

Abstract
As Covid-19 affects millions of people worldwide, the global health care will encounter an increasing burden of the aftermaths of the disease. Evidence shows that up to a fifth of the patients develop fibrotic tissue in the lung. The SARS outbreak in the early 2000 resulted in chronic pulmonary fibrosis in a subset (around 4%) of the patients, and correlated to reduced lung function and forced expiratory volume (FEV). The similarities between corona virus infections causing SARS and Covid-19 are striking, except that the novel coronavirus, SARS-CoV-2, has proven to have an even higher communicability. This would translate into a large number of patients seeking care for clinical signs of pulmonary fibrosis, given that the Covid-19 pandemic has up till now (Sept 2020) affected around 30 million people. The SARS-CoV-2 is dependent on binding to the angiotensin converting enzyme 2 (ACE2), which is part of the renin-angiotensin system (RAS). Downregulation of ACE2 upon virus binding disturbs downstream activities of RAS resulting in increased inflammation and development of fibrosis. The poor prognosis and risk of developing pulmonary fibrosis are therefore associated with the increased expression of ACE2 in risk groups, such as obesity, heart disorders and aging, conferring plenty of binding opportunity for the virus and subsequently the internalization of ACE2, thus devoiding the enzyme from acting counter-inflammatory and antifibrotic. Identifying pathways that are associated with Covid-19 severity that result in pulmonary fibrosis may enable early diagnosis and individualized treatment for these patients to prevent or reduce irreversible fibrotic damage to the lung.
Keywords: Covid-19, Chronic pulmonary fibrosis, Angiotensin converting enzyme 2 (ACE2), Cytokines, Chemokines, Matrix metalloproteases, Extracellular matrix
Highlights
-
•
Covid-19 may lead to pulmonary fibrosis.
-
•
Urgency to monitor severe cases of Covid-19 longitudinally post-infection.
-
•
Convergent pathways in idiopathic pulmonary fibrosis and Covid-19.
-
•
Antifibrotic treatment in Covid-19.
1. Background
The 2019 Covid-19 pandemic caused by coronavirus SARS-CoV-2, has by September 2020 demanded close to one million people's lives, and affected 30 million people. In most patients, the infection is mild but with age, the risk of developing serious or fatal disease increases substantially in magnitudes of ten to up to 100-fold between patients below the age of 60 (0.38% mortality) and those older (up to 27% mortality) [1]. In fatal Covid-19 cases, diffuse alveolar damage (DAD) is prominent with fibrin exudates, hyaline membrane formation, hyperplasia and loss of the alveolar type II cells along with disruption of the basement membrane and thickening of the alveolar walls. This may lead to the collapse of the alveoli, edema and inflammation and cause fatal dyspnea. Emerging data show that up to 17% of Covid-19 patients develop pulmonary fibrosis [2]. Although mature fibrosis is not evident in most fatal cases, consolidations are associated with more severe disease developing over time [3]. Histologically, DAD is classified as acute, organizing or fibrosing, where fibrosing DAD is associated with longer duration of illness, hospitalization and mechanical ventilation [4]. The plausible risk of developing chronic pulmonary fibrosis over time in these patients needs to be considered, and therefore we aim to review up-to-date knowledge about this patient group. The severe acute respiratory syndrome (SARS) between 2002 and 2004, caused by a similar coronavirus (SARS-CoV), resulted in chronic pulmonary fibrosis in a small subset (around 4%) of the patients, which was evident 15 years after the original infection and correlated to reduced lung function and forced expiratory volume (FEV) [5]. It is still debatable whether signs of early fibrosis in the current Covid-19 pandemic foretell progress into pulmonary interstitial fibrosis disease or if it will resolve over time [6,7]. However, in light of the SARS epidemic outbreak in the early 2000, Covid-19 may in some susceptible patients develop into chronic pulmonary fibrosis [8], with high unmet clinical need for efficient treatment. Current research lag behind in identifying the patients susceptible for developing chronic pulmonary fibrosis although patient stratifications are starting to crystalize. Underlying chronic inflammation, and mitigation of the innate immune system, may underlie the cytokine storm seen in severe cases [9]. Still more research is needed to unravel which pathways are hi-jacked during Covid-19 to develop chronic pulmonary fibrosis, and this review aims to lean towards the knowledge built in the field of idiopathic pulmonary fibrosis (IPF) since striking resemblances exist.
1.1. The risk groups in Covid-19 and underlying mechanisms
The severity of Covid-19 varies widely in patients and risk increases with age, male gender, and people with underlying disorders such as cardiovascular disease, diabetes and obesity [[10], [11], [12]]. In fatal cases with Covid-19, patients have developed complications such as severe pneumonia, pulmonary edema, acute respiratory distress syndrome (ARDS), organ failure, and septic shock. Age and male gender have been shown to be independent risk factors for more severe disease development [13,14]. The alarming severe outcome in these groups of patients is partly caused by a “cytokine storm”, which calls for means to mitigate the inflammation. However, blocking single or several cytokines may be unfavourable in Covid-19 due to the virally induced lymphopenia and the abrogated type 1 interferon response seen in Covid-19. These phenotypes may hallmark a more severe Covid-19 infection, where the virus buys time to replicate by escaping the innate immune system. As reviewed by Quartuccio et al. several biological drugs, including anti-IL-6 and anti-IL-1 treatment, are promising in treating the cytokine storm, however, cautiousness is emphasized since quenching the immune response could result in a diminished viral clearance [9]. To prevent lung damage and fibrosis, it will be important to understand the underlying pathways that may be parallell but not always equal to virus clearance. Though, the thought to use novel antifibrotic therapies designed for IPF is appealing since several of them, for example molecules blocking intergrins and galectins in the TGF-beta pathway, also act to inhibit viral infection [8]. Yet another treatment in Covid-19 are PDE4 inhibitors (PDE4i). These have recently been suggested as promising molecules to beat the severe inflammation in Covid-19, acting to block infiltration of neutrophils, monocytes, and lymphocytes and to reduce inflammatory cytokine and chemokine production from these cells and from the lung epithelium [15].
The host immune response to the coronavirus infection differs between males and females and between young and old persons, which could be answering to the question why there is a discrepancy in the clinical outcome of SARS-CoV-2 infections [16]. The disparity in infection outcome between sexes may therefore be due to the enhanced innate and adaptive immune responses in females that could result in better defense against this virus [17]. Moreover, children are equally susceptible to the SARS-CoV-2, however, their immune system, being on higher alert due to common infections in childhood as well as vaccination protocols, result in a trained immune memory and may represent a cross protection against various pathogens including SARS-CoV-2. Children infected with the coronavirus also seem to maintain lymphocyte levels, which in adults are reduced and a clinical sign of bad prognosis. Lymphocytopenia is seen in 48% of adult Covid-19 patients [18].
1.2. The fibrotic link
As a result of the severe inflammatory response in serious Covid-19 disease, including a cytokine storm, regulatory pathways are activated to counteract and heal the damaged tissue. If imbalanced or prolonged, this results in a fibrotic response manifested as different patterns in computed tomography (CT) scans including interstitial thickening, ground glass opacities, irregular interface, coarse reticular pattern and parenchymal band [3,19]. Irregular interface and parenchymal band could predict early formation of pulmonary fibrosis and may contribute to the severity and fatality of the disease [3].
1.3. The consequences of ACE2 engagement in virus infection leading to fibrosis
The renin-angiotensin system (RAS) plays a key role in maintaining blood pressure homeostasis and salt balance, but is also critically involved in acute lung diseases such as ARDS. The RAS has been implicated in pulmonary fibrosis and especially the downstream actions of angiotensin I (Ang I), which is cleaved by angiotensin converting enzyme (ACE) into angiotensin II (Ang II). In this way, Ang II promotes inflammatory and fibrotic responses through the Ang II type 1 receptor (AT1R) [20]. The other arm in the RAS involves cleavage of Ang I into the Ang (1–9) petide by the angiotensin converting enzyme 2 (ACE2) and counteracts the ACE arm. Downstream events of Ang (1–9 ) results in a reduction of inflammation and fibrosis through the Mas receptor. In addition ACE2 also acts on Ang II by converting it into Ang (1–7) peptide, which further attenuates inflammation. The ACE2 thus functions as a negative regulator of the angiotensin system [20]. The involvement of the RAS system in Covid-19 infection is thoroughly reviewed by Furuhashi et al. [20]. The host entry of SARS-CoV-2 is mediated through binding to membrane-anchored ACE2, similar to SARS-CoV in SARS [21]. Further, the transmembrane protease serine 2 (TMPRSS2) and potentially related proteases e.g. ADAM17 promote SARS-CoV-2 entry by ACE2 cleavage, which increases viral uptake, in addition to activating the S protein of the virus for membrane fusion [21,22]. Soluble ACE2, circulating in the blood may on the other hand have a protective effect by binding the coronavirus, hindering endocytosis. In animal models of pulmonary fibrosis and in IPF (idiopathic pulmonary fibrosis) patients, the ACE2 is downregulated in the lung, thus deminishing the ACE2 pathway [23]. However, in critically ill Covid-19 patients, the ACE2 has been seen to be upregulated in affected lung tissue compared to adjacent healthy lung tissue, along with increased plasma levels of ACE2 [24]. Although increased soluble ACE2 may be beneficial with the downstream action of Ang-(1–7) counteracting inflammation and fibrosis, increased membrane bound ACE2, which is also upregulated in smokers, older people and diabetes, provides the virus with plenty of entry points [25,26]. Lessons could be learned from other organs, and in obesity and diabetes, as adipocytes from abdominal fat express ACE2, which may serve as a reservoir for the SARS-CoV-2, explaining these patients’ high risk of severe disease [27]. The poor prognosis and risk of developing pulmonary fibrosis are therefore associated with increased expression of ACE2 in risk groups, explained by enhanced binding opportunities for the virus and intervention with the ACE2 activity and its beneficial anti-inflammatory and antifibrotic effects. This further favors the ACE-/Ang II arm, pushing towards increased inflammation and fibrosis [28]. Thus, the RAS plays a critical role in the pathogenesis of Covid-19 and other lung diseases, illustrated in Fig. 1 to outline the molecular events involved in Covid-19 infection upon ACE2 binding and downstream events.
Fig. 1.
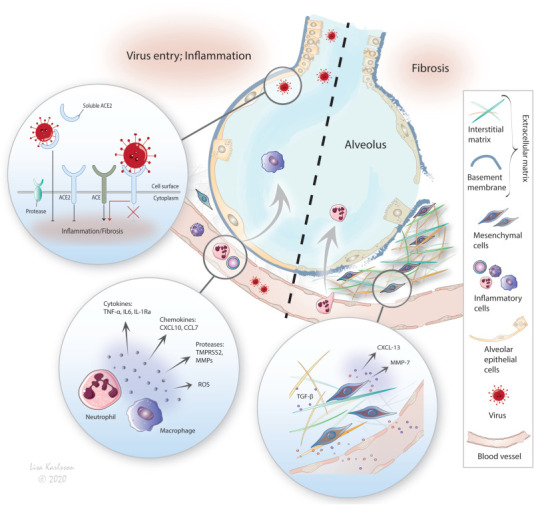
The SARS-CoV-2 depends on protease cleavage by TMPRSS2 of the ACE2 receptor and the spike protein of the virus for efficient cell invasion. The infection results in downregulation (endocytosis) of the ACE2 diminishing its anti-inflammatory and anti-fibrotic arm in the renin-angiotensin-system resulting in increased inflammation and subsequently fibrosis. ACE (angiotensin converting enzyme), TNF (tumor necrosis factor), IL (interleukin), CXCL, CCL (chemokines), TMPRSS2 (transmembrane protease serine 2), MMP (matrix metalloprotease).
1.4. Effector molecules that may contribute to fibrosis
Given the altered balance of the RAS in Covid-19 and in IPF [29], more similarities are shared between these diseases such as increased levels of VEGF and IL-6. Mechanistically, the ACE-/Ang II arm is activated upon increased IL-6 and reactive oxygen species (ROS). This onsets the TGF-beta pathway, which in turn is known to be highly involved in fibrosis [30]. In fact, IL-6 and IL-1 are known regulators of the inflammatory and also fibrotic response in IPF, also seen in Covid-19 patients [8] potentially contributing to the severe cytokine storm seen in these patients. In severe/fatal cases of Covid-19, the inflammatory mediators IP-10 (CXCL10), MCP-3 (CCL7) and IL-1 RA were clearly upregulated. Moreover, increased CXCL10 and another chemotactile cytokine MCP-2 (CCL8) have also been linked to ARDS for activating various immune cells and counteracting pulmonary fibrosis [31,32]. Interestingly, in our studies using an ex vivo model mimicking swift cellular responses in IPF [33], we found increased expression of CXCL13 (unpublished data), a chemokine usually expressed by a subset of T helper cells exposed to antigens and that has B cell chemoattractant properties. CXCL13 is identified as a predictive marker for IPF [34]. Also matrix metallopeptidase 7 (MMP7), which has also been suggested as a biomarker for IPF, was distinguished in our model. In line with these findings, coronaviruses depend on host MMPs and other proteases such as TMPRSS2, for cell infiltration, survival, and replication [21,35], further implicating converging pathways in pulmonary fibrosis and Covid-19.
1.5. The chronic inflammation confers fibrosis
It is established that chronic inflammation affects the tissue microenvironment and indeed, the massive inflammation seen in severe Covid-19 induces fibroblastic activation and increased production of extracellular matrix components, MMPs and tissue inhibitors of MMPs (TIMPs), which serve to counteract the damages inflicted by inflammation including ROS, proteases and other effector molecules. The wound healing response includes thickening of the parenchyma and induction of fibrosis in Covid-19, a process that alters the biomechanics of the lung and therefore lung function. Prolonged, these events may cause chronic pulmonary fibrosis similar to IPF, showing a stiff tissue with less compliance [36]. Importantly, the fibrosis renders altered biomechanical properties as more fibrosis increases tissue stiffness, although inhomogeneously, throughout the lung [37]. Therefore it will be important to also study the effect of altered biomechanics alongside with other parameters. In line with this, cells closely sense and respond to their surrounding milieu [38,39], where the stiff IPF lung tissue [40] is found to induce specific cellular responses and thus being highly involved in disease progression [33,41]. This self-propagating cellular answer towards the stiffer microenvironment induces enhanced deposition of extracellular matrix proteins, including deposition of glycosaminoglycans (GAGs). We have shown that there is significant change in amount and structure of GAGs in IPF [42], specifically, highly sulfated heparan sulphate was found in the border zone between highly fibrotic areas and more normal structures of the distal lung. Changes in GAGs will in turn affect the retaining and release of growth factors e.g. TNF-alpha, IL-6 and TGF-beta and their activities, which in turn contribute to altered cellular activity and further propagate fibrosis.
2. Conclusion
The risk factors for developing poor prognosis for Covid-19 include obesity, heart disorders and aging and a common denominator for these groups is chronic low grade inflammation with augmented baseline of IL-6 and TNF alpha [[43], [44], [45]]. Therefore, we conclude the importance of identifying subgroups of Covid-19 patients that have a high risk of developing chronic structural changes ultimately leading to pulmonary fibrosis. Identifying pathways, which are associated with Covid-19 severity, that results in pulmonary fibrosis may enable early diagnosis and individualized treatment for these patients to prevent or reduce irreversible fibrotic damage to the lung. Key to understanding the convergent disease mechanisms in pulmonary fibrosis and Covid-19, is the development of clinical translatable ex vivo models as well as a robust animal model. Promisingly, Israelow et al. reported on a platform for rapidly testing prophylactics and novel therapies in a mouse model that present a robust SARS-CoV-2 infection following inoculation with patient authentic virus [46]. This gives promise to solve some enigmas relating to Covid-19.
Declaration of competing interest
There is no conflict of interest of any of the authors.
Acknowledgement
This research was funded by the Swedish Heart-Lung Foundation, grant number20140293, the Swedish foundation for Strategic Research, grant number SBE13-0130, the Swedish research Council, grant number 2016-01190, the Royal Physiographic Society of Lund, the Olle Engkvist, the Crafoord Foundation, the Greta and John Kock Foundation, the Alfred Österlund Foundation, the Åke och Inger Bergkvist foundation, the Medical Faculty of Lund University and ALF.
List of abbreviations
- DAD
diffuse alveolar damage
- SARS
severe acute respiratory syndrome
- FEV
forced expiratory volume
- ARDS
acute respiratory distress syndrome
- CT
computed tomography
- RAS
renin-angiotensin system
- Ang I
angiotensin I
- ACE
angiotensin converting enzyme
- Ang II
angiotensin II
- AT1R
Ang II type 1 receptor
- ACE2
angiotensin converting enzyme 2
- TMPRSS2
transmembrane protease serine 2
- IPF
idiopathic pulmonary fibrosis
- ROS
reactive oxygen species
- CXCL10
inflammatory mediator IP-10
- CCL7
inflammatory mediator MCP-3
- CCL8
chemotactile cytokine MCP-2
- MMPs
matrix metallopeptidases
- TIMPs
tissue inhibitors of MMPs
- GAGs
glycosaminoglycans
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Authors' information
Jenny Wigén compiled the article including reference, evaluation, and writing; Anna Löfdahl contributed with references and writing regarding idiopathic pulmonary fibrosis, Linda Elowsson Rendin contributed with references and writing regarding idiopathic pulmonary fibrosis and biomechanics, Leif Bjermer contributed to medical evaluation of the references chosen, Gunilla Westergren-Thorsson contributed with scientific evaluation of the article.
Disclosure statement and conflict of interest
LB has during the last three years attended advisory board and o given lectures for the following companies: Airsonett, ALK, Astrazeneca, Boehringer. Chiesi, GlaxoSmithklein, Novartis, Sanofi Genentech and TEVA, outside the submitted work. GWT reports grants from Cantargia and Boeringer-Ingelheim, non-financial support from Astra-Zeneca, all three are, however, outside the submitted work. The other authors have nothing to disclose.
References
- 1.Verity R., Okell L.C., Dorigatti I. Estimates of the severity of coronavirus disease 2019: a model-based analysis. Lancet Infect. Dis. 2020;20(6):669–677. doi: 10.1016/S1473-3099(20)30243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Y., Guan H., Zhou S. Initial CT findings and temporal changes in patients with the novel coronavirus pneumonia (2019-nCoV): a study of 63 patients in Wuhan, China. Eur. Radiol. 2020;30:3306–3309. doi: 10.1007/s00330-020-06731-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye Z., Zhang Y., Wang Y. Chest CT manifestations of new coronavirus disease 2019 (COVID-19): a pictorial review. Eur. Radiol. 2020;30:4381–4389. doi: 10.1007/s00330-020-06801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y., Wu J., Wang S. Progression to fibrosing diffuse alveolar damage in a series of 30 minimally invasive autopsies with COVID-19 pneumonia in Wuhan, China. Histopathology. 2020 doi: 10.1111/his.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang P., Li J., Liu H. Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: a 15-year follow-up from a prospective cohort study. Bone Res. 2020;8:8. doi: 10.1038/s41413-020-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan Y., Guan H., Zhou S. Initial CT findings and temporal changes in patients with the novel coronavirus pneumonia (2019-nCoV): a study of 63 patients in Wuhan, China. Eur. Radiol. 2020;30:3306–3309. doi: 10.1007/s00330-020-06731-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan F, Ye T, Sun P, et al. Time course of lung changes on chest CT during recovery from 2019 novel coronavirus (COVID-19) pneumonia. Radiology 2020: 200370. [DOI] [PMC free article] [PubMed]
- 8.George P.M., Wells A.U., Jenkins R.G. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med. 2020;8(8):807–815. doi: 10.1016/S2213-2600(20)30225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quartuccio L., Semerano L., Benucci M. Urgent avenues in the treatment of COVID-19: targeting downstream inflammation to prevent catastrophic syndrome. Joint Bone Spine. 2020;87:191–193. doi: 10.1016/j.jbspin.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu C., Chen X., Cai Y. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020;180(7):934–943. doi: 10.1001/jamainternmed.2020.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sohrabi C., Alsafi Z., O'Neill N. World Health Organization declares global emergency: a review of the 2019 novel coronavirus (COVID-19) Int. J. Surg. 2020;76:71–76. doi: 10.1016/j.ijsu.2020.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian S., Xiong Y., Liu H. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod. Pathol. 2020 doi: 10.1038/s41379-020-0536-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D., Yin Y., Hu C. Clinical course and outcome of 107 patients infected with the novel coronavirus, SARS-CoV-2, discharged from two hospitals in Wuhan, China. Crit. Care. 2020;24:188. doi: 10.1186/s13054-020-02895-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen N., Zhou M., Dong X. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridgewood C., Damiani G., Sharif K. Rationale for evaluating PDE4 inhibition for mitigating against severe inflammation in COVID-19 pneumonia and beyond. Isr. Med. Assoc. J. 2020;22:335–339. [PubMed] [Google Scholar]
- 16.Li M.Y., Li L., Zhang Y. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty. 2020;9:45. doi: 10.1186/s40249-020-00662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaillon S., Berthenet K., Garlanda C. Sexual dimorphism in innate immunity. Clin. Rev. Allergy Immunol. 2019;56:308–321. doi: 10.1007/s12016-017-8648-x. [DOI] [PubMed] [Google Scholar]
- 18.Sungnak W., Huang N., Becavin C. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu M., Liu Y., Xu D. Prediction of the development of pulmonary fibrosis using serial thin-section CT and clinical features in patients discharged after treatment for COVID-19 pneumonia. Korean J. Radiol. 2020;21:746–755. doi: 10.3348/kjr.2020.0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furuhashi M., Moniwa N., Takizawa H. Potential differential effects of renin-angiotensin system inhibitors on SARS-CoV-2 infection and lung injury in COVID-19. Hypertens. Res. 2020;43:837–840. doi: 10.1038/s41440-020-0478-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann M., Kleine-Weber H., Schroeder S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280 e278. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heurich A., Hofmann-Winkler H., Gierer S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014;88:1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X., Molina-Molina M., Abdul-Hafez A. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008;295:L178–L185. doi: 10.1152/ajplung.00009.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H., Shang W., Liu Q. Infection; China: 2020. Clinical Characteristics of 194 Cases of COVID-19 in Huanggang and Taian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalan R., Bornstein S.R., El-Armouche A. The ACE-2 in COVID-19: foe or friend? Horm. Metab. Res. 2020;52:257–263. doi: 10.1055/a-1155-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leung J.M., Yang C.X., Tam A. ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur. Respir. J. 2020;55 doi: 10.1183/13993003.00688-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruglikov I.L., Scherer P.E. The role of adipocytes and adipocyte-like cells in the severity of COVID-19 infections. Obesity. 2020;28(7):1187–1190. doi: 10.1002/oby.22856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tseng Y.H., Yang R.C., Lu T.S. Two hits to the renin-angiotensin system may play a key role in severe COVID-19. Kaohsiung J. Med. Sci. 2020;36(6):389–392. doi: 10.1002/kjm2.12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan W.S.D., Liao W., Zhou S. Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr. Opin. Pharmacol. 2018;40:9–17. doi: 10.1016/j.coph.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Eid R.A., Alkhateeb M.A., El-Kott A.F. A high-fat diet rich in corn oil induces cardiac fibrosis in rats by activating JAK2/STAT3 and subsequent activation of ANG II/TGF-1beta/Smad3 pathway: the role of ROS and IL-6 trans-signaling. J. Food Biochem. 2019;43 doi: 10.1111/jfbc.12952. [DOI] [PubMed] [Google Scholar]
- 31.Keane M.P., Belperio J.A., Moore T.A. Neutralization of the CXC chemokine, macrophage inflammatory protein-2, attenuates bleomycin-induced pulmonary fibrosis. J. Immunol. 1999;162:5511–5518. [PubMed] [Google Scholar]
- 32.Jiang D., Liang J., Campanella G.S. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J. Clin. Invest. 2010;120:2049–2057. doi: 10.1172/JCI38644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elowsson Rendin L., Lofdahl A., Ahrman E. Matrisome properties of scaffolds direct fibroblasts in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20164013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han B.K., Kuzin I., Gaughan J.P. Baseline CXCL10 and CXCL13 levels are predictive biomarkers for tumor necrosis factor inhibitor therapy in patients with moderate to severe rheumatoid arthritis: a pilot, prospective study. Arthritis Res. Ther. 2016;18:93. doi: 10.1186/s13075-016-0995-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips J.M., Gallagher T., Weiss S.R. Neurovirulent murine coronavirus JHM.SD uses cellular zinc metalloproteases for virus entry and cell-cell fusion. J. Virol. 2017;91 doi: 10.1128/JVI.01564-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marinelli J.P., Levin D.L., Vassallo R. Quantitative assessment of lung stiffness in patients with interstitial lung disease using MR elastography. J. Magn. Reson. Imag. 2017;46:365–374. doi: 10.1002/jmri.25579. [DOI] [PubMed] [Google Scholar]
- 37.Melo E., Cardenes N., Garreta E. Inhomogeneity of local stiffness in the extracellular matrix scaffold of fibrotic mouse lungs. J. Mech. Behav. Biomed. Mater. 2014;37:186–195. doi: 10.1016/j.jmbbm.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 38.Vogel V. Unraveling the mechanobiology of extracellular matrix. Annu. Rev. Physiol. 2018;80:353–387. doi: 10.1146/annurev-physiol-021317-121312. [DOI] [PubMed] [Google Scholar]
- 39.Asano S., Ito S., Takahashi K. Matrix stiffness regulates migration of human lung fibroblasts. Phys. Rep. 2017;5 doi: 10.14814/phy2.13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haak A.J., Tan Q., Tschumperlin D.J. Matrix biomechanics and dynamics in pulmonary fibrosis. Matrix Biol. 2018;73:64–76. doi: 10.1016/j.matbio.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parker M.W., Rossi D., Peterson M. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Invest. 2014;124:1622–1635. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hallgren O., Nihlberg K., Dahlback M. Altered fibroblast proteoglycan production in COPD. Respir. Res. 2010;11:55. doi: 10.1186/1465-9921-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kern L., Mittenbuhler M.J., Vesting A.J. Obesity-induced TNFalpha and IL-6 signaling: the missing link between obesity and inflammation-driven liver and colorectal cancers. Cancers. 2018:11. doi: 10.3390/cancers11010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maggio M., Guralnik J.M., Longo D.L. Interleukin-6 in aging and chronic disease: a magnificent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2006;61:575–584. doi: 10.1093/gerona/61.6.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markousis-Mavrogenis G., Tromp J., Ouwerkerk W. The clinical significance of interleukin-6 in heart failure: results from the BIOSTAT-CHF study. Eur. J. Heart Fail. 2019;21:965–973. doi: 10.1002/ejhf.1482. [DOI] [PubMed] [Google Scholar]
- 46.Israelow B., Song E., Mao T. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J. Exp. Med. 2020:217. doi: 10.1084/jem.20201241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.