrate, TCA cycle, and amino acid metabolites following the U-13C6-glucose isotope switch, respectively. Most r values were greater than 0.75 (mean = 0.90), showing the trend estimation to be reliable. Along with the relative abundance of metabolites, the relative metabolic flux was obtained, which was used to investigate altered metabolism in response to the Myc oncogene. Based on fR, Figure 6B showed that Myc-On and Myc-Off cells were clearly separated in the PCA score plot. The total variance explained by PC1 and PC2 was 99.8%. Table 2 shows metabolites with significant relative flux difference between Myc-On and Myc-Off cells, with p < 0.05 and FC > 2 or < 0.5. In addition, we annotated 35 of these 46 metabolites in IPath Pathways (Figure 6C, the changes were not quantitatively marked). Significant metabolites (p < 0.05 and FC < 0.5) with downregulated flux in Myc-On cells were acetyl-l-glutamine, aconitate, alanine, citrate, cytidine, dCDP, fructose, and sarcosine. Significant metabolites (p < 0.05 and FC > 2) with upregulated flux in Myc-On cells were adenosine, adenosyl-l-homocysteine, ADP, AMP, ATP, fructose 6-phosphate, G16BP, GDP, glutamine, reduced glutathione, hydroxyproline, l-ascorbic acid, NAD, NADH, serine, UDP, UDP-GlcNAc, and xylitol. Notably, metabolites with relative flux present only in Myc-On cells were 1-methyladenosine, acetylcarnitine, oxidized glutathione, glycine, and NADP; metabolites with relative flux present only in Myc-Off cells were 2-hydroxyglutaric acid, 3-phosphoglyceric acid, and N-acetylneuraminic acid.
Table 2.
Metabolites with Significant Relative Flux Difference between Myc-On and Myc-Off Cells
| metabolites | Myc-On | Myc-Off | p | FCa |
|---|---|---|---|---|
| acetyl-l-glutamine | 16.7 ± 0.9 | 57.8 ± 8.3 | 0.002 | 0.29 |
| aconitate | 76.0 ± 9.2 | 468.9 ± 97.4 | 0.004 | 0.16 |
| alanine | 67416.3 ± 3769.3 | 492002.7 ± 63432.7 | 0.0009 | 0.14 |
| citrate | 1168.3 ± 65.6 | 7488.0 ± 1385.5 | 0.003 | 0.16 |
| cytidine | 10.6 ± 1.1 | 878.0 ± 212.3 | 0.004 | 0.01 |
| dCDP | 47.3 ± 2.8 | 512.2 ± 49.6 | 0.0003 | 0.09 |
| fructose | 1025.9 ± 87.8 | 2142.1 ± 127.4 | 0.00002 | 0.48 |
| sarcosine | 68655.1 ± 4338.6 | 196243.0 ± 11649.0 | 0.00005 | 0.35 |
| adenosine | 20.2 ± 3.2 | 0.2 ± 0.2 | 0.001 | 99.15 |
| adenosyl-l-homocysteine | 406.3 ± 52.0 | 18.0 ± 10.4 | 0.0005 | 22.53 |
| ADP | 8212.1 ± 794.4 | 236.0 ± 31.7 | 0.0003 | 34.80 |
| AMP | 8363.1 ± 601.3 | 148.3 ± 13.3 | 0.0001 | 56.40 |
| ATP | 2574.2 ± 245.6 | 364.6 ± 39.1 | 0.0003 | 7.06 |
| fructose 6-phosphate | 13758.9 ± 1573.1 | 1751.5 ± 282.4 | 0.0005 | 7.86 |
| G16BP | 13206.5 ± 949.9 | 4038.6 ± 1093.9 | 0.00002 | 3.27 |
| GDP | 400.0 ± 30.3 | 32.6 ± 11.7 | 0.00003 | 12.27 |
| glutamine | 73176.0 ± 6580.4 | 23324.5 ± 2489.7 | 0.0002 | 3.14 |
| reduced glutathione | 13195.2 ± 1356.1 | 979.4 ± 996.1 | 0.00001 | 13.47 |
| hydroxyproline | 14657.2 ± 1208.7 | 4784.8 ± 348.3 | 0.0002 | 3.06 |
| l-ascorbic acid | 492.3 ± 16.7 | 167.8 ± 16.1 | 0.0000001 | 2.93 |
| NAD | 2814.0 ± 77.3 | 566.5 ± 31.2 | 0.0000008 | 4.97 |
| NADH | 19.7 ± 3.0 | 3.1 ± 0.6 | 0.001 | 6.39 |
| serine | 1426.0 ± 57.6 | 707.6 ± 48.2 | 0.000002 | 2.02 |
| UDP | 3266.4 ± 186.5 | 98.1 ± 15.6 | 0.00005 | 33.28 |
| UDP-GlcNAc | 106.7 ± 6.2 | 30.3 ± 2.5 | 0.00002 | 3.52 |
| xylitol | 336.1 ± 17.6 | 92.3 ± 6.0 | 0.00002 | 3.64 |
| 1-methyladenosine | 10.4 ± 1.2 | |||
| acetylcarnitine | 11929.6 ± 1467.6 | |||
| oxidized glutathione | 4575.3 ± 280.3 | |||
| glycine | 239.2 ± 12.5 | |||
| NADP | 1.3 ± 0.2 | |||
| 2-hydroxyglutaric acid | 107.9 ± 11.8 | |||
| 3-phosphoglyceric acid | 5568.8 ± 1099.5 | |||
| N-acetylneuraminic acid | 330.7 ± 159.0 |
FC: Myc-On/Myc-Off
To further assess the robustness of CIT-MS measurement, enrichment results for a few representative metabolites were compared with those using GC-MS (Table 3). The deviation between the two methods was calculated to determine measurement accuracy: (ECIT − EGC-MS)/ECIT, where ECIT is the enrichment value obtained from CIT, and EGC-MS is from GC-MS. Samples were collected at different time points from Myc-On cells post U-13C6-glucose labeling. Then, each sample was divided into two parts: one for GC-MS analysis and the other for CIT-MS analysis. As can be seen, CIT-MS is highly consistent with traditional MFA using GC-MS.
Table 3.
Deviation between CIT-MS and Traditional MFA Using GC-MS
| metabolite | 24 h | 48 h | 72 h | 120 h |
|---|---|---|---|---|
| serine | 0.11 | 0.05 | 0.13 | 0.02 |
| alanine | 0.03 | 0.08 | 0.07 | 0.07 |
| asparagine | 0.14 | 0.02 | 0.16 | 0.06 |
| lactate | 0.08 | 0.07 | 0.06 | 0.05 |
| succinate | 0.15 | 0.25 | 0.11 | 0.03 |
| glutamate | 0.03 | 0.05 | 0.10 | 0.07 |
DISCUSSION
MFA plays an important role in systems biology research as a highly useful technique to determine dynamic metabolic reactions in the metabolome.1,19,32,35,40,70–72 However, metabolite coverage in MFA is limited to a relatively small number of metabolites, mostly of central carbon metabolism. In addition, MS detection in MFA requires optimal sensitivity, since isotopic peaks tend to exponentially decrease with m/z. While a number of previous approaches have been developed,26,27,34,70,73 broad and reliable detection of cellular flux currently remains a challenge.
In this study, we have described an innovative CIT-MS strategy to enable broad MFA (Figure 1) for the reliable and routine measurement of biological systems such as cell models. CIT-MS is based on LC-MS/MS detection, since it provides excellent sensitivity for detecting exponentially decaying isotopic signals. In addition, large-scale LC-MS/MS has been established to measure levels of up to ~1,900 metabolites/features.50,51 Toward turning this approach into a practical tool in MFA, we have developed and optimized LC-MS/MS data collection and analysis approaches, isotopic peak identification methods, and computational algorithms. Starting with a list of 310 metabolites for which all (unlabeled) chemical standards were available in-house, the isotopic MRMs were assembled depending on the number of carbon atoms (for 13C-tracking) in the precursor and product ions. After data collection, MID and ME were calculated to correct for natural abundance. Similar to KFP,25–28 our novel approach was developed to study flux but was able to derive relative flux, which is highly useful in comparative MFA. Taking this pilot study for example, accurate quantitation of 46 metabolites will cost a lot of time, effort, and expense; for best quantitation, stable isotope labeled internal standards (iSTDs) are often required, but many iSTDs are not commercially available and/or very expensive. In contrast, the approach described herein does not require absolute quantitation, which significantly reduces experimental efforts in terms of both time and complexity. Notably, our method still well meets the requirement to investigate flux changes, when comparing biological systems under different status. In an initial validation, our results indicated excellent CIT-MS performance in MFA as determined by glucose isotopologue standards (Figure 2). Cumulatively, these data showed that CIT-MS has good analytical performance for reliable and broad MFA.
In a proof-of-principle study using Myc-On and Myc-Off Tet21N cells, 142 of 310 unlabeled metabolites were detected. We further found that 80 metabolites were able to be enriched by U-13C6-glucose (Figure 3), most of which achieved steady state within 96 h of isotopic culturing (Figures 4 and S2–S4). The PCA score plot showed Myc-On and Myc-Off cells to have dramatically different enrichment profiles under the steady state (Figure 5A). Univariate testing showed significant (p < 0.05) enrichment differences in 37 metabolites, nine of which also had FC > 2 or < 0.5 (Figure 5B). These findings suggest our novel CIT-MS method to be highly applicable and scientifically relevant to Myc(oncogene)-related metabolic reprogramming.40,61,65,66
We further applied CIT-MS in a pseudosteady state MFA study. Within 27 h of isotopic culturing, we determined 46 metabolites for which disappearance of the unlabeled form followed an exponential decay function. Based on fR values, Myc-On and Myc-Off cells clearly had differential flux profiles under the pseudosteady state (Figure 6). In particular, 8 metabolites had significantly downregulated flux in Myc-On cells (p < 0.05 and FC < 0.5), and 18 metabolites were significantly upregulated (p < 0.05 and FC > 2) (Table 2). Interestingly, 5 and 3 metabolites had relative flux present only in Myc-On and Myc-Off cells, respectively. Notably, some of these metabolites were rarely connected to the Myc oncogene or more broadly, cancer.
A primary argument for CIT-MS over existing MFA approaches is broader coverage of the metabolome. For instance, a comparison of various methods for flux analysis of Myc-induced metabolic reprogramming in B-cells showed isotopically nonstationary 13C flux analysis to achieve maximum resolution, although enrichment could only be determined for 18 amino acids.40 A recent comprehensive 13C-MFA of E. coli metabolism using traditional GC-MS returned seven validated metabolites for analysis, all representative of central carbon metabolism.71 In contrast, 142 targeted metabolites were reliably detected using our proposed method; while 80 were found to be enriched, and 46 were successfully fitted to exponential decay. Therefore, CIT-MS provides increased metabolome coverage for routine flux analysis, a readily apparent benefit to current techniques. In addition, some studies have proposed methods for high-resolution (HR) 13C MFA.74–79 CIT-MS can be used as a complementary approach to HR 13C MFA, and based on MRM detection, it provides unique advantages of high sensitivity and specificity which are highly useful to access low-abundance metabolites and isotopic peaks.80
While detailed biological interpretation is out of the scope of this study, our MFA results from Tet21N cells fit well with previous studies. As is well-known, Myc causes significant alterations in global gene and protein expression, and activation of the Myc oncogene facilitates proliferation through metabolic reprogramming.36,40,61,65,66 Under the steady state, we found that significant metabolites in Myc-On cells all had low enrichment compared to Myc-Off cells, especially amino acid and TCA cycle metabolites (Figure 5B). Additionally, fR values of many TCA cycle metabolites were decreased in Myc-On cells under the pseudosteady state, while glutamine flux was increased in these cells (Table 2). These results indicate that Myc-On cells are less dependent on glucose in the metabolome. In fact, previous studies showed that Myc-induced metabolic reprogramming resulted in glutamine addiction, despite the abundant supply of glucose. In addition, glutamine addiction was correlated with Myc-induced redirection of glucose carbon away from mitochondria, as a result of the Warburg effect.66,81,82 Furthermore, it would be very interesting to carry out further biological validation of the significant metabolites in Figure 5B and Table 2, especially those present only in one group of cells (Myc-On or Myc-Off).
The major limitation to implementation of CIT-MS is that the whole process is not fully automated yet. To monitor the full list of MRMs using LC-MS/MS, 7 injections are needed for each sample: 5 and 2 in positive and negative ionizationmode, respectively, using the same collision energy and cell accelerator voltage for both polarities. Currently, we are making significant efforts to the analytical process to accommodate automation and assist with the deconvolution/computation process. Eventually, the CIT-MS method will be simple to conduct, incorporating an add-in package into the commercial software. A second limitation is the realized MFA coverage. In theory, CIT-MS is applicable to any metabolite detectable by LC-MS/MS; we started with ~310 metabolites in the detection panel of our large-scale LC-MS/MS assay that are of strong biological significance.45,48,49,51–57 In this proof-of-principle project using Myc-On/Off cells as an example, although a marked improvement was seen compared to previous studies,40,71 only 142 of 310 targeted metabolites were detected, and flux estimates were measured for only 46 of those metabolites. While this may be inherently due to Myc-biology,36,40,65,66,81 CIT-MS generally requires a careful selection of metabolites and metabolic pathways to target.
CONCLUSION
In summary, we have developed a novel CIT-MS method for systemic and broad MFA and have successfully applied it for the discovery of altered metabolite enrichment and flux in cell metabolic reprogramming. CIT-MS is able to cover flux of a variety of metabolites, including some previously less-studied ones. Relative flux provides a simple approach to compare flux of a metabolite from different sample groups, which is sufficient in many biological studies. CIT-MS is innovative and powerful and carries the advantages of broad coverage, easy implementation, fast throughput, and, more importantly, high fidelity and accuracy. In addition, our approach can be easily expanded to include other tracers (such as 15N-tracers), a wide range of other metabolites, and various biological systems, such as gut microbiome, animal models, and humans. Therefore, it is expected that the broader impacts of CIT-MS are multifaceted and include analytical technique developments that will ultimately provide a large number of scientists with highly reliable and robust methods and tools for characterizing metabolic flux in complex biological systems.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH (1R01ES030197-01, 1P01HL146369-01A1) and the College of Health Solutions at Arizona State University.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c01767.
Table S3, regression analysis of Figure 2 data; Figure S1, pathway view of flux metabolites; Figure S2, enriched carbohydrate metabolites; Figure S3, enriched TCA cycle metabolites; Figure S4, enriched amino acid metabolites; Figure S5, exponential fitting of time-course nucleotide data; Figure S6, exponential fitting of time-course carbohydrate data; Figure S7, exponential fitting of time-course TCA cycle data; and Figure S8, exponential fitting of time-course amino acid data (PDF)
The authors declare no competing financial interest.
Contributor Information
Xiaojian Shi, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Scottsdale, Arizona 85259, United States.
Bowei Xi, Department of Statistics, Purdue University, West Lafayette, Indiana 47907, United States.
Paniz Jasbi, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Scottsdale, Arizona 85259, United States.
Cassidy Turner, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Scottsdale, Arizona 85259, United States.
Yan Jin, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Scottsdale, Arizona 85259, United States.
Haiwei Gu, Arizona Metabolomics Laboratory, College of Health Solutions, Arizona State University, Scottsdale, Arizona 85259, United States.
REFERENCES
- (1).Deberardinis RJ; Thompson CB Cell 2012, 148 (6), 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rabinowitz JD; White E Science 2010, 330 (6009), 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Buescher JM; Antoniewicz MR; Boros LG; Burgess SC; Brunengraber H; Clish CB; DeBerardinis RJ; Feron O; Frezza C; Ghesquiere B; Gottlieb E; Hiller K; Jones RG; Kamphorst JJ; Kibbey RG; Kimmelman AC; Locasale JW; Lunt SY; Maddocks ODK; Malloy C; Metallo CM; Meuillet EJ; Munger J; Nöh K; Rabinowitz JD; Ralser M; Sauer U; Stephanopoulos G; St-Pierre J; Tennant DA; Wittmann C; Vander Heiden MG; Vazquez A; Vousden K; Young JD; Zamboni N; Fendt SM Curr. Opin. Biotechnol 2015, 34, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hsu PP; Sabatini DM Cell 2008, 134 (5), 703–707. [DOI] [PubMed] [Google Scholar]
- (5).Ridaura VK; Faith JJ; Rey FE; Cheng J; Duncan AE; Kau AL; Griffin NW; Lombard V; Henrissat B; Bain JR; Muehlbauer MJ; Ilkayeva O; Semenkovich CF; Funai K; Hayashi DK; Lyle BJ; Martini MC; Ursell LK; Clemente JC; Van Treuren W; Walters WA; Knight R; Newgard CB; Heath AC; Gordon JI Science 2013, 341 (6150), 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Escobedo N; Oliver G Cell Metab. 2017, 26 (4), 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cunnane S; Nugent S; Roy M; Courchesne-Loyer A; Croteau E; Tremblay S; Castellano A; Pifferi F; Bocti C; Paquet N; Begdouri H; Bentourkia M; Turcotte E; Allard M; Barberger-Gateau P; Fulop T; Rapoport SI Nutrition 2011, 27 (1), 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Di Domenico F; Barone E; Perluigi M; Butterfield DA Antioxid. Redox Signaling 2017, 26 (8), 364–387. [DOI] [PubMed] [Google Scholar]
- (9).Ahn CH; Min SH; Lee D-H; Oh TJ; Kim KM; Moon JH; Choi SH; Park KS; Jang HC; Ha J; Sherman AS; Lim S J. Clin. Endocrinol. Metab 2017, 102 (8), 2905–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Matsumoto I; Kuhara T Mass Spectrom. Rev 1996, 15 (1), 43–57. [DOI] [PubMed] [Google Scholar]
- (11).Vander Heiden MG; Cantley LC; Thompson CB Science 2009, 324 (5930), 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kim JW; Dang CV Cancer Res. 2006, 66 (18), 8927–8930. [DOI] [PubMed] [Google Scholar]
- (13).Zamboni N; Fendt SM; Rühl M; Sauer U Nat. Protoc 2009, 4 (6), 878–892. [DOI] [PubMed] [Google Scholar]
- (14).Chokkathukalam A; Kim DH; Barrett MP; Breitling R; Creek DJ Bioanalysis 2014, 6 (4), 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bodamer OAF; Halliday D Arch. Dis. Child 2001, 84 (5), 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sellers K; Fox MP; Ii MB; Slone SP; Higashi RM; Miller DM; Wang Y; Yan J; Yuneva MO; Deshpande R; Lane AN; Fan TWM. J. Clin. Invest 2015, 125 (2), 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Antoniewicz MR Curr. Opin. Biotechnol 2013, 24 (6), 973–978. [DOI] [PubMed] [Google Scholar]
- (18).Fan TWM; Lane AN J. Biomol. NMR 2011, 49 (3–4), 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Fan TW-M; Lane AN The Handbook of Metabolomics; Higashi RM, Ed.; Springer: New York, 2012; Vol. 17, DOI: 10.1007/978-1-61779-618-0. [DOI] [Google Scholar]
- (20).Le A; Lane AN; Hamaker M; Bose S; Gouw A; Barbi J; Tsukamoto T; Rojas CJ; Slusher BS; Zhang H; Zimmerman LJ; Liebler DC; Slebos RJC; Lorkiewicz PK; Higashi RM; Fan TWM; Dang CV Cell Metab. 2012, 15 (1), 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lane AN; Fan TWM; Xie Z; Moseley HNB; Higashi RM Anal. Chim. Acta 2009, 651 (2), 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lane AN; Fan TWM; Bousamra M; Higashi RM; Yan J; Miller DM OMICS 2011, 15 (3), 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dettmer K; Aronov PA; Hammock BD Mass Spectrom. Rev 2007, 26 (1), 51–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Want EJ; Nordström A; Morita H; Siuzdak G J. Proteome Res. 2007, 6 (2), 459–468. [DOI] [PubMed] [Google Scholar]
- (25).Yuan J; Fowler WU; Kimball E; Lu W; Rabinowitz JD Nat. Chem. Biol 2006, 2 (10), 529–530. [DOI] [PubMed] [Google Scholar]
- (26).Yuan J; Bennett BD; Rabinowitz JD Nat. Protoc 2008, 3 (8), 1328–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cobbold SA; Vaughan AM; Lewis IA; Painter HJ; Camargo N; Perlman DH; Fishbaugher M; Healer J; Cowman AF; Kappe SHI; Llinas M J. Biol. Chem 2013, 288 (51), 36338–36350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wu C; Xiong W; Dai J; Wu Q J. Phycol. 2016, 52 (1), 116–124. [DOI] [PubMed] [Google Scholar]
- (29).Winter G; Krömer JO Environ. Microbiol 2013, 15 (7), 1901–1916. [DOI] [PubMed] [Google Scholar]
- (30).Molero G; Aranjuelo I; Teixidor P; Araus JL; Nogúes S. Rapid Commun. Mass Spectrom 2011, 25 (5), 599–607. [DOI] [PubMed] [Google Scholar]
- (31).Klein S; Heinzle E Wiley Interdiscip. Rev. Syst. Biol. Med 2012, 4 (3), 261–272. [DOI] [PubMed] [Google Scholar]
- (32).Millard P; Letisse F; Sokol S; Portais J-C Bioinformatics 2012, 28 (9), 1294–1296. [DOI] [PubMed] [Google Scholar]
- (33).Nöh K; Wiechert W Appl Microbiol. Biotechnol 2011, 91 (5), 1247–1265. [DOI] [PubMed] [Google Scholar]
- (34).Mairinger T; Hann S Anal. Bioanal Chem 2017, 409 (15), 3713–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Noack S; Nöh K; Moch M; Oldiges M; Wiechert W J. Biotechnol 2011, 154 (2–3), 179–190. [DOI] [PubMed] [Google Scholar]
- (36).Carroll PA; Diolaiti D; McFerrin L; Gu H; Djukovic D; Du J; Cheng PF; Anderson S; Ulrich M; Hurley JB; Raftery D; Ayer DE; Eisenman RN Cancer Cell 2015, 27 (2), 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Du J; Rountree A; Cleghorn WM; Contreras L; Lindsay KJ; Sadilek M; Gu H; Djukovic D; Raftery D; Satrústegui J; Kanow M; Chan L; Tsang SH; Sweet IR; Hurley JB J. Biol. Chem 2016, 291 (9), 4698–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tedeschi PM; Johnson-Farley N; Lin H; Shelton LM; Ooga T; Mackay G; Van Den Broek N; Bertino JR; Vazquez A Cancer Metab. 2015, 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Suthers PF; Burgard AP; Dasika MS; Nowroozi F; Van Dien S; Keasling JD; Maranas CD Metab. Eng 2007, 9 (5–6), 387–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Murphy TA; Dang CV; Young JD Metab. Eng 2013, 15 (1), 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mosconi L Eur. J. Nucl. Med. Mol. Imaging 2005, 32 (4), 486–510. [DOI] [PubMed] [Google Scholar]
- (42).Cajka T; Fiehn O Anal. Chem 2016, 88 (1), 524–545. [DOI] [PubMed] [Google Scholar]
- (43).Buas MF; Gu H; Djukovic D; Zhu J; Drescher CW; Urban N; Raftery D; Li CI Gynecol. Oncol 2016, 140 (1), 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Fang N; Yu S; Adams SH; Ronis MJJ; Badger TM J. Lipid Res 2016, 57 (10), 1917–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jasbi P; Mitchell NM; Shi X; Grys TE; Wei Y; Liu L; Lake DF; Gu H J. Proteome Res 2019, 18 (7), 2791–2802. [DOI] [PubMed] [Google Scholar]
- (46).Lv W; Wang L; Xuan Q; Zhao X; Liu X; Shi X; Xu G Anal. Chem 2020, 92, 6043–6050. [DOI] [PubMed] [Google Scholar]
- (47).Wang Y; Liu F; Li P; He C; Wang R; Su H; Wan J-B Anal. Chim. Acta 2016, 927, 82–88. [DOI] [PubMed] [Google Scholar]
- (48).Jasbi P; Baker O; Shi X; Gonzalez LA; Wang S; Anderson S; Xi B; Gu H; Johnston CS Food Funct. 2019, 10 (11), 7343–7355. [DOI] [PubMed] [Google Scholar]
- (49).Jasbi P; Wang D; Cheng SL; Fei Q; Cui JY; Liu L; Wei Y; Raftery D; Gu H J. Chromatogr. B: Anal. Technol Biomed. Life Sci 2019, 1105, 26–37. [DOI] [PubMed] [Google Scholar]
- (50).Gu H; Zhang P; Zhu J; Raftery D Anal. Chem 2015, 87 (24), 12355–12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Shi X; Wang S; Jasbi P; Turner C; Hrovat J; Wei Y; Liu J ; Gu H Anal. Chem 2019, 91 (21), 13737–13745. [DOI] [PubMed] [Google Scholar]
- (53).Yuan M; Breitkopf SB; Yang X; Asara JM Nat. Protoc 2012, 7 (5), 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Roberts LD; Souza AL; Gerszten RE; Clish CB Curr. Protoc. Mol. Biol 2012, 98 (1), 30.2.1–30.2.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gooding JR; Jensen MV; Newgard CB Arch. Biochem. Biophys 2016, 589, 120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Shah SH; Kraus WE; Newgard CB Circulation 2012, 126 (9), 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Jang C; Chen L; Rabinowitz JD Cell 2018, 173 (4), 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Reaves ML; Rabinowitz JD Curr. Opin. Biotechnol 2011, 22 (1), 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wishart DS; Feunang YD; Marcu A; Guo AC; Liang K ; Vázquez-Fresno RV; Sajed T; Johnson D; Li C; Karu N; Sayeeda Z; Lo E; Assempour N; Berjanskii M; Singhal S; Arndt D; Liang Y; Badran H; Grant J; Serra-Cayuela A; Liu Y; Mandal R; Neveu V; Pon A; Knox C; Wilson M; Manach C; Scalbert A Nucleic Acids Res. 2018, 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Guijas C; Montenegro-Burke JR; Domingo-Almenara X; Palermo A; Warth B; Hermann G; Koellensperger G; Huan T; Uritboonthai W; Aisporna AE; Wolan DW; Spilker ME; Benton HP; Siuzdak G Anal. Chem 2018, 90 (5), 3156–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Liu J; Hanavan PD; Kras K; Ruiz YW; Castle EP; Lake DF; Chen X; O’Brien D; Luo H; Robertson KD; Gu H; Ho TH J. Proteome Res 2018, 18 (1), 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Gu H; Carroll PA; Du J; Zhu J; Neto FC; Eisenman RN; Raftery D Angew. Chem., Int. Ed 2016, 55 (50), 15646–15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Gu H; Du J; Carnevale Neto F; Carroll PA; Turner SJ; Chiorean EG; Eisenman RN; Raftery D Analyst 2015, 140 (8), 2726–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Chong J; Soufan O; Li C; Caraus I; Li S; Bourque G; Wishart DS; Xia J Nucleic Acids Res. 2018, 46 (W1), W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Creek DJ; Chokkathukalam A; Jankevics A; Burgess KEV; Breitling R; Barrett MP Anal. Chem 2012, 84 (20), 8442–8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Eberlin LS; Gabay M; Fan AC; Gouw AM; Tibshirani RJ; Felsher DW; Zare RN. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (29), 10450–10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Satoh K; Yachida S; Sugimoto M; Oshima M; Nakagawa T; Akamoto S; Tabata S; Saitoh K; Kato K; Sato S; Igarashi K; Aizawa Y; Kajino-Sakamoto R; Kojima Y; Fujishita T; Enomoto A; Hirayama A; Ishikawa T; Taketo MM; Kushida Y; Haba R; Okano K; Tomita M; Suzuki Y; Fukuda S; Aoki M; Soga T Proc. Natl. Acad. Sci. U. S. A 2017, 114 (37), E7697–E7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Ahn WS; Antoniewicz MR Metab. Eng 2013, 15 (1), 34–47. [DOI] [PubMed] [Google Scholar]
- (68).Alves TC; Pongratz RL; Zhao X; Yarborough O; Sereda S; Shirihai O; Cline GW; Mason G; Kibbey RG Cell Metab. 2015, 22 (5), 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Davis RJ; Gönen M; Margineantu DH; Handeli S; Swanger J; Hoellerbauer P; Paddison PJ; Gu H; Raftery D; Grim JE; Hockenbery DM; Margolin AA; Clurman BE Proc. Natl. Acad. Sci. U. S. A 2018, 115 (21), 5462–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).McCloskey D; Young JD; Xu S; Palsson BO; Feist AM Anal. Chem 2016, 88 (2), 1362–1370. [DOI] [PubMed] [Google Scholar]
- (71).Gonzalez JE; Long CP; Antoniewicz MR Metab. Eng 2017, 39, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Antoniewicz MR Exp. Mol. Med 2018, 50 (4), 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Balcells C; Foguet C; Tarragó-Celada J; de Atauri P; Marin S; Cascante M TrAC, Trends Anal. Chem 2019, 120, 115371. [Google Scholar]
- (74).Long CP; Antoniewicz MR Nat. Protoc 2019, 14 (10), 2856–2877. [DOI] [PubMed] [Google Scholar]
- (75).Wittmann C; Heinzle E Biotechnol. Bioeng 1999, 62 (6), 739–750. [DOI] [PubMed] [Google Scholar]
- (76).Schwaiger-Haber M; Hermann G; El Abiead Y; Rampler E; Wernisch S; Sas K; Pennathur S; Koellensperger G Anal. Bioanal. Chem 2019, 411 (14), 3103–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Llufrio EM; Cho K; Patti GJ Nat. Protoc 2019, 14 (7), 1970–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Long CP; Au J; Gonzalez JE; Antoniewicz MR Metab. Eng 2016, 38, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Hu Y; Zheng Q; Wanek W Anal. Chem 2017, 89 (17), 9192–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Choi J; Antoniewicz MR Front. Microbiol 2019, 10, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Gao P; Tchernyshyov I; Chang TC; Lee YS; Kita K; Ochi T; Zeller KI; De Marzo AM; Van Eyk JE; Mendell JT; Dang CV Nature 2009, 458 (7239), 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Altman BJ; Stine ZE; Dang CV Nat. Rev. Cancer 2016, 16 (10), 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.