

Abstract
Fc gamma receptors (FcγRs) translate antigen recognition by immunoglobulin G (IgG) into various immune responses. A better understanding of this key element of immunity promises novel insights into mechanisms of (auto-/allo-)immune diseases and more rationally designed antibody-based drugs. Glycosylation on both IgG and FcγR impacts their interaction dramatically. Regarding FcγR glycosylation profiling, major analytical challenges are associated with the presence of multiple glycosylation sites in close proximity and large structural heterogeneity. To address these challenges, we developed a straightforward and comprehensive analytical methodology to map FcγRIIIb glycosylation in primary human cells. After neutrophil isolation and immunoprecipitation, glycopeptides containing a single site each were generated by a dual-protease in-gel digestion. The complex mixture was resolved by liquid chromatography–tandem mass spectrometry (LC-MS/MS) providing information on the level of individual donors. In contrast to recently published alternatives for FcγRIIIb, we assessed its site-specific glycosylation in a single LC-MS/MS run and simultaneously determined the donor allotype. Studying FcγRIIIb derived from healthy donor neutrophils, we observed profound differences as compared to the soluble variant and the homologous FcγRIIIa on natural killer cells. This method will allow assessment of differences in FcγRIII glycosylation between individuals, cell types, subcellular locations, and pathophysiological conditions.
Introduction
Binding of immunoglobulin G (IgG) to Fc gamma receptors (FcγRs) initiates and regulates important immune responses.1,2 Therefore, FcγRs play a key role in homeostasis and under many pathological conditions.3,4 This is widely exploited for therapeutic purposes, for example, with monoclonal antibodies or polyclonal intravenous IgG.5−7 The key interaction between IgG and FcγRs is heavily regulated by the proteoform distribution of either binding partner, e.g., through post-translational modifications. The impact of IgG proteoforms has been extensively studied in the last decades.8,9 Subclass, allotype, and glycosylation, especially fucosylation of IgG impacts FcγR binding.10−12 FcγR-mediated IgG effector functions are partially regulated by varying FcγR expression on different immune cells.13 For example, FcγRIIIb or CD16b is exclusively expressed as a monomeric protein mainly on granulocytes, while the homologous FcγRIIIa or CD16a is expressed on NK cells, monocytes, macrophages, and dendritic cells. Unlike all other transmembrane FcγRs, the human FcγRIIIb is GPI-anchored and lacks a transmembrane and cytoplasmic signaling domain. FcγRIIIb is an 233 amino acid protein, which consists of an N-terminal signal peptide (18 amino acids) cleaved during protein processing and two domains in the extracellular region.14 These domains share 97% sequence homology with FcγRIIIa.15 Despite the homology, FcγRIIIa binds IgG stronger than FcγRIIIb due to a single amino acid difference, G129 versus D129.16 FcγRIIIb seems to affect signaling of other Fc receptors by accumulating in lipid rafts, which are enriched in kinases (Src) and are required for ITAM phosphorylation and signaling.17 Given the relatively high expression levels of FcγRIIIb on neutrophils with 120 000–300 000 copies per cell,18 and the dominance of neutrophils among white blood cells, FcγRIIIb can be considered to be the most abundant FcγR in circulation. Known functions include activation of neutrophil degranulation, cell adhesion, calcium mobilization, and neutrophil tethering to soluble immune complexes.19−21
Despite recent advances, the role of FcγR proteoforms is only poorly understood.14 Allotypes lead to differentially active proteoforms. There are three known allotypes of FcγRIIIb, neutrophil antigen 1 (NA1) and 2 (NA2), and SH (SH being uncommon). They vary in their affinity to IgG and capacity to induce phagocytosis of IgG-opsonized targets.22 The two major FcγRIIIb allotypes, NA1 and NA2, differ in four amino acids, resulting in four and six potential N-glycosylation sites for NA1 (N38, N74, N162, and N169) and NA2 (N38, N45, N64, N74, N162, and N169), respectively.23 Five of the potential N-glycosylation sites are conserved between FcγRIIIa and FcγRIIIb, namely N38, N45, N74, N162, and N169. The vast majority of glycan data on FcγRIII are derived from the endogenous FcγRIIIa of NK cells24 and monocytes25 as well as recombinant protein expressed in human embryonic kidney (HEK) cells, Chinese hamster ovary (CHO) cells,26 Baby Hamster Kidney (BHK) cells,27 and murine cell line NS0.28 Both variants, endogenous as well as recombinant FcγRIIIa/b are modified by N-glycans, but only for recombinant FcγRIIIa a single O-glycan modification has been detected.26 FcγR glycosylation strongly impacts the interaction with IgG. Some receptor glycans have direct glycan–glycan and glycan–protein interactions with bound IgG.29,30 Deglycosylation of site N162 (unique to FcγRIIIa and FcγRIIIb), not only strongly increases the affinity of FcγRIIIa to IgG but also alleviates its sensitivity to IgG core fucosylation.31,32 Furthermore, differences in FcγRIIIb glycosylation in different cell models have been shown to impact IgG binding.33,34 The available studies underline the importance of FcγR glycosylation, but can only sketch a very rough picture of its functional impact.
A prominent reason for this lack of functional understanding is the limited data available on FcγR glycosylation of primary human cells.14 While the great heterogeneity of proteoforms, especially in FcγRIIIb, was already apparent in studies from the 1990s,18 glycomics studies on primary human cells only became possible in recent years.35 However, owing to the great complexity and differential functional impact of glycosylation sites, only site-specific glycoproteomics studies can characterize FcγR glycosylation to the necessary extent. Recent studies on healthy volunteers revealed FcγRIIIa glycosylation of NK cells24 and monocytes36 and FcγRIIIb glycosylation of neutrophils.37 Therein, cells were purified by negative selection with magnetic beads, followed by immunoprecipitation of FcγR. Additionally, the soluble FcγRIIIb,38 which originates from shedding from neutrophils upon their activation, has been studied in plasma. The purified receptor from all sources was analyzed by bottom-up glycoprotein analysis/glycoproteomics following protease cleavage with chymotrypsin and/or endoproteinase Glu-C (GluC).26 Targeted analysis of FcγRIIIb using immunoprecipitation (IP) offers important advantages because it increases the depth of analysis, and hence the number of identified and quantified glycoforms. Untargeted glycoprofiling strategies are advantageous as they provide a glimpse of the glycosylation of many proteins. Although they may overcome potential proteoform biases in antibody-specificity for IP, the increased sample complexity and resulting co-elution of (glyco-)peptides in such approaches introduces significant biases due to ion suppression and undersampling.39,40
Although groundbreaking, the previous two studies on site-specific glycosylation of FcγRIIIb had some limitations.37,38 They relied on two independent proteolytic cleavages, thus necessitating multiple liquid chromatography–mass spectrometry (LC-MS) runs to cover the whole receptor. Yagi et al., focused on soluble FcγRIIIb whose function is largely unknown.38 They used pooled blood from multiple donors, loosing inter-donor variability. Washburn et al., analyzed only three of the six potential N-glycosylation sites, but accumulated strong data on inter-donor variability of 50 donors.37 Nonetheless, we still know very little about the functional and clinical impact of FcγR glycosylation to prefer a method focusing only on certain glycosylation sites. Other existing strategies covering all sites of FcγRIIIb or FcγRIIIa are quite complicated and difficult to apply to clinical investigations where eventually large numbers of samples need to be detected in a robust way.24,38
Here, we present a method for the site-specific analysis of all glycosylation sites of FcγRIIIb in a single LC-MS/MS experiment. Using this, we identified and relatively quantified neutrophil-derived FcγRIIIb glycosylation individually for multiple donors. Additionally, our approach allowed a qualitative overview of site occupancy and the determination of donor allotypes. This was enabled by avoiding glycopeptide enrichment, which also promises more robustness. Additionally, interferences from endogenous IgG and from a leaking capture antibody are avoided by a simple nonreducing SDS-PAGE step. Additionally, our method is generic for FcγRIIIa and FcγRIIIb, making it potentially applicable to a wide range of leukocytes. Moving toward clinical investigations of FcγR glycosylation will be essential for a complete understanding of the (patho-)physiological role of IgG-FcγR interactions. Our methodology presents a uniquely suited starting point, as it is unprecedented in its ability to simultaneously cover individual donor, subclass, allotype, cell and site differences of FcγRIII glycosylation comprehensively.
Experimental Section
Buffers and Reagents
Trizma hydrochloride, Tris(hydroxymethyl)aminomethane, Protease Inhibitor Cocktail (Set V, EDTA-Free), phenylmethylsulfonyl fluoride (PMSF), ethylenediaminetetraacetic acid (EDTA), and glycerol were obtained from Sigma-Aldrich (Steinheim, Germany). Di-sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O), potassium dihydrogen phosphate (KH2PO4), and NaCl were obtained from Merck (Darmstadt, Germany). CARIN lysis buffer (pH 8.0) was prepared in-house with 20 mM Tris-HCl, 137 mM NaCl, 10 mM EDTA, 0.1 M NaF, 1% NP-40, and 10% glycerol. Protease Inhibitor Cocktail and PMSF were added to prevent unwanted proteolysis. Phosphate buffered saline (PBS, 0.035 M, pH 7.6) was prepared in-house with 5.7 g/L Na2HPO4, 2H2O, 0.5 g/L KH2PO4, and 8.5 g/L NaCl. The Coomassie stain was prepared in-house according to Candiano et al.41 using Coomassie Blue G-250 (Sigma-Aldrich). SDS-PAGE reagents were of the NuPAGE series (ThermoFisher Scientific) and included: LDS Sample Buffer (4×) (nonreducing loading buffer), a PageRuler Prestained Protein Ladder (protein standard), 4–12% Bis-Tris gel, and 4-morpholinepropanesulfonic acid (MOPS) SDS running buffer.
Materials
GluC (Staphyloccoccus aureus Protease V8) and chymotrypsin were obtained from Worthington Biochemical Corp. ( Lakewood, USA). Ultrapure deionized water (MQ) was generated using a Q-Gard 2 system (Millipore, Amsterdam, The Netherlands). MS grade acetonitrile (ACN) was acquired from Biosolve B.V. (Valkenswaard, The Netherlands). Iodoacetamide (IAA), dithiothreitol (DTT), ethylenediaminetetraacetic acid (EDTA), analytical grade formic acid (FA), and LC-MS grade water were obtained from Sigma-Aldrich (Steinheim, Germany). More information on buffers and reagent can be found in the Supporting Information. FcγRIIIs were immunoprecipitated from the neutrophil cell lysate using a mouse anti-CD16 monoclonal IgG2a antibody (Ref M9087, Clone CLB-FcR gran/1, 5D2, Sanquin, Amsterdam, The Netherlands). Prior to use, antibodies were labeled with biotin.
Antibody Biotinylation
At first, the antibodies were buffer exchanged from Tris buffer to PBS buffer using the Zeba spin protocol (ThermoFisher Scientific, Rockford, IL), as amine-containing buffers may interfere with biotinylation. Subsequently, the Z-link Sulfo-NHS-Biotinylation protocol (ThermoFisher Scientific) was followed. The level of biotin incorporation was measured by HABA Assay (ThermoFisher Scientific).
Neutrophil Cell Isolation and FcγRIIIb Purification
Neutrophils were isolated from whole blood of three healthy volunteers as described previously.42 Briefly, the blood was collected from healthy donors in tubes coated with spray dried EDTA for anticoagulation (VACUETTE TUBE 9 mL K3E K3EDTA, Greiner Bio-One, Amsterdam, The Netherlands). Mononuclear leukocytes and platelets were removed by centrifugation (2000 rpm, 18 min, 25 °C) using a Ficoll gradient with a specific density of 1.077 g/mL (Ficoll-Paque PLUS, GE Healthcare, Freiburg, Germany). Erythrocytes were subsequently lysed with isotonic NH4Cl solution at 4 °C. With this standard method, a high degree of depletion of other cell types is obtained and neutrophils are isolated with a typical purity of over 95%. The isolated neutrophils were washed two times with 1 mL of cold PBS. The cells were counted on a CASY automated cell counter (Thermo Fisher Scientific, Rockford, IL) followed by centrifugation (1200 rpm, 4 min, 4 °C). The neutrophils were resuspended in CARIN buffer at a final concentration of 50 × 106 cells/mL (Table S1). The cells were then incubated on ice for 5–10 min. Finally, cell lysates were sonicated for 30 s at 20 kHz. The cell lysates were centrifuged at 13 000g for 15 min at 4 °C. The cellular debris pellet was discarded. The total protein content of the supernatant was measured with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Rockford, IL). For FcγRIII identification and purification, respectively, an amount of ∼3 and ∼16 million primary human neutrophils from healthy donors was used for immunoprecipitation (Table S1). Neutrophil proteins (100 or 500 μg) were incubated, while rotating, with 5 or 25 μg of biotinylated antibodies in total volume of 1 mL of CARIN lysis buffer overnight at 4 °C. A rough estimate of the antibody/FcγRIII ratio was 2500:1. High Capacity Streptavidin Agarose Resin beads (Thermo) (10 μL) were washed twice with 1 mL of CARIN buffer and incubated with the preformed FcγRIII-anti-CD16 immune complexes for 1 h at 4 °C under rotation. The beads were centrifuged for 2 min at 2500g, supernatant was removed, and washed four times with 1 mL of CARIN buffer. Finally, FcγRIII was eluted from the beads two times with 150 μL of 200 mM FA. The eluates were then dried by vacuum centrifugation at 60 °C for 2 h.
SDS-PAGE
The immunoprecipitation of FcγRIIIb from 100 μg of total neutrophil proteins was evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The dried samples were resuspended in 20 μL of nonreducing loading buffer, incubated at 95 °C for 5 min and applied to a 4–12% Bis-Tris gel. The migration was performed at a constant voltage of 200 V for 50 min in 4-morpholinepropanesulfonic acid (MOPS) SDS running buffer. Gels were stained using Commassie blue. The presence of CD16 was confirmed by western blotting, using an anti-CD16 mouse monoclonal IgG1 conjugated to horseradish peroxidase (DJ130c, sc-20052 HRP, Lot #D2617, Santa Cruz Biotech; see also Supporting Information).
In-Gel Proteolytic Digestion and LC-MS Glycopeptide Analysis
For the in-gel LC-MS workflow, 500 μg of total neutrophil proteins were used for immunoprecipitation. Subsequently, SDS-PAGE was performed at a constant voltage of 200 V for only 15 min. The obtained gels were silver stained (SilverQuest Staining Kit, Invitrogen) and the protein of interest was cut out from the gel. Excised bands were subjected to in-gel digestion with endoproteinase GluC and chymotrypsin, which was performed on a Proteineer DP digestion robot (Bruker, Bremen, Germany).26,43,44 Reduction with 10 mM DTT was followed by alkylation with 50 mM IAA45 and 3 wash and shrink cycles with 25 mM ammonium bicarbonate (pH 8.4) and neat acetonitrile, respectively. The shrunken gel bands were soaked for 45 min at 10 °C in 25 mM ammonium bicarbonate, containing both proteases at a concentration of 12.8 ng/mL each. Excess protease solution was removed, followed by overnight digestion at 37 °C. Peptides were extracted with 100 μL of H2O/acetonitrile/formic acid (50:50:1), lyophilized and dissolved in solvent A (3% ACN/95% water containing 0.1% FA (v/v)) prior to injection. Liquid chromatography–tandem mass sprectometry (LC-MS/MS) analysis was performed on a nanoLC-MS system equipped with an Easy nLC 1000 gradient HPLC system (Thermo, Bremen, Germany) and an Orbitrap Fusion LUMOS mass spectrometer (Thermo). Prior to sample injection, the peptides and glycopeptides extracted from the bands were lyophilized and dissolved in solvent A (water containing 0.1% FA (v/v)). The samples were then loaded onto an in-house packed C18 precolumn (100 μm × 15 mm; Reprosil-Pur C18-AQ 3 μm, Dr. Maisch, Ammerbuch, Germany) and separated on a homemade analytical nanoLC column (30 cm × 75 μm; Reprosil-Pur C18-AQ 3 μm). The digested (glyco-)peptides were eluted using a linear gradient from 10 to 40% solvent B (80% ACN/20% water containing 0.1% FA (v/v)) over 20 min, followed by a column washing step with solvent B to 100% (at 25 min), and reconditioning with solvent A for 12 min. The nanoLC column was drawn to a tip of ∼5 μm and acted as the electrospray needle of the MS source. The Orbitrap Fusion LUMOS mass spectrometer was operated in data-dependent MS/MS (top-20 mode) with the collision energy set at 32% normalized collision energy (NCE) and the MS/MS spectrum was recorded on the Orbitrap. The MS1 full scan spectra were acquired within a mass range m/z of 400–1500 and MS/MS was set to auto (i.e., depending on the m/z of the selected precursor ion). The resolution setting for MS1 scans was 12 × 104 and an AGC target value of 4 × 104 for an accumulation time of maximum 50 ms. Dynamic exclusion duration was 10 s with a single repeat count, and charge states in the range 1–5 were included for MS/MS. The resolution of MS/MS scans was 3 × 104 with an AGC target of 5 × 104 with the maximum fill time of 60 ms. MS/MS spectra were generated from precursors isolated with the quadrupole with an isolation width of 1.2 Da. During acquisition, a product ion trigger was set on the HexNAc oxonium ion at m/z 204.087. Upon the detection of the oxonium ion, three additional data-dependent MS/MS scans of the same precursor were executed in the ion-routing multipole with higher energy collisional dissociation (HCD) collision energies of 32, 37, and 42% NCE, respectively, at an AGC target of 5 × 105 with a maximum fill time of 200 ms. In addition, the acquisition of collision-induced dissociation (CID) spectra in the linear ion trap was performed for the precursor and recorded at 35% NCE. The presented results on protein identification, coverage, and purity are from a standard data-dependent HCD run (without exclusive m/z 204.087 triggering). For the identification of glycopeptides in these runs, MS/MS spectra containing the specific HexNAc oxonium ion at m/z 204.087 (HexNAc, [C8H14NO5]+) were extracted from the raw data and written to an .mgf file using in-house routines. This filtering step also ensured to only include spectra containing the HexNAc oxonium ion with a strong signal (among top 30 peaks).
Identification and Quantification of Site-Specific Glycosylation
Initially, LC-MS/MS data were processed with Byonic (Protein Metrics, Cupertino, CA v3.2-38).46 MS/MS spectra were searched against an extensive human proteome database combined with a predefined glycan list (Table S2). Glycopeptide identification with Byonic scores below 150 were removed from the analysis. Digestion specificity was set as nonspecific (slowest), allowing for both specific and miscleaved peptides. Glycosylation was set as a common modification and other modifications were anticipated upon prevalence: Glycan modification/+ [glycan composition] Da @NGlycan | common1; carbamidomethyl/+57.021 Da @C | fixed; oxidation/+15.995 Da @M | common2; acetyl/+42.011 Da @Protein N-term | rare1. The Byonic search allowed one common modification and one rare modification per peptide. For more parameters refer to Table S3. Byonic identifications were manually verified and extended using Xcalibur (Thermo). Table S4 gives an overview of the identification level of individual glycoforms (Byonic, manual MS/MS, manual MS). Regarding the manual identification, MS1 sum spectra were generated—around the retention times reported by Byonic—and searched for expected monosaccharide differences. This was done per combination of unique peptide backbone and number of sialic acids, the main two retention time determinants. Some sialic acid variants were inferred, improving the identification of multisialylated glycan compositions. Annotation of MS1 spectra was based on the precursor mass with a tolerance of ±0.05 Th. Manual MS/MS interpretation was based on, first the clear presence of an m/z corresponding to a peptide or peptide+HexNAc fragment ion of a previously identified glycosylated sequence and second the presence of a dominating pattern of oxonium ions. During the manual interpretation, differences in (glyco-)peptide sequences between the FcγRIIIb allotypes, NA1, and NA2, were taken into consideration.
For automatic alignment, integration, and extraction of LC-MS data, the in-house software LacyTools (Version 1.1.0-alpha) was used as described previously.47 As an input, the raw data files were converted to mzXML files and a list of identified glycopeptides along with their retention times was created. For the area integration of the sum spectra, the following settings were applied: sum spectrum resolution of 100, extraction mass window of 0.07 Th, extraction time window of 15 s, percentage of the theoretical isotopic pattern of 95%, min charge stage 2, and max charge stage 4. This resulted in integrated signal intensities for each glycopeptides per charge stage (from [M+2H]2+ to [M+4H]4+). After extraction, the analytes were curated from the identification list if the average mass error was outside ±20 ppm and the isotopic pattern deviated more than 20% from the theoretical one. This resulted in the exclusion of signals of all doubly charged glycopeptides and inclusion of signals of triply and some quadruply charged analytes. All included charge stage signals for the same glycopeptide were summed, the absolute intensities were corrected for the fraction of isotopes integrated and used for relative quantification. Total area normalization per glycosylation site was used for relative quantitation. The intensities of the iron adduct ([M+FeIII]3+) and ammonia adduct ([M+2H+NH4]3+) signals were significant. Hence, the relative quantification was performed on extracted areas of protonated, iron adduct, and ammonia adduct peaks. The N-glycosylation site occupancy was determined as the fraction of all glycopeptide signals in the sum of glycopeptide and nonglycosylated peptide signals.
Results and Discussion
FcγRIIIb Purification and Identification
Similar to previous reports,48 the western blot showed a smear from 50 to 80 kDa, confirming the presence of FcγRIIIb with its abundant and diverse glycosylation pattern (Figure 1). A comparison of total cell lysate, flow-through, and eluate demonstrates the efficacy of the purification. Nonreducing SDS-PAGE allowed separation of FcγRIIIb from interferences derived, for example, from the capture antibody or endogenous IgG (Figure S1). This is simple and preferable to affinity removal of IgG which may lose specific FcγR proteoforms due to high affinity interactions.24 In the eluate, the FcγRIIIb could not be detected by Coomassie blue staining (LOD ca. 25 ng for well-defined bands). This indicated a recovery value below 4–20%, determined by the percentage ratio of the eluted FcγRIIIb (<25 ng) to the theoretical amount of FcγRIIIb on the neutrophil surface (160–630 ng). This is consistent with the western blot, where the eluate fraction is significantly lower, as determined from neutrophil signals corresponding to 1/4 of the expected optical density at 100%. Despite low recovery, the purification scheme resulted in sufficient material for LC-MS(/MS) analysis after in-gel digestion. Furthermore, MS/MS analysis of the purified protein yielded a sequence coverage of approximately 80% and did not indicate any major interferences in the relevant gel band (Table S5). Thus, a strong enrichment of FcγRIIIb from neutrophil lysate was achieved.
Figure 1.
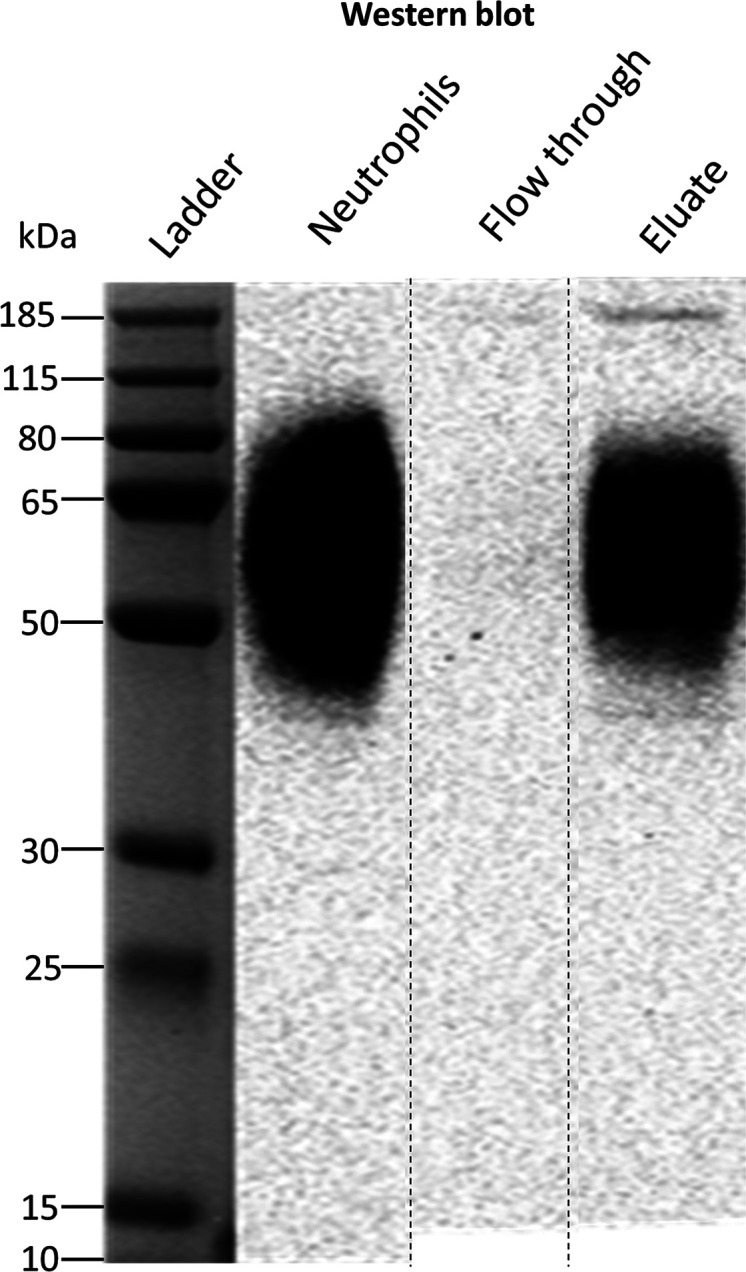
Western blot of human neutrophil lysate before and after FcγRIII immunoprecipitation. The identity of FcγRIIIb was confirmed with an anti-CD16 antibody. The flow-through lanes show the diluted unbound fraction of the immunoprecipitation, while the eluate lanes present the purified FcγRIIIb protein. The neutrophils lanes represent ∼25 μg of total protein content from the neutrophil cell lysate from donor 2. Apparent differences in the western blot are due to concentration differences. The weaker signal of extremely low and high MW proteoforms is lost at lower concentrations after IP. The cropping area is indicated by dashed lines. The complete gel and blot are presented in Figures S1 and S2.
Coverage and Protein Identity
Isolated FcγRIIIb was subjected to an in-gel endoproteinase GluC and chymotrypsin treatment prior to LC-MS(/MS) analysis.26 This approach was sufficient to generate and identify unique peptides for all six glycosylation motifs. FHN45ESLISSQASSY (NES), FIDAATVN64DSGEY (NDS), RCQTN74LSTLSDPVQLE (NLS), CRGLVGSKN162VSSE (NVS), and TVN169ITITQGLAV (NIT) were found in human neutrophils (Table S6), while SPEDN38ESQW was detected only in the recombinant receptor (Figures S3 and S4). An overview of FcγRIIIb glycopeptides is depicted in Figure 2. Our method required only one LC-MS/MS run to obtain full coverage of all glycosylation sites.
Figure 2.

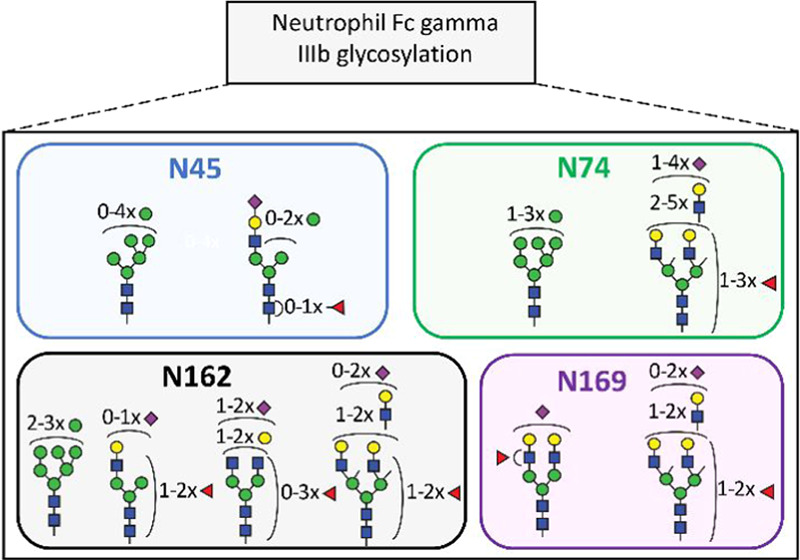
Schematic representation of site-specific N-glycosylation of endogenous FcγRIIIb NA1 and NA2 from human neutrophils. N-glycosylation sites are denoted in bold, blue letters. All determined and occupied glycopeptides are depicted in green with their corresponding set of glycans. The peptide with an unoccupied N-glycosylation site (site N64 NA2) is marked in brown, while the sequence of the peptide containing site N38, that was only detected in recombinant receptor, is indicated in red. Amino acids in yellow show the sequence variations between NA1 and NA2.
FcγRIIIb showed a lower electrophoretic mobility for Donor 1, suggesting the less glycosylated NA1 allotype (Figure S2). The allotype can be determined by proteomics,37 which was achieved simultaneously with glycoprofiling. The assignments were based on the intensity ratios of allele-specific peptides: for example, FHN45ENLISSQASSY (NEN) and FIDAATVD64DSGEY (DDS) for NA1, and FHN45ESLISSQASSY (NES) and FIDAATVN64DSGEY (NDS) for NA2 (Table S7). Indeed, Donor 1 was a NA1/NA2 individual, with a 1:5 ratio of NA2 N45 oligomannose glycopeptides (NES/(NES+NEN)). Relative quantitation may be biased by reduced ionization efficiency of glycopeptides compared to nonglycosylated peptides. However, a similar ratio (1:4) for the unoccupied NA2 N64 peptide (NDS/(DDS+NDS)) was observed. Thus, the 1:5/1:4 ratio likely reflects the real expression levels between NA2 and NA1, potentially caused by naturally occurring gene copy number variation of FcγRIIIb.49 Donor 2 and 3 were NA2 homozygous, since they contained approximately 98% of FHN45ESLISSQASSY and FIDAATVN64DSGEY peptides (Table S7). The residual 2% were explained by peptides sequences shared between FcγRIIIb NA1 and FcγRIIIa (FHN45ENLISSQASSY, FIDAATVD64DSGEY, and FIDAATVD64DSGEYR). We cannot exclude that these signals may be derived from contamination by NK cells, macrophages, and/or monocytes. Additionally, deamidation of site N64 may contribute to the D64 peptide signals, but does not explain the N45 signals. Importantly, at these low levels, the impact of FcγRIIIa glycopeptides on FcγRIIIb glycosylation profiling can be neglected. Therefore, co-isolation is preferred as it enables the use of the same sample preparation protocol for FcγRIIIa dominated cell types.
Neither peptides nor glycopeptides corresponding to the glycosylation site N38 were observed in our LC-MS/MS data of healthy donors. Of note, we were able to detect complex glycans occupying site N38 on recombinant FcγRIIIb (Figures S3 and S4). Yagi et al., previously reported on site N38 large, highly elaborated glycan structures in the range of m/z 1400–2000.38 The identified features on recombinant FcγRIIIb possess on average less antennae which are also less processed.
Glycopeptide Identification
10 N-glycan compositions at N45, 15 at N74, 30 at N162, and 6 at N169 were identified. Of these 61 compositions, 36 glycoforms were confirmed by tandem mass spectrometry (Table S4). Based on glycopeptide fragmentation data, mass accuracy, isotopologue pattern, and biosynthetic pathways, we propose the N-glycan structures shown in Figures 2 and S5. Numerous structural isomers could be present for the same N-glycan composition. We identified multiple isomeric structures, but were not able to resolve them quantitatively. We confirmed the presence of sialic acid by the diagnostic ion at m/z 292.103. Antennary fucosylation was confirmed by the presence of a B-ion at m/z 512.19750 [hexose+N-acetylhexosamine+fucose+H]+. In contrast, core fucosylation was indicated by the formation of the ion assigned as [peptide+N-acetylhexosamine+fucose+H]+. N-glycans containing N-acetyllactosamine (LacNAc) repeats, were indicated by signals at m/z 731.272 [N-acetylhexosamine2+hexose2+H]+.
Two novel (N74 and N169) and three already described (N45, N64, and N162) N-glycosylation sites of neutrophil-derived FcγRIIIb were identified. Per site, the nature and number of glycans differ, and among other things, reflect the extend of biosynthetic processing. The glycan heterogeneity ranges from oligomannose type glycans to highly processed complex type glycans with LacNAc extensions. Three N-glycosylation sites, N45, N162, and N169, were found to be fully occupied. Glycosylated and nonglycosylated forms of the peptide containing site N74 indicated partial occupancy. This peptide was estimated to be glycosylated at 80, 64, and 58% for donor 1, 2, and 3, respectively. Site N64 was known to be unoccupied37 and indeed corresponding peptides were exclusively nonglycosylated. Molecular dynamics simulations of the highly homologous FcγRIIIa (V158 allotype) have shown intramolecular interactions between the peptide backbone residues 60–70 and glycans at N45, which may explain the preference for anunoccupied site N64.30 Moreover, this intramolecular interaction may inhibit enzymatic N-glycan processing in the Golgi, explaining the restricted processing at site N45. The occupancy of site N64 appears to be the most prominent difference between neutrophil-bound and soluble FcγRIIIb (Table S8); the former unoccupied, while the latter displayed highly branched glycans on site N64.38 Different glycosylation profiles of resting neutrophil-bound FcγRIIIb and soluble FcγRIIIb, released by activated neutrophils,23 may warrant further study.
MS spectra obtained for sites N162 and N45 with annotation of the major glycoforms are given in Figures 3 and S6. For site N162, complex di- and tri-antennary glycans were found accompanied by a small percentage of oligomannose glycans. Site N45 mainly showed oligomannosidic glycans with a significant fraction of hybrid and complex structures. N74 predominantly elaborated as di- to tetra-antennary complex glycans with a small amount of oligomannose type glycans. N169 was found to exclusively carry di- and tri-antennary complex glycans.
Figure 3.

MS sum spectrum (retention time 10.2–14.4 min; 94 spectra) showing the major glycoforms of the N162 site. Interestingly, ammonia adducts were exclusively observed for glycopeptides carrying an oligomannose glycan, while iron adducts were detected for both oligomannose and complex structures. For more details on MS/MS spectrum see Figure S9. *: N162 glycopeptides with a miscleaved peptide backbone; #: unidentified glycopeptide (z = 2); and $: iron adducts [M+FeIII]3+. NVS: CRGLVGSKN162VSSE peptide backbone.
Site-Specific Quantification of FcγIIIb N-Glycans from Human Neutrophils
The 61 identified glycopeptides were targeted for relative quantification in a site-specific manner (Figure 4). FcγRIIIb glycosylation of the three healthy donors displayed very similar glycosylation patterns (Figure S7). Derived glycosylation traits—complexity, number of fucoses per glycan, and number of sialic acids per glycan—were calculated to facilitate the comparison of different sites (Figure S8) and with other studies (Table S9).
Figure 4.

Site-specific, relative quantification of FcγRIIIb glycoforms. (a) 10 different glycan compositions were found on the glycosylation site N45, (b) 11 compositions on N74, (c) 29 compositions on N162, and (d) 6 compositions on N169. Mean and SD of three donors are shown. Bar colors indicate glycan classes: green, oligomannose type; orange, hybrid type; blue, neutral complex; and purple, sialylated complex, with one (light), two (medium), or three (dark) sialic acids per glycan.
As shown in Figure 4a and Table S8, site N45 was decorated with 86% oligomannose type glycans (M6, M7, M8, and M9). Remaining were 7% hybrid (M4A1G1S1, M5A1G1S1, and FM4A1G1S1) and 8% complex (A1G1S1, FA1G1S1) type glycans, with and without core fucose. In contrast to soluble FcγRIIIb, containing only oligomannose N-glycans,37 the neutrophil-bound N45 contains a significant amount of sialylated hybrid and monoantennary complex N-glycans.
Site N162 contained 98% of sialylated, complex, mono- to tri-antennary glycans, with some evidence of LacNAc repeats, and 2% of oligomannose glycans (Figure 4c). The derived traits revealed a high level of galactosylation and fucosylation, which indicate a high expression of both galactosyltransferases and fucosyltransferases (Figure S8). On average, 1.3 fucoses per glycan were displayed on N162 glycans. Diagnostic ions provided the evidence for both core and antennary fucosylation. Some monofucosylated compositions (H5N4F1S1, H5N4F1) were detected as both core and antennary fucosylated isomers. In comparison to the other glycosylation sites, N162 possesses the lowest amount of sialic acids per glycan (0.9), which implies poorer accessibility for sialyltransferases.
On site N74, sialylated, fucosylated, and complex N-glycans with multiple LacNAc extensions represent the largest group (86%) of structures (Figure 4b). Fragmentation of these glycans resulted in the formation of the oxonium ion of m/z 657.237, assigned as [N-acetylglucosamine+galactose+N-acetylneuraminic acid]+ (Table S4). No evidence for the presence of oligosialylated antennae was seen in the fragments indicating that the tri-sialylated structures are at least tri-antennary. Even though N74 is not directly involved in antibody binding,51 there are some speculations regarding LacNAc repetition in cell activation regulation through the modulation of receptor clustering.52 This site is characterized by the highest numbers of sialic acids (2.1) and fucoses (1.5) per glycan, which implies high accessibility for sialyltransferases and fucosyltranserases.
Site N169 is mainly occupied by a diantennary and monosialylated (H5N4F1S1) structure (Figure 4d). Interestingly, core fucosylation at N169 was reported for soluble FcγRIIIb, whereas we only observed evidence for antennary fucosylation (Table S4). In general, as depicted in Figure S8, N-glycans modifying N169 were fully galactosylated and exhibited a moderate number of sialic acids (1.1) and fucoses (1.1) per glycan.
Overall, we confirmed the presence of antennary fucosylated glycans for three N-glycosylation sites, namely N74, N162, and N169 (Table S4). N74 and N169 were annotated as predominantly occupied by antennary fucosylated structures, lacking evidence for core fucosylation. In contrast, glycans at N162 presented a mixture of both core and antennary fucosylated isomers. In addition to complex type species, oligomannose structures (M8, M9) complement the repertoire of N74 (14%) and N162 (2%). For N74, oligomannose structures are being reported for the first time. Interestingly, LacNAc repeats were also detected for some N162 glycans (Table S4). Generally, the presence of oligomannose type glycoforms is not expected among highly processed glycopeptides. However, it is consistent with a recent glycomics study on FcγRIIIa24 and with a recent study mapping of subcellular glycans during the cell maturation in healthy human neutrophils,53 where different stages of N-glycan processing within one site may reflect the developmental stage of granulopoiesis.53N-glycan processing is initially influenced by the protein expression and precursor availability. Differential processing of the sites, however, is influenced by transferase accessibility which seems to correlate with solvent accessibility. The more exposed an N-glycosylated asparagine residue is, the more processed its glycans generally are.54 Additionally, the large biosynthetic gap between the oligomannose type glycans and the large (at least partially) tetra-antennary glycans may indicate different subcellular fractions. Considering the overall glycosylation pattern of FcγRIIIb in neutrophils, we estimated that nearly all glycans of FcγRIIIb were fully galactosylated, indicating a high activity of galactosyltransferases. Consequently, the partially sialylated and core fucosylated glycans suggest moderate activity of sialyltransferases and α1,6-fucosyltransferases. Finally, the high average number of fucose per glycan and MS/MS data for antennary fucosylation provide evidence for a high activity of α1,2, α1,3, or α1,4 fucosyltransferases in the neutrophils (Figure S8b, Table S9).
Comparison of Site-Specific N-Glycosylation of FcγRIIIb
Sites N74 and N169 of neutrophil-bound FcγRIIIb are profiled for the first time. Glycosylation profiles of sites N45 and N162, both qualitatively and quantitatively, were highly consistent with the study of Washburn et al. (Tables S8 and S9).37 A minor difference is the observation of oligomannose N-glycans at site N162 in our study.
Recently, a report on the site-specific glycosylation of human NK cell FcγRIIIa has been published.24 Major glycosylation differences on sites N45, N74, and N169 clearly distinguish the two FcγRIII isoforms (Tables S8 and S9). FcγRIIIb showed less processing than FcγRIIIa on N45, containing mainly oligomannose glycans, while FcγRIIIa expressed mainly hybrid type glycans. The most pronounced differences were observed in the levels of antennary fucosylation. FcγRIIIb had on average more than one fucose per glycan, up to 1.5 on N74. Moreover, only antennary fucosylation was observed for monofucosylated glycans at site N169. In stark contrast, glycoforms of FcγRIIIa are reported to be almost exclusively core fucosylated, except small amounts of antennary fucosylation on sites N74 and N162. This hints at a higher activity of α1,2; α1,3; or α1,4 fucosyltransferases in the neutrophils versus NK cells. N-glycans at N162 showed the highest similarity between the two receptor isotypes. This observation suggests that the glycosylation profiles of this functionally relevant site are conserved among FcγRIII isoforms and cells. However, sialylation of N162 appeared lower in neutrophil FcγRIIIb than in NK cell FcγRIIIa with 0.9 and 1.3 sialic acids per glycan at N162, respectively.
Notably, glycosylation differences influence FcγRIII properties and function.34 Thus, for in vitro studies it is important to use an expressing system that confers an endogenous-like glycosylation profile. Among all four mammalian systems producing differently processed N-glycans (HEK293, CHO, BHK, and NS0),26−28 CHO cells constitute a good expression vehicle, where highly branched N74 glycans in FcγRIII were carrying LacNAc repetitions. However, to produce antennary fucosylation, prevalent in FcγRIIIb, the HEK293 system constitutes a better vehicle. Additionally, glycoengineering allows to create specific FcγRIIIb structures for functional studies.55,56 The present data indicate that neutrophil-derived FcγRIIIb N-glycosylation is rather consistent between healthy individuals, but significantly differs from soluble FcγRIIIb, recombinant or serum-derived and NK cell-derived FcγRIIIa profiles.
Conclusions
In this study, we describe a straightforward and comprehensive site-specific profiling of FcγRIIIb N-glycosylation with a resolution of a single donor. However, by design the approach may be applicable to many different leukocytes.
The observed differences between the plasma-derived and the neutrophil-derived FcγRIIIb demonstrate a significant biological diversity. It would be of great interest to compare FcγRIIIb glycosylation profiles of subcellular fractions or FcγRIIIb from resting neutrophils to soluble receptors in the same donor. The source and impact of the simultaneous similarity, site N162, and dissimilarity, other sites, between FcγRIIIb and FcγRIIIa glycosylation warrants further study.
Additional isomer differentiation would also be desirable, but would likely need a separation method with a higher degree of isomer separation.
We believe that a throughput optimized adaptation of the presented approach could be used for defining glycan signatures of FcγRIII under different pathophysiological conditions in various cell types or even subcellular compartments. This would reveal a yet hidden layer of regulation of antibody-mediated (auto-)immune responses. However, sensitivity should be further improved for such aims. A better understanding of glycosylation as an additional layer of regulation of FcγR activity is likely to improve the performance of antibody-based therapeutic interventions and provide clinical markers for personalized medicine in the long run.
Acknowledgments
We thank Gillian Dekkers for expressing the recombinant receptors used in the initial development of the method. This research was supported by the European Union (Glycosylation Signatures for Precision Medicine project, GlySign, Grant No. 722095), and the Netherlands Organization for Scientific Research (NWO) via Vernieuwingsimpuls Veni (Project No. 722.016.008) and Medium Investment (to P.A.V.; 91116004; partly financed by ZonMw) grants.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c02342.
Experimental details on the Western blot; Table S1 Donor information; Table S3 Byonic search parameters; Figure S1 Non-reducing SDS-PAGE of IP eluate; Figure S2 Western blot of donors; Table S5 Byonic output; Table S6 Site-specific peptides; Figure S3 Recombinant FcγRIIIb NA1; Figure S4 Recombinant FcγRIIIb NA2; Table S7 Allelic (glyco-)peptide sequences; Figure S5 Glycosylation site comparison; Table S8 Cell- and subclass-specific glycosylation; Figure S6 Sum spectra N45 glycopeptides; Figure S7 Relative quantitation per donor; Figure S8 Derived traits; Table S9 Site-specific derived glycosylation traits; Figure S9 Stepping-energy HCD MS/MS spectrum; and Table S10 Derived trait calculation (PDF). Table S2 Glycan composition list for Byonic search; and Table S4 Details glyocpeptide identification, including evidence of glycan isomers (XLSX). Raw LC-MS/MS data on neutrophil-derived FcγRIIIb; .fasta files containing protein sequence of FcγRIIIb (NA1 and NA2 allotype) and FcγRIIIa (158V and 158F);(PDF)
and the full dataset on relative quantitation of glycosylation of recombinant FcγRIIIb have been deposited elsewhere (MassIVE, identifier MSV000085913) (XLSX)
Author Contributions
∥ I.W. and T.S. contributed equally to this work.
The authors declare the following competing financial interest(s): I.W. is employed by Genos Ltd. and E.L.G is employed by Pepscope Ltd. No potential conflict of interest was reported by the remaining authors.
Supplementary Material
References
- Nimmerjahn F.; Ravetch J. V. Nat. Rev. Immunol. 2008, 8, 34–47. 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- Kapur R.; Einarsdottir H. K.; Vidarsson G. Immunol. Lett. 2014, 160, 139–144. 10.1016/j.imlet.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Ben Mkaddem S.; Benhamou M.; Monteiro R. C. Front. Immunol. 2019, 10, 811. 10.3389/fimmu.2019.00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S.; Woof J. M.; Hart S. P.; Dransfield I. Clin. Exp. Immunol. 2009, 157, 244–254. 10.1111/j.1365-2249.2009.03980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth P. M.; Pietersz G. A. Nat. Rev. Drug Discovery 2012, 11, 311–331. 10.1038/nrd2909. [DOI] [PubMed] [Google Scholar]
- Hudis C. A. N. Engl. J. Med. 2007, 357, 39–51. 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- Schwab I.; Nimmerjahn F. Nat. Rev. Immunol. 2013, 13, 176–189. 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- Cobb B. A. Glycobiology 2020, 30, 202–213. 10.1093/glycob/cwz065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan N.; Falck D.; Wuhrer M. Glycobiology 2020, 30, 226–240. 10.1093/glycob/cwz048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidarsson G.; Dekkers G.; Rispens T. Front. Immunol. 2014, 5, 520. 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y.; Barb A. W. Glycobiology 2020, 30, 214–225. 10.1093/glycob/cwz068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Taeye S. W.; Bentlage A.; Mebius M. M.; Meesters J. I.; Lissenberg S.; Falck D.; Sénard T.; Salehi N.; Wuhrer M.; Schuurman J.; Labrijn A. F.; Rispens T.; Vidarsson G. Front. Immunol. 2020, 11, 740. 10.3389/fimmu.2020.00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales C. Front. Immunol. 2017, 8, 280. 10.3389/fimmu.2017.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes J. M.; Cosgrave E. F.; Struwe W. B.; Wormald M.; Davey G. P.; Jefferis R.; Rudd P. M. Curr. Top. Microbiol. Immunol. 2014, 382, 165–199. [DOI] [PubMed] [Google Scholar]
- Bruhns P.; Iannascoli B.; England P.; Mancardi D. A.; Fernandez N.; Jorieux S.; Daeron M. Blood 2009, 113, 3716–3725. 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- Roberts J. T.; Barb A. W. J. Biol. Chem. 2018, 293, 19899–19908. 10.1074/jbc.RA118.005273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara S.; Amon L.; Luhr J. J.; Nimmerjahn F.; Dudziak D.; Lux A. Front. Immunol. 2020, 11, 1320. 10.3389/fimmu.2020.01320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizinga T. W.; Kleijer M.; Tetteroo P. A.; Roos D.; von dem Borne A. E. Blood 1990, 75, 213–217. 10.1182/blood.V75.1.213.213. [DOI] [PubMed] [Google Scholar]
- Coxon A.; Cullere X.; Knight S.; Sethi S.; Wakelin M. W.; Stavrakis G.; Luscinskas F. W.; Mayadas T. N. Immunity 2001, 14, 693–704. 10.1016/S1074-7613(01)00150-9. [DOI] [PubMed] [Google Scholar]
- Chen K.; Nishi H.; Travers R.; Tsuboi N.; Martinod K.; Wagner D. D.; Stan R.; Croce K.; Mayadas T. N. Blood 2012, 120, 4421–4431. 10.1182/blood-2011-12-401133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Jonsson F. Front. Immunol. 2019, 10, 1958. 10.3389/fimmu.2019.01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon J. E.; Edberg J. C.; Kimberly R. P. J. Clin. Invest. 1990, 85, 1287–1295. 10.1172/JCI114566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizinga T. W.; de Haas M.; Kleijer M.; Nuijens J. H.; Roos D.; von dem Borne A. E. J. Clin. Invest. 1990, 86, 416–423. 10.1172/JCI114727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K. R.; Nott J. D.; Barb A. W. Mol. Cell. Proteomics 2019, 18, 2178–2190. 10.1074/mcp.RA119.001607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts J. T.; Patel K. R.; Barb A. W. Mol. Cell. Proteomics 2020, 19, 362–374. 10.1074/mcp.RA119.001733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeck A.; Pohlentz G.; Schlothauer T.; Peter-Katalinic J.; Regula J. T. J. Proteome Res. 2011, 10, 3031–3039. 10.1021/pr1012653. [DOI] [PubMed] [Google Scholar]
- Kawasaki N.; Okumoto T.; Yamaguchi Y.; Takahashi N.; Fridman W.; Sautès-Fridman C.; Yagi H.; Kato K. J. Glycomics Lipidomics 2014, 4, 2153-0637. [Google Scholar]
- Cosgrave E. F.; Struwe W. B.; Hayes J. M.; Harvey D. J.; Wormald M. R.; Rudd P. M. J. Proteome Res. 2013, 12, 3721–3737. 10.1021/pr400344h. [DOI] [PubMed] [Google Scholar]
- Ferrara C.; Grau S.; Jager C.; Sondermann P.; Brunker P.; Waldhauer I.; Hennig M.; Ruf A.; Rufer A. C.; Stihle M.; Umana P.; Benz J. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12669–12674. 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi G. P.; Falconer D. J.; Barb A. W. Biochemistry 2017, 56, 3174–3177. 10.1021/acs.biochem.7b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata-Koyama M.; Iida S.; Okazaki A.; Mori K.; Kitajima-Miyama K.; Saitou S.; Kakita S.; Kanda Y.; Shitara K.; Kato K.; Satoh M. Glycobiology 2008, 19, 126–134. 10.1093/glycob/cwn110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers G.; Bentlage A. E. H.; Plomp R.; Visser R.; Koeleman C. A. M.; Beentjes A.; Mok J. Y.; van Esch W. J. E.; Wuhrer M.; Rispens T.; Vidarsson G. Mol. Immunol. 2018, 94, 54–60. 10.1016/j.molimm.2017.12.006. [DOI] [PubMed] [Google Scholar]
- Hayes J. M.; Frostell A.; Karlsson R.; Muller S.; Martin S. M.; Pauers M.; Reuss F.; Cosgrave E. F.; Anneren C.; Davey G. P.; Rudd P. M. Mol. Cell. Proteomics 2017, 16, 1770–1788. 10.1074/mcp.M117.066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi G. P.; Barb A. W. J. Biol. Chem. 2018, 293, 16842–16850. 10.1074/jbc.RA118.004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K. R.; Roberts J. T.; Subedi G. P.; Barb A. W. J. Biol. Chem. 2018, 293, 3477–3489. 10.1074/jbc.RA117.001207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts J. T.; Patel K. R.; Barb A. W. Mol. Cell. Proteomics 2020, 19, 362–374. 10.1074/mcp.RA119.001733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn N.; Meccariello R.; Duffner J.; Getchell K.; Holte K.; Prod’homme T.; Srinivasan K.; Prenovitz R.; Lansing J.; Capila I.; Kaundinya G.; Manning A. M.; Bosques C. J. Mol. Cell. Proteomics 2019, 18, 534–545. 10.1074/mcp.RA118.001142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H.; Takakura D.; Roumenina L. T.; Fridman W. H.; Sautes-Fridman C.; Kawasaki N.; Kato K. Sci. Rep. 2018, 8, 2719 10.1038/s41598-018-21145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley N. M.; Hebert A. S.; Westphall M. S.; Coon J. J. Nat. Commun. 2019, 10, 1311 10.1038/s41467-019-09222-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Lin Y. H.; Dingess K. A.; Mank M.; Stahl B.; Heck A. J. R. J. Proteome Res. 2020, 19, 1941–1952. 10.1021/acs.jproteome.9b00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiano G.; Bruschi M.; Musante L.; Santucci L.; Ghiggeri G. M.; Carnemolla B.; Orecchia P.; Zardi L.; Righetti P. G. Electrophoresis 2004, 25, 1327–1333. 10.1002/elps.200305844. [DOI] [PubMed] [Google Scholar]
- Kuijpers T. W.; Tool A. T.; van der Schoot C. E.; Ginsel L. A.; Onderwater J. J.; Roos D.; Verhoeven A. J. Blood 1991, 78, 1105–1111. 10.1182/blood.V78.4.1105.1105. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Wilm M.; Vorm O.; Mann M. Anal. Chem. 1996, 68, 850–858. 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Tomas H.; Havlis J.; Olsen J. V.; Mann M. Nat. Protoc. 2006, 1, 2856–2860. 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Gundry R. L.; White M. Y.; Murray C. I.; Kane L. A.; Fu Q.; Stanley B. A.; Van Eyk J. E. Curr. Protoc. Mol. Biol. 2009, 10.25.1–10.25.23. 10.1002/0471142727.mb1025s88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M.; Kil Y. J.; Becker C. Curr. Protoc. Bioinf. 2012, 13.20.1–13.20.14. 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen B. C.; Falck D.; de Haan N.; Hipgrave Ederveen A. L.; Razdorov G.; Lauc G.; Wuhrer M. J. Proteome Res. 2016, 15, 2198–2210. 10.1021/acs.jproteome.6b00171. [DOI] [PubMed] [Google Scholar]
- Galon J.; Moldovan I.; Galinha A.; Provost-Marloie M. A.; Kaudewitz H.; Roman-Roman S.; Fridman W. H.; Sautes C. Eur. J. Immunol. 1998, 28, 2101–2107. . [DOI] [PubMed] [Google Scholar]
- Breunis W. B.; van Mirre E.; Geissler J.; Laddach N.; Wolbink G.; van der Schoot E.; de Haas M.; de Boer M.; Roos D.; Kuijpers T. W. Hum. Mutat. 2009, 30, E640–650. 10.1002/humu.20997. [DOI] [PubMed] [Google Scholar]
- Wuhrer M.; Deelder A. M.; van der Burgt Y. E. Mass Spectrom. Rev. 2011, 30, 664–680. 10.1002/mas.20337. [DOI] [PubMed] [Google Scholar]
- Sondermann P.; Huber R.; Oosthuizen V.; Jacob U. Nature 2000, 406, 267–273. 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- Demetriou M.; Granovsky M.; Quaggin S.; Dennis J. W. Nature 2001, 409, 733–739. 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan V.; Dieckmann R.; Loke I.; Tjondro H.; Chatterjee S.; Bylund J.; Thaysen-Andersen M.; Karlsson N. G.; Karlsson-Bengtsson A. J. Biol. Chem. 2020, 12648–12660. 10.1074/jbc.RA120.014011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaysen-Andersen M.; Packer N. H. Glycobiology 2012, 22, 1440–1452. 10.1093/glycob/cws110. [DOI] [PubMed] [Google Scholar]
- Hayes J. M.; Wormald M. R.; Rudd P. M.; Davey G. P. J. Inflammation Res. 2016, 9, 209–219. 10.2147/JIR.S121233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner M. J.; Shah S. R.; Saeui C. T.; Ariss R.; Yarema K. J. Front. Immunol. 2018, 9, 2485. 10.3389/fimmu.2018.02485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.