

Graphical abstract

Keywords: Polyfluoroflavones, Imidazole, Nucleophilic aromatic substitution of fluorine, Flavone-5-hydroxyflavone and flavone-coumarin rearrangements, Antiviral activity
Highlights
-
•
Synthesis of the new 2-aryl-4-oxo-4H-polyfluorochromen-3-carboxylates as analogs of natural methoxy-containing flavones.
-
•
The alternative flavone-5-hydroxyflavone and flavone-coumarin rearrangements of polyfluoroflavones in basic aqueous acetonitrile medium.
-
•
New 7-(1H-imidazol-1-yl)-substituted polyfluorinated flavones and coumarins.
-
•
Imidazolyl-functionalized flavones as high prospective antiviral agents.
Abstract
A simple and convenient method for the synthesis of new methyl 2-(4-methoxyphenyl)- and 2-(3,4-dimethoxyphenyl)-4-oxo-4H-polyfluorochromen-3-carboxylates as analogs of natural methoxy-containing flavones is proposed. As a result of their directed modification under basic conditions, 7-imidazolyl-substituted derivatives were obtained. In aqueous-organic medium under basic conditions, 5,6,7,8-tetrafluoro-3-(methoxycarbonyl)flavones were transformed into 6,8-difluoro-5-hydroxy-7-(1H-imidazol-1-yl)-3-(methoxycarbonyl)flavones as a result of flavone-5-hydroxyflavone rearrangement, while 6,7,8-trifluorinated analogs underwent a flavone-coumarin rearrangement to give 6,8-difluoro-3-(hydroxyarylidene)-7-(1H-imidazol-1-yl)coumarins under the same conditions. Acid hydrolysis of methyl polyfluoroflavone-3-carboxylates allowed to obtain 2-aryl-4H-polyfluorochromen-4-ones. Evaluation of the antiviral activity of the synthesized compounds against influenza A (H1N1) and Coxsackie B3 viruses showed that 2-(3,4-dimethoxyphenyl)-5,6,8-trifluoro-7-(1H-imidazol-1-yl)-4H-chromene-4-one has the most promising properties.
1. Introduction
Taking into account modern realities, it is safe to say that one of the important tasks of medical chemistry is the development of new effective antiviral drugs. Influenza and a number of other clinically similar viral infections are among the most widespread diseases, which annually account for up to 90 % cases of registered respiratory infections. Influenza viruses of type A are considered to be one of the most dangerous, since the features of their genome determine its high variability, leading to both evading the immune response and selection of drug-resistant strains of the virus. The pharmaceutical market currently offers a limited range of internationally recognized anti-influenza drugs. For the treatment of outpatients with acute uncomplicated influenza for the 2019–2020 season [1], drugs with the structure of substituted pyrans, such as zanamivir and laninamivir, are recommended.
In addition, in recent years, there has been a trend towards activation of infectious diseases caused by enteroviruses. The specificity of enterovirus infections is the polymorphism of clinical manifestations, long-term viral shedding, widespread asymptomatic forms, as well as the lack of specific prevention and treatment methods, which makes enterovirus infections practically uncontrollable. One of the main causative agents of enterovirus infections are Coxsackie viruses which are part of the B enterovirus group (CVB) and members of the picornavirus family (Picornaviridae) [2]. Particularly dangerous is the Coxsackie virus B3, which can cause heart diseases, such as myocarditis [3]. Despite numerous efforts, no specific drugs have yet been developed against pathogens of Picornaviridae family [4].
The objects of our study are polyfluorine-containing 2-aryl-4H-chromen-4-ones, which are included in a large group of flavonoids – promising scaffolds in medicinal chemistry [5]. Flavone derivatives are known for their wide spectrum of biological activity, for example, antiaggregative [6], cholinergic [7], antispasmodic and analeptic [8], antitumor [9], and antimicrobial [10]. The works aimed at searching for effective antiviral drugs among natural and chemically modified flavones are promising [11], since the presence of the pyran cycle in these compounds determines their great potential as antiviral agents. Antiviral drug flacoside was obtained on the basis of plant raw materials [12].
It is worth noting that the transformations of fluorine-containing compounds are of growing interest among researchers, due to the presence of electron-withdrawing fluorine atoms in organic molecules, which not only significantly changes their physical and chemical properties, offering new synthetic possibilities, but also varies the biological action spectrum [13].
In this work, 2-(4-methoxyphenyl)- and 2-(3,4-dimethoxyphenyl)-substituted methyl 4-oxo-4H-polyfluorochromen-3-carboxylates 4a,b, 5a,b (Scheme 1 ) were synthesized for the first time as fluorine-containing analogs of natural 3'-methoxy- and 3',4'-dimethoxy-containing flavones: acacetin, pectolinarigenin, eupatilin, nobiletin, etc. [14]. The possibility of obtaining imidazolyl-functionalized derivatives on their basis as potential antiviral agents was also studied, since among the substituted imidazoles, compounds with a different spectrum of antiviral action were found [15]. Besides, the imidazole fragment is present in the successfully used antiviral drug imidazolyl ethanamide pentandioic acid (ingavirin), which is effective against influenza A, B, parainfluenza, adenovirus, respiratory syncytial virus; and in preclinical studies against coronavirus, enteroviruses, including Coxsackie virus, etc. [16].
Scheme 1.

Synthesis of flavones 4a,b, 5a,b and their reactions with imidazole.
2. Results and discussion
For the synthesis of new 4'-methoxy- and 3',4'-dimethoxy-containing 3-(methoxycarbonyl)polyfluoroflavones 4a,b, 5a,b a well-proven one-pot method [10a] was used based on acylation of 3-aryl-3-oxopropanoates 1a,b activated with magnesium methoxide, which were obtained from commercially available methoxy-substituted acetophenones, with polyfluorobenzoyl chlorides 2, 3 (Scheme 1). The formed polyfluoroflavones 4a,b, 5a,b having methoxyl groups in the aryl substituent can be considered as analogs of natural methoxy-containing flavones and precursors of hydroxy-substituted flavones.
For the introduction of an imidazole residue into the synthesized polyfluoroflavones 4a,b and 5a,b it is convenient to use SNAr reactions, because such systems are characterized by selective substitution of the fluorine atom at the activated C7 position [17]. However, as our previous experience shows [10a,18], the result of the reaction of polyfluorochromen-4-ones with N-nucleophiles depends on the nature of the reagents and conditions used. Since, in addition to nucleophilic substitution reactions, the pyrone ring opening with the formation of substituted enamino esters is possible, and in case of 3-(alkoxycarbonyl)-substituted derivatives, a chromone – coumarin rearrangement can be realized.
In the reaction of 2-(3,4-dimethoxyphenyl)-substituted polyfluoroflavones 4a, 5a with imidazole in polar MeCN in the presence of Cs2CO3 and DIPEA the corresponding 7-(1H-imidazol-1-yl)-3-methoxycarbonylpolyfluoroflavones 6a, 7a were obtained with moderate yields (up to 44 %). Nevertheless, under these conditions, nucleophilic substitution of fluorine was accompanied by the side formation of a mixture of unidentified products, which was confirmed by TLC control. The reaction of 2-(4-methoxyphenyl)-substituted flavones 4b, 5b with imidazole in the same conditions led to 7-imidazolyl-substituted flavones 6b, 7b (Scheme 1), however, for a complete conversion of the starting fluorine-containing substrates, a base replacement was required, namely, CaH2 used instead of Cs2CO3 and DIPEA, while the process time increased from 14−16 h to 24 h.
To increase the yield of the target 7-substituted products, the reaction of flavones 4a,b and 5a,b with imidazole was also carried out in a mixture of MeCN and 0.1 M carbonate buffer (Na2CO3 / NaHCO3, pH ∼ 9.7). It was expected that the use of a basic aqueous-organic medium in the presence of a base would promote the reaction of the nucleophile at the C7 center of the flavone, due to the high solvating ability of water. However, under these conditions, water acted as the second nucleophilic reagent, and besides the reaction route depended on the structure of the initial polyfluoroflavone 4, 5. Thus, the reaction of tetrafluoroflavones 4а,b with imidazole in a basic aqueous-organic medium resulted in 6,8-difluoro-5-hydroxy-7-(1H-imidazol-1-yl)-3-(methoxycarbonyl)flavones 8a,b, while from the reaction of trifluoro-containing analogs 5a,b, 6,8-difluoro-7-(1H-imidazol-1-yl)chromen-2,4(3H)-diones 9a,b were isolated under similar conditions (Scheme 1).
It can be assumed that the formation of products 8a,b and 9a,b occurs as a result of the pyrone ring opening of the starting flavones 4a,b and 5a,b due to the attack of the hydroxide ion at the C2 center and the formation of tricarbonyl intermediates A, A', which undergo alternative routes of cyclization into new chromenones, depending on the presence of an ortho-fluorine atom in the polyfluoroaromatic fragment. This assumption was confirmed by experimental data on the example of tetrafluoroflavone 4a and its trifluorinated analog 5a transformations into the corresponding flavone 10a and coumarin 11a under heating in a mixture of MeCN and 0.1 M carbonate buffer (pH ∼ 9.7) for 6 h (Scheme 2 ) compared to longer 24 -h process of formation of imidazole-substituted analogs 8a and 9a (Scheme 1).
Scheme 2.

Alternative routes of recyclization of tetrafluoro- and trifluoro-substituted flavones 4a and 5a in basic aqueous acetonitrile medium.
It is obvious that the formation of coumarin 11a occurs by the only possible intramolecular cyclocondensation of the intermediate A' due to the attack of the phenolate anion of the trifluoroaryl residue on the carbonyl atom of the ester fragment. The isolation of coumarin 11a with a good preparative yield indicates a high selectivity of the occurring flavone-coumarin rearrangement. During the formation of 6,7,8-trifluoro-5-hydroxyflavone 10а, the tetrafluoro-containing intermediate A undergoes an alternative cyclization pathway, accompanied by the elimination of hydrogen fluoride, due to the intramolecular SNAr substitution of the ortho-fluorine atom in the tetrafluoroaryl fragment by the enolate anion at the aroyl substituent. In our opinion, the preference for this direction is determined by the fact that the oxygen atom of the less electron-negative non-fluorinated aroyl fragment has the higher electron-density.
However, this flavone-5-hydroxyflavone rearrangement is not the only route in this reaction, as evidenced by the formation of a mixture of unidentified products confirmed by TLC and the moderate yield of the product 10а.
An attempt to obtain fluorine-containing analogs of natural hydroxy-substituted flavones by using classical methods of ethers hydrolysis [19] did not give the desired result, since flavones 4a, 6a underwent only decarboxylation, leading to flavones 12, 13 (Scheme 3 ).
Scheme 3.

Decarboxylation of flavones 4a, 6a.
In order to find new antiviral agents, we evaluated the effect of obtained flavones against the strain of influenza type A/Puerto Rico/8/34 (H1N1) on the MDCK cell line and Coxsackie virus B3 on the Vero cell line (Table 1 ) using ribavirin as a positive control.
Table 1.
Antiviral activity of polyfluoroflavones.
| Compound | Influenza virus A/Puerto Rico/8/34 (H1N1), MDCK cell line |
Coxsackie virus B3, Vero cell line |
||||
|---|---|---|---|---|---|---|
| CC50, μM* | IC50, μM** | SI# | CC50, μM | IC50, μM | SI# | |
| 4a | 109 ± 8 | 49 ± 6 | 2 | 153 ± 11 | >61 | 3 |
| 5а | 508 ± 32 | 32 ± 4 | 16 | >2538 | 635 ± 42 | 4 |
| 6a | 61 ± 4 | 13 ± 3 | 5 | 91 ± 6 | >65 | 1 |
| 7а | >679 | >679 | 1 | 568 ± 38 | >226 | 3 |
| 8b | 629 ± 41 | 39 ± 5 | 16 | n/t1 | n/t | n/t |
| 13 | >746 | 25 ± 4 | 30 | 761 ± 60 | 29 ± 4 | 26 |
| ribavirin | >2130 | 36 ± 4 | 59 | >2130 | 34 ± 4 | >63 |
50 % cytotoxic concentration, the concentration resulting in death of 50 % of cells in culture.
50 % inhibiting concentration, the concentration resulting in decrease of virus’ production by 50 % comparing to control.
selectivity index.
n/t – not tested.
While studying the cytotoxicity on the MDCK cell line, it was found that tetrafluoroflavone 4а exhibits higher toxicity than its trifluorine-containing analog 5а; a similar pattern is observed for imidazolyl-substituted trifluoro- and difluoroflavones 6a, 7a (Table 1). Replacement of fluorine at the C5 position with a hydroxyl group also leads to a decrease of cytotoxicity in the case of 6,8-difluoro-5-hydroxy-7-(1H-imidazol-1-yl)flavone 8b. Thus, a decrease in the number of fluorine atoms in flavones has a positive effect on their toxicity. In addition, the absence of a methoxycarbonyl moiety at the C3 position also reduces the cytotoxicity of 7-imidazolyltriflavone 13.
The inhibitory activity against the influenza A virus of unsubstituted tetra- and trifluoroflavones 4a and 5a is similar and close to that of ribavirin, but taking into account the greater toxicity of tetrafluoro-derivative 4a, the selectivity index (SI) of trifluoroflavone 5a is eight times higher than its tetrafluorinated analog 4a. The introduction of an imidazolyl substituent at the C7 position of flavones leads to a decrease of IC50 for trifluoroflavone 6а as compared to the starting compound 4а, but to a significant increase for the difluorinated derivative 7а. Taking into account the CC50’s of these compounds, the SI of flavone 6а is five times higher than that for compound 7a and about three times higher than that of the flavone 4a. Flavone 8b, which has imidazole and hydroxyl substituents, shows inhibitory activity at the level of ribavirin, and its SI is 16. The decarboxylated product 13 has a combination of low toxicity and high effective concentration, resulting in its SI equal to 30.
In general, out of six analyzed compounds, three have a selectivity index of more than 10. This trend indicates the prospects for further expansion of chemical libraries and the study of compounds of this series as potential anti-influenza drugs.
We also studied the inhibitory activity of compounds 4а, 5а, 6а, 7а and 13 against a phylogenetically unrelated virus of the Picornaviridae family – Coxsackie virus B3 – in a Vero cell culture (Table 1). Along with this, the same patterns of cytotoxic properties by these compounds in relation to another cell line Vero were noted. However, only one of the five analyzed substances – decarboxylated flavone 13 – demonstrated antiviral properties against the Coxsackie virus B3. Considering that it was the compound that showed the maximum activity against the phylogenetically unrelated influenza virus, it should be said that further structure optimization of this group as potential broad-spectrum antiviral agents could be interesting.
3. Conclusion
In this work a simple and convenient method for the preparation of new methyl 2-(4-methoxyphenyl)- and 2-(3,4-dimethoxyphenyl)-4-oxo-4H-polyfluorochromen-3-carboxylates as analogs of natural methoxy-containing flavones is proposed. It was found that the result of the reactions of the synthesized 3-(methoxycarbonyl)polyfluoroflavones with imidazole depends on the structure of their fluorinated nucleus and on the reaction conditions. 7-Imidazolyl-substituted 3-(methoxycarbonyl)polyfluoroflavones are formed as a result of nucleophilic aromatic substitution of the fluorine atom under basic conditions. Aqueous-organic medium under basic conditions promote the transformation of tetrafluoroflavones into 6,8-difluoro-5-hydroxy-7-imidazolyl-3-(methoxycarbonyl)flavones, and trifluoro-containing analogs into 6,8-difluoro-3-(hydroxyarylidene)-7-imidazolylcoumarins. It was shown that these reactions are realized due to the pyrone ring opening of flavones under the action of hydroxyl ion as the second nucleophilic reagent, while the direction of further intramolecular cyclization is determined by the presence or absence of an ortho-fluorine atom in the polyfluorinated residue. Therefore, in the case of tetrafluorine-containing derivatives flavone-5-hydroxyflavone rearrangement occurs, and for trifluorinated analogs – flavone-coumarin one.
Acid hydrolysis of 3-(methoxycarbonyl)polyfluoroflavones resulted in their decarboxylation with formation of 2-(4-methoxyphenyl)- and 2-(3,4-dimethoxyphenyl)-4H-polyfluorochromen-4-ones, which are suitable for further functionalization at the C3 position.
Evaluation of the antiviral activity of synthesized compounds against influenza A (H1N1) and Coxsackie B3 viruses showed the promise of further study of modified polyfluoroflavones as potential antiviral drugs.
4. Experimental section
4.1. Chemistry: general information
All the reagents except fluorine-containing benzoyl chlorides and flavones are commercially available and used without further purification. Solvents were prepared according to standard methods of purification. The Nuclear magnetic resonance spectra (NMR) of the synthesized compounds were recorded on a Bruker DRX-400 and Bruker AVANCE III 500 spectrometers (1H, 400.13 (DRX400) and 500.13 (AV500) MHz, 13C, 100.6 MHz, SiMe4 as an internal standard, 19F, 376.44 (DRX400) and 470.52 (AV500) MHz, C6F6 as internal standard (chemical shifts were converted to CCl3F)). IR spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer using a diffuse reflectance attachment (DRA) in the range of 4000–400 cm– 1. High-resolution mass spectra (HRMS) were recorded on a Bruker MaXis Impact HD mass spectrometer (ESI). Elemental (C, H, N) analysis was performed on a Perkin Elmer PE 2400 Series II CHNS-O EA 1108 elemental analyzer. Melting points were measured on a Stuart SMP3 in open capillaries and are uncorrected. The reaction progress was monitored by TLC on ALUGRAM XTRA SIL G/ UV254 plates. Column chromatography was performed on Alfa Aesar silica gel 60 (0.063–0.2 mm).
The starting 3-oxoesters 1a,b were synthesized by a known procedure [20]. Methyl 2-aryl-4-oxo-4H-polyfluorochromene-3-carboxylates 4a,b, 5a,b were obtained by a recently described technique [10a].
4.2. Synthesis of methyl 2-aryl-4-oxo-4H-polyfluorochromen-3-carboxylates 4a,b, 5a,b
In a three-necked flask, equipped with a reflux condenser and a drop funnel, several iodine crystals and 0.12 g (5 mmol) of magnesium turnings were added, heated with stirring until the vigorous sublimation of iodine, then 1 mL (25 mmol) of MeOH and a few drops of CCl4 were added. While vigorous hydrogen evoluation, 20 mL of absolute toluene was added to the mixture. Then 5 mmol of corresponding 3-aryl-3-oxopropanoate 1a,b was added in portions while stirring. The mixture was refluxed until the magnesium turnings were completely dissolved, and then cooled to room temperature. The solvent was removed in vacuo till the mass thickening, then 20 mL of absolute toluene was added. Polyfluorobenzoyl chloride 2, 3 (5 mmol) in 10 mL of absolute toluene was added to the mixture dropwise with stirring at the room temperature. The mixture was stirred for 1 h at room temperature, and then 2 h while heating at 80 °C. Then mixture was poured into 100 mL 0.5 M HCl and stirred. An organic layer was separated and dried over anhydrous Mg2SO4. The solvent was removed in vacuo, and the residue was crystallized fractionally from toluene.
4.2.1. Methyl 5,6,7,8-tetrafluoro-2-(3,4-dimethoxyphenyl)-4-oxo-4H-chromene-3-carboxylate (4a)
was synthesized from 1.19 g (5 mmol) of methyl 3-(3,4-dimethoxyphenyl)-3-oxopropanoate 1a and 1.15 g (5 mmol) of pentafluorobenzoyl chloride 2. Yield 1.65 g (80 %), white powder, mp 197–199 °C. IR: ν 3015, 2973, 2941, 2851 (C-H), 1735 (CO2Me), 1652 (C = O), 1514, 1497, 1435, 1370 (C = C, C–H), 1032, 1016 (C–F) cm–1. 1H NMR (400.13 MHz, (CD3)2SO): δ 3.78, 3.80, 3.87 (9H, all s, 3 OMe), 7.20–7.35 (3H, m, C6H3) ppm. 13C NMR (125.8 MHz, (CD3)2SO): δ 52.9 (s, C-18), 55.6, 55.8 (both s, C-15, 16), 109.7 (d, J =7.7 Hz, C-4a), 110.4 (s, C-14), 112.0 (s, C-11), 117.0 (s, C-3), 121.7 (s, C-10), 121.8 (s, C-9), 137.1 (m, C-5), 137.2 (m, C-6), 141.1 (m, C-8a), 142.9 (m, C-8), 144.4 (m, C-7), 148.8 (s, C-13), 152.3 (s, C-12), 161.2 (s, C-2), 164.3 (s, C-17), 170.9 (s, C-4) ppm. 19F NMR (376.44 MHz, (CD3)2SO): δ -161.79, -158.62, -148.65, -144.79 (4 F, all m) ppm. Anal. calcd. for C19H12F4O6: C, 55.35; H, 2.93; found: C, 55.63; H, 3.10.
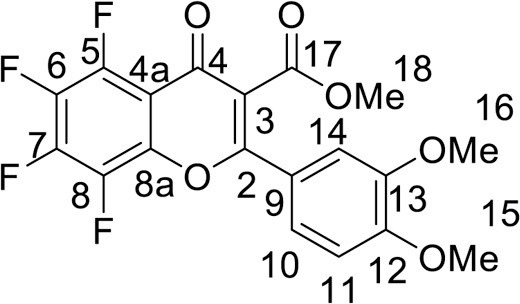
4.2.2. Methyl 5,6,7,8-tetrafluoro-2-(4-methoxyphenyl)-4-oxo-4H-chromene-3-carboxylate (4b)
was synthesized from 1.04 g (5 mmol) of methyl 3-(4-methoxyphenyl)-3-oxopropanoate 1b and 1.15 g (5 mmol) of pentafluorobenzoyl chloride 2. Yield 1.47 g (77 %), pink powder, mp 152–154 °C. IR: ν 3089, 2984, 2964, 2937, 2844 (C-H), 1740 (CO2Me), 1649 (C = O), 1589, 1514, 1497, 1443, 1374 (C = C, C–H), 1026 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.77, 3.87 (6H, both s, 2 OMe), 7.18–7.69 (4H, m, C6H4) ppm. 13C NMR (125.8 MHz, (CD3)2SO): δ 52.9 (s, C-15), 55.6 (s, C-13), 109.7 (d, J =8.0 Hz, C-4a), 114.8 (s, C-11), 116.7 (s, C-9), 121.6 (s, C-3), 129.6 (s, C-10), 136.7 (m, C-5), 137.5 (m, C-6), 141.1 (m, C-8a), 143.2 (m, C-8), 144.6 (m, C-7), 161.2 (s, C-2), 162.5 (s, C-12), 164.3 (s, C-14), 170.9 (s, C-4) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -161.87, -158.83, -148.66, -144.84 (4 F, all m) ppm. Anal. calcd. for C18H10F4O5: C, 56.56; H, 2.64; found: C, 56.46; H, 2.69.
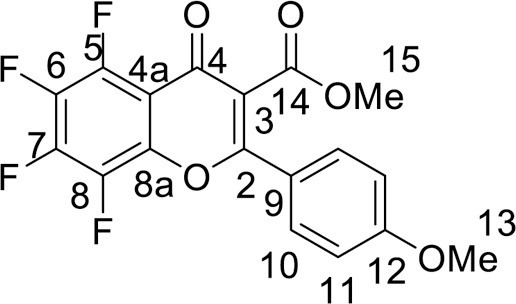
4.2.3. Methyl 6,7,8-trifluoro-2-(3,4-dimethoxyphenyl)-4-oxo-4H-chromene-3-carboxylate (5a)
was synthesized from 1.19 g (5 mmol) of methyl 3-(3,4-dimethoxyphenyl)-3-oxopropanoate 1a and 1.06 g (5 mmol) of tetrafluorobenzoyl chloride 3. Yield 1.26 g (64 %), white powder, mp 192–195 °C. IR: ν 3013, 2989, 2941, 2851 (C-H), 1732 (CO2Me), 1645, 1632 (C = O), 1513, 1484, 1392 (C = C, C–H), 1036, 1036, 1018 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.77, 3.81, 3.87 (9H, all s, 3 OMe), 7.20–7.36 (3H, m, C6H3), 7.90 (1H, m, H, C-5) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -151.50 (1 F, d, J =18.4 Hz), -150.40, -136.62 (2 F, both m) ppm. Anal. calcd. for C19H13F3O6: C, 57.88; H, 3.32; found: C, 58.11; H, 3.34.
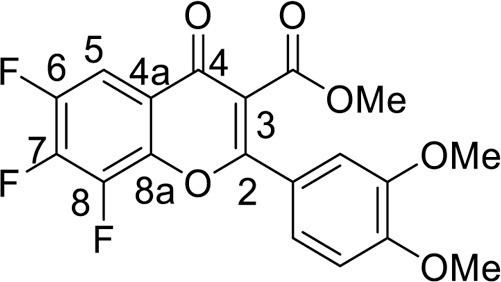
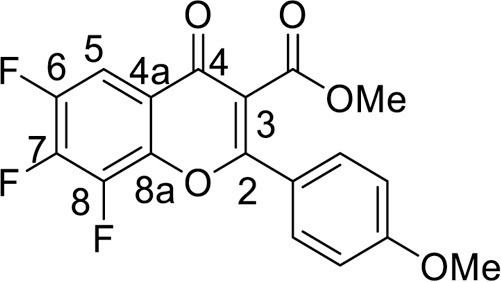
4.2.4. Methyl 6,7,8-trifluoro-2-(4-methoxyphenyl)-4-oxo-4H-chromene-3-carboxylate (5b)
was synthesized from 1.04 g (5 mmol) of methyl 3-(4-methoxyphenyl)-3-oxopropanoate 1b and 1.06 g (5 mmol) of tetrafluorobenzoyl chloride 3. Yield 1.27 g (70 %), white powder, mp 177–179 °C. IR: ν 3071, 2964, 2851 (C–H), 1741 (CO2Me), 1648, 1636 (C = O), 1511, 1485, 1433 (C = C, C–H), 1048, 1018 (C–F) cm−1. 1H NMR (400.13 MHz, (CD3)2SO): δ 3.76, 3.87 (6H, both s, 2 OMe), 7.18–7.70 (4H, m, C6H4), 7.90 (1H, d.d.d, J HF = 9.9, 8.0, 2.0 Hz, H, C-5) ppm. 19F NMR (376.44 MHz, (CD3)2SO): δ -151.71, -150.42, -136.69 (3 F, all m) ppm. Anal. calcd. for C18H11F3O5: C, 59.35; H, 3.04; found: C, 59.20; H, 3.17.
4.3. General procedures for the synthesis of compounds 6-9
4.3.1. Method A
2-(3,4-Dimethoxyphenyl)-4-oxo-4H-polyfluorochromene-3-carboxylate 4a or 5a (0.5 mmol), imidazole (68 mg, 1 mmol), Cs2CO3 (163 mg, 0.5 mmol) and DIPEA 65 mg (0.5 mmol) were suspended in 10 mL of MeCN. The reaction mixture was heated at 80 °C. The reaction progress was monitored by TLC. At the end of the reaction, the mixture was diluted with 50 mL of H2O, extracted with DCM, 2 × 10 mL. Organic layer was separated, the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel, using DCM.
4.3.2. Method B
2-(4-Methoxyphenyl)-4-oxo-4H-polyfluorochromen-3-carboxylate 4b or 5b (0.5 mmol), imidazole (34 mg, 0.5 mmol) and CaH2 (21 mg 0.5 mmol) were suspended in 10 mL of MeCN. The reaction mixture was heated at 80 °C under inert atmosphere of argon. The reaction progress was monitored by TLC. At the end of the reaction, organic phase was separated from CaH2, the solvent was removed in vacuo. The residue was crystallized from 50 % EtOH (v/v) and then purified by column chromatography on silica gel, using DCM-Et2O mixture (1÷1 v/v).
4.3.3. Method C
2-Aryl-4-oxo-4H-polyfluorochromen-3-carboxylate 4, 5 (0.5 mmol), imidazole (68 mg, 1 mmol) and DIPEA 65 mg (0.5 mmol) were suspended in a mixture of 5 mL of MeCN and 5 mL of 0.1 M carbonate buffer. The reaction mixture was heated at 80 °C. The reaction progress was monitored by TLC. At the end of the reaction, the mixture was diluted with 50 mL of H2O, extracted with DCM, 2 × 10 mL. Organic layer was separated; the solvent was removed in vacuo. The residue was purified by column chromatography on silica gel, using DCM.
4.3.3.1. Methyl 5,6,8-trifluoro-2-(3,4-dimethoxyphenyl)-7-(1H-imidazol-1-yl)-4-oxo-4H-chromene-3-carboxylate (6a) was synthesized according to the method A
Yield 78 mg (34 %), yellow powder, mp 218–220 °C (dec). IR: ν 3135, 3056, 2937, 2847 (C-H), 1742 (CO2Me), 1651 (C = O), 1517, 1488, 1377 (C–N, C = C, C–H), 1021 (C–F) cm–1. 1H NMR (400.13 MHz, (CD3)2SO): δ 3.80, 3.81, 3.87 (9H, all s, 3 OMe), 7.21–7.37 (3H, m, C6H3), 7.38, 7.75, 8.38 (3H, all s, imidazole-1-yl) ppm. 13C NMR (125.8 MHz, (CD3)2SO): δ 53.0 (s, C-18), 55.6, 55.8 (both s, C-15, 16), 110.4 (s, C-14), 112.1 (s, C-11), 112.7 (d, J =8.9 Hz, C-4a), 117.2 (s, C-3), 120.1 (m, C-19), 121.1 (br.s, C-21), 121.7 (s, C-10), 121.8 (s, C-9), 128.8 (s, C-20), 138.2 (s, C-7), 140.7 (m, C-6), 140.8 (m, C-5), 140.8 (m, C-8), 143.6 (m, C-8a), 148.8 (s, C-13), 152.4 (s, C-12), 161.3 (s, C-2), 164.4 (s, C-17), 171.0 (br.s, C-4) ppm. 19F NMR (376.44 MHz, (CD3)2SO): δ -148.82, -145.22, -144.02 (3 F, all m) ppm. HRMS (ESI), m/z: calcd. for C22H15F3N2O6 [M+H]+ 461.0955; found 461.0953.
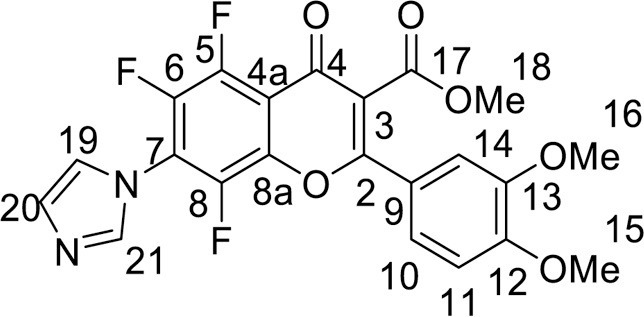
4.3.3.2. Methyl 5,6,8-trifluoro-2-(4-methoxyphenyl)-7-(1H-imidazol-1-yl)-4-oxo-4H-chromene-3-carboxylate (6b) was synthesized according to the method B
Yield 95 mg (44 %), yellow powder, mp 218–220 °C (dec). IR: ν 3129, 2950, 2842 (C-H), 1738 (CO2Me), 1646 (C = O), 1513, 1485, 1368 (C–N, C = C, C–H), 1022 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.78, 3.87 (6H, both s, 2 OMe), 7.19–7.70 (4H, m, C6H4), 7.27, 7.67, 8.16 (3H, all s, imidazole-1-yl) ppm. 13C NMR (125.76 MHz, (CD3)2SO): δ 52.9 (s, C-15), 55.6 (s, C-13), 112.5 (d, J =8.8 Hz, C-4a), 114.9 (s, C-11), 116.9 (s, C-16), 120.3 (m, C-3), 120.7 (s, C-9), 121.8 (s, C-17), 129.6 (s, C-10), 138.2 (s, C-18), 140.7 (m, C-7), 141.3 (m, C-6), 140.9 (m, C-5), 142.9 (m, C-8), 144.6 (m, C-8a), 161.4 (s, C-2), 162.6 (s, C-12), 164.3 (s, C-14), 170.9 (s, C-4) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -148.04, -144.34 (2 F, both m), -143.51 (1 F, d, J =14.9 Hz) ppm. Anal. calcd. for C21H13F3N2O5*1/4 H2O: C, 58.00; H, 3.13; N, 6.44; found: C, 57.87; H, 3.06; N, 6.13.
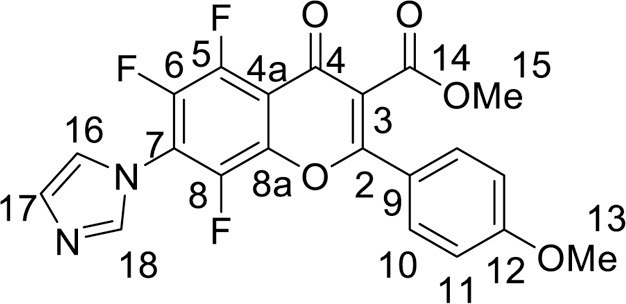
4.3.3.3. Methyl 6,8-difluoro-2-(3,4-dimethoxyphenyl)-7-(1H-imidazol-1-yl)-4-oxo-4H-chromene-3-carboxylate (7a) was synthesized according to the method A
Yield 97 mg (44 %), yellow powder, mp 248–250 °C. IR: ν 3142, 3121, 2975, 2847 (C-H), 1731 (CO2Me), 1634 (C = O), 1579, 1563, 1474, 1397 (C–N, C = C, C–3004, 2937, 2845 (C-H), 1741 (CO2Me), 1636 (C = O), 1516, 1477, 1363, 1265 (C–N, C = C, C–H), 1018, 1003 (C–F) cm–1. 1H NMR (400.13 MHz, (CD3)2SO): δ 3.79, 3.81, 3.87 (9H, all s, 3 OMe), 7.20–7.35 (3H, m, C6H3), 7.24, 7.63, 8.12 (3H, all s, imidazole-1-yl), 7.91 (1H, d.d, J = 9.4, 1.6 Hz, H, C-5) ppm. 19F NMR (376.44 MHz, (CD3)2SO): δ -137.19 (1 F, br.s), -123.24 (1 F, d, J =9.3 Hz) ppm. Anal. calcd. for C22H16F2N2O6: C, 59.73; H, 3.65; N, 6.33; found: C, 59.43; H, 3.69; N, 6.19.
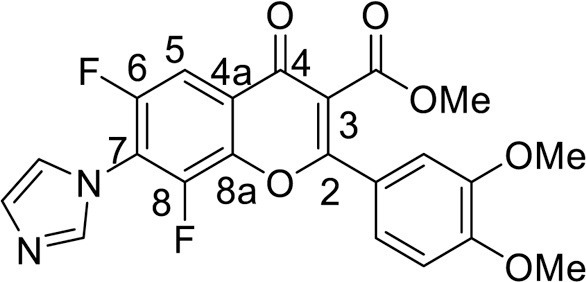
4.3.3.4. Methyl 6,8-difluoro-2-(4-methoxyphenyl)-7-(1H-imidazol-1-yl)-4-oxo-4H-chromene-3-carboxylate (7b) was synthesized according to the method B
Yield 101 mg (49 %), yellow powder, mp 178–180 °C. IR: ν 3142, 3121, 2975, 2847 (C-H), 1731 (CO2Me), 1634 (C = O), 1579, 1563, 1474, 1397 (C–N, C = C, C–H), 1014, 999 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.78, 3.87 (6H, both s, 2 OMe), 7.19–7.72 (4H, m, C6H4), 7.24, 7.63, 8.11 (3H, all s, imidazole-1-yl), 7.92 (1H, d.d, J = 9.6, 1.7 Hz, H, C-5) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -136.44 (1 F, br.s), -122.30 (1 F, d, J =9.1 Hz) ppm. Anal. calcd. for C21H14F2N2O5: C, 61.17; H, 3.42; N, 6.79; found: C, 61.31; H, 3.38; N, 6.71.
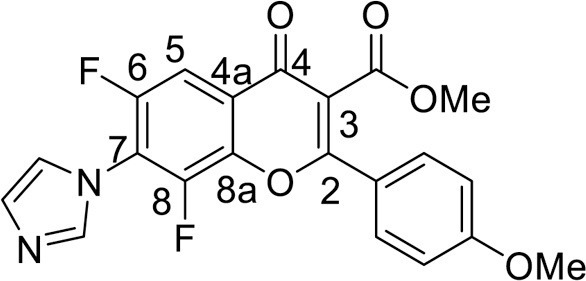
4.3.3.5. Methyl 6,8-difluoro-5-hydroxy-7-(1H-imidazol-1-yl)-2-(4-methoxyphenyl)-4-oxo-4H-chromene-3-carboxylate (8a) was synthesized according to the method C
Yield 87 mg (38 %), pale yellow powder, mp 225–228 °C. IR: ν 3175, 3120, 2980 (C-H), 1728 (CO2Me), 1652, 1622 (C = O), 1515, 1488, 1385 (C–N, C = C, C–H), 993 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.80, 3.81, 3.88 (9H, all s, 3 OMe), 7.22–7.8 (3H, m, C6H3, H, imidazole-1-yl), 7.63, 8.12 (2H, both br.s, imidazole-1-yl), 11.86 (1H, s, OH) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -154.08 (1 F, br.s), -150.70 (1 F, s) ppm. Anal. calcd. for C22H16F2N2O7: C, 57.65; H, 3.52; N, 6.11; found: C, 57.44; H, 3.606; N, 5.89.
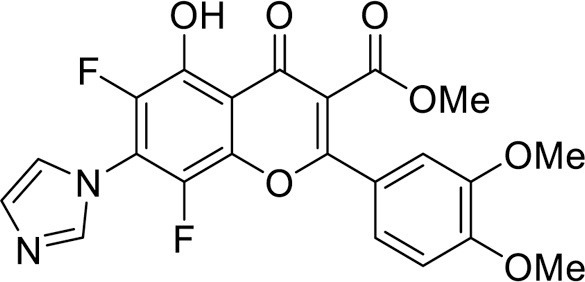
4.3.3.6. Methyl 6,8-difluoro-5-hydroxy-7-(1H-imidazol-1-yl)-2-(4-methoxyphenyl)-4-oxo-4H-chromene-3-carboxylate (8b) was synthesized according to the method C
Yield 75 mg (35 %), yellow powder, mp 203–205 °C. IR: ν 3135, 3120, 2949 (C-H), 1743 (CO2Me), 1662 (C = O), 1514, 1488, 1392 (C–N, C = C, C–H), 1026 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.80, 3.87 (6H,both s, 2 OMe), 7.20–7.72 (4H, m, C6H4), 7.24, 7.64, 8.12 (3H, all s, imidazole-1-yl), 11.89 (1H, s, OH) ppm. 13C NMR (125.76 MHz, (CD3)2SO): δ 53.0 (s, C-15), 55.6 (s, C-13), 109.2 (d, J =3.9 Hz, C-4a), 114.9 (m, C-16), 114.9 (s, C-11), 120.9 (s, C-3), 121.9 (s, C-9), 129.4 (s, C-17), 129.9 (s, C-10), 135.7 (br.s, C-6), 137.6 (br.s, C-8), 138.2 (s, C-18), 139.9 (m, C-5), 140.2 (br.s, C-7), 142.7 (m, C-8a), 162.8 (s, C-12), 163.7 (s, C-2), 163.9 (s, C-14), 178.2 (br.s, C-4) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -154.03 (1 F, br.s), -150.72 (1 F, d, J FF =5.7 Hz) ppm. HRMS (ESI), m/z: calcd. for C21H14F2N2O6 [M−H]− 427.0747; found 427.0748. Anal. calcd. for C21H14F2N2O6: C, 58.88; H, 3.29; N, 6.54; found: C, 58.68; H, 3.28; N, 6.32.
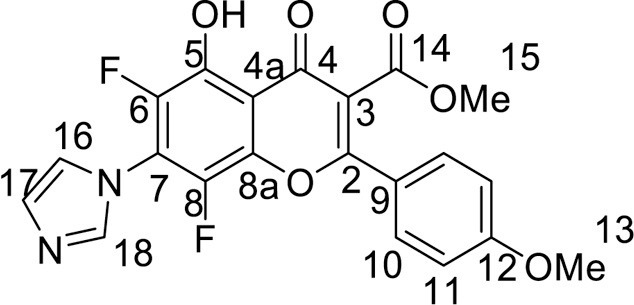
4.3.3.7. 3-[(3,4-Dimethoxyphenyl)(hydroxy)methylene]-6,8-difluoro-7-(1H-imidazol-1-yl)-2H-chromene-2,4(3 H)-dione (9a) was synthesized according to the method C
Yield 105 mg (48 %), yellow powder, mp 246–248 °C. IR: ν 3510 (O-H), 3006, 2940 (C-H), 1705, 1630, 1596, 1576 (C = O), 1513, 1404, 1267 (C–N, C = C, C–H), 1017 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.78, 3.82 (6H, both s, 2 OMe), 6.95–7.70 (3H, m, C6H3), 7.40 (1H, d.d, J = 8.3, 1.7 Hz, H, C-5), 7.84, 8.07, 9.32 (3H, all s, imidazole-1-yl) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -138.94, -128.56 (2 F, both m) ppm. Anal. calcd. for C21H14F2N2O6: C, 58.88; H, 3.29; N, 6.54; found: C, 58.58; H, 3.28; N, 6.32.
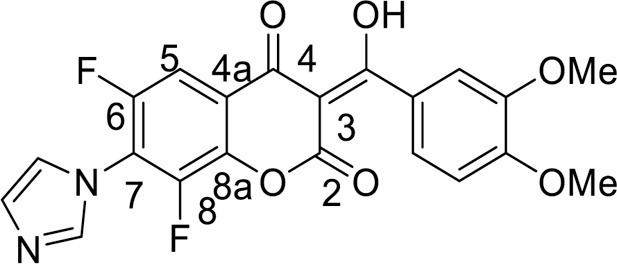
4.3.3.8. 6,8-Difluoro-3-[hydroxy(4-methoxyphenyl)methylene]-7-(1H-imidazol-1-yl)-2H-chromene-2,4(3 H)-dione (9b) was synthesized according to the method C
Yield 105 mg (53 %), white powder, mp 238–240 °C. IR: ν 3510 (O-H), 3006, 2940 (C-H), 1705, 1630, 1596, 1576 (C = O), 1513, 1404, 1267 (C–N, C = C, C–H), 1017 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.82 (3H, s, OMe), 6.95–7.77 (4H, m, C6H4), 7.67 (1H, d, J =9.6 Hz, H, C-5), 7.85, 8.07, 9.33 (3H, all s, imidazole-1-yl) ppm. 13C NMR (125.76 MHz, (CD3)2SO): δ 55.3 (s, C-14), 101.0 (br.s, C-3), 105.9 (m, C-15), 113.3 (s, C-12), 114.7 (m, C-4a), 123.6 (m, C-5), 124.5 (s, C-10), 131.0 (s, C-11), 131.8 (s, C-16), 138.1 (s, C-17), 138.8 (m, C-8a), 143.6 (m, C-7), 149.3 (m, C-8), 151.3 (m, C-6), 160.0 (s, C-13), 162.4 (s, C-2), 168.1 (s, C-4), 192.6 (s, C-9) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -138.94, -128.56 (2 F, both m) ppm. Anal. calcd. for C20H12F2N2O5*½H2O: C, 58.97; H, 3.22; N, 6.88; found: C, 59.14; H, 3.36; N, 6.96.
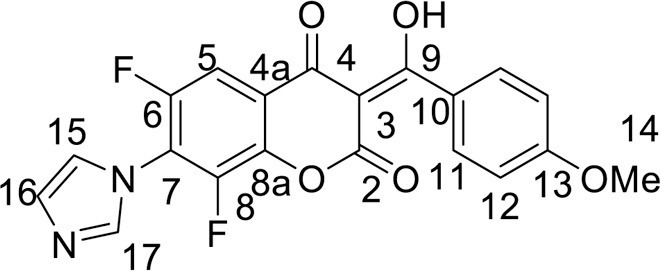
4.3.3.9. Methyl 2-(3,4-dimethoxyphenyl)-6,7,8-trifluoro-5-hydroxy-4-oxo-4H-chromene-3-carboxylate (10a)
Flavone 4a 210 mg (0.5 mmol), was suspended in 10 mL of a MeCN and 0.1 M carbonate buffer mixture (1÷1, v/v, pH ∼ 9.7). The reaction mixture was heated at 80 °C for 6 h. At the end of the reaction, the solvent was removed in vacuo. The precipitate was washed with H2O and crystallized from EtOH. Yield 102 mg (50 %), yellow powder, mp 273–277 °C (dec). 1H NMR (400.13 MHz, (CD3)2SO): δ 3.70, 3.79, 3.84 (9H, all s, 3 OMe), 7.14–7.26 (3H, m, C6H3) ppm. 19F NMR (376.44 MHz, (CD3)2SO): δ -187.38, -168.90, -156.62 (3 F, all m) ppm. Anal. calcd. for C19H13F3O7: C, 55.62; H, 3.19; found: C, 55.17; H, 3.12.
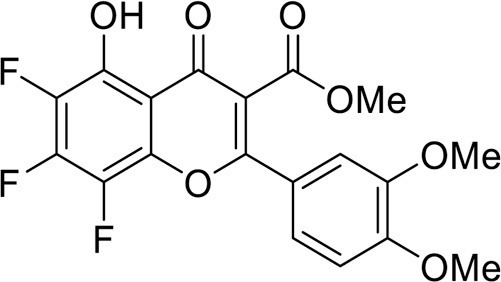
4.3.3.10. Methyl 2-(3,4-dimethoxyphenyl)-6,7,8-trifluoro-5-hydroxy-4-oxo-4H-chromene-3-carboxylate (11a) obtained according to 10a from flavone 5a 197 mg (0.5 mmol)
Yield 152 mg (80 %), white powder, mp >350 °C (dec). IR: ν 3047, 2971, 2941, 2843 (C–H), 1687 (C = O), 1575, 1517, 1480, 1402 (C–N, C = C, C–H), 996 (C–F) cm–1. 1H NMR (400.13 MHz, (CD3)2SO): δ 3.76, 3.80 (6H, both s, 2 OMe), 6.91–7.36 (3H, m, C6H3), 7.49 (1H, s, H, C-5) ppm. 13C NMR (125.76 MHz, (CD3)2SO): δ 55.4, 55.6 (both s, C-16, 17), 99.0 (s, C-3), 106.0 (d.d, J = 18.9, 2.9 Hz, C-5), 110.5 (s, C-15), 111.2 (s, C-12), 119.5 (d, J =2.9 Hz, C-4a), 123.6 (s, C-11), 132.9 (s, C-10), 138.2 (m, C-8), 139.5 (m, C-8a), 140.8 (m, C-7), 145.3 (m, C-6), 148.1 (s, C-17), 151.8 (s, C-16), 160.9 (s, C-2), 170.8 (s, C-4), 193.9 (s, C-9) ppm. 19F NMR (376.44 MHz, (CD3)2SO): δ -156.96, -154.31, -142.68 (3 F, all m) ppm. Anal. calcd. for C18H11F3O6: C, 56.85; H, 2.92; found: C, 56.56; H, 2.80.
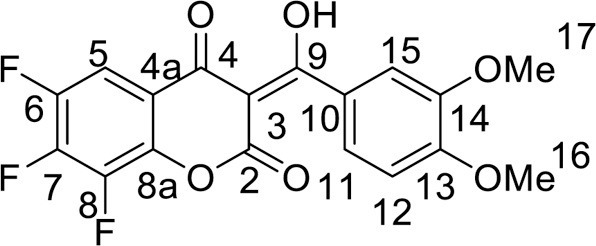
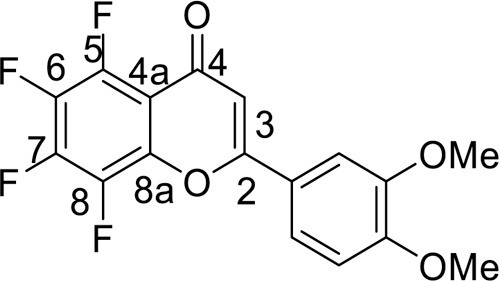
4.3.3.11. 5,6,7,8-Tetrafluoro-2-(3,4-dimethoxyphenyl)-4H-chromene-4-one (12)
Flavone 4a (0.5 mmol), was dissolved in 2 mL of EtOH, then 8 mL of 60 % H2SO4 (w/w) was slowly added to a mixture while stirring. The reaction mixture was heated at 80 °C for 2 h. At the end of the reaction, the mixture was diluted with 50 mL of H2O and cooled. The precipitate was filtered off and purified by column chromatography on silica gel, using CHCl3. Yield 181 mg (92 %), yellow powder, mp 249–250 °C. IR: ν 3066, 2985, 2957, 2848 (C-H), 1649, 1632 (C = O), 1513, 1492, 1364, 1338 (C = C, C–H), 992 (C–F) cm–1. 1H NMR (400.13 MHz, (CDCl3): δ 3.98 (6H, s, 2 OMe), 6.69–7.57 (3H, m, C6H3), 7.35 (1H, s, H, C-3) ppm. 19F NMR (376.44 MHz, (CDCl3): δ -160.78, -158.49, -148.22, -142.66 (4 F, all m) ppm. Anal. calcd. for C17H10F4O4: C, 57.64; H, 2.85; found: C, 57.86; H, 2.73.
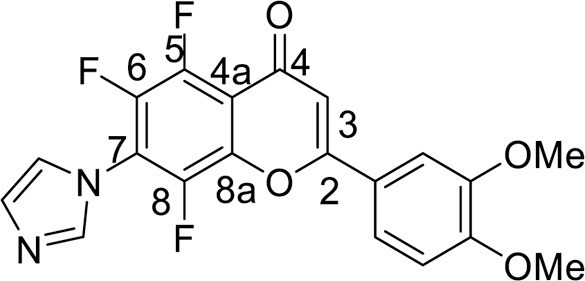
4.3.3.12. 5,6,8-Trifluoro-2-(3,4-dimethoxyphenyl)-7-(1H-imidazol-1-yl)-4H-chromene-4-one (13)
Polyfluorochromen-3-carboxylate 6a (0.5 mmol), was dissolved in 2 mL of EtOH, then 8 mL of 60 % H2SO4 (w/w) was slowly added to a mixture while stirring. The reaction mixture was heated at 80 °C for 2 h. At the end of the reaction, the mixture was diluted with 50 mL of H2O and cooled. The precipitate was filtered off and crystallized from acetone. Yield 181 mg (90 %), yellow powder, mp 266–268 °C. IR: ν 3081, 2941 (C-H), 1649 (C = O), 1516, 1495, 1366 (C–N, C = C, C–H), 995 (C–F) cm–1. 1H NMR (500.13 MHz, (CD3)2SO): δ 3.86, 3.87 (6H, both s, 2 OMe), 7.18–7.68 (3H, m, C6H3), 7.25 (1H, s, H, C-3), 7.81, 8.05, 9.20 (3H, all s, imidazole-1-yl) ppm. 19F NMR (470.52 MHz, (CD3)2SO): δ -149.57 (1 F, d, J =18.7 Hz), -145.75 (1 F, m), -143.62 (1 F, d, J =14.0 Hz) ppm. Anal. calcd. for C20H13F3N2O4: C, 59.71; H, 3.26; N, 6.96; found: C, 59.59; H, 3.12; N, 6.78.
 |
4.4. Biological assay
4.4.1. Toxicity studies
Microtetrazolium test [21] was used to study cytotoxicity of the compounds. Briefly, series of two-fold dilutions of each compound (300−4 μg/ mL) in MEM were prepared. MDCK cells (ATCC CCL-34) were incubated for 48 h at 37 °C in 5% CO2 in the presence of the dissolved substances. The degree of destruction of the cell monolayer was then evaluated in the microtetrazolium test (MTT). The cells were washed twice with saline, and a solution of 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (ICN Biochemicals Inc., Aurora, Ohio) (0.5 mg/ mL) in phosphate-buffered saline was added to the wells. After 1 h incubation, the wells were washed and the formazan residue dissolved in DMSO (0.1 mL per well). The optical density of wells was then measured on a Multiscan FC photometer (Thermo Scientific, USA) at wavelength of 540 nm and plotted against concentration of the compounds. Each concentration was tested in three parallels. The 50 % cytotoxic dose (CC50) of each compound (i.e., the compound concentration that causes the death of 50 % cells in a culture, or decreasing the optical density twice as compared to the control wells) was calculated from the data obtained. Values of CC50 obtained in microgram/mL were then calculated into micromoles.
4.4.2. Cell protection assay
The compounds in appropriate concentrations were added to MDCK or Vero cells (0.1 mL per well). MDCK cells were further infected with A/Puerto Rico/8/34 (H1N1) influenza virus (m.o.i 0,01), and Vero cells were infected with either Coxsackie B3 virus (m.o.i. 0.01). Plates were incubated for 72 h at 36 °C at 5% CO2. After that, cell viability was assessed by MTT test as described above. The cytoprotective activity of compounds was considered as their ability to increase the values of OD comparing to control wells (with virus only, no drugs). Based on the results obtained, the values of IC50, i.e. concentration of compounds that result in 50 % cells protection were calculated using GraphPad Prism software. Values of IC50 obtained in microgram/mL were then calculated into micromoles. For each compound the value of selectivity index (SI) was calculated as ratio of CC50 to IC50.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by the Russian Foundation for Basic Research (RFBR) and the Ministry of Industry and Science of the Sverdlovsk Region (project No. 20-43-660011).
The equipment of the CCU “Spectroscopy and analysis of organic compounds” of the Postovsky Institute of Organic Synthesis UD RAS was used in this work.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.jfluchem.2020.109657.
Appendix A. Supplementary data
The following is Supplementary data to this article:
References
- 1.Uyeki T.M., Bernstein H.H., Bradley J.S., Englund J.A., File T.M., Fry A.M., Gravenstein S., Hayden F.G., Harper S.A., Hirshon J.M., Ison M.G., Johnston B.L., Knight S.L., McGeer A., Riley L.E., Wolfe C.R., Alexander P.E., Pavia A.T. Clinical practice guidelines by the infectious diseases society of America: 2018 update on diagnosis, treatment, chemoprophylaxis, and institutional outbreak management of seasonal influenza. Clin. Infect. Dis. 2019;68:895–902. doi: 10.1093/cid/ciy874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simmonds P., Gorbalenya A.E., Harvala H., Hovi T., Knowles N.J., Lindberg A.M., Oberste M.S., Palmenberg A.C., Reuter G., Skern T., Tapparel C., Wolthers K.C., Woo P.C.Y., Zell R. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch. Virol. 2020;165:793–797. doi: 10.1007/s00705-019-04520-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Crom S.C.M., Rossen J.W.A., van Furth A.M., Obihara C.C. Enterovirus and parechovirus infection in children: a brief overview. Eur. J. Pediatr. 2016;175:1023–1029. doi: 10.1007/s00431-016-2725-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shahani L., Ariza-Heredia E.J., Chemaly R.F. Antiviral therapy for respiratory viral infections in immunocompromised patients. Exp. Rev. Anti-Infect. Ther. 2017;15:401–415. doi: 10.1080/14787210.2017.1279970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Singh M., Kaur M., Silakari O. Flavones: an important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014;84:206–239. doi: 10.1016/j.ejmech.2014.07.013. [DOI] [PubMed] [Google Scholar]; (b) Kitamura K., Ando Y., Matsumoto T., Suzuki K. Total synthesis of aryl C-glycoside natural products: strategies and tactics. Chem. Rev. 2018;118:1495–1598. doi: 10.1021/acs.chemrev.7b00380. [DOI] [PubMed] [Google Scholar]; (c) Silva L.N., Zimmer K.R., Macedo A.J., Trentin D.S. Plant natural products targeting bacterial virulence factors. Chem. Rev. 2016;116:9162–9236. doi: 10.1021/acs.chemrev.6b00184. [DOI] [PubMed] [Google Scholar]
- 6.Correia-da-Silva M., Sousa E., Duarte B., Marques F., Carvalho F., Cunha-Ribeiro L.M., Pinto M.M.M. Flavonoids with an oligopolysulfated moiety: a new class of anticoagulant agents. J. Med. Chem. 2011;54:95–106. doi: 10.1021/jm1013117. [DOI] [PubMed] [Google Scholar]
- 7.(a) Singh H., Singh J.V., Gupta M.K., Singh P., Sharma S., Nepali K., Bedi P.M.S. Benzoflavones as cholesterol esterase inhibitors: synthesis, biological evaluation and docking studies. Bioorg. Med. Chem. Lett. 2017;27:850–854. doi: 10.1016/j.bmcl.2017.01.020. [DOI] [PubMed] [Google Scholar]; (b) Wei Y., Peng A.-I., Weng L. Ma, Peng G., Du Y., Tang J. Synthesis and biological evaluation of phosphorylated flavonoids as potent and selective inhibitors of cholesterol esterase. Eur. J. Med. Chem. 2014;74:751–758. doi: 10.1016/j.ejmech.2013.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Torres-Piedra M., Figueroa M., Hernández-Abreu O., Ibarra-Barajas M., Navarrete-Vázquez G., Estrada-Soto S. Vasorelaxant effect of flavonoids through calmodulin inhibition: ex vivo, in vitro, and in silico approaches. Bioorg. Med. Chem. 2011;19:542–546. doi: 10.1016/j.bmc.2010.10.063. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zeidner J.F., Foster M.C., Blackford A.L., Litzow M.R., Morris L.E., Strickland S.A., Lancet J.E., Bose P., Levy M.Y., Tibes R., Gojo I., Gocke C.D., Rosner G.L., Little R.F., Wright J.J., Doyle L.А., Smith B.D., Karp J.E. Final results of a randomized multicenter phase II study of alvocidib, cytarabine, and mitoxantrone versus cytarabine and daunorubicin (7+3) in newly diagnosed high-risk acute myeloid leukemia (AML) Leuk. Res. 2018;72:92–95. doi: 10.1016/j.leukres.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cui J., Li S. Structure, chemistry and pharmacology of naphthoflavones. Mini-Rev. Med. Chem. 2013;13:1357–1368. doi: 10.2174/1389557511313090010. [DOI] [PubMed] [Google Scholar]; (c) Gilbert E.R., Liu D. Flavonoids influence epigenetic-modifying enzyme activity: structure-function relationships and the therapeutic potential for caner. Curr. Med. Chem. 2010;17:1756–1768. doi: 10.2174/092986710791111161. [DOI] [PubMed] [Google Scholar]
- 10.(a) Shcherbakov K.V., Artemyeva M.A., Burgart Ya.V., Evstigneeva N.P., Gerasimova N.A., Zilberberg N.V., Kungurov N.V., Saloutin V.I., Chupakhin O.N. Transformations of 3-acyl-4H-polyfluorochromen-4-ones under the action of amino acids and biogenic amines. J. Fluorine Chem. 2019;226 doi: 10.1016/j.jfluchem.2019.109354. [DOI] [Google Scholar]; (b) Sagrera G., Bertucci A., Vazques A., Seoane G. Synthesis and antifungal activities of natural and synthetic biflavonoids. Bioorg. Med. Chem. 2011;19:3060–3073. doi: 10.1016/j.bmc.2011.04.010. [DOI] [PubMed] [Google Scholar]; (c) Bykov V.A., Dubinskaya V.A., Rebrov L.B., Mineeva M.F., Skuridin S.G., Evdokimov Yu.M. Comples approach to investigation of the mechanisms of action of antimicrobial and antiviral drugs. Pharm. Chem. J. 2008;42:105–110. doi: 10.1007/s11094-008-0081-2. [DOI] [Google Scholar]; (d) Desideri N., Conti C., Mastromarino P., Mastropaolo F. Synthesis and anti-rhinovirus activity of 2-styrilchromones. Antiviral Chem. Chemoter. 2000;11:373–381. doi: 10.1177/095632020001100604. [DOI] [PubMed] [Google Scholar]
- 11.(a) Chintakrindi A.S., Gohil D.J., Chowdhary A.S., Kanyalkar M.A. Desighn, synthesis and biological evaluation of substituted flavones and aurones as potential anti-influenza agents. Bioorg. Med. Chem. 2020;28 doi: 10.1016/j.bmc.2019.115191. [DOI] [PubMed] [Google Scholar]; (b) Zhong M., Wang H.-Q., Yan H.-Y., Wu S., Gu Z.-Y., Li Y.-H. Santin inhibits influenza A virus replication through regulating MAPKs and NF-kB pathways. J. Asian Nat. Prod. Res. 2019;21:1205–1214. doi: 10.1080/10286020.2018.1520221. [DOI] [PubMed] [Google Scholar]; (c) Seong R.-K., Kim J.-A., Shin O.S. Wogonin a flavonoid isolated from Scutellaria baicalensis has anti-viral activities against influenza infection via modulation of AMPK pathways. Acta Virol. 2018;62:78–85. doi: 10.4149/av_2018_109. [DOI] [PubMed] [Google Scholar]; (d) Dayem A.A., Choi H.Y., Kim Y.B., Cho S.-G. Antiviral effect of methylated flavonol isorhamnetin against influenza. PLoS One. 2015;10 doi: 10.1371/journal.pone.0121610. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Martin-Benlloch X., Haid S., Novodomska A., Rominger F., Pietschmann T., Davioud-Charvet E., Elhabiri M. Physicochemical properties govern the activity of potent antiviral flavones. ACS Omega. 2019;4:4871–4887. doi: 10.1021/acsomega.8b033324. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Puranik N.V., Rani R., Singh V.A., Tomar S., Puntambekar H.M., Srivastava P. Evaluation of the antiviral potential of halogenated dihydrorugosaflavonoids and molecular modeling with nsP3 protein of Chikungunya virus (CHIKV) ACS Omega. 2019;4:20335–20345. doi: 10.1021/acsomega.9b02900. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Xu J.-J., Wu X., Li M.-M., Li G.-Q., Yang Y.-T., Luo H.-J., Huang W.-H., Chung H.Y., Ye W.-C., Wang G.-C., Li Y.-L. Antiviral activity of polymethoxylated flavones from “Guangchenpi”, the edible and medicinal pericarps of Citrus reticulate ‘Chachi’. J. Agric. Food Chem. 2014;62:2182–2189. doi: 10.1021/jf404310y. [DOI] [PubMed] [Google Scholar]; (h) Martin-Benlloch X., Elhabiri M., Lanfranchi D.A., Davioud-Charvet E. A practical and ecomomical high-yielding, six-step sequence synthesis of flavone: application to the multigram-scale synthesis of ladanein. Org. Process Res. Dev. 2014;18:613–617. doi: 10.1021/op4003642. [DOI] [Google Scholar]; (i) Grienke U., Braun H., Seidel N., Kirchmair J., Richter M., Krumholz A., von Grafenstein S., Liedl K.R., Schmidtke M., Rollinger J.M. Comuter-guided approach to acsess the anti-influenza activity of licorice constituencies. J. Nat. Prod. 2013;77:563–570. doi: 10.1021/np400817j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Souza J., Molfetta F.A., Honório K.M., Santos R.H.A., da Silva A.B.F. A study on the picornavirus activity of flavonoid compounds (flavones) by using quantum chemical and chemometric methods. J. Chem. Inf. Comput. Sci. 2004;44:1153–1161. doi: 10.1021/ci030384n. [DOI] [PubMed] [Google Scholar]
- 12.Mashkovski M.D. fourteenth ed. Novaya Volna; Moscow: 2002. Lekarstvennie Sredstva (in Russian) [Google Scholar]
- 13.(a) Johnson B.M., Shu Y.-Z., Zhuo X., Meanwell N.A. Metabolic and pharmaceutical aspects of fluorinated compounds. J. Med. Chem. 2020;63:6315–6386. doi: 10.1021/acs.jmedchem.9b01877. [DOI] [PubMed] [Google Scholar]; (b) Zhou Y., Wang J., Gu Z., Wang S., Zhu W., Aceña J.L., Soloshonok V.A., Izawa K., Liu H. Nex generation of fluorine-containing pharmaceuticals, compounds currently in phase II-III clinical trials of major pharmaceutical companies: new structural trends and therapeutic areas. Chem. Rev. 2016;116:422–518. doi: 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]; (c) Gillis E.P., Eastman K.J., Hill M.D., Donnelly D.J., Meanwell N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- 14.Harborne J.B., Marby T.J., Marby H. Springer; Boston: 1975. The Flavonoids. [DOI] [Google Scholar]
- 15.(a) Dong J., Chen S., Li R., Chu W., Jiang H., Ling Y., Yang Z., Hu W. Imidazol-based pinanamine derivatives: discovery of dual inhibitors of the wild-type and drug-resistant mutant of the influenza A virus. Eur. J. Med. Chem. 2016;108:605–615. doi: 10.1016/j.ejmech.2015.12.013. [DOI] [PubMed] [Google Scholar]; (b) Sharma D., Narasimhan B., Kumar P., Judge V., Narang R., de Clercq E., Balzarini J. Synthesis, antimicrobial and antiviral evaluation of substituted imidazole derivatives. Eur. J. Med. Chem. 2009;44:2347–2353. doi: 10.1016/j.ejmech.2008.08.010. [DOI] [PubMed] [Google Scholar]; (c) Saudi M., Zmurko J., Kaptein S., Rosenski J., Neyts J., van Aerschot A. Synthesis and evaluation of imidazole-4,5-pyrazine-2,3-dicarboxamides targeting dengue and yellow fever virus. Eur. J. Med. Chem. 2014;87:529–539. doi: 10.1016/j.ejmech.2014.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zarubaev V.V., Garshinina A.V., Kalinina N.A., Shtro A.A., Belyaevskaya S.V., Slita A.V., Nebolsin V.E., Kisilev O.I. Activity of ingavirin (6-[2-(1H-imidazol-4-yl)ethylamino]-5-oxo-hexanoic acid) against human respiratory viruses in in vivo experiments. Pharmaceuticals. 2011;4:1518–1534. doi: 10.3390/ph4121518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shcherbakov K.V., Burgart Ya.V., Saloutin V.I., Chupakhin O.N. Polyfluorine-containing 4H-chromen-4-ones: synthesis and transformations. Russ. Chem. Bull. 2016;65:2151–2162. doi: 10.1007/s11172-016-1563-0. [DOI] [Google Scholar]
- 18.(a) Shcherbakov K.V., Burgart Ya.V., Saloutin V.I., Chupakhin O.N. Modification of polyfluoro-containing 3-(ethoxycarbonyl)flavones by biogenic amines and amino acids. Curr. Org. Synth. 2018;15:707–714. doi: 10.2174/1570179415666180405120706. [DOI] [Google Scholar]; (b) Shcherbakov K.V., Bazhin D.N., Burgart Ya.V., Saloutin V.I. Novel 3-acetyl-2-methyl-polyfluorochromones in reaction with amines and esters of amino acids. Chem. Heterocycl. Compd. 2015;51:961–968. doi: 10.1007/s10593-016-1805-y. [DOI] [Google Scholar]; (c) Shcherbakov K.V., Burgart Ya.V., Saloutin V.I. Features of reactions of polyfluorinated ethyl 4-oxo-2-phenyl-4H-chromene-3-carboxylates with N-nucleophiles. Russ. J. Org. Chem. 2013;49:719–729. doi: 10.1134/S1070428013050151. [DOI] [Google Scholar]
- 19.Ranu B.C., Bhar S. Dealkylation of ethers. Org. Prep. Proc. Int. 1996;28:371–409. doi: 10.1080/00304949609356549. [DOI] [Google Scholar]
- 20.Galli U., Ciraolo E., Massaroti A., Margaria J.P., Sorba G., Hirsch E., Tron G.C. The Guareschi pyridine scaffold as a voluable platform for the identification of selective PI3K inhibitors. Molecules. 2015;20:17275–17287. doi: 10.3390/molecules200917275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.