

Abstract
Objectives
This phase I study aimed to evaluate the antitumor effect and safety of programmed death‐ligand‐1 (PD‐L1)–targeting autologous chimeric antigen receptor T (CAR‐T) cells for patients with non‐small cell lung cancer (NSCLC).
Methods
Programmed death‐ligand‐1–specific CAR‐T cells were generated using lentiviral transduction. Four patients with NSCLC were recruited, but only one patient was finally involved. CAR‐T cells were infused on three different days (total dose during therapy, 1 × 106 CAR‐T cells kg−1 body weight). The date on which the patient received the first CAR‐T cell infusion was designated as Day 0.
Results
Circulating CAR‐T cells accounted for 3.30% of the patient’s peripheral blood T cells detected by FACS analysis during the first follow‐up (Day +29). The chest CT scan showed subtle tumor shrinkage (stable disease). On Day +43, the patient developed pyrexia without any known causes and dyspnoea that rapidly deteriorated to respiratory failure in 3 days. The chest X‐ray and CT scan showed bilateral extensive pulmonary infiltration in addition to the tumor silhouette on the left upper lung. The interleukin (IL)‐6 levels in serum dramatically increased (> 100‐fold). The patient was immediately transferred to the ICU where he received oxygen and intravenous infusions of tocilizumab and methylprednisolone. His symptoms rapidly improved and the pulmonary inflammation gradually resolved.
Conclusion
The clinical manifestations and test findings for this patient with NSCLC might represent unique clinical manifestations of solitary organ damage secondary to PD‐L1–specific CAR‐T cell therapy. The differential diagnosis, underlying mechanism and prevention and treatment strategies for such complications have also been discussed.
Keywords: CAR‐T, IL‐6, NSCLC, PD‐L1, pulmonary toxicity
We studied the circulating PD‐L1–specific CAR‐T cells in a patient with non‐small cell lung cancer. Circulating PD‐L1–specific CAR‐T cells accounted for 3.30% of the patient’s peripheral blood T cells detected by FACS in the first follow‐up (Day +29), and a chest CT scan showed subtle shrinkage of tumors (stable disease). However, the patient was immediately transferred to the ICU on Day +47 because he deteriorated quickly into respiratory failure in three days. This case might represent a unique clinical manifestation of solitary organ damage secondary to PD‐L1–specific CAR‐T cell therapy.

Introduction
Adoptive T‐cell therapy (ATC) involving CD19‐specific chimeric antigen receptor T (CAR‐T) cells has been highly successful in the treatment of hematopoietic malignancies. 1 , 2 In such approaches, CAR‐T cells are genetically engineered to express CAR molecules that usually comprise an artificial fusion protein composed of an extracellular antigen‐binding motif, transmembrane domain and intracellular T‐cell activation signalling moieties (CD28, 4‐1BB and CD3zeta). 3 , 4 Although therapy involving CD19‐specific CAR‐T cells has provided up to 92% complete response in adult patients with relapsed or refractory B‐cell acute lymphoblastic leukaemia (ALL), the use of CAR‐T cells for treating malignant solid tumors involves several challenges. One of the major limitations is the identification of tumor‐specific surface antigens for CAR, in addition to issues such as the capability of CAR‐T cells to home to tumor tissues and overcoming the tumor immunosuppressive environment. 5
Cancer immunity mediated by T cells is closely regulated by inhibitory pathways and activating signals. 6 , 7 Programmed death‐ligand‐1 (PD‐L1)/programmed death‐1 (PD‐1) is one of the main immune checkpoints. Cancer cells show induced or constitutive expression of PD‐L1, which can bind to its receptor PD‐1 expressed on T cells; this leads to the downregulation of T‐cell effector functions, proliferation and survival, thus contributing to the evasion of host immune attacks. 8 , 9 Blockade of this interaction by therapeutic anti‐PD‐1 or anti‐PD‐L1 antibodies could relieve the inhibition of T cells and restore tumor‐specific T‐cell functions, which has implications for clinical treatment. 9 , 10 Remarkable progress has been achieved because of the development and approval of PD‐L1/PD‐1‐blocking monoclonal antibodies for the management of non‐small cell lung cancer (NSCLC). 11 However, in a low percentage (< 20%) of patients with NSCLC, treatment responses to these interventions are accompanied by severe adverse reactions such as autoimmune disorders, leading to lethal cardiotoxicity and interstitial pneumonitis. 12 , 13
Programmed death‐ligand‐1 is expressed not only on tumor cells but also on myeloid‐derived suppressive cells (MDSCs) in the tumor environment, further complicating immune tolerance. 9 , 14 However, only trace PD‐L1 RNA levels have been detected in vital organs, such as the heart, lung, kidney and liver. 15 We envisioned that PD‐L1–specific CAR‐T cells would migrate to and be activated in tumor lesions and specifically kill PD‐L1–positive lung cancer cells and MDSCs, thus greatly improving the immunosuppressive tumor microenvironment and enhancing immunotherapeutic efficacy. The preclinical experiments also supported this hypothesis, and PD‐L1–specific CAR‐T cells showed potent in vitro and in vivo antitumor effects. However, the risk of the on‐target off‐tumor toxicities of human PD‐L1–specific CAR‐T cells could not be excluded in the mouse xenograft experiments because of the antigenic differences in mouse PD‐L1. Here, we report a case of severe delayed pulmonary toxicity that occurred following infusions of human PD‐L1–specific CAR‐T cells in our clinical trial. We have also elaborated on the differential diagnosis, underlying mechanisms and strategies for preventing or treating such complications.
Results
Human PD‐L1–specific CAR‐T cells
Transduction was performed with a multiplicity of infection (MOI) of 4.57:1 and was followed by 2 days of culture; the autologous T cells were frozen in 7.5% dimethyl sulfoxide freezing medium and liquid nitrogen until use for the patient. Transduction efficiency was determined by fluorescence‐activated cell sorting (FACS) analysis of an aliquot of transduced T cells stained with biotinylated PD‐L1:human Fc fusion protein and allophycocyanin (APC)‐labelled streptavidin. The transduction efficiency was approximately 20% because of low MOI (Figure 1). To assess the functional activity of PD‐L1–specific CAR‐T cells, transduced T cells were co‐cultured with the PD‐L1–positive lung cancer cell lines H1975 and H3122. Compared to the control, that is CD38‐specific CAR‐T cells, PD‐L1 CAR‐T cells were found to be significantly cytotoxic towards PD‐L1–positive H1975 and H3122 cells. Moreover, PD‐L1 CAR‐T cells were more potent than PD1 fusion receptor (PD1‐CD28‐CD3zeta) T cells, indicating the strong killing effect of PD‐L1 CAR‐T cells on lung cancer (Figure 2).
Figure 1.
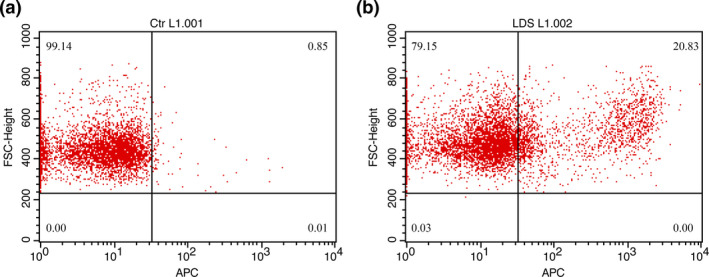
FACS results for autologous PD‐L1 CAR‐T cells. (a) Non‐transduced T cells were used as control. (b) Transduced T cells. Both a and b were stained with biotinylated PD‐L1::human Fc fusion protein and APC‐labelled streptavidin. Approximately 20% of the transduced cells were CAR‐positive T cells.
Figure 2.
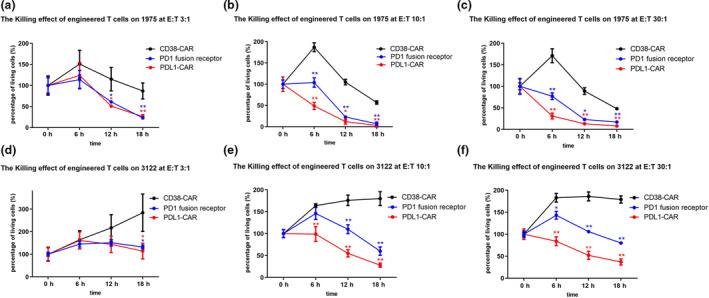
Cytotoxic effects of PD‐L1 CAR transduced T cells on lung carcinoma cells, as compared to the effects of PD‐1 fusion receptor T cells. The cell‐killing effects of PD‐1 fusion receptor (PD1‐CD28Z‐CD3zeta) T cells and PD‐L1 CAR‐T cells were investigated after co‐incubation with lung cancer cells in different effector to target ratios. CAR‐T cell killing by CD38 CAR cells, PD‐1 fusion receptor T cells and PD‐L1 CAR‐T cells were investigated after co‐incubation with lung cancer cells in different effector to target ratios (E:T = 3:1, 10:1 and 30:1, respectively). n = 3. (a–c) H1975 cells; (d–f) H3122 cells. CD38‐specific CAR‐T cells were included as the control. PD‐L1 CAR‐T cells were significantly more potent than PD‐1 fusion receptor T cells. One‐way ANOVA and the Student’s t‐test were used. *P‐value < 0.05;**P‐value < 0.01.
Patient evaluation
The patient was a 61‐year‐old man with a 40‐year history of heavy smoking. He had been diagnosed 4 years earlier with advanced lung adenocarcinoma of the left upper lobe with atelectasis. On Day −12, the maximum tumor diameter was found to be 78.6 mm on performing computed tomography (CT) imaging of the mediastinal window (Figure 3a), and the Eastern Cooperative Oncology Group (ECOG) performance status was estimated as 1. Pathological analysis confirmed the diagnosis of NSCLC; the tumor was a poorly differentiated adenocarcinoma (Figure 4a) with a proliferative rate of 40% (percentage of Ki67‐positive area). The PD‐L1 expression levels, as determined by immunohistochemistry (IHC) with the Ventana rabbit monoclonal antibody SP142, were heterogeneous; approximately 20% of the tumor tissue was positive for PD‐L1 (Figure 4b and e), whereas the surrounding normal lung tissues were negative (Figure 4c and f). Genetic analysis of the biopsy sample revealed that there were no mutations in epidermal growth factor receptor (EGFR), but there was a highly prevalent mutation in Kras (G12F). The patient received multiple rounds of combined chemotherapy (pemetrexed disodium, nedaplatin, docetaxel, gemcitabine and carboplatin), but the disease still progressed. He started experiencing hoarseness in May 2018 and found it difficult to speak; his speech deteriorated gradually. The patient has refused all antitumor therapies since then and has not received checkpoint blockade therapy either.
Figure 3.
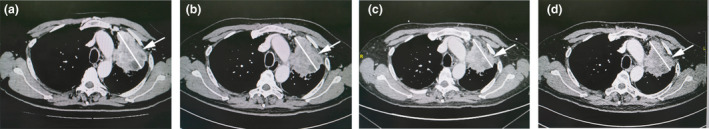
CT imaging of the mediastinal window of the patient. The results obtained on four representative days are shown. (a) Day −12 before the patient was administered CAR‐T cells: maximum tumor diameter, 78.6 mm; (b) Day +29, that is when the patient came for the first follow‐up: the maximum tumor diameter, 73.3 mm (indicating stable disease [SD]); (c) Day +49, that is 2 days after the patient was admitted because of ALL/ARDS and respiratory failure: maximum tumor diameter, 70.2 mm (SD); (d) Day +64, that is when the patient recovered from ALI/ARDS: maximum tumor diameter, 72.8 mm.
Figure 4.
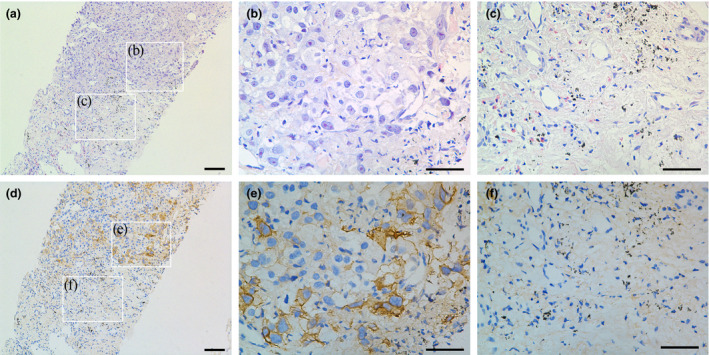
Pathological examination of the patient’s lung tumor via core needle biopsy. (a) Representative section of H.E. staining showing adenocarcinoma. Scale bar: 100 µm. (b, c) High‐power observations of tumor and non‐tumor regions in a. Scale bar: 50 μm. (d) Corresponding section of IHC staining. (e, f) High‐power observations of PD‐L1 expression in tumor and non‐tumor regions in d. Tumor cells were positive for PD‐L1 expression, whereas the adjacent non‐malignant lung tissue was negative. Scale bar: 50 µm.
Clinical course of human PD‐L1–specific CAR‐T cell treatment
After the patient underwent lymphodepletion treatments with fludarabine (25 mg m‐2) and cyclophosphamide (250 mg m−2) for three consecutive days, he was intravenously administered infusions of PD‐L1–specific CAR‐T cells on three different days (1 × 105 kg−1 on Day 0, 3 × 105 kg−1 on Day +3 and 6 × 105 kg−1 on Day +7). The infusions were initiated on 12 October 2018, which was designated as Day 0 in our clinical trial protocol. The CAR‐T cells were administered as planned, without any adverse reactions. The patient was discharged from hospital as per schedule (Supplementary figure 2). Subsequently, he developed pyrexia (38–39°C) without any known causes on Day +21; the pyrexia lasted for 2 days and was accompanied by intermittent dyspnoea. The patient recovered soon after using an over‐the‐counter nonsteroidal anti‐inflammatory drug (NSAID) at his physician’s direction. The patient came back on Day +29 for a scheduled follow‐up. His condition was stable, and there was no exacerbation of any signs or symptoms; his chest CT scan indicated subtle tumor shrinkage. The maximum tumor diameter was 73.3 mm, indicating stable disease (SD; Figure 3b). FACS analysis detected PD‐L1–specific CAR‐T cells in peripheral blood, which accounted for 3.30% of his peripheral blood T cells (Figure 5c and d), looks odd on its own on this line indicating high CAR‐T cell proliferation after infusion in relation to his absolute lymphocyte counts (Supplementary table 1).
Figure 5.
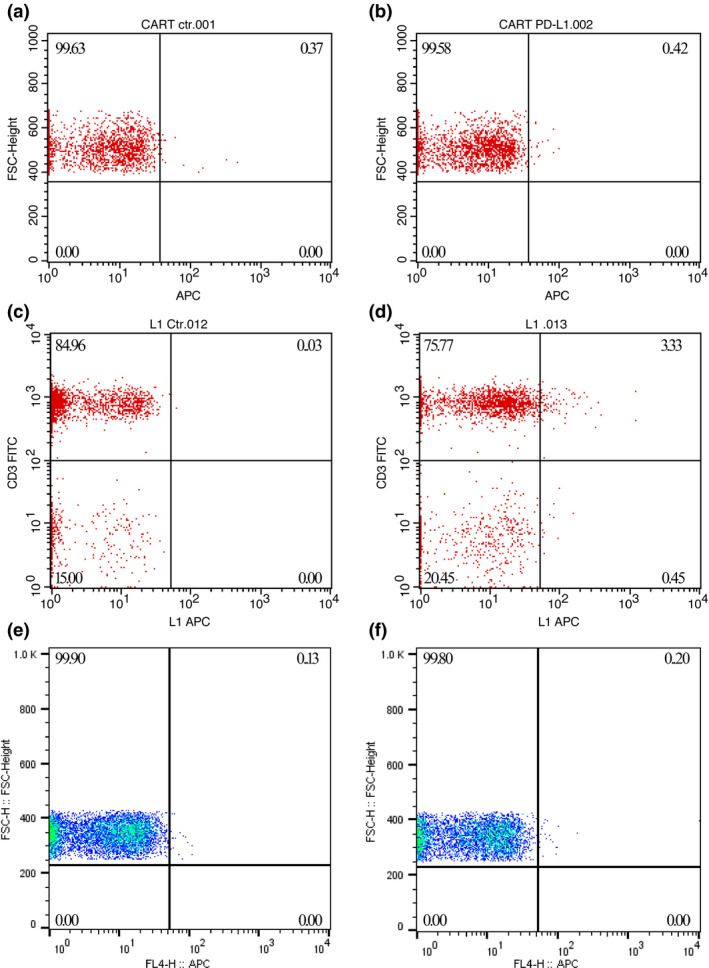
PD‐L1 CAR‐T cells proliferated greatly after transfusion in the patient’s circulation.(upper panels) No PD‐L1 CAR‐T cells were detected on Day +11, when the patient was discharged from the hospital after CAR‐T cell infusions. (a) Patient’s PBMCs stained with APC‐labelled streptavidin alone. (b) Patient’s PBMCs stained with both biotinylated PD‐L1::human Fc fusion protein and APC‐labelled streptavidin. (middle panels) Approximately 3.30% of the total T cells were CAR positive on Day +29. (c) Patient’s PBMCs stained with FITC CD3 and APC‐labelled streptavidin. (d) Patient’s PBMCs stained with FITC CD3 and both biotinylated PD‐L1::human Fc fusion protein and APC‐labelled streptavidin. (lower panels) PD‐L1 CAR‐T cells were undetectable after the patient developed ALI/ARDS on Day +48. (e, f) The staining patterns were the same as those in a and b, respectively.
The patient experienced pyrexia and dyspnoea again on Day +47 without any upper respiratory symptoms. The dyspnoea rapidly worsened, and the patient was rushed to the intensive care unit (ICU) in 3 days because of respiratory failure along with hypoxaemia and hypocapnia (PaO2, 57 mmHg; PaCO2, 32 mmHg) and arterial blood oxygen saturation of 91%. The chest X‐ray showed extensive bilateral pulmonary infiltration in addition to the silhouette of the upper left lung tumors (Figure 6a). However, except for tachycardia, the patient maintained stable circulatory functions, and his electrocardiogram (ECG) showed no S‐T segment abnormalities, except for sinus tachycardia. The serum cytokine assays showed that only the interleukin (IL)‐6 and C‐reactive protein (CRP) levels dramatically increased (1301.95 pg mL−1 and 83.46 mg L−1, respectively; Figure 7 and Table 1), while CAR‐engineered T cells could not be detected in the peripheral blood after tocilizumab and corticosteroid administration (Figure 5e and f). To confirm the cytokine levels, the patient’s sample was also sent to another third‐party laboratory for further analysis, which showed consistent results (Supplementary table 2).
Figure 6.
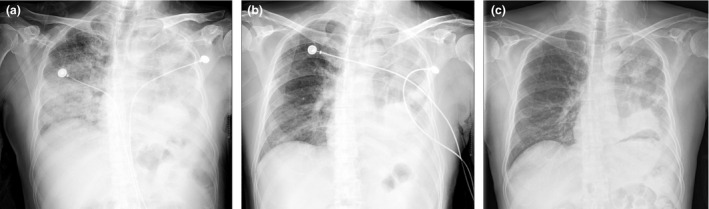
Bilateral extensive pulmonary infiltrates evolved with treatment. Representative chest X‐ray images obtained at the following time points have been provided: (a) when the patient developed ARDS and was admitted to the ICU on Day +47; (b) at 1 week after administration of corticosteroids and the IL‐6 receptor blocker on Day +53; (c) after transfer out of the ICU on Day +57. At the onset of ALI/ARDS, extensive bilateral pulmonary infiltration was noted, in addition to the tumor and atelectasis in the left lung. The infiltrates rapidly resolved on treatment with the tumor and atelectasis present.
Figure 7.
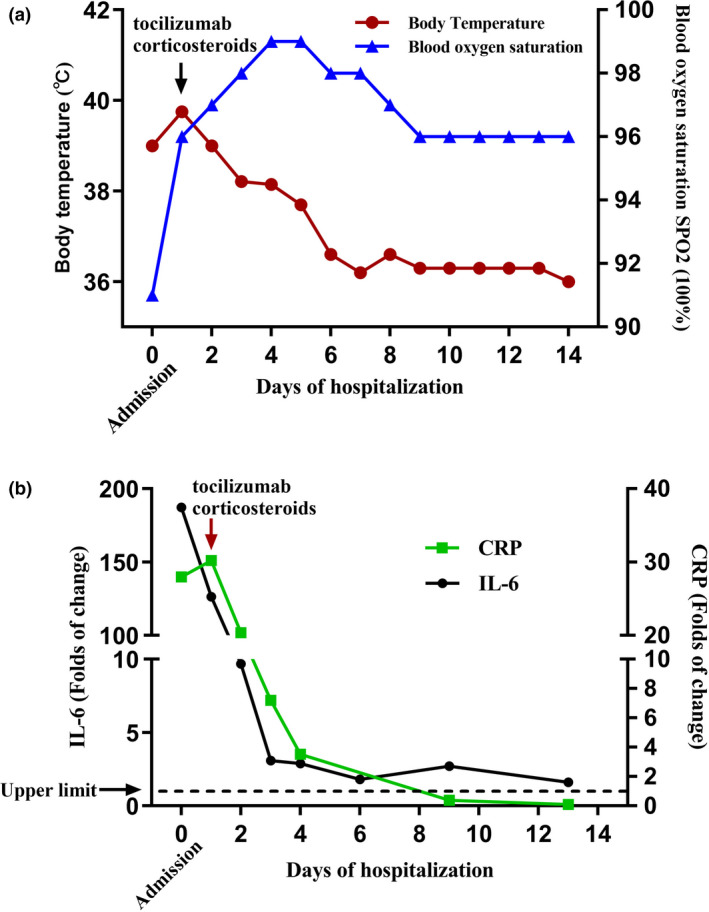
The patient’s condition rapidly improved after treatment. (a) The patient’s body temperature started decreasing, and his blood oxygen saturation also immediately increased. (b) The patient’s serum cytokine IL‐6 and CRP levels immediately decreased.
Table 1.
Serum cytokine levels analysed at Sun Yat‐sen University Cancer Center
| Cytokines | Sampling date | |||||||
|---|---|---|---|---|---|---|---|---|
| Day +48 | Day +49 | Day +50 | Day +51 | Day +54 | Day +56 | Day +60 | Normal range | |
| IL‐2 (pg mL−1) | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | 0.00–5.71 |
| IL‐4 (pg mL−1) | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | 0.00–2.80 |
| IL‐6 (pg mL−1) | 1301.9↑ | 99.75↑ | 31.84↑ | 29.82↑ | 18.68↑ | 27.93↑ | 16.52↑ | 0.00–10.30 |
| IL‐10 (pg mL−1) | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | 0.00–4.91 |
| TNF (pg mL−1) | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | 0.00–4.60 |
| IFN‐γ (pg mL−1) | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | < 2.50 | 0.00–7.42 |
In the ICU, the patient was administered maximal 70% oxygen via a high‐flow nasal cannula. Tocilizumab (8 mg kg−1 during the first consecutive 3 days) and methylprednisolone (16 mg kg−1 on the first 2 days and 3.5 mg kg−1 on the third day) were administered intravenously during the following 3 days. Subsequently, the corticosteroid (dexamethasone) dose was gradually tapered. As routine practice, the antibiotics/antifungal agents, cefoperazone and sulbactam, were also administered in the first week, followed by ceftriaxone sodium and fluconazole in the next week for suspected infections. The dyspnoea and pyrexia rapidly decreased, blood oxygen saturation increased (Figure 7a), and IL‐6 levels showed a steep decline (Figure 7b). The pulmonary infiltrates gradually resolved (Figure 6b and c), and chest CT imaging showed that the tumors shrank further (70.2 mm; Day +49, 2 days after being transferred to the ICU), which indicated SD (Figure 3c). However, the patient developed rashes on the left side of his face and in his left eye and experienced sharp pain at 9 days (Day +56) after administration of large doses of corticosteroids. He was diagnosed as having herpes zoster by an ophthalmologist, following which he was treated with intravenous infusions of an antiviral drug (acyclovir) for 10 days and an immune booster (thymalfasin). His skin lesions dramatically ameliorated, but the pain persisted. The patient was discharged in stable condition after 18‐day hospitalisation (Day +65) and continued to be treated as an outpatient for his shingles.
During the course of these severe adverse events, the patient also presented with characteristic manifestations of acute lung injury (ALI)/acute respiratory distress syndrome (ARDS), which can be caused by a heterogeneous group of disorders with similar clinical and radiological features. 16 The prognosis and therapeutic options greatly vary depending on the cause; therefore, correct diagnosis is crucial for ALI/ARDS management. We retrospectively reviewed the clinical features of the patient and his therapeutic response to various intervention modalities. The patient had acute onset of pyrexia (< 39°C) and dyspnoea and the level of only one cytokine (IL‐6) increased; there were no circulatory and neural dysfunctions. These findings could not support typical cytokine release syndrome (CRS)/cytokine storm (CS). 17 The onset of symptoms, response to large doses of corticosteroids and benign clinical outcome were essentially inconsistent with idiopathic/acute interstitial pneumonitis (AIP), while the laboratory results, that is complete blood count and microbial testing results (Supplementary table 1), did not support a diagnosis of infectious pneumonia. Therefore, we speculated that this episode could be reasonably considered as solitary organ damage caused by the toxic effects of PD‐L1–specific CAR‐T cell therapy on normal pulmonary tissue because of elevated PD‐L1 expression (on‐target off‐tumor effects). However, this could not be confirmed via pathological analysis such as lung biopsy or bronchoscopy alveolar lavage.
Discussion
In addition to overcoming the hostile tumor microenvironment, identifying highly specific targets is one of the main challenges involved in the application of CAR‐T cell treatments to malignant solid tumors. 18 In spite of these difficulties, several cell surface molecules, such as Erb‐B2/Her2, carboxyl‐anhydrase‐IX (CAIX), mesothelin and EGFRVIII, were eventually chosen as antigens and were targeted by CAR‐T cells for treating malignant solid tumors; however, these approaches met with very limited success and on‐target off‐tumor toxicities were noted because of the expression of these molecules on normal tissues. 19 , 20 Using CAIX‐specific CAR‐T cells for treating CAIX‐expressing metastatic renal cell carcinoma (RCC) could lead to hepatotoxicity, indicated by increase in alanine aminotransferase, aspartate aminotransferase and total bilirubin levels. 20 This kind of finding would represent a typical case of on‐target off‐tumor toxicities of CAR‐engineered T cells, which was supported by liver biopsies of the affected patients. In an extreme case of adverse reaction caused by CAR‐T cells used for treating malignant solid tumors, adoptive transfer of Her2‐specific CAR‐T cells led to cardiopulmonary collapse and death of a female patient with Her2‐expressing metastatic colon cancer, which was hypothetically caused by low Erb‐B2 expression on normal lung tissue. 19 In this case, the Her2‐targeting receptor had been derived from trastuzumab, which is widely used in the treatment of Her2+ breast cancer without any severe adverse reactions, thus illustrating the great potency of receptor‐modified T cells. 19
PD‐L1/PD‐1 checkpoint–blocking monoclonal antibodies have been approved as the first‐line treatment for patients with NSCLC. However, the objective response rates in patients with NSCLC are not ideal, that is < 20%. 21 On the basis of the PD‐L1 expression levels on human vital organs (e.g. heart, lungs, liver and kidneys), we assumed that CAR‐T cells targeting PD‐L1 on lung cancer cells would be well tolerated and therefore constructed PD‐L1–binding single‐chain Fv fragments (scFv) from atezolizumab, a PD‐L1–blocking monoclonal antibody. In preclinical studies, we found that PD‐L1 scFv‐engineered CAR‐T cells could be specifically activated and effectively killed PD‐L1+ lung cancer cells. Although IHC did not show PD‐L1 expression in lung biopsy samples, PD‐L1 transcripts could still be detected at reads per kilobase of transcript per million reads mapped (RKPM) values of 3.599 ± 1.496 and 3.869 ± 0.755 in the heart and lungs, respectively. 15 Because of the concern that PD‐L1–specific CAR‐T cells might evoke on‐target off‐tumor toxicities, especially against the lungs and heart, we administered a low dose of CAR‐T cells (1 × 106 kg−1 body weight), which was provided in three separate infusions within 7 days. The infused PD‐L1–specific CAR‐T cells were found to substantially proliferate on Day +29. The patient developed ALI/ARDS on Day +47, which rapidly progressed to respiratory failure. Given the fact that the patient’s clinical presentation was not consistent with typical CRS/CS, AIP and infectious pneumonia, we speculated that the ALI/ARDS in this case would have resulted from human PD‐L1–specific CAR‐T cells attacking normal pulmonary tissue because of the elevated expression PD‐L1 expression levels observed, that is the on‐target off‐tumor effect. In contrast to typical CRS/CS, 22 only the IL‐6 level dramatically increased (> 100‐fold), while the other cytokine levels only showed mild increase (< 2‐fold). This salient feature also differed from those noted for acute liver damage and respiratory failure caused by CAIX‐specific and Erb‐B2–specific CAR‐T cells, respectively. 19 , 20 Thus, IL‐6 level analysis would provide a sensitive and specific test for diagnosing delayed pulmonary toxicity following PD‐L1–CAR‐T cell therapy; however, more clinical data are required to validate this.
It was unclear why the patient showed no signs or symptoms of cardiotoxicities, although the PD‐L1 RNA expression levels were similar in both the lungs and heart. We assumed that the infused PD‐L1–specific CAR‐T cells might accumulate and proliferate in the patient’s lungs before the development of pulmonary toxicity. In line with this, it has been previously reported that T cells accumulate in the lungs immediately following the infusion and then leak out to the peripheral organs, such as the liver and kidneys. 23 Moreover, PD‐L1 expression is highly dynamic and is strongly induced in normal tissues by interferon (IFN)‐γ through paracrine/autocrine action. 24 The patient would have had chronic pulmonary inflammation because of long‐term smoking. 25 The lungs would be in constant communication with the external environment; therefore, PD‐L1 expression could increase with mild insults and the ensuing inflammation. It would incur the attack of PD‐L1–specific CAR‐T cells, leading to aggravation of pulmonary inflammation. Thus, a vicious cycle of inflammation and lung injury because of PD‐L1–specific CAR‐T cells would be initiated, which would be the mechanism underlying this severe adverse event. Therefore, patients who smoke should be excluded from clinical trials involving PD‐L1–specific CAR‐T cells. In the case of the patient described here, the quality, proliferation and persistence of PD‐L1–specific CAR‐T cells, and not the expression efficiency (20%), determined their efficacy and delayed toxic effects.
Chest CT imaging performed during the first follow‐up showed SD according to RECIST criteria; however, the long‐term effects still need to be determined. PD‐L1–specific CAR‐T cells could still provide a useful alternative for treating advanced NSCLC if strategies are developed to prevent or treat pulmonary toxicity. It has been reported that the hepatotoxicity would be entirely prevented if the patients were administered CAIX‐blocking monoclonal antibody (G250) before initiating therapy. 20 Therefore, we will consider administering the FDA‐approved PD‐L1–blocking monoclonal antibody atezolizumab at 1–2 weeks after infusion of PD‐L1–specific CAR‐T cells in our subsequent clinical trials. To avoid confounding interpretation of the response to atezolizumab versus that to CAR‐T cells, we would recruit patients for whom atezolizumab therapy has already failed. In addition, the PD‐L1–blocking monoclonal antibody could be used to treat the on‐target toxicities caused by PD‐L1–specific CAR‐T cells, instead of using the IL‐6 receptor–blocking monoclonal antibody tocilizumab. In conclusion, the severe pulmonary toxicity that developed following PD‐L1–specific CAR‐T cell treatment in this study has led to many questions, but has also provided a different perspective to deliberate on for the application of CAR‐T cells to malignant solid tumor treatment.
Methods
Production of human PD‐L1–specific CAR‐T cells
The PD‐L1–specific CAR gene was cloned into a lentiviral vector and was composed of an extracellular PD‐L1–binding domain, CD28 transmembrane, a cytoplasmic domains and cytoplasmic CD3zeta chain (Supplementary figure 1). The PD‐L1–binding domain was composed of single‐chain variable fragments (scFv) that, as proof of concept, were derived from the existing monoclonal antibody atezolizumab. 26 The genetically modified T cells were produced by lentiviral transduction in accordance with the Good Manufacturing Practice guidelines of the China Food and Drug Administration. The transduced T cells passed multiple sterility assays and were also found to be negative for the presence of replication competent lentivirus by quantitative polymerase chain reaction before use. 27
Clinical study design
The clinical trial was initiated at Sun Yat‐sen University Cancer Center to investigate the safety and efficacy of PD‐L1–specific CAR‐T cells for the treatment of patients with advanced NSCLC. The protocol was reviewed and approved by the Institutional review board (IRB) and is available at ClinicalTrials.gov (NCT03330834). The major recruitment criteria included advanced disease (clinical stage IIIC or IV, based on UICC/AJCC criteria) refractory to standard therapy but with adequate performance status (ECOG score, of 0–1), and PD‐L1–positive tumor cells ≥ 10%, as determined using standard IHC (anti‐human PD‐L1 rabbit monoclonal antibody; 1:25 dilution; clone SP142; Spring Bioscience, Pleasanton, CA, USA). All the participating patients provided written informed consent.
As per the study design, after leukapheresis, the patients would receive non‐myeloablative leukodepletion chemotherapy consisting of cyclophosphamide (250 mg m‐2) and fludarabine (25 mg m‐2) on Days −4, −3 and −2 before the first administration (Day 0) of three intravenous infusions of PD‐L1–specific CAR‐T cells amounting to a total dose of 1 × 106 CAR‐T cells kg−1 of body weight. To ensure safety, the CAR‐T cells were intravenously infused at 1 × 105 kg−1 body weight on Day 0, 3 × 105 kg−1 body weight on Day +3 and 6 × 105 kg−1 body weight on Day +7. The patient would be discharged from hospital on Day +10 and would be followed up as per schedule (Supplementary figure 2). If the treatment was found to be safe and effective, the patient could be given another dose of PD‐L1–specific CAR‐T cells after 6 months, on request.
Flow cytometry analyses
Flow cytometry analysis was performed with the BD FACSCalibur system (BD Biosciences, Franklin Lakes, NJ, USA); the results were analysed with the FlowJo software. To evaluate PD‐L1 CAR expression, the transduced T cells were incubated with PD‐L1::human Fc fusion protein (4 ng/reaction; BPS, San Diego, CA, USA) on ice for 20 min and washed once with phosphate‐buffered saline (PBS); subsequently, they were incubated with APC‐labelled streptavidin (1:500 dilution; Biolegend, San Diego, CA, USA) on ice for 10 min. Next, the cells were washed with and resuspended in PBS and used for FACS analysis. CD3‐fluorescein isothiocyanate (FITC) was purchased from BD Bioscience.
Cytokine and cytotoxicity assays
The serum cytokine levels (IL‐2, IL‐4, IL‐6, IL‐10, tumor necrosis factor [TNF]‐α, and IFN‐γ) of the patient were analysed using ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions at Sun Yat‐sen University Cancer Center. The samples were simultaneously also analysed at a third‐party laboratory, Guangzhou KingMed Diagnostics Group Co., Ltd, to confirm the results. The in vitro cytotoxicity against lung carcinoma cells (H1975 and H3122) was determined using xCELLigence RTCA SP (ACEA Biosciences, San Diego, CA, USA) according to the manufacturer’s instructions. The control group for PD‐L1–specific CAR‐T cells was CD38‐specific CAR‐T cells. The potency of PD‐L1–specific CAR‐T cells was also compared with that of PD1 fusion receptor T cells.
Statistical analysis
Data have been presented in terms of mean ± standard deviation (SD) values. Data from different groups were analysed with by one‐way analysis of variance (ANOVA). GraphPad Prism 5 was used for statistical calculations. *P < 0.05 and **P < 0.01 were considered statistically significant.
Author contributions
Liming Xia: Funding acquisition.
Conflicts of interest
The authors declare no conflict of interest.
Patient consent
Written informed consent was obtained from all the subjects.
Ethics approval
The protocol was reviewed and approved by the Institutional Review Board (IRB) of Sun Yat‐sen University Cancer Center and is also available at ClinicalTrials.gov (NCT03330834).
Supporting information
{kind=link}
Acknowledgments
The authors thank Lin Hong and Xijie Fan from Yiyang Biotechnology Company, Ltd, Guangzhou, China, for their help in performing some experiments and collecting data. This study was funded by the National Key R&D Program of China (No. 2018YFC1313400), the National Natural Science Foundation of China (Nos. 81572865 and 81773110), the Science and Technology Planning Project of Guangdong Province, China (No. 2017B020227003), the Major Project of Cooperative Innovation of Health Care in Guangzhou (No. 201704020215), the National Natural Science Foundation Youth Foundation of China (No. 81803079) and the Guangdong Natural Science Foundation of China (No. 2018A030310237). This work was supported by District High Tech R&D Funds (No. 2017‐L179) and Guangzhou Yiyang Biotechnology Company, Ltd, Guangzhou, China.
Contributor Information
Liming Xia, Email: xiajch@sysucc.org.cn.
Li Zhang, Email: zhangli@sysucc.org.cn.
Jianchuan Xia, Email: xiajch@sysucc.org.cn.
References
- 1. Kochenderfer JN, Dudley ME, Carpenter RO et al Donor‐derived CD19‐targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013; 122: 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davila ML, Riviere I, Wang X et al Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6: 224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kochenderfer JN, Rosenberg SA. Treating B‐cell cancer with T‐cells expressing anti‐CD19 chimeric antigen receptors. Nat Rev Clin Oncol 2013; 10: 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maus MV, Grupp SA, Porter DL et al Antibody‐modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014; 123: 2625–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Long KB, Young RM, Boesteanu ACCAR et al T cell therapy of non‐hematopoietic malignancies: detours on the road to clinical success. Front Immunol 2018; 9: 2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018; 8: 1069–1086. [DOI] [PubMed] [Google Scholar]
- 7. Sanmamed MF, Chen LA. Paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell 2018; 175: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iwai Y, Hamanishi J, Chamotok K et al Cancer immunotherapies targeting the PD‐1 signaling pathway. J Biomed Sci 2017; 24: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong H, Strome SE, Salomao DR et al Tumor‐associated B7–H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8: 793–800. [DOI] [PubMed] [Google Scholar]
- 10. Carbognin L, Pilotto S, Milella M et al Differential activity of nivolumab, pembrolizumab and MPDL3280A according to the tumor expression of programmed death‐ligand‐1 (PD‐L1): sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS One 2015; 10: e0130142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meyers DE, Bryan PM, Banerji S et al Targeting the PD‐1/PD‐L1 axis for the treatment of non‐small‐cell lung cancer. Curr Oncol 2018; 25: e324–e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martins F, Sykiotis GP, Maillard M et al New therapeutic perspectives to manage refractory immune checkpoint‐related toxicities. Lancet Oncol 2019; 20: e54–e64. [DOI] [PubMed] [Google Scholar]
- 13. Wills B, Brahmer JR, Naidoo J. Treatment of complications from immune checkpoint inhibition in patients with lung cancer. Curr Treat Options Oncol 2018; 19: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis RJ, Moore EC, Clavijo PE et al Anti‐PD‐1 efficacy can be enhanced by inhibition of myeloid‐derived suppressor cells with a selective inhibitor of PI3Kδ/γ. Cancer Res 2017; 77: 2607–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fagerberg L, Hallström BM, Oksvold P et al Analysis of the human tissue‐specific expression by genome‐wide integration of transcriptomics and antibody‐based proteomics. Mol Cell Proteomics 2014; 13: 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steinberg KP, Hudson LD. Acute lung injury and acute respiratory distress syndrome. The clinical syndrome. Clin Chest Med 2000; 21: 401–417. [DOI] [PubMed] [Google Scholar]
- 17. Lee DW, Santomasso BD, Locke FL et al ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 2019; 25: 625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T‐cell therapy. Nat Biotechnol 2013; 31: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morgan RA, Yang JC, Kitano M et al Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ErbB2. Mol Ther 2010; 18: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lamers CH, Sleijfer S, van Steenbergen S et al Treatment of metastatic renal cell carcinoma with CAIX CAR‐engineered T cells: clinical evaluation and management of on‐target toxicity. Mol Ther 2013; 21: 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hendriks LEL, Henon C, Auclin E et al Outcome of patients with non‐small cell lung cancer and brain metastases treated with checkpoint inhibitors. J Thorac Oncol 2019; 14: 1244–1254. [DOI] [PubMed] [Google Scholar]
- 22. Brudno JN, Kochenderfer JN. Recent advances in CAR T‐cell toxicity: mechanisms, manifestations and management. Blood Rev 2019; 34: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ritchie DS, Neeson PJ, Khot A et al Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther 2013; 21: 2122–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD‐L1 checkpoint. Immunity 2018; 48: 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thomsen M, Ingebrigtsen TS, Marott JL et al Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA 2013; 309: 2353–2361. [DOI] [PubMed] [Google Scholar]
- 26. Socinski MA, Jotte RM, Cappuzzo F et al Atezolizumab for first‐line treatment of metastatic nonsquamous NSCLC. N Eng J Med 2018; 378: 2288–2301. [DOI] [PubMed] [Google Scholar]
- 27. Skrdlant LM, Armstrong RJ, Keidaisch BM et al Detection of replication competent lentivirus using a qPCR assay for VSV‐G. Mol Ther Methods Clin Dev 2017; 8: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials