

Abstract
Wolfram syndrome is a rare genetic spectrum disorder characterized by insulin-dependent diabetes mellitus, optic nerve atrophy and progressive neurodegeneration and ranges from mild to severe clinical symptoms. There is currently no treatment to delay, halt, or reverse the progression of Wolfram syndrome, raising the urgency for innovative therapeutics for this disease. Here, we summarize our vision for developing novel treatment strategies and achieving a cure for Wolfram syndrome-spectrum disorder.
Keywords: Wolfram syndrome, endoplasmic reticulum, WFS1, gene therapy, regenerative therapy
Clinical manifestations of Wolfram syndrome
Wolfram syndrome is a rare, monogenic life-threatening disease caused largely by mutations in the Wolfram syndrome 1 (WFS1) gene or, in a small fraction of patients, pathogenic variants in the CDGSH iron sulfur domain protein 2 (CISD2) gene [1]. While insulin-dependent diabetes mellitus, optic nerve atrophy and neurodegeneration are cardinal features of this disease, many patients also develop other symptoms, ranging from hearing loss and endocrine deficiencies to neurologic and psychiatric conditions [1]. Accordingly, recent clinical and genetic findings have revealed that Wolfram syndrome is best characterized as a spectrum disorder. Most Wolfram syndrome patients carry two recessive pathogenic variants in the WFS1 gene. The disease begins to manifest at around ages 6 and 10, with the clinical onset of insulin-dependent diabetes mellitus and optic nerve atrophy, respectively. Diabetes insipidus, neurogenic bladder, obstructive sleep apnea and deafness may also develop in the next two decades of life, along with symptoms of brainstem and cerebellar atrophy, such as dysphagia, ataxia and central sleep apnea [1–4]. Some pathogenic variants of WFS1, particularly dominant variants, cause deafness or diabetes alone [5, 6]. Other dominant WFS1 variants give rise to deafness and optic nerve atrophy or autosomal dominant congenital cataracts [7, 8]. Recently, Dr. Hattersley’s team and ours identified several dominant de novo WFS1 variants associated with a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts [9]. Consequently, WFS1 is a locus of broad interest to various disease processes, highlighting the need for therapeutics targeting the gene towards potential treatements for Wolfram syndrome-spectrum disorders.
Wolfram syndrome is well-recognized as a prototype of endoplasmic reticulum (ER) disorder [10]. The ER plays a critical role in the proper folding of secretory and membrane proteins, calcium homeostasis and lipid biosynthesis. In Wolfram syndrome, dysregulation of calcium homeostasis and misfolding of pathogenic WFS1 variants causes ER stress in pancreatic β-cells, neurons, retinal ganglion cells and oligodendrocytes, resulting in the dysfunction and degeneration of affected tissues. ER dysfunction can also alter mitochondrial dynamics, thereby contributing to neuropsychiatric aspects of this disease [11]. Wolfram syndrome is thus a systemic disorder caused by ER dysfunction with no treatments available to delay, halt or reverse disease progression. Nevetheless, numerous avenues of therapeutic development are currently under study, heralding the possibility of new treatments on the horizon (Figure 1). Below we discuss the ongoing efforts on the development of novel treatments for Wolfram syndrome.
Figure 1. Current and potential therapeutic Strategies for Wolfram Syndrome.
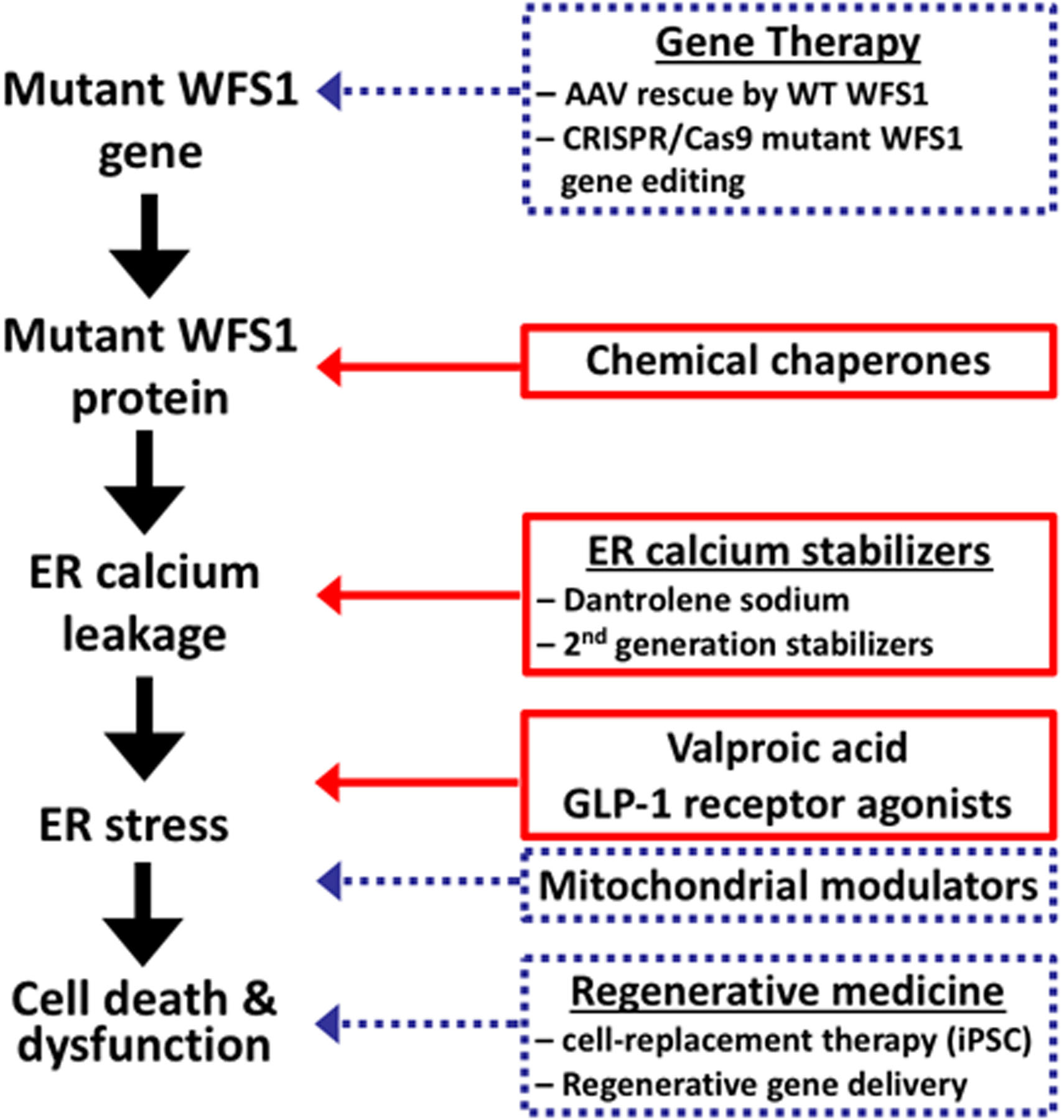
Schematic shows various pathophysiological steps in development of Wolfram syndrome. The discussed therapies are shown at the step that they target. Therapies currently in clinical trials and being developed pre-clinically are in solid red and dashed blue boxes respectively.
Chemical chaperones
A common molecular signature of Wolfram syndrome is cellular stress caused by the expression of mutant WFS1 proteins derived from pathogenic WFS1 alleles. To resolve this, we are testing chemical chaperones that can optimize the structure of mutant WFS1 proteins. Chemical chaperones, such as 4-phenylbutyric acid (PBA) and tauroursodeoxycholic acid (TUDCA), are drugs that are known to mitigate ER stress by rescuing or stabilizing the native conformation of mutant WFS1 proteins, thereby reducing protein aggregation and ER stress [12] (Figure 1). Such designer chemical chaperones hold the potential to delay Wolfram syndrome disease progression by reducing the misfolded WFS1 protein load in affected cell types, thereby salvaging remaining tissue function.
ER Calcium Stabilizers
To target calcium dyshomeostasis for the treatment of Wolfram syndrome, we are developing and repurposing putative ER calcium stabilizers. We recently discovered, for example, that dantrolene sodium, an Food and Drug Administration (FDA)-approved drug for malignant hyperthermia and muscle spasm, can act as an ER calcium stabilizer by targeting ER calcium transporters, more specifically ryanodine receptors. Dantrolene sodium can suppress cell death and dysfunction in neuronal and β-cell mouse models of Wolfram syndrome, as well as in induced pluripotent stem cell (iPSC) models of this disease [13] (Figure 1). These results prompted us to initiate an ongoing phase 1b/2 clinical trial of dantrolene sodium to assess its safety and tolerability in pediatric and adult patients with Wolfram syndrome (Clinical Trial Numberi :NCT02829268).
We are also now developing novel ER calcium stabilizers tailored for Wolfram together with the National Center for Advancing Translational Sciences (NCATS)ii. These second-generation ER calcium stabilizers also target ER calcium transporters, but are intended to be safer, more potent and have greater bioavailibility to the central nervous system (CNS) and eyes than dantrolene sodium.
Targeting ER stress
An interesting molecular target for therapeutic development is p21, which plays a role in cell proliferation and survival after ER stress. Valproic acid, a well-known mood stabilizer, has been shown to increase the expression of p21 and confer protection against cell death in a cell of Wolfram syndrome [14]. Interestingly, valproic acid also induces WFS1 expression and modulates the ER stress response [15]. A phase 2 double-blind, placebo-controlled drug repurposing trial is currently underway to evaluate valproic acid in patients with Wolfram syndrome (Clinical Trial Number: NCT03717909).
GLP-1 receptor agonists are another promising treatment for preventing ER stress-mediated cell death in Wolfram syndrome. It has been previously shown that GLP-1 can suppress apoptosis in cell models of Wolfram syndrome [13]. Now, data from two rodent models of Wolfram syndrome and one patient case report confirm that GLP-1 receptor agonists may improve diabetes in Wolfram syndrome [16, 17]. Further pre-clinical and clincal studies are needed to assess the broader efficacy of GLP-1 agonists in patients with Wolfram syndrome.
Potential other approaches
Interestingly, recent reports indicate that WFS1 loss-of-function is associated with mitochondrial dysfunction [11, 18]. This likely stems from ER calcium leakage/ER stress causing mitochondrial calcium overload, which results in aberrant organellar function and decreased ATP production. In light of these reports, mitochondrial modulators aimed at restoring mitochondrial function may merit further investigation in the context of Wolfram syndrome, as they may be able to delay neurodegeneration by reducing neuronal dysfunction.
Gene Therapy
One of the approaches to provide a cure to Wolframs syndrome could be via gene therapy. Using adeno-associated viral (AAV) systems, wildtype WFS1 could be transferred into the retinal ganglion cells of patients with Wolfram syndrome (Figure 1) to supplement the production of correct protein in the human body. We also envision using CRISPR/CAS9 gene editing technology for replacing pathogenic WFS1 variants with wildtype WFS1 alleles in Wolfram syndrome patients’ iPSCs and then create iPSC-derived organoids to further determine whether this approach can be used in combination with regenerative cell-replacement efforts (discussed below).
Regenerative Medicine
Given the deleterious effects of chronic ER stress on specific cell types in Wolfram syndrome, there is a dire need for regenerative medicine efforts aimed at replacing these damaged tissues. More specifically, there is a need for replacing pancreatic β-cells and retinal ganglion cells in patients, as defects in these cell types have the greatest impact on patients’ quality of life. To this end, regenerative therapy options using iPSCs have been developed [19]. iPSCs from patients with Wolfram syndrome and their respective siblings and/or parents have been generated [13] which could potentially be differentiated into neural progenitor cells, retinal ganglion cells, oligodentrocytes and pancreatic β-cells for therapeutic testing and molecular investigation. These iPSCs-derived cell types may one day be used for cell-based replacement therapy.
In addition to developing cell-replacement therapeutic strategies, the regenerative properties of factors such as mesencephalic astrocyte-derived neurotrophic factor (MANF), can be tested on tissues especially sensitive to WFS1 loss-of-function. MANF has been shown to activate proliferation of primary islets and confer protection against ER stress-mediated cell death [20]. A therapeutic strategy could be to directly deliver MANF to neurons, pancreatic β-cells and retinal ganglion cells via AAV systems, with the goal of suppressing neurodegeneration and improving β-cell mass, glucose tolerance and visual acuity. Further safety and efficacy studies will be required to optimize delivery, minimize adverse effects and maximize therapeutic benefit.
Concluding remarks and future perspectives
Wolfram syndrome is a rare genetic disorder with more than 200 pathogenic variants reported in association with disease. Clinical and genetic heterogeneity, as well as variable expressivity, pose a challenge for designing effective therapies in this population. We should therefore aim to design personalized treatments for Wolfram syndrome patients in the future. The first step towards this goal is to stratify patients based on their genetics. Genetic testing based on next-generation sequencing (NGS) technology, including exome and genome sequencing, has the ability to do this by identifying DNA variants that highly correlate with each patient’s clinical signs and symptoms. Routine use of NGS-based genetic testing will not only facilitate patient counseling by medical geneticists and genetic counselors, but also serve as a first step towards designing personalized treatments for patients with Wolfram syndrome.
Notably, although Wolfram syndrome is an ultra-rare genetic disorder, its constituent medical components (e.g. diabetes mellitus, deafness, and retinal degeneration) and underlying ER pathophysiology are not as rare. Consequently, novel treatments designed for this ultra-rare disorder may have broader implications for more common medical conditions related to ER stress and dysfunction. Thus, by leveraging the tools and therapeutic efforts targeting Wolfram syndrome, we may very well identify novel treatment modalities for more prevalent disorders such as diabetes mellitus and neurodegenerative diseases.
Acknowledgments
This work was partly supported by the grants from the National Institutes of Health/NIDDK (DK112921, DK113487, DK020579), National Institutes of Health/NCATS (TR002065), JDRF (2-SRA-2016-233-S-N) and philanthropic supports from the Unravel Wolfram Syndrome Fund, the Silberman Fund, the Stowe Fund, the Snow Foundation, the Eye Hope Foundation, the Ellie White Foundation for the Rare Genetic Disorders, and the Feiock Fund to F. Urano. The authors thank all the members of the Washington University Wolfram Syndrome Study and Research Clinic for their support (https://wolframsyndrome.dom.wustl.edu) and all the participants in the Wolfram syndrome International Registry and Clinical Study, Research Clinic, and Clinical Trials for their time and efforts.
Footnotes
Conflict of interests statement
F. Urano received research funding from Eli Lilly and Amarantus BioScience for the development of MANF-based regenerative therapy. He also received chemical compounds from Amylyx Pharmaceuticals, Mitochon Pharmaceuticals, Aetas Pharma, and National Center for Advancing Translational Sciences for the development small molecule-based therapies targeting ER stress.
Resources
References
- 1.Urano F (2016) Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr Diab Rep 16 (1), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett TG et al. (1995) Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 346 (8988), 1458–63. [DOI] [PubMed] [Google Scholar]
- 3.Hershey T et al. (2012) Early Brain Vulnerability in Wolfram Syndrome. PLoS One 7 (7), e40604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshall BA et al. (2013) Phenotypic characteristics of early Wolfram syndrome. Orphanet J Rare Dis 8 (1), 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesperance MM et al. (2003) Mutations in the Wolfram syndrome type 1 gene (WFS1) define a clinical entity of dominant low-frequency sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 129 (4), 411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnycastle LL et al. (2013) Autosomal dominant diabetes arising from a wolfram syndrome 1 mutation. Diabetes 62 (11), 3943–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rendtorff ND et al. (2011) Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am J Med Genet A 155A (6), 1298–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berry V et al. (2013) Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet 21 (12), 1356–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Franco E et al. (2017) Dominant ER Stress-Inducing WFS1 Mutations Underlie a Genetic Syndrome of Neonatal/Infancy-Onset Diabetes, Congenital Sensorineural Deafness, and Congenital Cataracts. Diabetes 66 (7), 2044–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonseca SG et al. (2010) Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 120 (3), 744–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cagalinec M et al. (2016) Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLoS Biol 14 (7), e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shang L et al. (2014) β-Cell Dysfunction Due to Increased ER Stress in a Stem Cell Model of Wolfram Syndrome. Diabetes 63 (3), 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu S et al. (2014) A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci U S A 111 (49), E5292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gharanei S et al. (2013) Vacuolar-type H+-ATPase V1A subunit is a molecular partner of Wolfram syndrome 1 (WFS1) protein, which regulates its expression and stability. Hum Mol Genet 22 (2), 203–17. [DOI] [PubMed] [Google Scholar]
- 15.Kakiuchi C et al. (2009) Valproate, a mood stabilizer, induces WFS1 expression and modulates its interaction with ER stress protein GRP94. PLoS ONE 4 (1), e4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toots M et al. (2018) Preventive treatment with liraglutide protects against development of glucose intolerance in a rat model of Wolfram syndrome. Sci Rep 8 (1), 10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondo M et al. (2018) Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia 61 (10), 2189–2201. [DOI] [PubMed] [Google Scholar]
- 18.Angebault C et al. (2018) ER-mitochondria cross-talk is regulated by the Ca(2+) sensor NCS1 and is impaired in Wolfram syndrome. Sci Signal 11 (553). [DOI] [PubMed] [Google Scholar]
- 19.Urano F (2014) Wolfram syndrome iPS cells: the first human cell model of endoplasmic reticulum disease. Diabetes 63 (3), 844–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindahl M et al. (2014) MANF is indispensable for the proliferation and survival of pancreatic beta cells. Cell Rep 7 (2), 366–75. [DOI] [PMC free article] [PubMed] [Google Scholar]